
Genomic Prediction across Structured Hybrid Populations and Environments in Maize

 , ,
, , 
 , and
, and
Abstract
1. Introduction
2. Results
2.1. Phenotypic Data Analysis
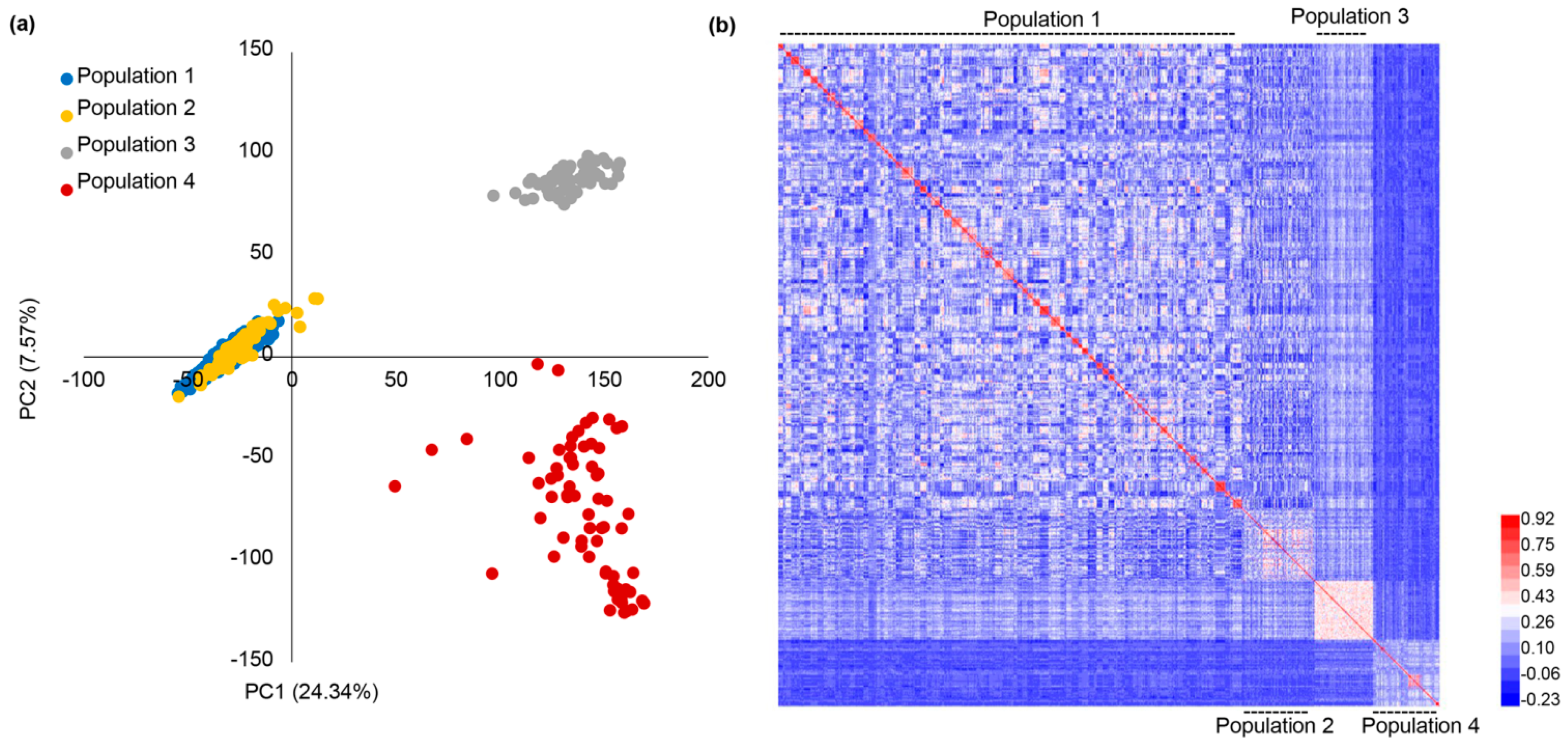
2.2. The Genetic Relationships of Multiple Populations
2.3. GP across Different Populations
2.4. GP across Different Environments
3. Discussion
4. Material and Methods
4.1. Plant Materials
4.2. Phenotype Evaluation and Phenotypic Data Analysis
4.3. Genotyping and Genotypic Data Analysis
4.4. GP Model
4.5. GP across Environments
4.6. GP across Different Populations
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tilman, D.; Balzer, C.; Hill, J.; Befort, B.L. Global food demand and the sustainable intensification of agriculture. Proc. Natl. Acad. Sci. USA 2011, 108, 20260–20264. [Google Scholar] [CrossRef]
- Gorjanc, G.; Jenko, J.; Hearne, S.J.; Hickey, J.M. Initiating maize pre-breeding programs using genomic selection to harness polygenic variation from landrace populations. BMC Genom. 2016, 17, 30. [Google Scholar] [CrossRef]
- Rutkoski, J.E.; Heffner, E.L.; Sorrells, M.E. Genomic selection for durable stem rust resistance in wheat. Euphytica 2011, 179, 161–173. [Google Scholar] [CrossRef]
- Yu, X.Q.; Li, X.R.; Guo, T.T.; Zhu, C.S.; Wu, Y.Y.; Mitchell, S.E.; Roozeboom, K.L.; Wang, D.H.; Wang, M.L.; Pederson, G.A.; et al. Genomic prediction contributing to a promising global strategy to turbocharge gene banks. Nat. Plants 2016, 2, 16150. [Google Scholar] [CrossRef]
- Heffner, E.L.; Lorenz, A.J.; Jannink, J.L.; Sorrells, M.E. Plant breeding with genomic selection: Gain per unit time and cost. Crop Sci. 2010, 50, 1681–1690. [Google Scholar] [CrossRef]
- Combs, E.; Bernardo, R. Accuracy of genomewide selection for different traits with constant population size, heritability, and number of markers. Plant Genome 2013, 6. [Google Scholar] [CrossRef]
- Crossa, J.; Perez, P.; Hickey, J.; Burgueno, J.; Ornella, L.; Ceron-Rojas, J.; Zhang, X.; Dreisigacker, S.; Babu, R.; Li, Y.; et al. Genomic prediction in CIMMYT maize and wheat breeding programs. Heredity 2014, 112, 48–60. [Google Scholar] [CrossRef]
- Liu, X.G.; Wang, H.W.; Wang, H.; Guo, Z.F.; Xu, X.J.; Liu, J.C.; Wang, S.H.; Li, W.X.; Zou, C.; Prasanna, B.M.; et al. Factors affecting genomic selection revealed by empirical evidence in maize. Crop J. 2018, 6, 341–352. [Google Scholar] [CrossRef]
