
Inhibition of SERPINE1 Function Attenuates Wound Closure in Response to Tissue Injury: A Role for PAI-1 in Re-Epithelialization and Granulation Tissue Formation


Abstract
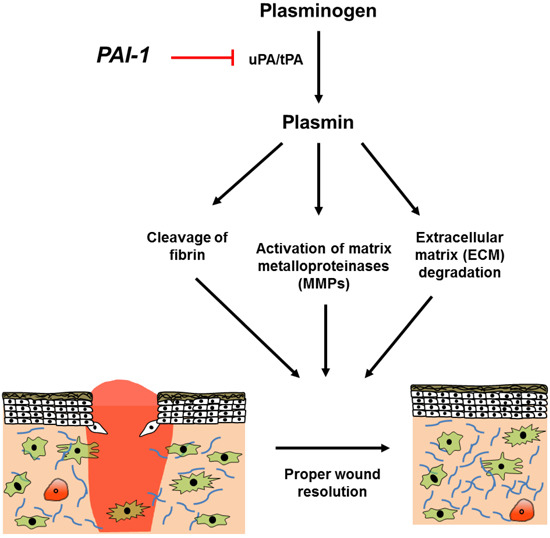
:
{kind=link}
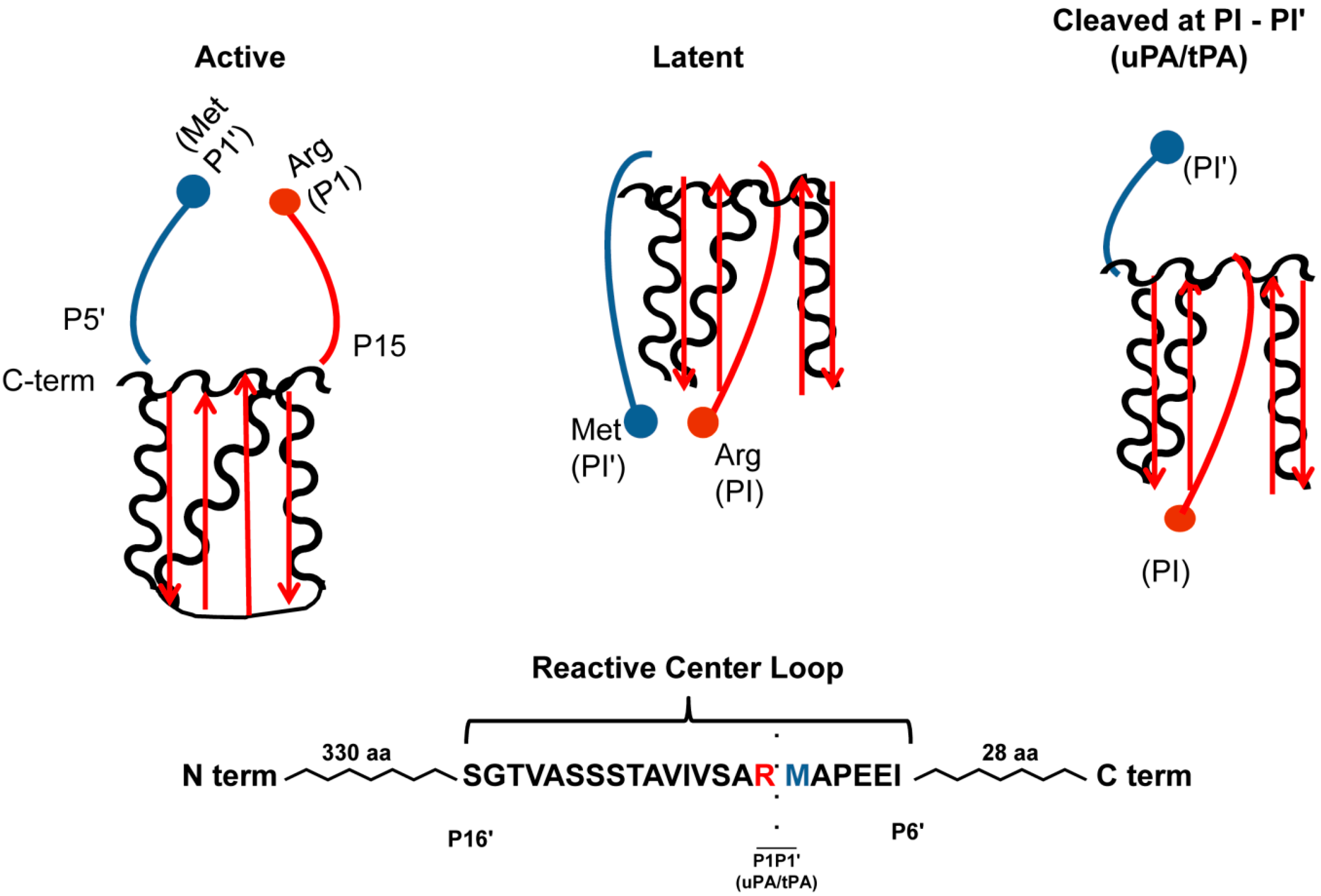{kind=link}
{kind=link}
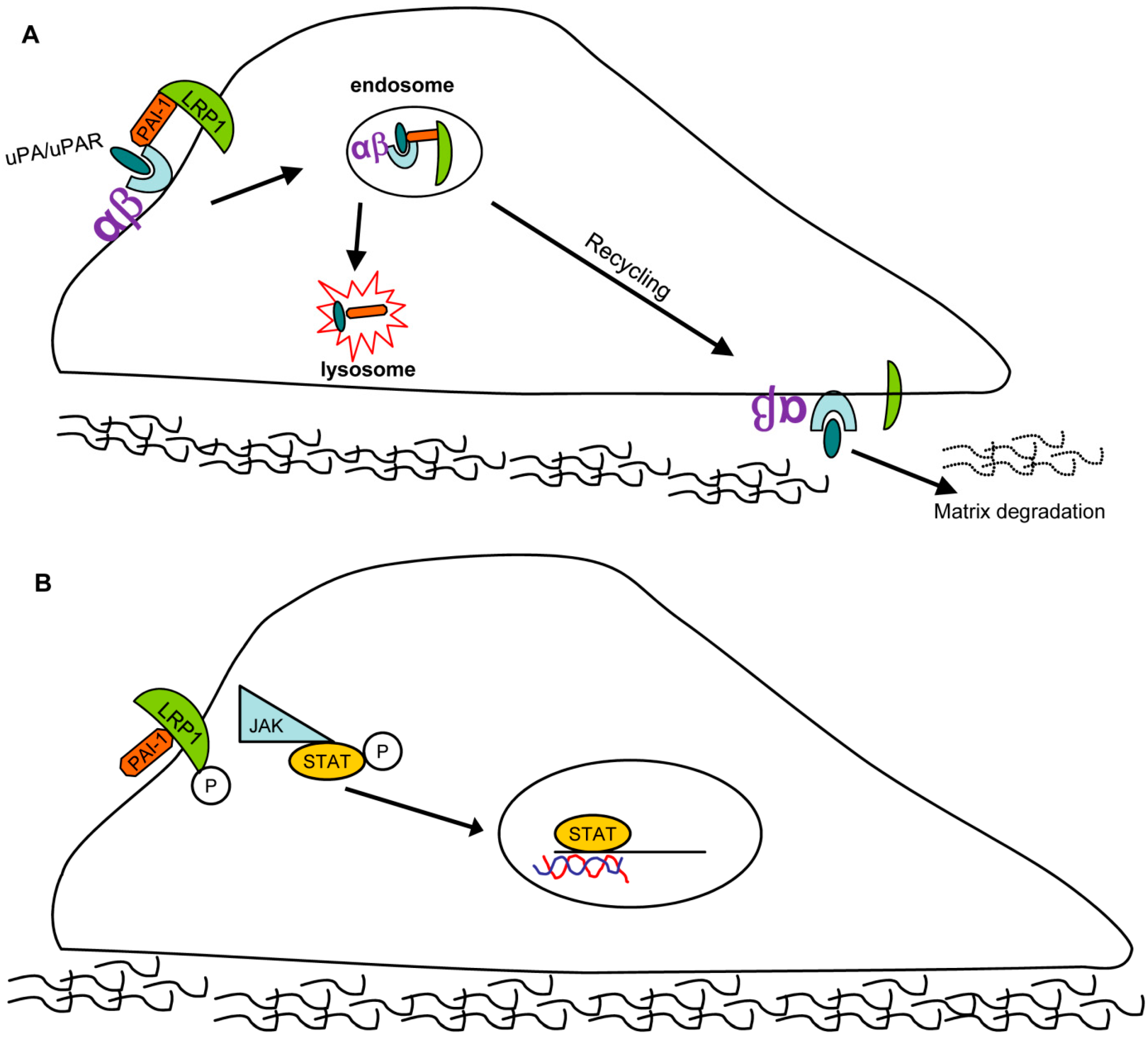{kind=link}
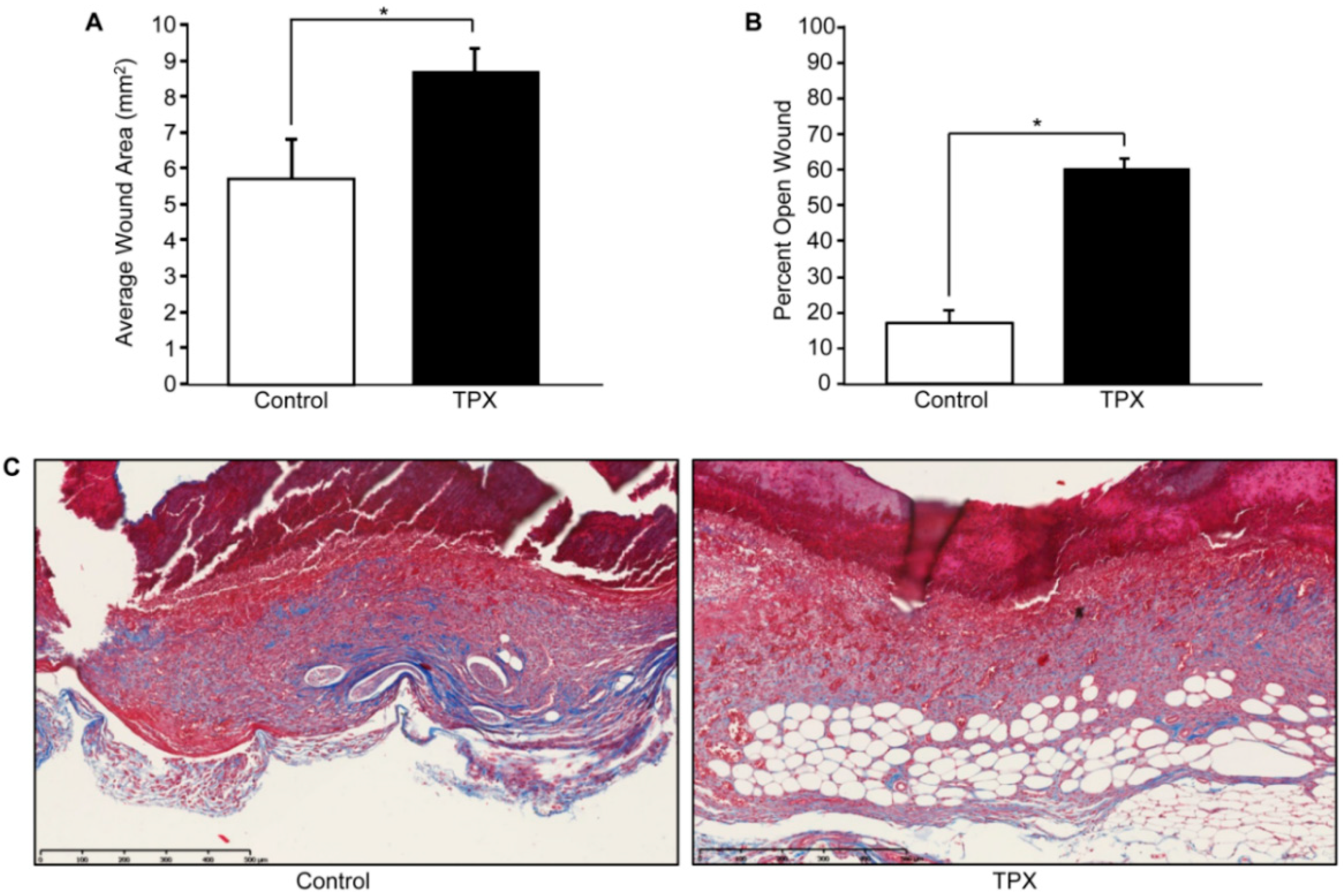{kind=link}
{kind=link}
1. Background
2. Structure and Chemical Antagonists
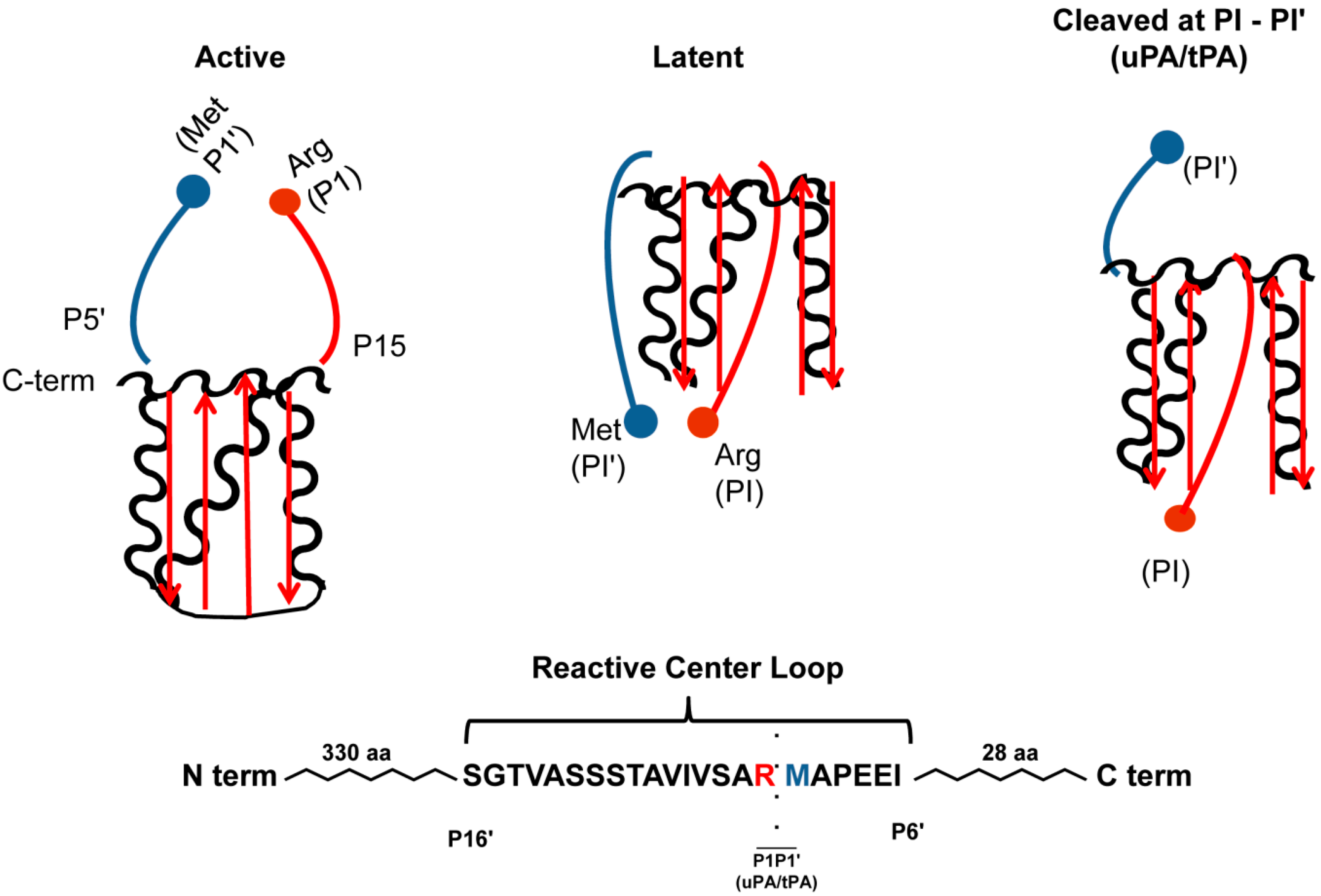
3. PAI-1 in Cellular Mechanisms of Disease Progression
4. PAI-1 as a Multifunctional Signaling “Ligand”
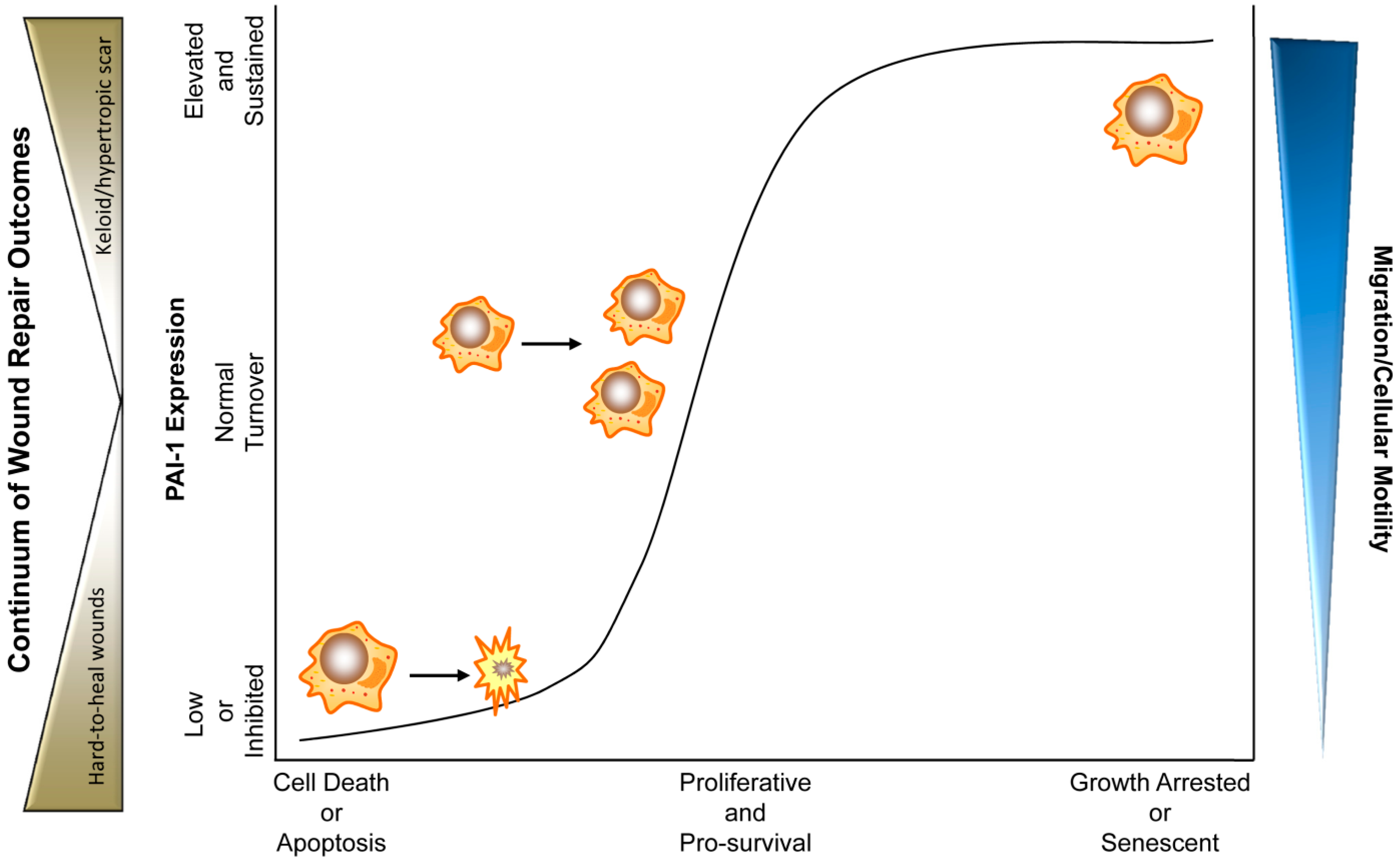
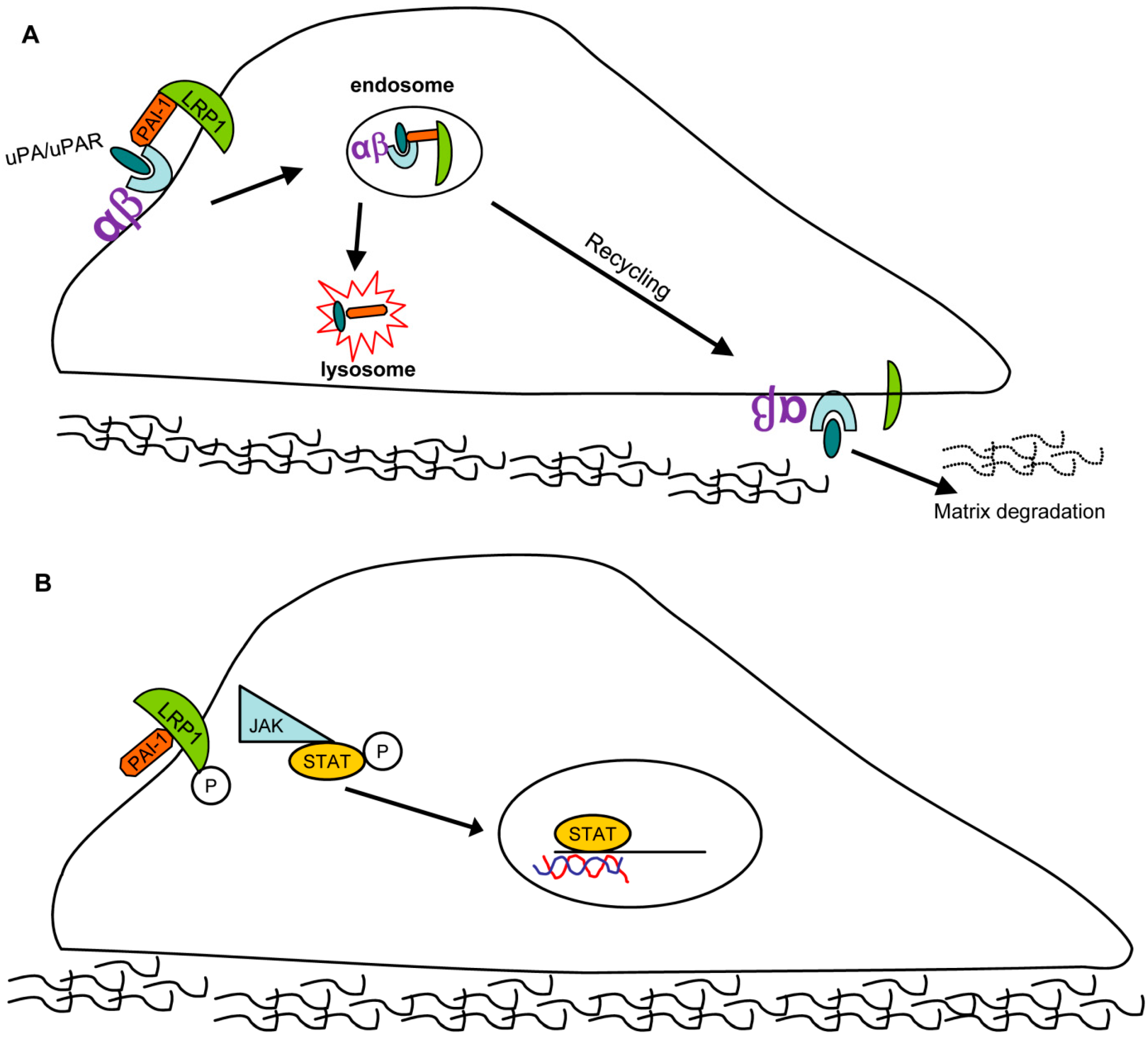
5. PAI-1 in Physiological and Pathophysiological Cutaneous Wound Healing
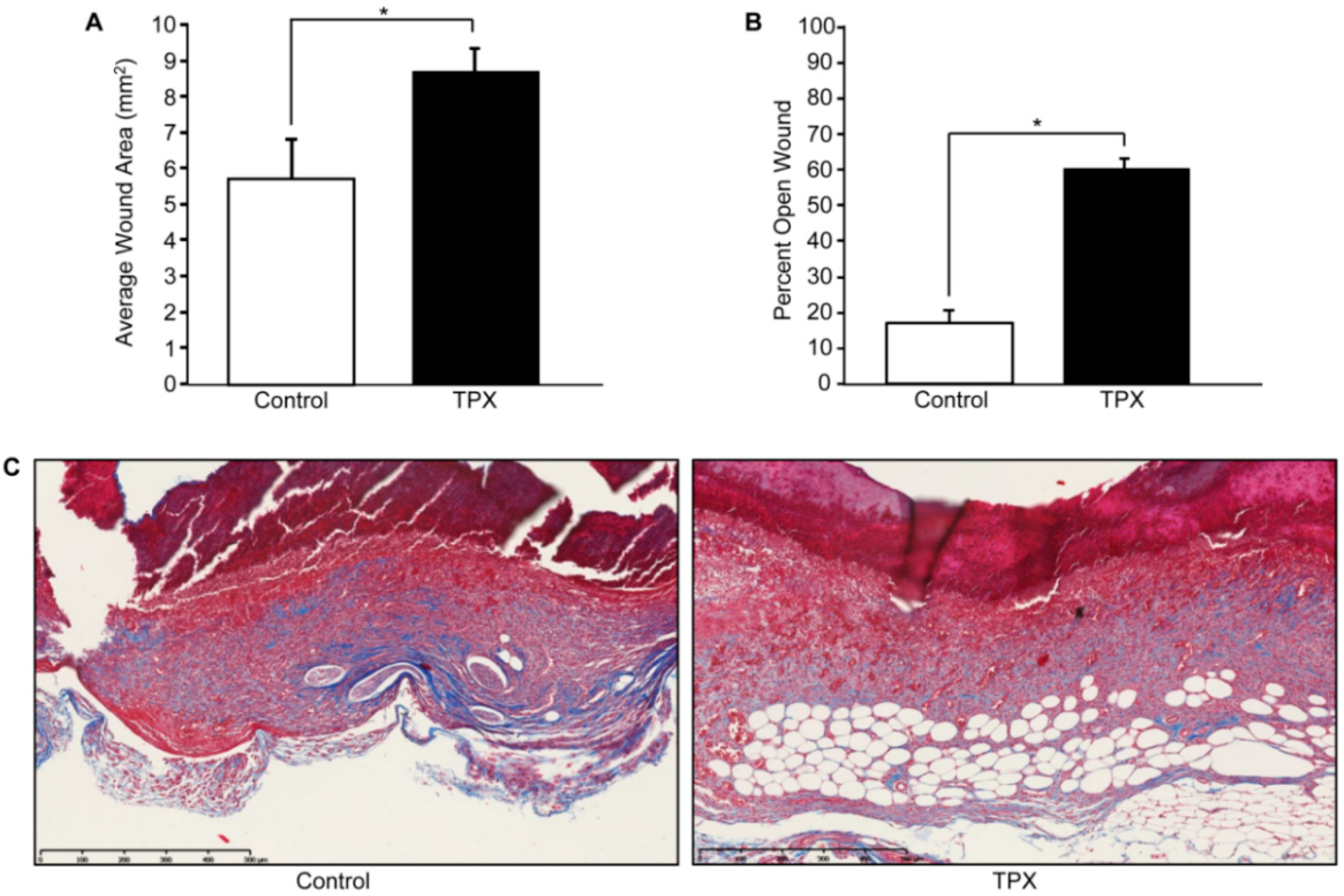
6. Conclusions and Therapeutic Considerations
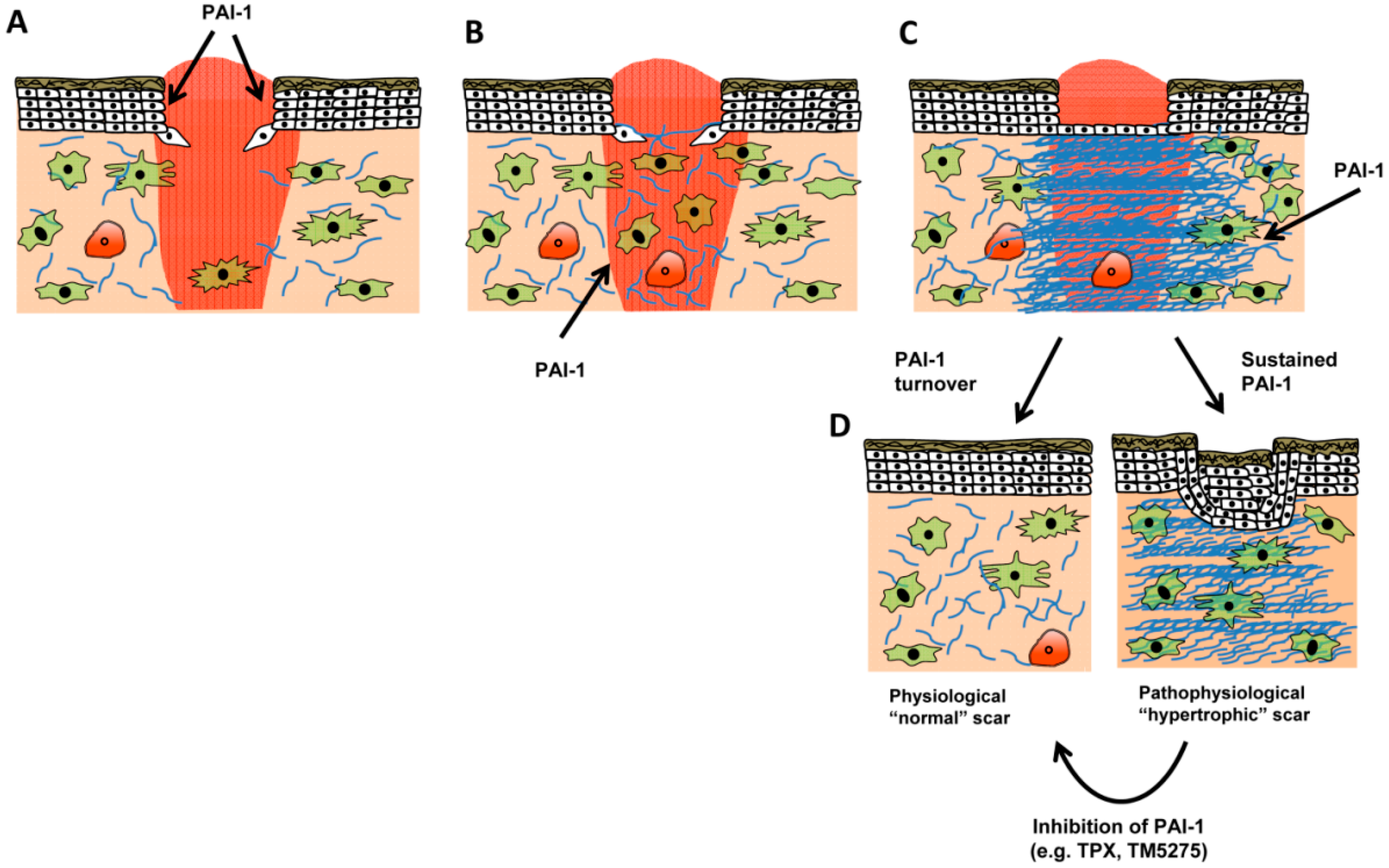
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Eri ckson, L.A.; Ginsberg, M.H.; Loskutoff, D.J. Detection and partial characterization of an inhibitor of plasminogen activator in human platelets. J. Clin. Investig. 1984, 74, 1465–1472. [Google Scholar] [CrossRef] [PubMed]
- Dellas, C.; Loskutoff, D.J. Historical analysis of PAI-1 from its discovery to its potential role in cell motility and disease. Thromb. Haemost. 2005, 93, 631–640. [Google Scholar] [PubMed]
- Mottonen, J.; Strand, A.; Symersky, J.; Sweet, R.M.; Danley, D.E.; Geoghegan, K.F.; Gerard, R.D.; Goldsmith, E.J. Structural basis of latency in plasminogen activator inhibitor-1. Nature 1992, 355, 270–273. [Google Scholar] [CrossRef] [PubMed]
- Levin, E.G.; Santell, L. Conversion of the active to latent plasminogen activator inhibitor from human endothelial cells. Blood 1987, 70, 1090–1098. [Google Scholar] [PubMed]
- Lindahl, T.L.; Sigurdardottir, O.; Wiman, B. Stability of plasminogen activator inhibitor 1 (PAI-1). Thromb. Haemost. 1989, 62, 748–751. [Google Scholar] [PubMed]
- Hekman, C.M.; Loskutoff, D.J. Endothelial cells produce a latent inhibitor of plasminogen activators that can be activated by denaturants. J. Biol. Chem. 1985, 260, 11581–11587. [Google Scholar] [PubMed]
- Lambers, J.W.; Cammenga, M.; Konig, B.W.; Mertens, K.; Pannekoek, H.; van Mourik, J.A. Activation of human endothelial cell-type plasminogen activator inhibitor (PAI-1) by negatively charged phospholipids. J. Biol. Chem. 1987, 262, 17492–17496. [Google Scholar] [PubMed]
