

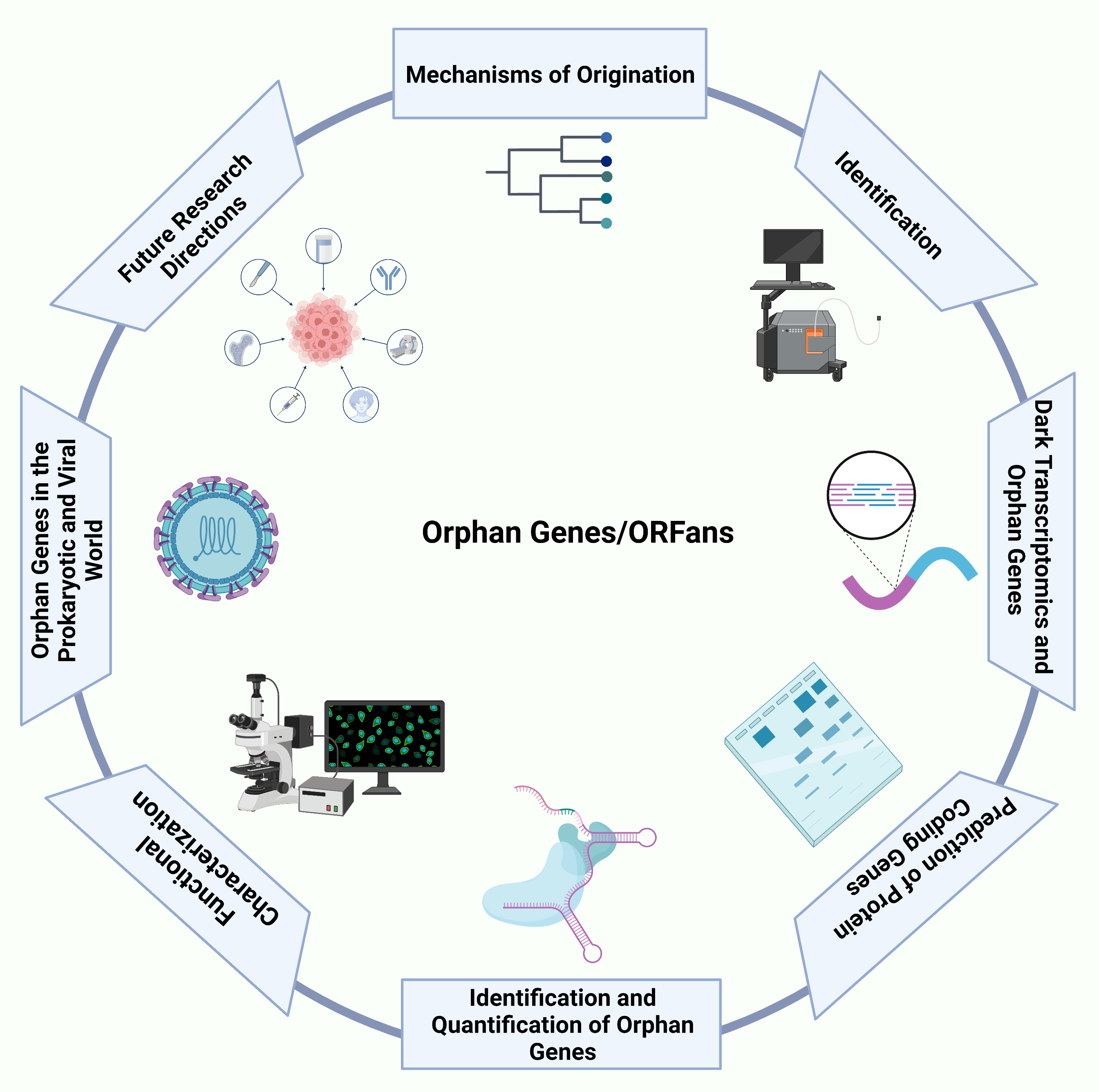
The Lost and Found: Unraveling the Functions of Orphan Genes


Abstract

1. Introduction
2. Mechanisms of OG Origination
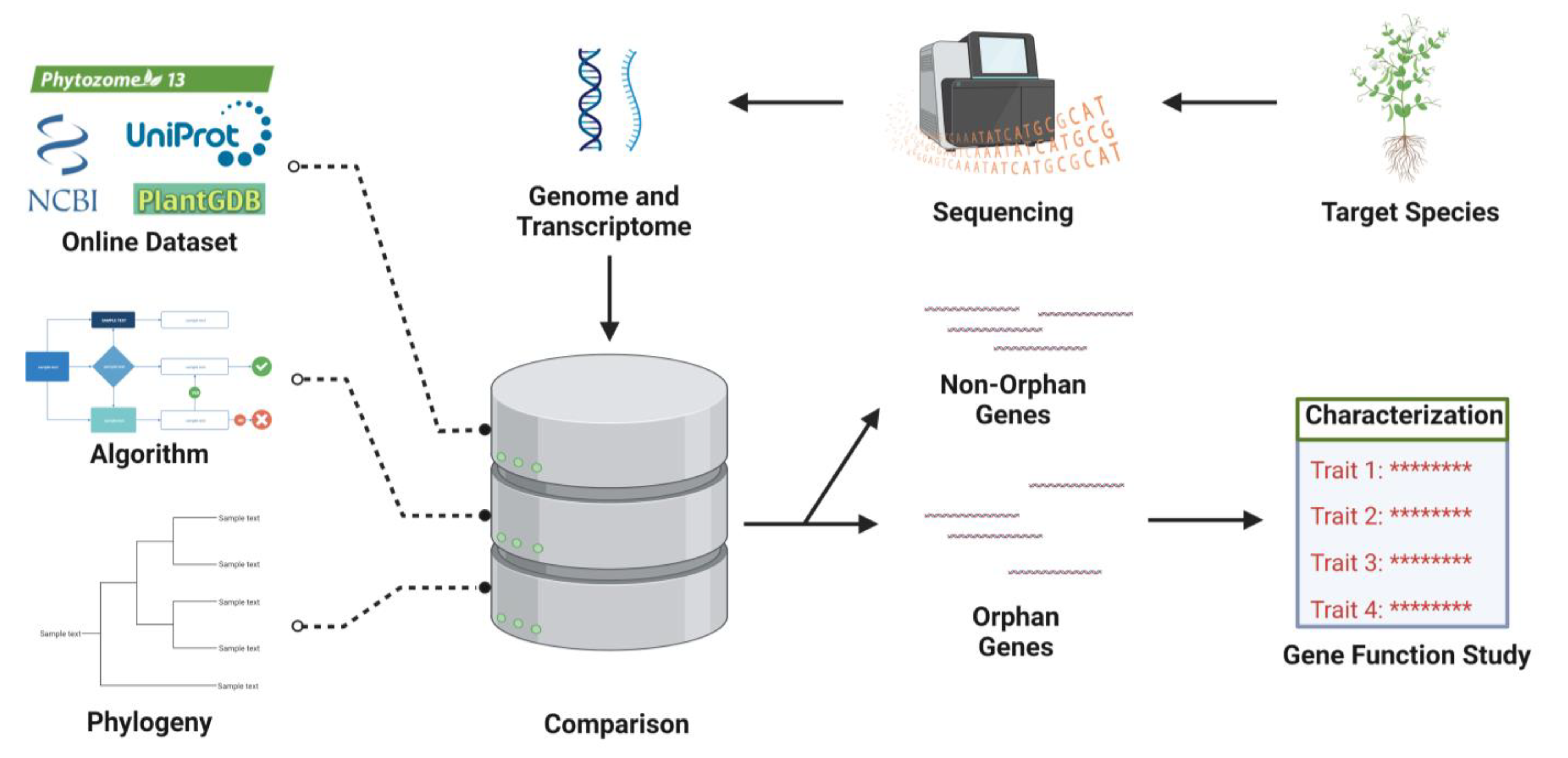
3. Identification of OGs
3.1. Methods of Orphan Genes Discovery
3.1.1. BLAST
3.1.2. Phylostratigraphy
3.1.3. ORFan-Finder
3.2. Orphan Genes Databases
3.2.1. NCBI
3.2.2. ORFanID
3.2.3. POGD
3.2.4. TOGD
3.2.5. ORFanage
3.3. Screening of OGs
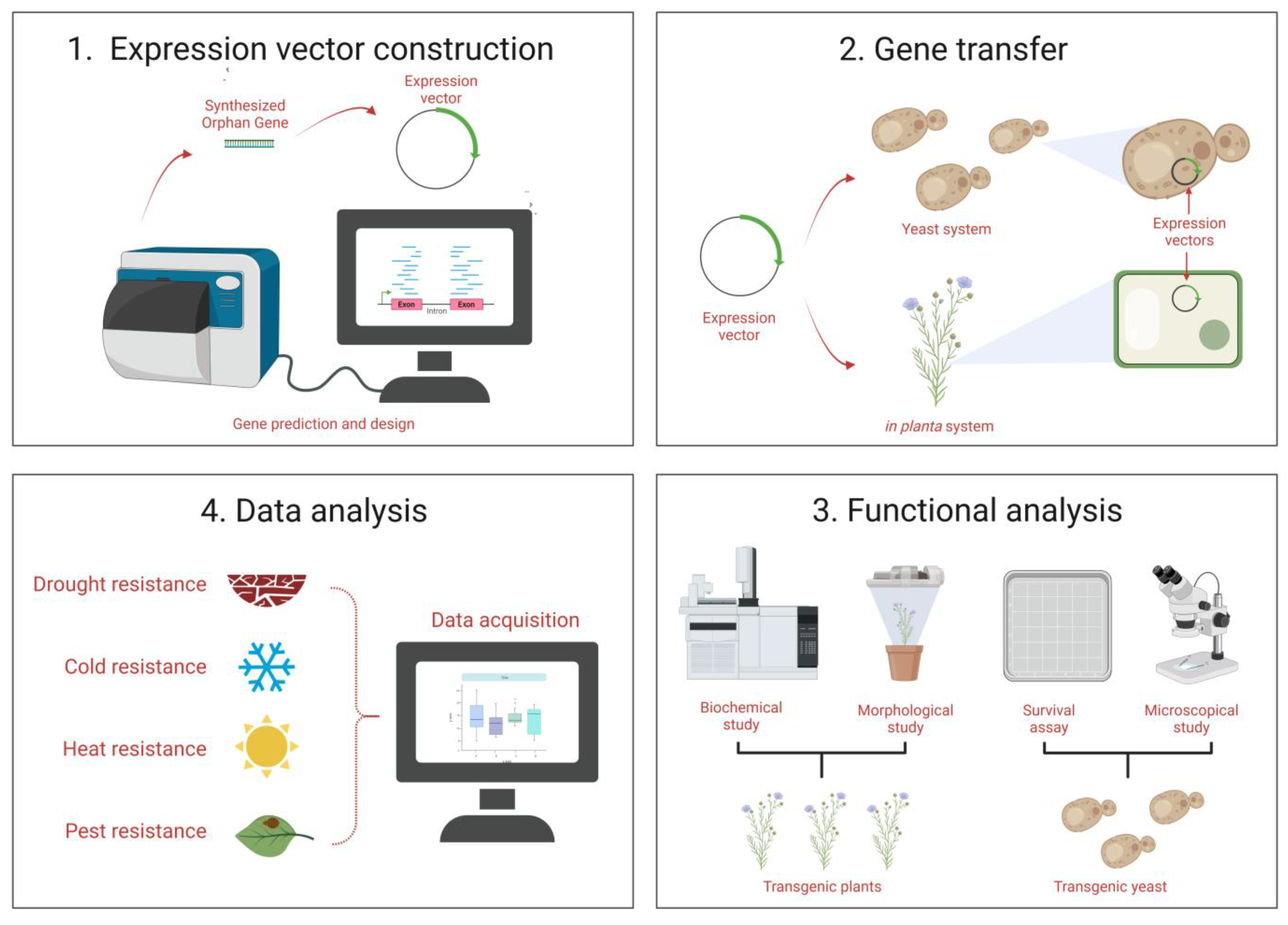
4. Functional Characterization
4.1. Characterization Based on Functionality of OGs
4.2. Genetic Basis and Morphological Level
5. Role of OGs in the Prokaryotic and Viral World
6. Role of Dark Transcriptomics in OG Evolution
7. Future Directions for Orphan Genes Research
7.1. Functionality Prediction
7.2. Comparative Genomics
7.3. Tissue-Specific Expression
7.4. Gene Expression and Knockdown Experiments
7.5. Evolutionary History
8. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tautz, D.; Domazet-Lošo, T. The evolutionary origin of orphan genes. Nat. Rev. Genet. 2011, 12, 692–702. [Google Scholar] [CrossRef] [PubMed]
- Toll-Riera, M.; Bosch, N.; Bellora, N.; Castelo, R.; Armengol, L.; Estivill, X.; Mar Alba, M. Origin of primate orphan genes: A comparative genomics approach. Mol. Biol. Evol. 2009, 26, 603–612. [Google Scholar] [CrossRef] [PubMed]
- Mayer, M.G.; Rödelsperger, C.; Witte, H.; Riebesell, M.; Sommer, R.J. The orphan gene dauerless regulates dauer development and intraspecific competition in nematodes by copy number variation. PLoS Genet. 2015, 11, e1005146. [Google Scholar] [CrossRef] [PubMed]
- Khalturin, K.; Hemmrich, G.; Fraune, S.; Augustin, R.; Bosch, T.C. More than just orphans: Are taxonomically-restricted genes important in evolution? Trends Genet. 2009, 25, 404–413. [Google Scholar] [CrossRef] [PubMed]
- Tanvir, R.; Ping, W.; Sun, J.; Cain, M.; Li, X.; Li, L. AtQQS orphan gene and NtNF-YC4 boost protein accumulation and pest resistance in tobacco (Nicotiana tabacum). Plant Sci. 2022, 317, 111198. [Google Scholar] [CrossRef]
- Neme, R.; Tautz, D. Phylogenetic patterns of emergence of new genes support a model of frequent de novo evolution. BMC Genom. 2013, 14, 1–13. [Google Scholar] [CrossRef]
- Zhang, W.; Gao, Y.; Long, M.; Shen, B. Origination and evolution of orphan genes and de novo genes in the genome of Caenorhabditis elegans. Sci. China Life Sci. 2019, 62, 579–593. [Google Scholar] [CrossRef]
- Ma, D.; Lai, Z.; Ding, Q.; Zhang, K.; Chang, K.; Li, S.; Zhao, Z.; Zhong, F. Identification, characterization and function of orphan genes among the current Cucurbitaceae genomes. Front. Plant Sci. 2022, 13. [Google Scholar] [CrossRef]
- Sun, W.; Zhao, X.-W.; Zhang, Z. Identification and evolution of the orphan genes in the domestic silkworm, Bombyx mori. FEBS Lett. 2015, 589, 2731–2738. [Google Scholar] [CrossRef]
- Arendsee, Z.W.; Li, L.; Wurtele, E.S. Coming of age: Orphan genes in plants. Trends Plant Sci. 2014, 19, 698–708. [Google Scholar] [CrossRef]
- Singh, U.; Wurtele, E.S. Genetic novelty: How new genes are born. eLife 2020, 9, e55136. [Google Scholar] [CrossRef]
- Basile, W.; Sachenkova, O.; Light, S.; Elofsson, A. High GC content causes orphan proteins to be intrinsically disordered. PLoS Comput. Biol. 2017, 13, e1005375. [Google Scholar] [CrossRef]