- Riedelsheimer, C.; Endelman, J.B.; Stange, M.; Sorrells, M.E.; Jannink, J.L.; Melchinger, A.E. Genomic predictability of interconnected biparental maize populations. Genetics 2013, 194, 493–503. [Google Scholar] [CrossRef]
- Li, D.D.; Wang, P.X.; Gu, R.L.; Fu, J.J.; Xu, Z.X.; Lyle, D.; Peng, Y.L.; Wang, G.Y.; Zhang, H.W. Genetic relatedness and the ratio of subpopulation-common alleles are related in genomic prediction across structured subpopulations in maize. Plant Breeding 2019, 138, 802–809. [Google Scholar] [CrossRef]
- Xu, S.Z.; Zhu, D.; Zhang, Q.F. Predicting hybrid performance in rice using genomic best linear unbiased prediction. Proc. Natl. Acad. Sci. USA 2014, 111, 12456–12461. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.H.; Yang, S.H.; Gong, J.Y.; Zhao, Y.; Feng, Q.; Gong, H.; Li, W.J.; Zhan, Q.L.; Cheng, B.Y.; Xia, J.H.; et al. Genomic analysis of hybrid rice varieties reveals numerous superior alleles that contribute to heterosis. Nat. Commun. 2015, 6, 6258. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, N.; Tai, S.S.; Wang, W.S.; Mansueto, L.; Palis, K.; Fuentes, R.R.; Ulat, V.J.; Chebotarov, D.; Zhang, G.Y.; Li, Z.K. SNP-Seek database of SNPs derived from 3000 rice genomes. Nucleic Acids Res. 2015, 43, D1023–D1027. [Google Scholar] [CrossRef]
- Cui, Y.R.; Li, R.D.; Li, G.W.; Zhang, F.; Zhu, T.T.; Zhang, Q.F.; Ali, J.; Li, Z.K.; Xu, S.Z. Hybrid breeding of rice via genomic selection. Plant Biotechnol. J. 2019, 18, 57–67. [Google Scholar] [CrossRef]
- Wang, C.L.; Chen, Y.H.; Ku, L.X.; Wang, T.G.; Sun, Z.H.; Cheng, F.F.; Wu, L.C. Mapping QTL associated with photoperiod sensitivity and assessing the importance of QTL x environment interaction for flowering time in maize. PLoS ONE 2010, 5, e14068. [Google Scholar] [CrossRef]
- Hu, X.M.; Wang, G.H.; Du, X.M.; Zhang, H.W.; Xu, Z.X.; Wang, J.; Chen, G.; Wang, B.; Li, X.H.; Chen, X.J.; et al. QTL analysis across multiple environments reveals promising chromosome regions associated with yield-related traits in maize under drought conditions. Crop J. 2021. [Google Scholar] [CrossRef]
- Ly, D.; Hamblin, M.; Rabbi, I.; Melaku, G.; Bakare, M.; Gauch, H.G.; Okechukwu, R.; Dixon, A.G.O.; Kulakow, P.; Jannink, J.L. Relatedness and genotype x environment interaction affect prediction accuracies in genomic selection: A study in cassava. Crop Sci. 2013, 53, 1312–1325. [Google Scholar] [CrossRef]
- Lopez-Cruz, M.; Crossa, J.; Bonnett, D.; Dreisigacker, S.; Poland, J.; Jannink, J.L.; Singh, R.P.; Autrique, E.; de los Campos, G. Increased prediction accuracy in wheat breeding trials using a marker × environment interaction genomic selection model. G3-Genes Genom. Genet. 2015, 5, 569–582. [Google Scholar] [CrossRef]