- Declerck, P.J.; de Mol, M.; Vaughan, D.E.; Collen, D. Identification of a conformationally distinct form of plasminogen activator inhibitor-1, acting as a noninhibitory substrate for tissue-type plasminogen activator. J. Biol. Chem. 1992, 267, 11693–11696. [Google Scholar] [PubMed]
- Patston, P.A.; Gettins, P.; Beechem, J.; Schapira, M. Mechanism of serpin action: Evidence that C1 inhibitor functions as a suicide substrate. Biochemistry 1991, 30, 8876–8882. [Google Scholar] [CrossRef] [PubMed]
- Gils, A.; Declerck, P.J. Proteinase specificity and functional diversity in point mutants of plasminogen activator inhibitor 1. J. Biol. Chem. 1997, 272, 12662–12666. [Google Scholar] [CrossRef] [PubMed]
- Aertgeerts, K.; de Bondt, H.L.; de Ranter, C.J.; Declerck, P.J. Mechanisms contributing to the conformational and functional flexibility of plasminogen activator inhibitor-1. Nat. Struct. Biol. 1995, 2, 891–897. [Google Scholar] [CrossRef] [PubMed]
- Simone, T.M.; Higgins, P.J. Low molecular weight antagonists of plasminogen activator inhibitor-1: Therapeutic potential in cardiovascular disease. Mol. Med. Ther. 2012, 1, 101. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Eren, M.; Vaughan, D.E.; Schleimer, R.P.; Cho, S.H. A plasminogen activator inhibitor-1 inhibitor reduces airway remodeling in a murine model of chronic asthma. Am. J. Respir. Cell Mol. Biol. 2012, 46, 842–846. [Google Scholar] [CrossRef] [PubMed]
- Krause, M.P.; Moradi, J.; Nissar, A.A.; Riddell, M.C.; Hawke, T.J. Inhibition of plasminogen activator inhibitor-1 restores skeletal muscle regeneration in untreated type 1 diabetic mice. Diabetes 2011, 60, 1964–1972. [Google Scholar] [CrossRef] [PubMed]
- Lijnen, H.R.; Alessi, M.C.; Frederix, L.; Collen, D.; Juhan-Vague, I. Tiplaxtinin impairs nutritionally induced obesity in mice. Thromb. Haemost. 2006, 96, 731–737. [Google Scholar] [PubMed]
- Schalkwijk, C.G.; Stehouwer, C.D. PAI-1 inhibition in obesity and the metabolic syndrome: A promising therapeutic strategy. Thromb. Haemost. 2006, 96, 698–699. [Google Scholar] [PubMed]
- Leik, C.E.; Su, E.J.; Nambi, P.; Crandall, D.L.; Lawrence, D.A. Effect of pharmacologic plasminogen activator inhibitor-1 inhibition on cell motility and tumor angiogenesis. J. Thromb. Haemost. 2006, 4, 2710–2715. [Google Scholar] [CrossRef] [PubMed]