- Vakirlis, N.; Carvunis, A.-R.; McLysaght, A. Synteny-based analyses indicate that sequence divergence is not the main source of orphan genes. eLife 2020, 9, e53500. [Google Scholar] [CrossRef]
- Fellner, L.; Simon, S.; Scherling, C.; Witting, M.; Schober, S.; Polte, C.; Schmitt-Kopplin, P.; Keim, D.A.; Scherer, S.; Neuhaus, K. Evidence for the recent origin of a bacterial protein-coding, overlapping orphan gene by evolutionary overprinting. BMC Evol. Biol. 2015, 15, 1–14. [Google Scholar] [CrossRef]
- Heinen, T.J.; Staubach, F.; Häming, D.; Tautz, D. Emergence of a new gene from an intergenic region. Curr. Biol. 2009, 19, 1527–1531. [Google Scholar] [CrossRef]
- Ziko, L.; Saqr, A.-H.A.; Ouf, A.; Gimpel, M.; Aziz, R.K.; Neubauer, P.; Siam, R. Antibacterial and anticancer activities of orphan biosynthetic gene clusters from Atlantis II Red Sea brine pool. Microb. Cell Factories 2019, 18, 1–16. [Google Scholar] [CrossRef]
- Luhua, S.; Ciftci-Yilmaz, S.; Harper, J.; Cushman, J.; Mittler, R. Enhanced tolerance to oxidative stress in transgenic Arabidopsis plants expressing proteins of unknown function. Plant Physiol. 2008, 148, 280–292. [Google Scholar] [CrossRef]
- Khalturin, K.; Anton-Erxleben, F.; Sassmann, S.; Wittlieb, J.; Hemmrich, G.; Bosch, T.C.G. A novel gene family controls species-specific morphological traits in Hydra. PLoS Biol. 2008, 6, e278. [Google Scholar] [CrossRef]
- Li, J.; Singh, U.; Bhandary, P.; Campbell, J.; Arendsee, Z.; Seetharam, A.S.; Wurtele, E.S. Foster thy young: Enhanced prediction of orphan genes in assembled genomes. Nucleic Acids Res. 2022, 50, e37. [Google Scholar] [CrossRef]
- Qi, M.; Zheng, W.; Zhao, X.; Hohenstein, J.D.; Kandel, Y.; O’Conner, S.; Wang, Y.; Du, C.; Nettleton, D.; MacIntosh, G.C. QQS orphan gene and its interactor NF-YC 4 reduce susceptibility to pathogens and pests. Plant Biotechnol. J. 2019, 17, 252–263. [Google Scholar] [CrossRef]
- Blanco-Melo, D.; Venkatesh, S.; Bieniasz, P.D. Origins and evolution of tetherin, an orphan antiviral gene. Cell Host Microbe 2016, 20, 189–201. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Zhan, Z.; Li, H.; Dong, X.; Cheng, F.; Piao, Z. Brassica rapa orphan genes largely affect soluble sugar metabolism. Hortic. Res. 2020, 7, 181. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.-M.; Pu, X.-J.; Zhou, S.-Z.; Li, P.; Luo, T.; Chen, Z.-X.; Chen, S.-L.; Liu, L. Orphan gene PpARDT positively involved in drought tolerance potentially by enhancing ABA response in Physcomitrium (Physcomitrella) patens. Plant Sci. 2022, 319, 111222. [Google Scholar] [CrossRef]
- Che, R.; Tong, H.; Shi, B.; Liu, Y.; Fang, S.; Liu, D.; Xiao, Y.; Hu, B.; Liu, L.; Wang, H. Control of grain size and rice yield by GL2-mediated brassinosteroid responses. Nat. Plants 2015, 2, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Chen, S.; Feng, A.; Su, J.; Wang, W.; Feng, J.; Chen, B.; Zhang, M.; Yang, J.; Zeng, L. Xa7, a small orphan gene harboring promoter trap for AvrXa7, leads to the durable resistance to Xanthomonas oryzae pv. oryzae. Rice 2021, 14, 1–16. [Google Scholar] [CrossRef]
- Perochon, A.; Jianguang, J.; Kahla, A.; Arunachalam, C.; Scofield, S.R.; Bowden, S.; Wallington, E.; Doohan, F.M. TaFROG encodes a Pooideae orphan protein that interacts with SnRK1 and enhances resistance to the mycotoxigenic fungus Fusarium graminearum. Plant Physiol. 2015, 169, 2895–2906. [Google Scholar] [CrossRef]
