
Genetic Improvement in Sunflower Breeding—Integrated Omics Approach

 , ,
, , 

Abstract
1. Introduction
2. Molecular Omics Profiling
2.1. Genomics—Pangenomics
2.2. Epigenomics
3. Gene Function Translation
3.1. Transcriptomics
3.2. Proteomics
3.3. Metabolomics
3.4. Phenomics
4. Integrated Omics Approach—Systems Biology
5. Conclusions and Prospects
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sedeek, K.E.M.; Mahas, A.; Mahfouz, M. Plant Genome Engineering for Targeted Improvement of Crop Traits. Front. Plant Sci. 2019, 10, 114. [Google Scholar] [CrossRef] [PubMed]
- Varshney, R.K.; Sinha, P.; Singh, V.K.; Kumar, A.; Zhang, Q.; Bennetzen, J.L. 5Gs for crop genetic improvement. Curr. Opin. Plant Biol. 2020, 56, 190–196. [Google Scholar] [CrossRef] [PubMed]
- Scossa, F.; Alseekh, S.; Fernie, A.R. Integrating multi-omics data for crop improvement. J. Plant Physiol. 2021, 257, 153352. [Google Scholar] [CrossRef] [PubMed]
- Fischer, T.; Byerlee, D.; Edmeades, G. Crop Yields and Global Food Security: Will Yield Increase Continue to Feed the World; ACIAR Monograph: Canberra, Australia, 2014; p. 634. [Google Scholar]
- Li, H.; Rasheed, A.; Hickey, L.T.; He, Z. Fast-Forwarding Genetic Gain. Trends Plant Sci. 2018, 23, 184–186. [Google Scholar] [CrossRef]
- Pal, U.; Patra, R.; Sahoo, N.; Bakhara, C.; Panda, M. Effect ofrefining on quality and composition of sunflower oil. J. Food Sci. Technol. 2015, 52, 4613–4618. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Choudhary, S.; Pandey, A.; Khan, M.K.; Thomas, G. Sunflower oil: Efficient oil source for human consumption. Emergent Life Sci. Res. 2015, 1, 1–3. [Google Scholar]
- Jocković, M.; Cvejić, S.; Jocić, S.; Marjanović-Jeromela, A.; Miladinović, D.; Jocković, B.; Miklič, V.; Radić, V. Evaluation of sunflower hybrids in multi-environmental trial (MET). Turk. J. Field Crop. 2019, 24, 202–210. [Google Scholar] [CrossRef]
- Imerovski, I.; Dedić, B.; Cvejić, S.; Miladinović, D.; Jocić, S.; Owens, G.L.; Kočiš Tubić, N.; Rieseberg, L.H. BSA-seq mapping reveals major QTL for broomrape resistance in four sunflower lines. Mol. Breed. 2019, 39, 41. [Google Scholar] [CrossRef]
- Balliau, T.; Durufle, H.; Blanchet, N.; Blein-Nicolas, M.; Langlade, N.B.; Zivy, M. Proteomic data from leaves of twenty-four sunflower genotypes under water deficit. OCL 2021, 28, 12. [Google Scholar] [CrossRef]
- Xu, Y.; Li, P.; Zou, C.; Lu, Y.; Xie, C.; Zhang, X.; Prasanna, B.M.; Olsen, M.S. Enhancing genetic gain in the era of molecular breeding. J. Exp. Bot. 2017, 68, 2641–2666. [Google Scholar] [CrossRef]
- Voss-Fels, K.P.; Cooper, M.; Hayes, B.J. Accelerating crop genetic gains with genomic selection. Theor. Appl. Genet. 2019, 132, 669–686. [Google Scholar] [CrossRef] [PubMed]
- Yadav, S.; Jackson, P.; Wei, X.; Ross, E.M.; Aitken, K.; Deomano, E.; Atkin, F.; Hayes, B.J.; Voss-Fels, K.P. Accelerating Genetic Gain in Sugarcane Breeding Using Genomic Selection. Agronomy 2020, 10, 585. [Google Scholar] [CrossRef]
- Xu, Y.; Liu, X.; Fu, J.; Wang, H.; Wang, J.; Huang, C.; Prasanna, B.M.; Olsen, M.S.; Wang, G.; Zhang, A. Enhancing Genetic Gain through Genomic Selection: From Livestock to Plants. Plant Commun. 2020, 1, 100005. [Google Scholar] [CrossRef] [PubMed]
- Finkel, E. With phenomics plant scientists hope to shift breeding into overdrive. Science 2009, 325, 380–381. [Google Scholar] [CrossRef]
- Cobb, J.N.; Declerck, G.; Greenberg, A.; Clark, R.; McCouch, S. Next-generation phenotyping: Requirements and strategies for enhancing our understanding of genotype-phenotype relationships and its relevance to crop improvement. Theor. Appl. Genet. 2013, 126, 867–887. [Google Scholar] [CrossRef]
- Carnielli, C.M.; Winck, F.V.; Leme, A.F.P. Functional annotation and biological interpretation of proteomics data. Biochem. Biophys. Acta Prot. Proteom. 2015, 1854, 46–54. [Google Scholar] [CrossRef]