- Crandall, D.L.; Quinet, E.M.; el Ayachi, S.; Hreha, A.L.; Leik, C.E.; Savio, D.A.; Juhan-Vague, I.; Alessi, M.C. Modulation of adipose tissue development by pharmacological inhibition of PAI-1. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 2209–2215. [Google Scholar] [CrossRef] [PubMed]
- Lijnen, H.R.; Alessi, M.C.; van Hoef, B.; Collen, D.; Juhan-Vague, I. On the role of plasminogen activator inhibitor-1 in adipose tissue development and insulin resistance in mice. J. Thromb. Haemost. 2005, 3, 1174–1179. [Google Scholar] [CrossRef] [PubMed]
- Weisberg, A.D.; Albornoz, F.; Griffin, J.P.; Crandall, D.L.; Elokdah, H.; Fogo, A.B.; Vaughan, D.E.; Brown, N.J. Pharmacological inhibition and genetic deficiency of plasminogen activator inhibitor-1 attenuates angiotensin II/salt-induced aortic remodeling. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 365–371. [Google Scholar] [CrossRef] [PubMed]
- Izuhara, Y.; Takahashi, S.; Nangaku, M.; Takizawa, S.; Ishida, H.; Kurokawa, K.; van Ypersele de Strihou, C.; Hirayama, N.; Miyata, T. Inhibition of plasminogen activator inhibitor-1: Its mechanism and effectiveness on coagulation and fibrosis. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 672–677. [Google Scholar] [CrossRef] [PubMed]
- Izuhara, Y.; Yamaoka, N.; Kodama, H.; Dan, T.; Takizawa, S.; Hirayama, N.; Meguro, K.; van Ypersele de Strihou, C.; Miyata, T. A novel inhibitor of plasminogen activator inhibitor-1 provides antithrombotic benefits devoid of bleeding effect in nonhuman primates. J. Cereb. Blood Flow Metab. 2010, 30, 904–912. [Google Scholar] [CrossRef] [PubMed]
- Ichimura, A.; Matsumoto, S.; Suzuki, S.; Dan, T.; Yamaki, S.; Sato, Y.; Kiyomoto, H.; Ishii, N.; Okada, K.; Matsuo, O.; et al. A small molecule inhibitor to plasminogen activator inhibitor 1 inhibits macrophage migration. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 935–942. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.T.; Vayalil, P.K.; Miyata, T.; Hagood, J.; Liu, R.M. Therapeutic value of small molecule inhibitor to plasminogen activator inhibitor-1 for lung fibrosis. Am. J. Respir. Cell Mol. Biol. 2012, 46, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Gorlatova, N.V.; Cale, J.M.; Elokdah, H.; Li, D.; Fan, K.; Warnock, M.; Crandall, D.L.; Lawrence, D.A. Mechanism of inactivation of plasminogen activator inhibitor-1 by a small molecule inhibitor. J. Biol. Chem. 2007, 282, 9288–9296. [Google Scholar] [CrossRef] [PubMed]