- Duarte, K.E.; Vieira, N.G.; Rego, E.; Martins, P.K.; Ribeiro, A.P.; Cunha, B.A.; Molinari, H.B.C.; Kobayashi, A.K.; de Sousa, C.A.; Marraccini, P. Analysis of the CcUNK8 orphan gene from Coffea canephora in Genetic Transformation of Setaria viridis. In Proceedings of the 11th Solanaceae Conference: Book of Abstracts, Porto Seguro, Brazil, 2–6 November 2014. [Google Scholar]
- Moon, H.; Jeong, A.-R.; Kwon, O.-K.; Park, C.-J. Oryza-Specific Orphan Protein Triggers Enhanced Resistance to Xanthomonas oryzae pv. oryzae in Rice. Front. Plant Sci. 2022, 13, 859375. [Google Scholar] [CrossRef]
- Li, T.-P.; Zhang, L.-W.; Li, Y.-Q.; You, M.-S.; Qian, Z. Functional analysis of the orphan genes Tssor-3 and Tssor-4 in male Plutella xylostella. J. Integr. Agric. 2021, 20, 1880–1888. [Google Scholar] [CrossRef]
- Abu, S.M. Characterization of Lipid Phosphate Phosphatase Genes and in Planta Expressed Orphan Genes in the Rice Blast Fungus. 2015. Available online: https://s-space.snu.ac.kr/bitstream/10371/119491/1/000000026097.pdf (accessed on 8 February 2023).
- Vieira, N.; Duarte, K.; Martins, P.; Ribeiro, A.; Da Cunha, B.; Molinari, H.; Kobayashi, A.; Marraccini, P.; Andrade, A. The coffee gene orphanage: S01P14. In Biotic and Abiotic Stress Tolerance in Plants: The Challenge for the 21st Century: Book of Abstracts of the CIBA 2013; EMBRAPA: Brasília, Brazil, 2013. [Google Scholar]
- Ni, F.; Qi, J.; Hao, Q.; Lyu, B.; Luo, M.-C.; Wang, Y.; Chen, F.; Wang, S.; Zhang, C.; Epstein, L. Wheat Ms2 encodes for an orphan protein that confers male sterility in grass species. Nat. Commun. 2017, 8, 15121. [Google Scholar] [CrossRef]
- Mofatto, L.S.; Carneiro, F.D.A.; Vieira, N.G.; Duarte, K.E.; Vidal, R.O.; Alekcevetch, J.C.; Cotta, M.G.; Verdeil, J.-L.; Lapeyre-Montes, F.; Lartaud, M. Identification of candidate genes for drought tolerance in coffee by high-throughput sequencing in the shoot apex of different Coffea arabica cultivars. BMC Plant Biol. 2016, 16, 1–18. [Google Scholar] [CrossRef]
- Perochon, A.; Váry, Z.; Malla, K.B.; Halford, N.G.; Paul, M.J.; Doohan, F.M. The wheat SnRK1α family and its contribution to Fusarium toxin tolerance. Plant Sci. 2019, 288, 110217. [Google Scholar] [CrossRef]
- Kapulkin, W.J. Retroviral origins of the Caenorhabditis elegans orphan gene F58H7. 5. bioRxiv 2016, 073510. [Google Scholar] [CrossRef]
- Loper, J.; Bruck, D.; Pechy-Tarr, M.; Maurhofer, M.; Keel, C.; Gross, H. Genomics of secondary metabolite production by Pseudomonas fluorescens Pf-5. In Proceedings of the American Society for Microbiology Conference, San Diego, CA, USA, 1 May 2007. [Google Scholar]
- Wang, D.; Calla, B.; Vimolmangkang, S.; Wu, X.; Korban, S.S.; Huber, S.C.; Clough, S.J.; Zhao, Y. The orphan gene ybjN conveys pleiotropic effects on multicellular behavior and survival of Escherichia coli. PLoS ONE 2011, 6, e25293. [Google Scholar] [CrossRef]
- Fellner, L.; Bechtel, N.; Witting, M.A.; Simon, S.; Schmitt-Kopplin, P.; Keim, D.; Scherer, S.; Neuhaus, K. Phenotype of htgA (mbiA), a recently evolved orphan gene of Escherichia coli and Shigella, completely overlapping in antisense to yaaW. FEMS Microbiol. Lett. 2014, 350, 57–64. [Google Scholar] [CrossRef]
- Andaluz, E.; Coque, J.J.R.; Cueva, R.; Larriba, G. Sequencing of a 4.3 kbp region of chromosome 2 of Candida albicans reveals the presence of homologues of SHE9 from Saccharomyces cerevisiae and of bacterial phosphatidylinositol-phospholipase C. Yeast 2001, 18, 711–721. [Google Scholar] [CrossRef]