- Lobos, G.A.; Camargo, A.V.; del Pozo, A.; Araus, J.L.; Ortiz, R.; Doonan, J.H. Editorial: Plant phenotyping and phenomics for plant breeding. Front. Plant Sci. 2017, 8, 2181. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, O.; Urrutia, M.; Berton, T.; Bernillon, S.; Deborde, C.; Jacob, D.; Maucourt, M.; Maury, P.; Durufle, H.; Gibon, Y.; et al. Metabolomic characterization of sunflower leaf allows discriminating genotype groups or stress levels with a minimal set of metabolic markers. Metabolomics 2019, 15, 56. [Google Scholar] [CrossRef]
- Mascher, M.; Schreiber, M.; Scholz, U.; Graner, A.; Reif, J.C.; Stein, N. Genebank genomics bridges the gap between the conservation of crop diversity and plant breeding. Nat. Genet. 2019, 51, 7, 1076–1081. [Google Scholar] [CrossRef]
- Salvi, S.; Tuberosa, R. The crop QTLome comes of age. Curr. Opin. Biotechnol. 2015, 32, 179–185. [Google Scholar] [CrossRef]
- Milner, S.G.; Jost, M.; Taketa, S.; Mazon, E.R.; Himmelbach, A.; Oppermann, M.; Weise, S.; Knupffer, H.; Basterrechea, M.; Konig, P. Genebank genomics highlights the diversity of a global barley collection. Nat. Genet. 2019, 51, 319–326. [Google Scholar] [CrossRef]
- Lu, K.; Wei, L.; Li, X.; Wang, Y.; Wu, J.; Liu, M.; Zhang, C.; Chen, Z.; Xiao, Z.; Jian, H.; et al. Whole-genome resequencing reveals Brassica napus origin and genetic loci involved in its improvement. Nat. Commun. 2019, 10, 1154. [Google Scholar] [CrossRef] [PubMed]
- Rieseberg, L.H.; Choi, H.; Chan, R.; Spore, C. Genomic map of a diploid hybrid species. Heredity 1993, 70, 285. [Google Scholar] [CrossRef]
- Gedil, M.A.; Wye, C.; Berry, S.T.; Seger, B.; Peleman, J.; Jones, R.; Leon, A.; Slabauh, M.B.; Knapp, S.J. An integrated restriction fragment lenght polimorphism-amplified fragment length polimorphism linkage map for cultivated sunflower. Genome 2001, 44, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.K.; Tang, S.; Slabaugh, M.B.; Heesacker, A.; Cole, G.; Herring, M.; Soper, J.; Han, F.; Chu, W.-C.; Webb, D.M.; et al. Towards a saturated molecular genetic linkage map for sunflower. Crop Sci. 2003, 43, 367–387. [Google Scholar] [CrossRef]
- Tang, S.; Kishore, V.K.; Knapp, S.J. PCR-multiplexes for a genome-wide framework of simple sequence repeat marker loci in cultivated sunflower. Theor. Appl. Genet. 2003, 107, 6–19. [Google Scholar] [CrossRef] [PubMed]
- Badouin, H.; Gouzy, J.; Grassa, C.J.; Murat, F.; Staton, S.E.; Cottret, L.; Lelandais-Briere, C.; Owens, G.L.; Carrere, S.; Mayjonade, B.; et al. The sunflower genome provides insights into oil metabolism, flowering and Asterid evolution. Nature 2017, 546, 148–152. [Google Scholar] [CrossRef] [PubMed]
- Terzić, S.; Boniface, M.-C.; Marek, L.; Alvarez, D.; Baumann, K.; Gavrilova, V.; Joita-Pacureanu, M.; Sujatha, M.; Valkova, D.; Velasco, L.; et al. Gene banks for wild and cultivated sunflower genetic resources. OCL 2020, 27, 9. [Google Scholar] [CrossRef]
- Dimitrijević, A.; Horn, R. Sunflower Hybrid Breeding: From Markers to Genomic Selection. Front. Plant Sci. 2018, 8, 2238. [Google Scholar] [CrossRef]
- Tettelin, H.; Masignani, V.; Cieslewicz, M.J.; Donati, C.; Medini, D.; Ward, N.L.; Angiuoli, S.V.; Crabtree, J.; Jones, A.L.; Durkin, A.S.; et al. Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae: Implications for the microbial “pan-genome”. Proc. Natl. Acad. Sci. USA 2005, 102, 13950–13955. [Google Scholar] [CrossRef]
- Khan, A.W.; Garg, V.; Roorkiwal, M.; Golicz, A.A.; Edwards, D.; Varshney, R.K. Super-Pangenome by Integrating the Wild Side of a Species for Accelerated Crop Improvement. Trends Plant Sci. 2020, 25, 148–158. [Google Scholar] [CrossRef] [PubMed]
- Warschefsky, E. Back to the wilds: Tapping evolutionary adaptations for resilient crops through systematic hybridization with crop wild relatives. Am. J. Bot. 2014, 101, 1791–1800. [Google Scholar] [CrossRef] [PubMed]