- Elokdah, H.; Abou-Gharbia, M.; Hennan, J.K.; McFarlane, G.; Mugford, C.P.; Krishnamurthy, G.; Crandall, D.L. Tiplaxtinin, a novel, orally efficacious inhibitor of plasminogen activator inhibitor-1: Design, synthesis, and preclinical characterization. J. Med. Chem. 2004, 47, 3491–3494. [Google Scholar] [CrossRef] [PubMed]
- Simone, T.M.; Higgins, S.P.; Archambeault, J.; Higgins, C.E.; Ginnan, R.G.; Singer, H.; Higgins, P.J. A small molecule PAI-1 functional inhibitor attenuates neointimal hyperplasia and vascular smooth muscle cell survival by promoting PAI-1 cleavage. Cell. Signal. 2015. [Google Scholar] [CrossRef]
- Fedotov, S.; Iomin, A. Migration and proliferation dichotomy in tumor-cell invasion. Phys. Rev. Lett. 2007, 98, 118101. [Google Scholar] [CrossRef] [PubMed]
- Ploplis, V.A.; Balsara, R.; Sandoval-Cooper, M.J.; Yin, Z.J.; Batten, J.; Modi, N.; Gadoua, D.; Donahue, D.; Martin, J.A.; Castellino, F.J. Enhanced in vitro proliferation of aortic endothelial cells from plasminogen activator inhibitor-1-deficient mice. J. Biol. Chem. 2004, 279, 6143–6151. [Google Scholar] [CrossRef] [PubMed]
- Simone, T.M.; Higgins, C.E.; Czekay, R.P.; Law, B.K.; Archambeault, J.; Kutz, S.M.; Higgins, P.J. SERPINE1: A molecular switch in the proliferation/migration dichotomy in wound-“activated” keratinocytes. Adv. Wound Care 2014, 3, 281–290. [Google Scholar] [CrossRef]
- Providence, K.M.; Higgins, P.J. PAI-1 expression is required for epithelial cell migration in two distinct phases of in vitro wound repair. J. Cell. Physiol. 2004, 200, 297–308. [Google Scholar] [CrossRef] [PubMed]
- Providence, K.M.; White, L.A.; Tang, J.; Gonclaves, J.; Staiano-Coico, L.; Higgins, P.J. Epithelial monolayer wounding stimulates binding of USF-1 to an E-box motif in the plasminogen activator inhibitor type 1 gene. J. Cell Sci. 2002, 115, 3767–3777. [Google Scholar] [CrossRef] [PubMed]
- Freytag, J.; Wilkins-Port, C.E.; Higgins, C.E.; Higgins, S.P.; Samarakoon, R.; Higgins, P.J. PAI-1 mediates the TGF-β1+EGF-induced “scatter” response in transformed human keratinocytes. J. Investig. Dermatol. 2010, 130, 2179–2190. [Google Scholar] [CrossRef] [PubMed]