- Gueuné, H.; Durand, M.-J.; Thouand, G.; DuBow, M.S. The ygaVP genes of Escherichia coli form a tributyltin-inducible operon. Appl. Environ. Microbiol. 2008, 74, 1954–1958. [Google Scholar] [CrossRef]
- Zhuo, L.; Wan, T.-Y.; Pan, Z.; Wang, J.-N.; Sheng, D.-H.; Li, Y.-Z. A Dual-Functional Orphan Response Regulator Negatively Controls the Differential Transcription of Duplicate groEL s and Plays a Global Regulatory Role in Myxococcus. Msystems 2022, 7, e01056-21. [Google Scholar] [CrossRef]
- Gressler, M.; Zaehle, C.; Scherlach, K.; Hertweck, C.; Brock, M. Multifactorial induction of an orphan PKS-NRPS gene cluster in Aspergillus terreus. Chem. Biol. 2011, 18, 198–209. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Sakamoto, M.; Fujii, G.; Tsuiji, H.; Kenetaka, K.; Asaka, M.; Hirohashi, S. Overexpression of orphan G-protein–coupled receptor, Gpr49, in human hepatocellular carcinomas with β-catenin mutations. Hepatology 2003, 37, 528–533. [Google Scholar] [CrossRef]
- Ordonez, D.; Meenagh, A.; Gomez-Lozano, N.; Castano, J.; Middleton, D.; Vilches, C. Duplication, mutation and recombination of the human orphan gene KIR2DS3 contribute to the diversity of KIR haplotypes. Genes Immun. 2008, 9, 431–437. [Google Scholar] [CrossRef]
- Hartig, M.B.; Iuso, A.; Haack, T.; Kmiec, T.; Jurkiewicz, E.; Heim, K.; Roeber, S.; Tarabin, V.; Dusi, S.; Krajewska-Walasek, M. Absence of an orphan mitochondrial protein, c19orf12, causes a distinct clinical subtype of neurodegeneration with brain iron accumulation. Am. J. Hum. Genet. 2011, 89, 543–550. [Google Scholar] [CrossRef] [PubMed]
- Wiles, T.J.; Norton, J.P.; Smith, S.N.; Lewis, A.J.; Mobley, H.L.; Casjens, S.R.; Mulvey, M.A. A phyletically rare gene promotes the niche-specific fitness of an E. coli pathogen during bacteremia. PLoS Pathog. 2013, 9, e1003175. [Google Scholar] [CrossRef] [PubMed]
- Aubourg, S.; Picaud, A.; Kreis, M.; Lecharny, A. Structure and expression of three src2 homologues and a novel subfamily of flavoprotein monooxygenase genes revealed by the analysis of a 25 kb fragment from Arabidopsis thaliana chromosome IV. Gene 1999, 230, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Aubourg, S.; Boudet, N.; Kreis, M.; Lecharny, A. In Arabidopsis thaliana, 1% of the genome codes for a novel protein family unique to plants. Plant Mol. Biol. 2000, 42, 603–613. [Google Scholar] [CrossRef] [PubMed]
- Ulijasz, A.T.; Andes, D.R.; Glasner, J.D.; Weisblum, B. Regulation of iron transport in Streptococcus pneumoniae by RitR, an orphan response regulator. J. Bacteriol. 2004, 186, 8123–8136. [Google Scholar] [CrossRef]
- Jiang, M.; Li, X.; Dong, X.; Zu, Y.; Zhan, Z.; Piao, Z.; Lang, H. Research Advances and Prospects of Orphan Genes in Plants. Front. Plant Sci. 2022, 13, 947129. [Google Scholar] [CrossRef]
- Wilson, G.; Bertrand, N.; Patel, Y.; Hughes, J.; Feil, E.; Field, D. Orphans as taxonomically restricted and ecologically important genes. Microbiology 2005, 151, 2499–2501. [Google Scholar] [CrossRef]
- Daubin, V.; Ochman, H. Bacterial genomes as new gene homes: The genealogy of ORFans in E. coli. Genome Res. 2004, 14, 1036–1042. [Google Scholar] [CrossRef]
- Becker, B.; Hoef-Emden, K.; Melkonian, M. Chlamydial genes shed light on the evolution of photoautotrophic eukaryotes. BMC Evol. Biol. 2008, 8, 1–18. [Google Scholar] [CrossRef]