- Li, D.D.; Xu, Z.X.; Gu, R.L.; Wang, P.X.; Lyle, D.; Xu, J.L.; Zhang, H.W.; Wang, G.Y. Enhancing genomic selection by fitting large-effect SNPs as fixed effects and a genotype-by-environment effect using a maize BC1F3:4 population. PLoS ONE 2019, 14, e0223898. [Google Scholar] [CrossRef]
- Dias, K.O.D.; Gezan, S.A.; Guimaraes, C.T.; Nazarian, A.; Silva, L.D.E.; Parentoni, S.N.; Guimaraes, P.E.D.; Anoni, C.D.; Padua, J.M.V.; Pinto, M.D.; et al. Improving accuracies of genomic predictions for drought tolerance in maize by joint modeling of additive and dominance effects in multi-environment trials. Heredity 2018, 121, 24–37. [Google Scholar] [CrossRef]
- Massman, J.M.; Gordillo, A.; Lorenzana, R.E.; Bernardo, R. Genomewide predictions from maize single-cross data. Theor. Appl. Genet. 2013, 126, 13–22. [Google Scholar] [CrossRef]
- Hayes, B.J.; Bowman, P.J.; Chamberlain, A.C.; Verbyla, K.; Goddard, M.E. Accuracy of genomic breeding values in multi-breed dairy cattle populations. Genet. Sel. Evol. 2009, 41, 51. [Google Scholar] [CrossRef]
- Massman, J.M.; Jung, H.J.G.; Bernardo, R. Genomewide selection versus marker-assisted recurrent selection to improve grain yield and stover-quality traits for cellulosic ethanol in maize. Crop Sci. 2013, 53, 58–66. [Google Scholar] [CrossRef]
- Wang, P.X.; Zhang, H.W.; Lyle, D.; Li, D.D.; Wang, G.Y.; Pan, Q.C.; Wang, J.H. Bulk pollen pollination in maize for efficient construction of introgression populations with high genome coverage. Plant Breed. 2019, 138, 252–258. [Google Scholar] [CrossRef]
- Ma, J.; Zhang, D.F.; Cao, Y.Y.; Wang, L.F.; Li, J.J.; Lubberstedt, T.; Wang, T.Y.; Li, Y.; Li, H.Y. Heterosis-related genes under different planting densities in maize. J. Exp. Bot. 2018, 69, 5077–5087. [Google Scholar] [CrossRef]
- Song, W.; Shi, Z.; Xing, J.F.; Duan, M.X.; Su, A.G.; Li, C.H.; Zhang, R.Y.; Zhao, Y.X.; Luo, M.J.; Wang, J.D.; et al. Molecular mapping of quantitative trait loci for grain moisture at harvest in maize. Plant Breed. 2017, 136, 28–32. [Google Scholar] [CrossRef]
- Zhou, Z.Q.; Zhang, C.S.; Lu, X.H.; Wang, L.W.; Hao, Z.F.; Li, M.S.; Zhang, D.G.; Yong, H.J.; Zhu, H.Y.; Weng, J.F.; et al. Dissecting the genetic basis underlying combining ability of plant height related traits in maize. Front. Plant Sci. 2018. [Google Scholar] [CrossRef]