- Providence, K.M.; Kutz, S.M.; Staiano-Coico, L.; Higgins, P.J. PAI-1 gene expression is regionally induced in wounded epithelial cell monolayers and required for injury repair. J. Cell. Physiol. 2000, 182, 269–280. [Google Scholar] [CrossRef] [PubMed]
- Romer, J.; Lund, L.R.; Eriksen, J.; Ralfkiaer, E.; Zeheb, R.; Gelehrter, T.D.; Dano, K.; Kristensen, P. Differential expression of urokinase-type plasminogen activator and its type-1 inhibitor during healing of mouse skin wounds. J. Investig. Dermatol. 1991, 97, 803–811. [Google Scholar] [CrossRef] [PubMed]
- Kortlever, R.M.; Higgins, P.J.; Bernards, R. Plasminogen activator inhibitor-1 is a critical downstream target of p53 in the induction of replicative senescence. Nat. Cell Biol. 2006, 8, 877–884. [Google Scholar] [CrossRef] [PubMed]
- Kortlever, R.M.; Nijwening, J.H.; Bernards, R. Transforming growth factor-β requires its target plasminogen activator inhibitor-1 for cytostatic activity. J. Biol. Chem. 2008, 283, 24308–24313. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.P.; Wang, W.L.; Liu, J.; Li, W.B.; Bai, L.L.; Yuan, Y.D.; Song, S.X. Plasminogen activator inhibitor-1 promotes the proliferation and inhibits the apoptosis of pulmonary fibroblasts by Ca(2+) signaling. Thromb. Res. 2012, 131, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Fang, D.K.; Zhang, J.Q. Pregnane X receptor: A double-edged sword. Chin. Med. J. 2009, 122, 1333–1341. [Google Scholar] [PubMed]
- Bajou, K.; Peng, H.; Laug, W.E.; Maillard, C.; Noel, A.; Foidart, J.M.; Martial, J.A.; DeClerck, Y.A. Plasminogen activator inhibitor-1 protects endothelial cells from FasL-mediated apoptosis. Cancer Cell 2008, 14, 324–334. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Kelm, R.J., Jr.; Budd, R.C.; Sobel, B.E.; Schneider, D.J. Inhibition of apoptosis and caspase-3 in vascular smooth muscle cells by plasminogen activator inhibitor type-1. J. Cell. Biochem. 2004, 92, 178–188. [Google Scholar] [CrossRef]
- Fang, H.; Placencio, V.R.; Declerck, Y.A. Protumorigenic activity of plasminogen activator inhibitor-1 through an antiapoptotic function. J. Natl. Cancer Inst. 2012, 104, 1470–1484. [Google Scholar] [CrossRef] [PubMed]