- Schmid, K.J.; Aquadro, C.F. The evolutionary analysis of “orphans” from the Drosophila genome identifies rapidly diverging and incorrectly annotated genes. Genetics 2001, 159, 589–598. [Google Scholar] [CrossRef]
- Conant, G.C.; Wolfe, K.H. Turning a hobby into a job: How duplicated genes find new functions. Nat. Rev. Genet. 2008, 9, 938–950. [Google Scholar] [CrossRef]
- Lynch, M.; Katju, V. The altered evolutionary trajectories of gene duplicates. TRENDS Genet. 2004, 20, 544–549. [Google Scholar] [CrossRef]
- Albà, M.M.; Castresana, J. On homology searches by protein Blast and the characterization of the age of genes. BMC Evol. Biol. 2007, 7, 544–549. [Google Scholar] [CrossRef]
- Kaessmann, H. Origins, evolution, and phenotypic impact of new genes. Genome Res. 2010, 20, 1313–1326. [Google Scholar] [CrossRef]
- Jin, G.H.; Zhou, Y.L.; Yang, H.; Hu, Y.T.; Shi, Y.; Li, L.; Siddique, A.N.; Liu, C.N.; Zhu, A.D.; Zhang, C.J. Genetic innovations: Transposable element recruitment and de novo formation lead to the birth of orphan genes in the rice genome. J. Syst. Evol. 2021, 59, 341–351. [Google Scholar] [CrossRef]
- Nekrutenko, A.; Wadhawan, S.; Goetting-Minesky, P.; Makova, K.D. Oscillating evolution of a mammalian locus with overlapping reading frames: An XLαs/ALEX relay. PLoS Genet. 2005, 1, e18. [Google Scholar] [CrossRef]
- Cai, J.; Zhao, R.; Jiang, H.; Wang, W. De novo origination of a new protein-coding gene in Saccharomyces cerevisiae. Genetics 2008, 179, 487–496. [Google Scholar] [CrossRef]
- Knowles, D.G.; McLysaght, A. Recent de novo origin of human protein-coding genes. Genome Res. 2009, 19, 1752–1759. [Google Scholar] [CrossRef]
- Panchy, N.; Lehti-Shiu, M.; Shiu, S.-H. Evolution of gene duplication in plants. Plant Physiol. 2016, 171, 2294–2316. [Google Scholar] [CrossRef]
- Jiang, C.; Hei, R.; Yang, Y.; Zhang, S.; Wang, Q.; Wang, W.; Zhang, Q.; Yan, M.; Zhu, G.; Huang, P. An orphan protein of Fusarium graminearum modulates host immunity by mediating proteasomal degradation of TaSnRK1α. Nat. Commun. 2020, 11, 1–13. [Google Scholar] [CrossRef]
- Alba, M.M.; Castresana, J. Inverse relationship between evolutionary rate and age of mammalian genes. Mol. Biol. Evol. 2005, 22, 598–606. [Google Scholar] [CrossRef] [PubMed]
- Desjardins, C.A.; Gadau, J.; Lopez, J.A.; Niehuis, O.; Avery, A.R.; Loehlin, D.W.; Richards, S.; Colbourne, J.K.; Werren, J.H. Fine-scale mapping of the Nasonia genome to chromosomes using a high-density genotyping microarray. G3 Genes Genomes Genet. 2013, 3, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Yuan, Y.; Tao, Y.; Jia, H.; Ma, Z. Identification, characterization and expression analysis of lineage-specific genes within Triticeae. Genomics 2020, 112, 1343–1350. [Google Scholar] [CrossRef] [PubMed]
- Elbasir, A.; Moovarkumudalvan, B.; Kunji, K.; Kolatkar, P.R.; Mall, R.; Bensmail, H. DeepCrystal: A deep learning framework for sequence-based protein crystallization prediction. Bioinformatics 2019, 35, 2216–2225. [Google Scholar] [CrossRef]
- Lin, Y.; Afshar, S.; Rajadhyaksha, A.M.; Potash, J.B.; Han, S. A machine learning approach to predicting autism risk genes: Validation of known genes and discovery of new candidates. Front. Genet. 2020, 11, 500064. [Google Scholar] [CrossRef]
- Domazet-Lošo, T.; Brajković, J.; Tautz, D. A phylostratigraphy approach to uncover the genomic history of major adaptations in metazoan lineages. Trends Genet. 2007, 23, 533–539. [Google Scholar] [CrossRef]