- Zsogon, A.; Čermak, T.; Naves, E.R.; Notini, M.M.; Edel, K.H.; Weinl, S.; Freschi, L.; Voytas, D.F.; Kudla, J.; Peres, L.E.P. De novo domestication of wild tomato using genome editing. Nat. Biotechnol. 2018, 36, 1211–1216. [Google Scholar] [CrossRef]
- Li, Y.H.; Zhou, G.; Ma, J.; Jiang, W.; Jin, L.; Zhang, Z.; Guo, Y.; Zhang, J.; Sui, Y.; Zheng, L.; et al. De novo assembly of soybean wild relatives for pan-genome analysis of diversity and agronomic traits. Nat. Biotechnol. 2014, 32, 1045–1052. [Google Scholar] [CrossRef] [PubMed]
- Scossa, F.; Brotman, Y.; de Abreu, E.L.F.; Willmitzer, L.; Nikoloski, Z.; Tohge, T.; Fernie, A.R. Genomics-based strategies for the use of natural variation in the improvement of crop metabolism. Plant Sci. 2016, 242, 47–64. [Google Scholar] [CrossRef]
- Golicz, A.A.; Bayer, P.E.; Barker, G.C.; Edger, P.P.; Kim, H.; Martinez, P.A.; Chan, C.K.K.; Severn-Ellis, A.; McCombie, W.R.; Parkin, I.A.P.; et al. The pangenome of an agronomically important crop plant Brassica oleracea. Nat. Commun. 2016, 7, 13390. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Feng, Q.; Lu, H.; Li, Y.; Wang, A.; Tian, Q.; Zhan, Q.; Lu, Y.; Zhang, L.; Huang, T.; et al. Pan-genome analysis highlights the extent of genomic variation in cultivated and wild rice. Nat. Genet. 2018, 50, 278–284. [Google Scholar] [CrossRef] [PubMed]
- Haberer, G.; Kamal, N.; Bauer, E.; Gundlach, H.; Fischer, I.; Seidel, M.A.; Spannagl, M.; Marcon, C.; Ruban, A.; Urbany, C.; et al. European maize genomes highlight intraspecies variation in repeat and gene content. Nat. Genet. 2020, 52, 950–957. [Google Scholar] [CrossRef]
- Petek, M.; Zagorscak, M.; Ramsak, Z.; Sanders, S.; Tomaz, S.; Tseng, E.; Zouine, M.; Coll, A.; Gruden, K. Cultivar-specific transcriptome and pan-transcriptome reconstruction of tetraploid potato. Sci. Data 2020, 7, 249. [Google Scholar] [CrossRef] [PubMed]
- Hubner, S.; Bercovich, N.; Todesco, M.; Mandel, J.R.; Odenheimer, J.; Ziegler, E.; Lee, J.S.; Baute, G.J.; Owens, G.L.; Grassa, C.J.; et al. Sunflower pan-genome analysis shows that hybridization altered gene content and disease resistance. Nat. Plants 2019, 5, 54–62. [Google Scholar] [CrossRef]
- Shanker, A.; Venkateswarlu, B. Abiotic Stress in Plants: Mechanisms and Adaptations; IntechOpen: London, UK, 2011; p. 428. [Google Scholar] [CrossRef]
- Bräutigam, K.; Vining, K.J.; Lafon-Placette, C.; Fossdal, C.G.; Mirouze, M.; Marcos, J.G.; Fluchs, S.; Fraga, M.F.; Guevara, M.A.; Abarca, D.; et al. Epigenetic regulation of adaptive responses of forest tree species to the environment. Ecol. Evol. 2013, 3, 399–415. [Google Scholar] [CrossRef]
- Niederhuth, C.E.; Schmitz, R.J. Covering your bases: Inheritance of DNA methylation in plant genomes. Mol. Plant 2014, 7, 472–480. [Google Scholar] [CrossRef]
- Großkinsky, D.K.; Syaifullah, S.J.; Roitsch, T. Integration of multi-omics techniques and physiological phenotyping within a holistic phenomics approach to study senescence in model and crop plants. J. Exp. Bot. 2018, 69, 825–844. [Google Scholar] [CrossRef] [PubMed]
- Cortijo, S.; Wardenaar, R.; Colome-Tatche, M.; Gilly, A.; Etcheverry, M.; Labadie, K.; Caillieux, E.; Hospital, F.; Aury, J.-M.; Wincker, P.; et al. Mapping the epigenetic basis of complex traits. Science 2014, 343, 1145–1148. [Google Scholar] [CrossRef] [PubMed]
- Ingouff, M.; Selles, B.; Michaud, C.; Vu, T.M.; Berger, F.; Schorn, A.J.; Autran, D.; Van Durme, M.; Nowack, M.K.; Martienssen, R.A.; et al. Live-cell analysis of DNA methylation during sexual reproduction in Arabidopsis reveals context and sex-specific dynamics controlled by noncanonical RdDM. Genes Dev. 2017, 31, 72–83. [Google Scholar] [CrossRef]