- Gomes-Giacoia, E.; Miyake, M.; Goodison, S.; Rosser, C.J. Targeting plasminogen activator inhibitor-1 inhibits angiogenesis and tumor growth in a human cancer xenograft model. Mol. Cancer Ther. 2013, 12, 2697–2708. [Google Scholar] [CrossRef] [PubMed]
- Czekay, R.P.; Loskutoff, D.J. Unexpected role of plasminogen activator inhibitor 1 in cell adhesion and detachment. Exp. Biol. Med. 2004, 229, 1090–1096. [Google Scholar]
- Czekay, R.P.; Aertgeerts, K.; Curriden, S.A.; Loskutoff, D.J. Plasminogen activator inhibitor-1 detaches cells from extracellular matrices by inactivating integrins. J. Cell Biol. 2003, 160, 781–791. [Google Scholar] [CrossRef] [PubMed]
- Akkawi, S.; Nassar, T.; Tarshis, M.; Cines, D.B.; Higazi, A.A. LRP and alphavbeta3 mediate tPA activation of smooth muscle cells. Am. J. Physiol. 2006, 291, H1351–H1359. [Google Scholar]
- Czekay, R.P.; Wilkins-Port, C.E.; Higgins, S.P.; Freytag, J.; Overstreet, J.M.; Klein, R.M.; Higgins, C.E.; Samarakoon, R.; Higgins, P.J. PAI-1: An integrator of cell signaling and migration. Int. J. Cell Biol. 2011, 2011, 562481. [Google Scholar] [CrossRef] [PubMed]
- Winkles, J.A. The TWEAK-Fn14 cytokine-receptor axis: Discovery, biology and therapeutic targeting. Nat. Rev. Drug Discov. 2008, 7, 411–425. [Google Scholar] [CrossRef] [PubMed]
- Munoz-Garcia, B.; Madrigal-Matute, J.; Moreno, J.A.; Martin-Ventura, J.L.; Lopez-Franco, O.; Sastre, C.; Ortega, L.; Burkly, L.C.; Egido, J.; Blanco-Colio, L.M. TWEAK-Fn14 interaction enhances plasminogen activator inhibitor 1 and tissue factor expression in atherosclerotic plaques and in cultured vascular smooth muscle cells. Cardiovasc. Res. 2011, 89, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Providence, K.M.; Higgins, S.P.; Mullen, A.; Battista, A.; Samarakoon, R.; Higgins, C.E.; Wilkins-Port, C.E.; Higgins, P.J. SERPINE1 (PAI-1) is deposited into keratinocyte migration “trails” and required for optimal monolayer wound repair. Arch. Dermatol. Res. 2008, 300, 303–310. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.C.; Duszczyszyn, D.A.; Castellino, F.J.; Ploplis, V.A. Accelerated skin wound healing in plasminogen activator inhibitor-1-deficient mice. Am. J. Pathol. 2001, 159, 1681–1688. [Google Scholar] [CrossRef] [PubMed]
- Romer, J.; Bugge, T.H.; Pyke, C.; Lund, L.R.; Flick, M.J.; Degen, J.L.; Dano, K. Impaired wound healing in mice with a disrupted plasminogen gene. Nat. Med. 1996, 2, 287–292. [Google Scholar] [CrossRef] [PubMed]