- O’Toole, Á.N.; Hurst, L.D.; McLysaght, A. Faster evolving primate genes are more likely to duplicate. Mol. Biol. Evol. 2018, 35, 107–118. [Google Scholar] [CrossRef]
- Carvunis, A.-R.; Rolland, T.; Wapinski, I.; Calderwood, M.A.; Yildirim, M.A.; Simonis, N.; Charloteaux, B.; Hidalgo, C.A.; Barbette, J.; Santhanam, B. Proto-genes and de novo gene birth. Nature 2012, 487, 370–374. [Google Scholar] [CrossRef]
- Donoghue, M.T.; Keshavaiah, C.; Swamidatta, S.H.; Spillane, C. Evolutionary origins of Brassicaceae specific genes in Arabidopsis thaliana. BMC Evol. Biol. 2011, 11, 1–23. [Google Scholar] [CrossRef]
- Arendsee, Z.; Li, J.; Singh, U.; Bhandary, P.; Seetharam, A.; Wurtele, E.S. fagin: Synteny-based phylostratigraphy and finer classification of young genes. BMC Bioinform. 2019, 20, 440. [Google Scholar] [CrossRef]
- Casola, C. From de novo to “de nono”: The majority of novel protein-coding genes identified with phylostratigraphy are old genes or recent duplicates. Genome Biol. Evol. 2018, 10, 2906–2918. [Google Scholar] [CrossRef]
- Ekstrom, A.; Yin, Y. ORFanFinder: Automated identification of taxonomically restricted orphan genes. Bioinformatics 2016, 32, 2053–2055. [Google Scholar] [CrossRef][Green Version]
- Gao, Q.; Jin, X.; Xia, E.; Wu, X.; Gu, L.; Yan, H.; Xia, Y.; Li, S. Identification of orphan genes in unbalanced datasets based on ensemble learning. Front. Genet. 2020, 11, 820. [Google Scholar] [CrossRef]
- Li, J.; Singh, U.; Arendsee, Z.; Wurtele, E.S. Landscape of the dark transcriptome revealed through re-mining massive RNA-Seq data. Front. Genet. 2021, 12, 722981. [Google Scholar] [CrossRef]
- Benson, D.A.; Karsch-Mizrachi, I.; Lipman, D.J.; Ostell, J.; Wheeler, D.L. GenBank: Update. Nucleic Acids Res. 2004, 32, D23–D26. [Google Scholar] [CrossRef]
- Entwistle, S.; Li, X.; Yin, Y. Orphan genes shared by pathogenic genomes are more associated with bacterial pathogenicity. Msystems 2019, 4, e00290-18. [Google Scholar] [CrossRef]
- Gunasekera, R.S.; Raja, K.K.; Hewapathirana, S.; Galbadage, T.; Tundrea, E.; Gunasekera, V.; Nelson, P.A. ORFanID: A Web-Based Search Engine for the Discovery and Identification of Orphan and Taxonomically Restricted Genes. bioRxiv 2022, 2022-02. [Google Scholar] [CrossRef]
- Reinhardt, J.A.; Jones, C.D. Two rapidly evolving genes contribute to male fitness in Drosophila. J. Mol. Evol. 2013, 77, 246–259. [Google Scholar] [CrossRef][Green Version]
- Yao, C.; Yan, H.; Zhang, X.; Wang, R. A database for orphan genes in Poaceae. Exp. Ther. Med. 2017, 14, 2917–2924. [Google Scholar] [CrossRef][Green Version]
- Gao, Q.; Yan, H.; Xia, E.; Zhang, S.; Li, S. TOGD: A database of orphan genes in Triticum aestivum. Int. J. Agric. Biol. 2019, 22, 961–966. [Google Scholar]
- Siew, N.; Azaria, Y.; Fischer, D. The ORFanage: An ORFan database. Nucleic Acids Res. 2004, 32, D281–D283. [Google Scholar] [CrossRef] [PubMed]
- Hubley, R.; Finn, R.D.; Clements, J.; Eddy, S.R.; Jones, T.A.; Bao, W.; Smit, A.F.; Wheeler, T.J. The Dfam database of repetitive DNA families. Nucleic Acids Res. 2016, 44, D81–D89. [Google Scholar] [CrossRef] [PubMed]
- Apweiler, R.; Bairoch, A.; Wu, C.H.; Barker, W.C.; Boeckmann, B.; Ferro, S.; Gasteiger, E.; Huang, H.; Lopez, R.; Magrane, M. UniProt: The universal protein knowledgebase. Nucleic Acids Res. 2004, 32, D115–D119. [Google Scholar] [CrossRef] [PubMed]