- Kawakatsu, T.; Nery, J.R.; Castanon, R.; Ecker, J.R. Dynamic DNA methylation reconfiguration during seed development and germination. Genome Biol. 2017, 18, 171. [Google Scholar] [CrossRef] [PubMed]
- Dubrovina, A.S.; Kiselev, K.V. Age-associated alterations in the somatic mutation and DNA methylation levels in plants. Plant Biol. 2016, 18, 185–196. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Li, Y.; Duan, W.; Huang, F.; Hou, X. Cold acclimation alters DNA methylation patterns and confers tolerance to heat and increases growth rate in Brassica rapa. J. Exp. Bot. 2017, 68, 1213–1224. [Google Scholar] [CrossRef]
- Xu, J.; Zhou, S.; Gong, X.; Song, Y.; van Nocker, S.; Ma, F.; Guan, Q. Single-base methylome analysis reveals dynamic epigenomic differences associated with water deficit in apple. Plant Biotechnol. J. 2018, 16, 672–687. [Google Scholar] [CrossRef] [PubMed]
- Correa, R.L.; Sanz-Carbonell, A.; Kogej, Z.; Muller, S.Y.; Ambros, S.; Lopez-Gomollon, S.; Gomez, G.; Baulcombe, D.C.; Elena, S.F. Viral fitness determines the magnitude of transcriptomic and epigenomic reprogramming of defense responses in plants. Mol. Biol. Evol. 2020, 37, 1866–1881. [Google Scholar] [CrossRef] [PubMed]
- Čitaković, I.; Dedić, B.; Banović Deri, B.; Jocić, S.; Cvejić, S.; Radanović, A.; Jocković, M.; Samardžić, J.; Miladinović, D. Defensin expression in sunflower under combined broomrape—downy mildew attack. In Proceedings of the Impact of Chromatin Domains on Plant Phenotypes, El Escorial, Spain, 9–11 December 2019; Society for Experimental Biology: El Escoril, Spain, 2019. Abstract number P19.79. p. 51. [Google Scholar]
- Bolukbasi, E.; Aras, E.S. Determination of DNA Methylation Levels with CRED-RA Technique in the Genome of Sunflower Seedlings (Helianthus annuus L.) Subjected to Zinc Stress. Int. J. Environ. Agric. Biotechnol. 2016, 1, 438–444. [Google Scholar] [CrossRef]
- Kanoosh, O.A.; Alfalahi, A.O.; Jano, F.O.; Alawadi, H.F.N. Epigenetic role of dna methylation in hybrid vigor of cytoplasmic male sterility in sunflower. Pak. J. Bot. 2021, 53, 1. [Google Scholar] [CrossRef]
- Gallusci, P.; Dai, Z.; Génard, M.; Gauffretau, A.; Leblanc-Fournier, N.; Richard-Molard, C.; Vile, D.; Brunel-Muguet, S. Epigenetics for plant improvement: Current knowledge and modeling avenues. Trends Plant Sci. 2017, 22, 610–623. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, R.J.; Schultz, M.D.; Lewsey, M.G.; O’Malley, R.C.; Urich, M.A.; Libiger, O.; Schork, N.J.; Ecker, J.R. Transgenerational epigenetic instability is a source of novel methylation variants. Science 2011, 334, 369–373. [Google Scholar] [CrossRef] [PubMed]
- Raju, S.K.K.; Shao, M.R.; Sanchez, R.; Xu, Y.Z.; Sandhu, A.; Graef, G.; Mackenzie, S. An epigenetic breeding system in soybean for increased yield and stability. Plant Biotechnol. J. 2018, 16, 1836–1847. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Zuo, Y.; Zang, Y.; Wu, C.; Su, W.; Jin, W.; Yu, H.; An, Y.; Li, Q. Large-scale transcriptome comparison of sunflower genes responsive to Verticillium dahliae. BMC Genom. 2017, 18, 42. [Google Scholar] [CrossRef]
- Zhu, J.K. Abiotic stress signaling and responses in plants. Cell 2016, 167, 313–324. [Google Scholar] [CrossRef]
- Coolen, S.; Proietti, S.; Hickman, R.; Davial Olivas, N.H.; Huang, P.-P.; Van Verk, M.C.; Van Pelt, J.A.; Wittenberg, A.H.J.; De Vos, M.; Prins, M.; et al. Transcriptome dynamics of Arabidopsis during sequential biotic and abiotic stresses. Plant J. 2016, 86, 249–267. [Google Scholar] [CrossRef]
- Kang, W.H.; Yeom, S.I. Genome-wide Identification, classification, and expression analysis of the receptor-like protein family in tomato. Plant Pathol. J. 2018, 34, 435–444. [Google Scholar] [CrossRef]
- Cohen, S.P.; Leach, J.E. Abiotic and biotic stresses induce a core transcriptome response in rice. Sci. Rep. 2019, 6273. [Google Scholar] [CrossRef]