- Simone, T.M.; Longmate, W.M.; Law, B.K.; Higgins, P.J. Targeted inhibition of PAI-1 activity impairs epithelial migration and wound closure following cutaneous injury. Adv. Wound Care 2014. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Murphy, S.B.; Kishore, R.; Vaughan, D.E. Global gene expression profiling in PAI-1 knockout murine heart and kidney: Molecular basis of cardiac-selective fibrosis. PLoS One 2013, 8, e63825. [Google Scholar] [CrossRef] [PubMed]
- Tomasek, J.J.; Gabbiani, G.; Hinz, B.; Chaponnier, C.; Brown, R.A. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat. Rev. Mol. Cell Biol. 2002, 3, 349–363. [Google Scholar] [CrossRef] [PubMed]
- Hinz, B.; Phan, S.H.; Thannickal, V.J.; Galli, A.; Bochaton-Piallat, M.L.; Gabbiani, G. The myofibroblast: One function, multiple origins. Am. J. Pathol. 2007, 170, 1807–1816. [Google Scholar] [CrossRef] [PubMed]
- Desmouliere, A.; Gabbian, G. Apoptosis mediates the decrease in cellularity during the transition between granulation tissue and scar. Exp. Nephrol. 1995, 3, 134–139. [Google Scholar] [PubMed]
- Wipff, P.J.; Rifkin, D.B.; Meister, J.J.; Hinz, B. Myofibroblast contraction activates latent TGF-beta1 from the extracellular matrix. J. Cell Biol. 2007, 179, 1311–1323. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.K.; Vaughan, D.E. PAI-1 in tissue fibrosis. J. Cell. Physiol. 2012, 227, 493–507. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Zhang, Q.; Ann, D.K.; Akhondzadeh, A.; Duong, H.S.; Messadi, D.V.; Le, A.D. Increased vascular endothelial growth factor may account for elevated level of plasminogen activator inhibitor-1 via activating ERK1/2 in keloid fibroblasts. Am. J. Physiol. Cell Physiol. 2004, 286, C905–C912. [Google Scholar] [CrossRef] [PubMed]
- Tuan, T.L.; Wu, H.; Huang, E.Y.; Chong, S.S.; Laug, W.; Messadi, D.; Kelly, P.; Le, A. Increased plasminogen activator inhibitor-1 in keloid fibroblasts may account for their elevated collagen accumulation in fibrin gel cultures. Am. J. Pathol. 2003, 162, 1579–1589. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Ly, C.M.; Ko, C.Y.; Meyers, E.E.; Lawrence, D.A.; Bernstein, A.M. UPA binding to PAI-1 induces corneal myofibroblast differentiation on vitronectin. Investig. Ophthalmol. Vis. Sci. 2012, 53, 4765–4775. [Google Scholar] [CrossRef]