- Bates, D.; Machler, M.; Bolker, B.M.; Walker, S.C. Fitting linear mixed-effects models using lme4. J. Stat. Soft. 2015, 67, 1–48. [Google Scholar] [CrossRef]
- Meng, L.; Li, H.H.; Zhang, L.Y.; Wang, J.K. QTL IciMapping: Integrated software for genetic linkage map construction and quantitative trait locus mapping in biparental populations. Crop J. 2015, 3, 269–283. [Google Scholar] [CrossRef]
- Hallauer, A.R.; Carena, M.J.; Miranda Filho, J.B. Quantitative Genetics in Maize Breeding; Springer Science & Business Media: New York, NY, USA, 2010. [Google Scholar]
- Murray, M.G.; Thompson, W.F. Rapid isolation of high molecular-weight plant DNA. Nucleic Acids Res. 1980, 8, 4321–4325. [Google Scholar] [CrossRef]
- Xu, C.; Ren, Y.G.; Jian, Y.Q.; Guo, Z.F.; Zhang, Y.; Xie, C.X.; Fu, J.J.; Wang, H.W.; Wang, G.Y.; Xu, Y.B.; et al. Development of a maize 55 K SNP array with improved genome coverage for molecular breeding. Mol. Breed. 2017, 37, 20. [Google Scholar] [CrossRef]
- Browning, B.L.; Browning, S.R. A unified approach to genotype imputation and haplotype-phase inference for large data sets of trios and unrelated individuals. Am. J. Human Genet. 2009, 84, 210–223. [Google Scholar] [CrossRef]
- Wimmer, V.; Albrecht, T.; Auinger, H.J.; Schon, C.C. Synbreed: A framework for the analysis of genomic prediction data using R. Bioinformatics 2012, 28, 2086–2087. [Google Scholar] [CrossRef]
- Isidro-Sánchez, J.; Akdemir, D.; Montilla-Bascón, G. Genome-wide association analysis using R. Methods Mol. Biol. 2017, 1536, 189–207. [Google Scholar]
- Xu, S.Z. Mapping quantitative trait loci by controlling polygenic background effects. Genetics 2013, 195, 1209–1222. [Google Scholar] [CrossRef] [PubMed]
- Pérez, P.; de los campos, G. Genome-wide regression and prediction with the BGLR statistical package. Genetics 2014, 198, 483–495. [Google Scholar] [CrossRef] [PubMed]
- Crossa, J.; de los Campos, G.; Maccaferri, M.; Tuberosa, R.; Burgueño, J.; Pérez-Rodríguez, P. Extending the marker x environment interaction model for genomic-enabled prediction and genome-wide association analysis in durum wheat. Crop Sci. 2016, 56, 2193–2209. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Population | N | Mean (Kg) | SD (Kg) | Minimum (Kg) | Maximum (Kg) | Range (Kg) | CV (%) |
|---|---|---|---|---|---|---|---|
| Population 1 | 475 | 753.71 | 43.55 | 641.95 | 947.32 | 305.37 | 5.78 |
| Population 2 | 72 | 815.52 | 54.11 | 691.36 | 959.49 | 268.12 | 6.63 |
| Population 3 | 60 | 656.97 | 34.87 | 587.86 | 745.70 | 157.83 | 5.31 |