- Mattiello, L.; Begcy, K.; da Silva, F.R.; Jorge, R.A.; Menossi, M. Transcriptome analysis highlights changes in the leaves of maize plants cultivated in acidic soil containing toxic levels of Al(3+). Mol. Biol. Rep. 2014, 41, 8107–8116. [Google Scholar] [CrossRef]
- Nejat, N.; Ramalingam, A.; Mantri, N. Advances in Transcriptomics of Plants. Adv. Biochem. Eng. Biotechnol. 2018, 164, 161–185. [Google Scholar] [CrossRef]
- Ramu, V.S.; Paramananthan, A.; Ramegowda, V.; Mohan-Raju, B.; Udayakumar, M.; Senthil-Kumar, M. Transcriptome Analysis of Sunflower Genotypes with Contrasting Oxidative Stress Tolerance Reveals Individual-and Combined-Biotic and Abiotic Stress Tolerance Mechanisms. PLoS ONE 2016, 11, e0157522. [Google Scholar] [CrossRef]
- Kang, W.H.; Mi Sim, Y.; Koo, N.; Nam, J.Y.; Lee, J.; Kim, N.; Jang, H.; Kim, Y.M.; Yeom, S.I. Transcriptome profiling of abiotic responses to heat, cold, salt, and osmotic stress of Capsicum annuum L. Sci. Data 2020, 17, 17. [Google Scholar] [CrossRef] [PubMed]
- Bing, J.; Ling, Y.; An, P.; Xiao, E.; Li, C.; Song, B.; Wang, Z. Transcriptomic basis for salt tolerance and disease resistance of silverleaf sunflower revealed by Iso-seq and RNA-seq. Res. Sq. 2019. [Google Scholar] [CrossRef]
- Escalante, M.; Vigliocco, A.; Moschen, S.; Fernandez, P.; Heinz, R.; Garcia-Garcia, F.; Di Rienzo, J.A.; Andrade, A.; Alemano, S. Transcriptomic Analysis Reveals a Differential Gene Expression Profile Between Two Sunflower Inbred Lines with Different Ability to Tolerate Water Stress. Plant Mol. Biol. Rep. 2020, 38, 222–237. [Google Scholar] [CrossRef]
- Azodi, C.B.; Pardo, J.; VanBuren, R.; de Los Campos, G.; Shiu, S.H. Transcriptome based prediction of complex traits in maize. Plant Cell 2020, 32, 139–151. [Google Scholar] [CrossRef]
- Badouin, H.; Boniface, M.-C.; Pouilly, N.; Fuchs, A.-L.; Vear, F.; Langlade, N.B.; Gouzy, J.; Munos, S. Pooled Single-Molecule transcriptomics identifies a giant gene under balancing selection in sunflower. BioRxiv Prepr. 2021. [Google Scholar] [CrossRef]
- Škorić, D. Sunflower breeding for resistance to abiotic and biotic stresses. In Abiotic and Biotic Stress in Plants—Recent Advances and Future Perspectives; Shanker, A.K., Shanker, C., Eds.; IntechOpen: London, UK, 2016; pp. 585–635. [Google Scholar] [CrossRef]
- Jocić, S.; Miladinović, D.; Kaya, Y. Breeding and Genetics of Sunflower. In Sunflower: Chemistry, Production, Processing, and Utilization; Martínez-Force, E., Dunford, N.T., Salas, J.J., Eds.; AOCS Press: Urbana, IL, USA, 2015; pp. 1–26. [Google Scholar]
- Yang, F.; Jacobsen, S.; Jørgensen, H.J.L.; Collinge, D.B.; Svensson, B.; Finnie, C. Fusarium graminearum and its interactions with cereal heads: Studies in the proteomics era. Front. Plant Sci. 2013, 4, 37. [Google Scholar] [CrossRef] [PubMed]
- Gong, F.P.; Hu, X.L.; Wang, W. Proteomic analysis of crop plants under abiotic stress conditions: Where to focus our research? Front. Plant Sci. 2015, 6, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, P.; Abdel Latef, A.A.H.; Rasool, S.; Akram, N.A.; Ashraf, M.; Gucel, S. Role of Proteomics in Crop Stress Tolerance. Front. Plant Sci. 2016, 7, 1336. [Google Scholar] [CrossRef] [PubMed]
- Printz, B.; Sergeant, K.; Guignard, C.; Renaut, J. Physiological proteome study of sunflowers exposed to a polymetallic constraint. Proteomics 2013, 13, 1993–2015. [Google Scholar] [CrossRef]
- Lopes Júnior, C.A.; Barbosa, H.S.; Galazzi, R.M.; Koolen, H.H.F.; Gozzo, F.C.; Arruda, M.A. Evaluation of proteome alterations induced by cadmium stress in sunflower (Helianthus annuus L.) cultures. Ecotoxicol. Environ. Saf. 2015, 119, 170–177. [Google Scholar] [CrossRef]
- Ghaffari, M.; Toorchi, M.; Valizadeh, M.; Shakiba, M. Proteomic prospects for tolerance of sunflower (Helianthus annuus) to drought stress during the flowering stage. Crop Pasture Sci. 2017, 68, 457–465. [Google Scholar] [CrossRef]