- Kriventseva, E.V.; Tegenfeldt, F.; Petty, T.J.; Waterhouse, R.M.; Simao, F.A.; Pozdnyakov, I.A.; Ioannidis, P.; Zdobnov, E.M. OrthoDB v8: Update of the hierarchical catalog of orthologs and the underlying free software. Nucleic Acids Res. 2015, 43, D250–D256. [Google Scholar] [CrossRef] [PubMed]
- Emrich, S.J.; Barbazuk, W.B.; Li, L.; Schnable, P.S. Gene discovery and annotation using LCM-454 transcriptome sequencing. Genome Res. 2007, 17, 69–73. [Google Scholar] [CrossRef]
- Neuhaus, K.; Landstorfer, R.; Fellner, L.; Simon, S.; Schafferhans, A.; Goldberg, T.; Marx, H.; Ozoline, O.N.; Rost, B.; Kuster, B. Translatomics combined with transcriptomics and proteomics reveals novel functional, recently evolved orphan genes in Escherichia coli O157: H7 (EHEC). BMC Genom. 2016, 17, 1–16. [Google Scholar] [CrossRef]
- Prabh, N.; Rödelsperger, C. Are orphan genes protein-coding, prediction artifacts, or non-coding RNAs? BMC Bioinform. 2016, 17, 1–13. [Google Scholar] [CrossRef]
- Gaillochet, C.; Develtere, W.; Jacobs, T.B. CRISPR screens in plants: Approaches, guidelines, and future prospects. Plant Cell 2021, 33, 794–813. [Google Scholar] [CrossRef]
- Li, D.; Dong, Y.; Jiang, Y.; Jiang, H.; Cai, J.; Wang, W. A de novo originated gene depresses budding yeast mating pathway and is repressed by the protein encoded by its antisense strand. Cell Res. 2010, 20, 408–420. [Google Scholar] [CrossRef]
- Lopez, J.; Mukhtar, M.S. Mapping protein-protein interaction using high-throughput yeast 2-hybrid. In Plant Genomics; Humana Press: New York, NY, USA, 2017; pp. 217–230. [Google Scholar]
- Palmieri, N.; Kosiol, C.; Schlötterer, C. The life cycle of Drosophila orphan genes. eLife 2014, 3, e01311. [Google Scholar] [CrossRef]
- Armengaud, J. A perfect genome annotation is within reach with the proteomics and genomics alliance. Curr. Opin. Microbiol. 2009, 12, 292–300. [Google Scholar] [CrossRef]
- Dujon, B. The yeast genome project: What did we learn? Trends Genet. 1996, 12, 263–270. [Google Scholar] [CrossRef]
- Ohno, S. Evolution by Gene Duplication; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2013. [Google Scholar]
- Luhua, S.; Hegie, A.; Suzuki, N.; Shulaev, E.; Luo, X.; Cenariu, D.; Ma, V.; Kao, S.; Lim, J.; Gunay, M.B. Linking genes of unknown function with abiotic stress responses by high-throughput phenotype screening. Physiol. Plant. 2013, 148, 322–333. [Google Scholar] [CrossRef]
- Guo, Y.L. Gene family evolution in green plants with emphasis on the origination and evolution of a rabidopsis thaliana genes. Plant J. 2013, 73, 941–951. [Google Scholar] [CrossRef]
- Jiang, M.; Zhang, Y.; Yang, X.; Li, X.; Lang, H. Brassica rapa orphan gene BR1 delays flowering time in Arabidopsis. Front. Plant Sci. 2023, 14, 697. [Google Scholar] [CrossRef]
- Bolouri Moghaddam, M.R.; Van den Ende, W. Sweet immunity in the plant circadian regulatory network. J. Exp. Bot. 2013, 64, 1439–1449. [Google Scholar] [CrossRef]
- Luo, D.; Huguet-Tapia, J.C.; Raborn, R.T.; White, F.F.; Brendel, V.P.; Yang, B. The Xa7 resistance gene guards the rice susceptibility gene SWEET14 against exploitation by the bacterial blight pathogen. Plant Commun. 2021, 2, 100164. [Google Scholar] [CrossRef]
- Xiao, W.; Liu, H.; Li, Y.; Li, X.; Xu, C.; Long, M.; Wang, S. A rice gene of de novo origin negatively regulates pathogen-induced defense response. PLoS ONE 2009, 4, e4603. [Google Scholar] [CrossRef]