| Population 4 | 68 | 687.40 | 58.63 | 539.14 | 840.69 | 301.55 | 8.53 |
| Population | ANOVA | H2 | R2 (%) | |||||
|---|---|---|---|---|---|---|---|---|
| Source | DF | SS | MS | F | p | |||
| Population 1 | Rep/Env | 3 | 122,714.40 | 40,904.80 | 11.24 | 0.00 | 0.63 | 91.17 |
| Genotype | 474 | 5,459,187.50 | 11,517.27 | 3.17 | 0.00 | |||
| Environment | 2 | 39,391,468.00 | 19,695,734.00 | 5413.62 | 0.00 | |||
| G by E | 935 | 4,392,113.50 | 4697.45 | 1.29 | 0.00 | |||
| Error | 1314 | 4,780,569.00 | 3638.18 | |||||
| Population 2 | Rep/Env | 3 | 24,789.26 | 8263.09 | 2.23 | 0.09 | 0.66 | 85.47 |
| Genotype | 71 | 957,095.81 | 13,480.22 | 3.63 | 0.00 | |||
| Environment | 2 | 2,448,691.25 | 1,224,345.63 | 329.97 | 0.00 | |||
| G by E | 130 | 651,743.31 | 5013.41 | 1.35 | 0.03 | |||
| Error | 187 | 693,851.19 | 3710.43 | |||||
| Population 3 | Rep/Env | 3 | 3349.01 | 1116.34 | 0.34 | 0.80 | 0.62 | 84.52 |
| Genotype | 59 | 548,701.06 | 9300.02 | 2.83 | 0.00 | |||
| Environment | 2 | 1,797,173.38 | 898,586.69 | 273.06 | 0.00 | |||
| GE_interaction | 118 | 431,993.81 | 3660.96 | 1.11 | 0.27 | |||
| Error | 156 | 513,373.16 | 3290.85 | |||||
| Population 4 | Rep/Env | 3 | 15,394.80 | 5131.60 | 1.56 | 0.20 | 0.74 | 88.79 |
| Genotype | 67 | 1,310,953.50 | 19,566.47 | 5.95 | 0.00 | |||
| Environment | 2 | 2,757,321.25 | 1,378,660.63 | 419.16 | 0.00 | |||
| GE_interaction | 133 | 762,263.31 | 5731.30 | 1.74 | 0.00 | |||
| Error | 186 | 611,768.25 | 3289.08 | |||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, D.; Xu, Z.; Gu, R.; Wang, P.; Xu, J.; Du, D.; Fu, J.; Wang, J.; Zhang, H.; Wang, G. Genomic Prediction across Structured Hybrid Populations and Environments in Maize. Plants 2021, 10, 1174. https://doi.org/10.3390/plants10061174
Li D, Xu Z, Gu R, Wang P, Xu J, Du D, Fu J, Wang J, Zhang H, Wang G. Genomic Prediction across Structured Hybrid Populations and Environments in Maize. Plants. 2021; 10(6):1174. https://doi.org/10.3390/plants10061174
Chicago/Turabian StyleLi, Dongdong, Zhenxiang Xu, Riliang Gu, Pingxi Wang, Jialiang Xu, Dengxiang Du, Junjie Fu, Jianhua Wang, Hongwei Zhang, and Guoying Wang. 2021. "Genomic Prediction across Structured Hybrid Populations and Environments in Maize" Plants 10, no. 6: 1174. https://doi.org/10.3390/plants10061174
APA StyleLi, D., Xu, Z., Gu, R., Wang, P., Xu, J., Du, D., Fu, J., Wang, J., Zhang, H., & Wang, G. (2021). Genomic Prediction across Structured Hybrid Populations and Environments in Maize. Plants, 10(6), 1174. https://doi.org/10.3390/plants10061174