- Yang, C.; Xu, L.; Zhang, N.; Islam, F.; Song, W.; Hu, L.; Liu, D.; Xie, X.; Zhou, W. iTRAQ-based proteomics of sunflower cultivars differing in resistance to parasitic weed Orobanche cumana. Proteomics 2017, 17, 13–14. [Google Scholar] [CrossRef]
- Liu, Y.; Lu, S.; Wang, S.; Huang, L.; Guo, L. Proteomics: A powerful tool to study plant responses to biotic stress. Plant Methods 2019, 15, 135. [Google Scholar] [CrossRef] [PubMed]
- Aebersold, R.; Mann, M. Mass-spectrometric exploration of proteome structure and function. Nature 2016, 537, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Kosova, K.; Vitamvas, P.; Urban, M.O.; Prašil, I.T.; Renaut, J. Plant Abiotic Stress Proteomics: The Major Factors Determining Alterations in Cellular Proteome. Front. Plant Sci. 2018, 9, 122. [Google Scholar] [CrossRef] [PubMed]
- Damerval, C.; Maurice, A.; Josse, J.M.; DeVienne, D. Quantita-tive trait loci underlying gene product variation: A novel perspective for analyzing regulation of genome expression. Genetics 1994, 137, 289–301. [Google Scholar] [CrossRef]
- Mohayeji, M.; Capriotti, A.L.; Cavaliere, C.; Piovesana, S.; Samperi, R.; Stampachiacchiere, S.; Toorchi, M.; Lagana, A. Heterosis profile of sunflower leaves: A label free proteomics approach. J. Proteom. 2014, 99, 101–110. [Google Scholar] [CrossRef]
- Fiehn, O. Metabolomics—the link between genotypes and phenotypes. Plant Mol. Biol. 2002, 48, 155–171. [Google Scholar] [CrossRef]
- Sousa Silva, M.; Cordeiro, C.; Roessner, U.; Figueiredo, A. Editorial: Metabolomics in Crop Research—Current and Emerging Methodologies. Front. Plant Sci. 2019, 10, 1013. [Google Scholar] [CrossRef] [PubMed]
- Razaqq, A.; Sadia, B.; Raza, A.; Hameed, M.K.; Saleem, F. Metabolomics: A Way Forward for Crop Improvement. Metabolites 2019, 9, 303. [Google Scholar] [CrossRef]
- Jorge, T.F.; Mata, A.T.; António, C. Mass spectrometry as a quantitative tool in plant metabolomics. Philos. Trans. R Soc. A 2016, 374, 20150370. [Google Scholar] [CrossRef] [PubMed]
- Hamany Djande, C.Y.; Pretorius, C.; Tugizimana, F.; Piater, L.A.; Dubery, I.A. Metabolomics: A Tool for Cultivar Phenotyping and Investigation of Grain Crops. Agronomy 2020, 10, 831. [Google Scholar] [CrossRef]
- Ghaste, M.; Mistrik, R.; Shulaev, V. Applications of Fourier Transform Ion Cyclotron Resonance (FT-ICR) and Orbitrap Based High Resolution Mass Spectrometry in Metabolomics and Lipidomics. Int. J. Mol. Sci. 2016, 17, 816. [Google Scholar] [CrossRef]
- Wen, W.; Li, K.; Alseekh, S.; Omranian, N.; Zhao, L.; Zhou, Y.; Xiao, Y.; Jin, M.; Yang, N.; Liu, H.; et al. Genetic determinants of the network of primary metabolism and their relationships to plant performance in a maize recombinant inbred line population. Plant Cell 2015, 27, 1839–1856. [Google Scholar] [CrossRef]
- Hong, J.; Yang, L.; Zhang, D.; Shi, J. Plant metabolomics: An indispensable system biology tool for plant science. Int. J. Mol. Sci. 2016, 17, 767. [Google Scholar] [CrossRef] [PubMed]
- Peluffo, L.; Lia, V.; Troglia, C.; Maringolo, C.; Norma, P.; Escande, A.; Hopp, H.E.; Lytovchenko, A.; Fernie, A.R.; Heinz, R.; et al. Metabolic profiles of sunflower genotypes with contrasting response to Sclerotinia sclerotiorum infection. Phytochemistry 2010, 71, 70–80. [Google Scholar] [CrossRef] [PubMed]
- Spring, O.; Pfannstiel, J.; Klaiber, I.; Conrad, J.; Beifuss, U.; Apel, L.; Aschenbrenner, A.-K.; Zipper, R. The nonvolatile metabolome of sunflower linear glandular trichomes. Phytochemistry 2015, 119, 83–89. [Google Scholar] [CrossRef]
- Meyer, R.C.; Steinfath, M.; Lisec, J.; Becher, M.; Witucka-Wall, H.; Törjék, O.; Fiehn, O.; Eckardt, A.; Willmitzer, L.; Selbig, J.; et al. The metabolic signature related to high plant growth rate in Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 2007, 104, 4759–4764. [Google Scholar] [CrossRef]