- Pedroja, B.S.; Kang, L.E.; Imas, A.O.; Carmeliet, P.; Bernstein, A.M. Plasminogen activator inhibitor-1 regulates integrin alphavbeta3 expression and autocrine transforming growth factor β signaling. J. Biol. Chem. 2009, 284, 20708–20717. [Google Scholar] [CrossRef] [PubMed]
- Diebold, I.; Kraicun, D.; Bonello, S.; Gorlach, A. The “PAI-1 paradox” in vascular remodeling. Thromb. Haemost. 2008, 100, 984–991. [Google Scholar] [PubMed]
- Balsara, R.D.; Ploplis, V.A. Plasminogen activator inhibitor-1: The double-edged sword in apoptosis. Thromb. Haemost. 2008, 100, 1029–1036. [Google Scholar] [PubMed]
- Longmate, W.M.; Monichan, R.; Chu, M.L.; Tsuda, T.; Mahoney, M.G.; DiPersio, C.M. Reduced fibulin-2 contributes to loss of basement membrane integrity and skin blistering in mice lacking integrin alpha3beta1 in the epidermis. J. Investig. Dermatol. 2014, 134, 1609–1617. [Google Scholar] [CrossRef] [PubMed]
- Horowitz, J.C.; Rogers, D.S.; Simon, R.H.; Sisson, T.H.; Thannickal, V.J. Plasminogen activation induced pericellular fibronectin proteolysis promotes fibroblast apoptosis. Am. J. Respir. Cell Mol. Biol. 2008, 38, 78–87. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Simone, T.M.; Higgins, P.J. Inhibition of SERPINE1 Function Attenuates Wound Closure in Response to Tissue Injury: A Role for PAI-1 in Re-Epithelialization and Granulation Tissue Formation. J. Dev. Biol. 2015, 3, 11-24. https://doi.org/10.3390/jdb3010011
Simone TM, Higgins PJ. Inhibition of SERPINE1 Function Attenuates Wound Closure in Response to Tissue Injury: A Role for PAI-1 in Re-Epithelialization and Granulation Tissue Formation. Journal of Developmental Biology. 2015; 3(1):11-24. https://doi.org/10.3390/jdb3010011
Chicago/Turabian StyleSimone, Tessa M., and Paul J. Higgins. 2015. "Inhibition of SERPINE1 Function Attenuates Wound Closure in Response to Tissue Injury: A Role for PAI-1 in Re-Epithelialization and Granulation Tissue Formation" Journal of Developmental Biology 3, no. 1: 11-24. https://doi.org/10.3390/jdb3010011
APA StyleSimone, T. M., & Higgins, P. J. (2015). Inhibition of SERPINE1 Function Attenuates Wound Closure in Response to Tissue Injury: A Role for PAI-1 in Re-Epithelialization and Granulation Tissue Formation. Journal of Developmental Biology, 3(1), 11-24. https://doi.org/10.3390/jdb3010011