- Fernandez, O.; Urrutia, M.; Bernillon, S.; Giauffret, C.; Tardieu, F.; Le Gouis, J.; Langlade, N.; Charcosset, A.; Moing, A.; Gibon, Y. Fortune telling: Metabolic markers of plant performance. Metabolomics 2016, 12, 158. [Google Scholar] [CrossRef]
- Alseekh, S.; Bermudez, L.; De Haro, L.A.; Fernie, A.R.; Carrari, F. Crop metabolomics: From diagnostics to assisted breeding. Metabolomics 2018, 14, 148. [Google Scholar] [CrossRef] [PubMed]
- Gonzales Ibarra, A.A.; Wrobel, K.; Barrientos, E.Y.; Corrales Escobosa, A.R.; Donis, I.E.; Wrobel, K. Changes of Metabolomic Profile in Helianthus annuus under Exposure to Chromium (VI) Studied by capHPLC-ESI-QTOF-MS and MS/MS. Hindawi J. Anal. Methods Chem. 2017, 3568621. [Google Scholar] [CrossRef]
- Chaudhary, J.; Khatri, P.; Singla, P.; Kumawat, S.; Kumari, A.; Vinaykumar, R.; Vikram, A.; Vikram, S.K.; Kardile, H.; Kumar, R.; et al. Advances in Omics Approaches for Abiotic Stress Tolerance in Tomato. Biology 2019, 8, 90. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Zhang, Y.; Du, J.; Guo, X.; Wen, W.; Gu, S.; Wang, J.; Fan, J. Crop Phenomics: Current Status and Perspectives. Front. Plant Sci. 2019, 10, 714. [Google Scholar] [CrossRef]
- Houle, D.; Govindaraju, D.R.; Omholt, S. Phenomics: The next challenge. Nat. Rev. Genet. 2010, 11, 855–866. [Google Scholar] [CrossRef]
- White, J.W.; Andrade-Sanchez, P.; Gore, M.A.; Bronson, K.F.; Coelt, T.A.; Conley, M.M.; Feldmann, K.A.; French, A.N.; Heun, J.T.; Hunsaker, D.J.; et al. Field-based phenomics for plant genetics research. Field Crops Res. 2012, 133, 101–112. [Google Scholar] [CrossRef]
- Ubbens, J.R.; Stavness, I. Deep Plant Phenomics: A Deep Learning Platform for Complex Plant Phenotyping Tasks. Front. Plant Sci. 2017, 8, 8. [Google Scholar] [CrossRef]
- Tardieu, F.; Cabrera-Bosquet, L.; Pridmore, T.; Bennett, M. Plant Phenomics, From Sensors to Knowledge. Curr. Boil. 2017, 27, 770–783. [Google Scholar] [CrossRef] [PubMed]
- Hall, H.C.; Fakhrzadeh, A.; Luengo Hendriks, C.L.; Fischer, U. Precision automation of cell type classification and sub-cellular fluorescence quantification from laser scanning confocal images. Front. Plant Sci. 2016, 7, 119. [Google Scholar] [CrossRef]
- Laxman, R.H.; Hemamalini, P.; Bhatt, R.M.; Sadashiva, A.T. Non-invasive quantification of tomato (Solanum lycopersicum L.) plant biomass through digital imaging using phenomics platform. Indian J. Plant Physiol. 2018, 23, 369–375. [Google Scholar] [CrossRef]
- Ochogavia, A.C.; Gil, M.; Picardi, L.; Nestares, G. Precision phenotyping of imidazolinone-induced chlorosis in sunflower. Breed. Sci. 2014, 64, 416–421. [Google Scholar] [CrossRef][Green Version]
- Ortiz-Bustos, C.M.; Perez-Bueno, M.L.; Baron, M.; Molinero-Ruiz, L. Use of Blue-Green Fluorescence and Thermal Imaging in the early detection of sunflower infection by the root parasitic weed Orobanche cumana Wallr. Front. Plant Sci. 2017, 8, 833. [Google Scholar] [CrossRef]
- Radanović, A.; Galinski, A.; Miladinović, D.; Cvejić, S.; Jocić, S.; Terzić, S.; Nagel, K.; Fiorani, F. Root phenotyping of NS sunflower. In Proceedings of the 7th Balkan Botanical Congress, Novi Sad, Serbia, 10–14 September 2018; Institute of Botany and Botanical Garden Jevremovac: Belgrade, Serbia, 2018; p. 165. [Google Scholar]
- Zorić, M.; Cvejić, S.; Mladenović, E.; Jocić, S.; Babić, Z.; Marjanović Jeromela, A.; Miladinović, D. Digital Image Analysis Using FloCIA Software for Ornamental Sunflower Ray Floret Color Evaluation. Front. Plant Sci. 2020, 11, 584822. [Google Scholar] [CrossRef] [PubMed]
- Aliiev, E. Automatic Phenotyping Test of Sunflower Seeds. Helia 2020, 43, 51–66. [Google Scholar] [CrossRef]
- Westhues, M.; Schrag, T.A.; Heuer, C.; Thaller, G.; Utz, H.F.; Schipprack, W.; Thiemann, A.; Seifert, F.; Ehret, A.; Schlereth, A.; et al. Omics-based hybrid prediction in maize. Theor. Appl. Genet. 2017, 130, 1927–1939. [Google Scholar] [CrossRef] [PubMed]
- Budak, H.; Hussain, B.; Khan, Z.; Ozturk, N.Z.; Ullah, N. From genetics to functional genomics: Improvement in drought signaling and tolerance in wheat. Front. Plant Sci. 2015, 6, 1012. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Ning, F.; Zhang, Q.; Wu, X.; Wang, W. Enhancing Omics Research of Crop Responses to Drought under Field Conditions. Front. Plant Sci. 2017, 8, 174. [Google Scholar] [CrossRef]
- Aizat, W.M.; Goh, H.H.; Baharum, S.N. Omics Applications for Systems Biology; Springer International Publishing: Cham, Switzerland, 2018; p. 99. [Google Scholar] [CrossRef]
- Jamil, I.N.; Remali, J.; Azizan, K.A.; Nor Muhammad, N.A.; Arita, M.; Goh, H.-H.; Aizat, W.M. Systematic Multi-Omics Integration (MOI) Approach in Plant Systems Biology. Front. Plant Sci. 2020, 11, 944. [Google Scholar] [CrossRef]
- Moschen, S.; Bengoa Luoni, S.; Di Rienzo, J.A.; del Pilar, M.C.; Tohge, T.; Watanabe, M.; Hollmann, J.; Gonzales, S.; Rivarola, M.; Garcia-Garcia, F.; et al. Integrating transcriptomic and metabolomic analysis to understand natural leaf senescence in sunflower. Plant Biotechnol. J. 2016, 14, 719–734. [Google Scholar] [CrossRef]
- Moschen, S.; Luoni, S.B.; Di Rienzo, J.A.; del Pilar Caro, M.; Higgins, J.; Tohge, T.; Watanabe, M.; Gonzalez, S.; Rivarola, M.; Garcia-Garcia, F.; et al. Integration of transcriptomic and metabolic data reveals hub transcription factors involved in drought stress response in sunflower (Helianthus annuus L.). Plant Mol. Biol. 2017, 94, 549–564. [Google Scholar] [CrossRef] [PubMed]
- Lavarenne, J.; Guyomarc’h, S.; Sallaud, C.; Ganet, P.; Lucas, M. The Spring of Systems Biology-Driven Breeding. Trends Plant Sci. 2018, 23, 706–720. [Google Scholar] [CrossRef] [PubMed]
- Weckwerth, W.; Ghatak, A.; Bellaire, A.; Chaturvedi, P.; Varshney, R.K. PANOMICS meets germplasm. Plant Biotechnol. J. 2020, 18, 1507–1525. [Google Scholar] [CrossRef] [PubMed]



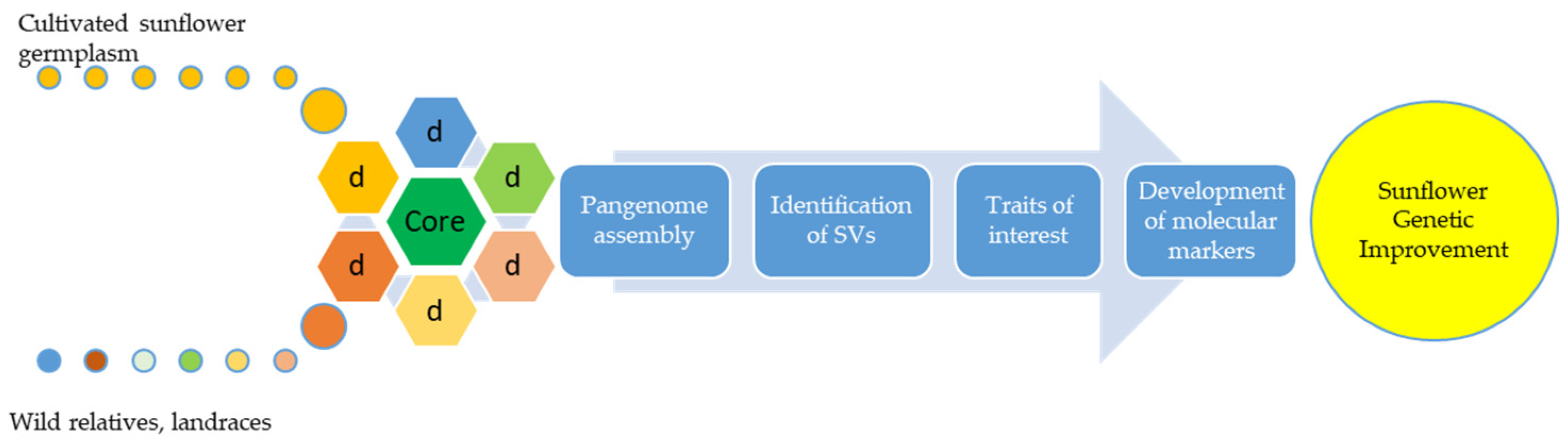{kind=link}
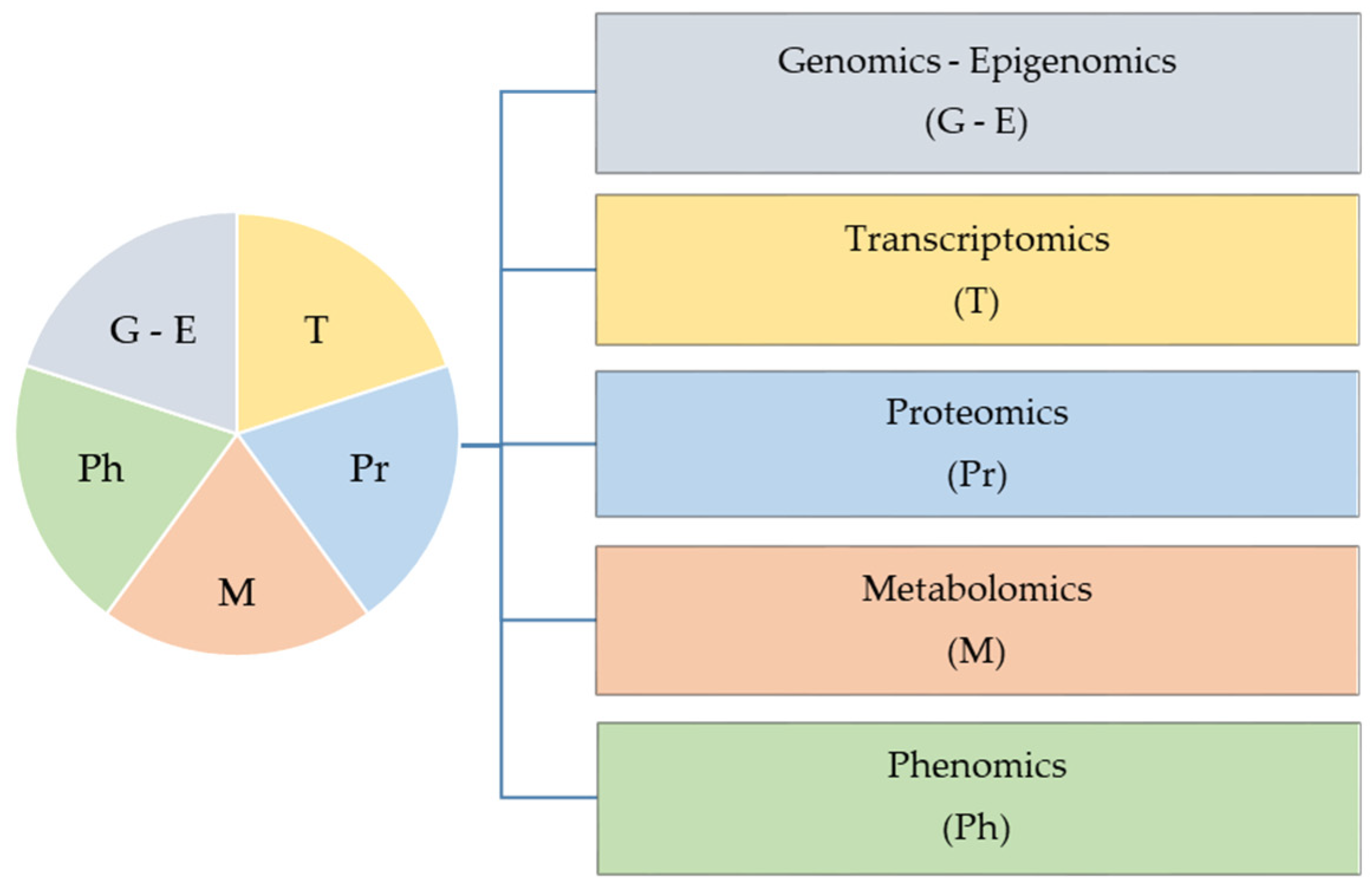{kind=link}
| Composition Type | Accessions | Strategy | Size | Reference |
|---|---|---|---|---|
| Genome | Inbred line XRQ | 102× sequencing coverage of the genome of the inbred line XRQ using 407 single-molecule real-time (SMRT) cells on the PacBio RS II platform. | 52,232 protein-coding genes 5803 spliced long non-coding RNAs | [28] |
| Pangenome | 493 sunflower accessions which include: 287 cultivated lines, 17 Native American landraces and 189 wild accessions representing 11 compatibile wild species | Pangenome assembled through de novo assembly of unmapped reads | 61,205 genes | [41] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jocković, M.; Jocić, S.; Cvejić, S.; Marjanović-Jeromela, A.; Jocković, J.; Radanović, A.; Miladinović, D. Genetic Improvement in Sunflower Breeding—Integrated Omics Approach. Plants 2021, 10, 1150. https://doi.org/10.3390/plants10061150
Jocković M, Jocić S, Cvejić S, Marjanović-Jeromela A, Jocković J, Radanović A, Miladinović D. Genetic Improvement in Sunflower Breeding—Integrated Omics Approach. Plants. 2021; 10(6):1150. https://doi.org/10.3390/plants10061150
Chicago/Turabian StyleJocković, Milan, Siniša Jocić, Sandra Cvejić, Ana Marjanović-Jeromela, Jelena Jocković, Aleksandra Radanović, and Dragana Miladinović. 2021. "Genetic Improvement in Sunflower Breeding—Integrated Omics Approach" Plants 10, no. 6: 1150. https://doi.org/10.3390/plants10061150
APA StyleJocković, M., Jocić, S., Cvejić, S., Marjanović-Jeromela, A., Jocković, J., Radanović, A., & Miladinović, D. (2021). Genetic Improvement in Sunflower Breeding—Integrated Omics Approach. Plants, 10(6), 1150. https://doi.org/10.3390/plants10061150