- Doares, S.H.; Narváez-Vásquez, J.; Conconi, A.; Ryan, C.A. Salicylic acid inhibits synthesis of proteinase inhibitors in tomato leaves induced by systemin and jasmonic acid. Plant Physiol. 1995, 108, 1741–1746. [Google Scholar] [CrossRef]
- Liu, J.; Fakhar, A.Z.; Pajerowska-Mukhtar, K.M.; Mukhtar, M.S. A TIReless battle: TIR domains in plant–pathogen interactions. Trends Plant Sci. 2022, 27, 426–429. [Google Scholar] [CrossRef]
- Mukhtar, M.S.; McCormack, M.E.; Argueso, C.T.; Pajerowska-Mukhtar, K.M. Pathogen tactics to manipulate plant cell death. Curr. Biol. 2016, 26, R608–R619. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Dong, X.; Lang, H.; Pang, W.; Zhan, Z.; Li, X.; Piao, Z. Mining of Brassica-specific genes (BSGs) and their induction in different developmental stages and under Plasmodiophora brassicae stress in Brassica rapa. Int. J. Mol. Sci. 2018, 19, 2064. [Google Scholar] [CrossRef] [PubMed]
- Muhammad Aslam, M.; Waseem, M.; Jakada, B.H.; Okal, E.J.; Lei, Z.; Saqib, H.S.A.; Yuan, W.; Xu, W.; Zhang, Q. Mechanisms of Abscisic Acid-Mediated Drought Stress Responses in Plants. Int. J. Mol. Sci. 2022, 23, 1084. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, A.; Adamo, M.; Crozet, P.; Margalha, L.; Confraria, A.; Martinho, C.; Elias, A.; Rabissi, A.; Lumbreras, V.; González-Guzmán, M. ABI1 and PP2CA phosphatases are negative regulators of Snf1-related protein kinase1 signaling in Arabidopsis. Plant Cell 2013, 25, 3871–3884. [Google Scholar] [CrossRef]
- Clamp, M.; Fry, B.; Kamal, M.; Xie, X.; Cuff, J.; Lin, M.F.; Kellis, M.; Lindblad-Toh, K.; Lander, E.S. Distinguishing protein-coding and noncoding genes in the human genome. Proc. Natl. Acad. Sci. USA 2007, 104, 19428–19433. [Google Scholar] [CrossRef]
- Cheng, H.; Aleman, T.S.; Cideciyan, A.V.; Khanna, R.; Jacobson, S.G.; Swaroop, A. In vivo function of the orphan nuclear receptor NR2E3 in establishing photoreceptor identity during mammalian retinal development. Hum. Mol. Genet. 2006, 15, 2588–2602. [Google Scholar] [CrossRef]
- Haider, N.B.; Demarco, P.; Nystuen, A.M.; Huang, X.; Smith, R.S.; Mccall, M.A.; Naggert, J.K.; Nishina, P.M. The transcription factor Nr2e3 functions in retinal progenitors to suppress cone cell generation. Vis. Neurosci. 2006, 23, 917–929. [Google Scholar] [CrossRef]
- Cheng, H.; Khan, N.W.; Roger, J.E.; Swaroop, A. Excess cones in the retinal degeneration rd7 mouse, caused by the loss of function of orphan nuclear receptor Nr2e3, originate from early-born photoreceptor precursors. Hum. Mol. Genet. 2011, 20, 4102–4115. [Google Scholar] [CrossRef]
- Li, C.-Y.; Zhang, Y.; Wang, Z.; Zhang, Y.; Cao, C.; Zhang, P.-W.; Lu, S.-J.; Li, X.-M.; Yu, Q.; Zheng, X. A human-specific de novo protein-coding gene associated with human brain functions. PLoS Comput. Biol. 2010, 6, e1000734. [Google Scholar] [CrossRef]
- Ehrlich, A.T.; Maroteaux, G.; Robe, A.; Venteo, L.; Nasseef, M.T.; van Kempen, L.C.; Mechawar, N.; Turecki, G.; Darcq, E.; Kieffer, B.L. Expression map of 78 brain-expressed mouse orphan GPCRs provides a translational resource for neuropsychiatric research. Commun. Biol. 2018, 1, 102. [Google Scholar] [CrossRef]
- Alamri, M.A.; Tahir ul Qamar, M.; Alabbas, A.B.; Alqahtani, S.M.; Alossaimi, M.A.; Azam, S.; Hashmi, M.H.; Rajoka, M.S.R. Molecular and Structural Analysis of Specific Mutations from Saudi Isolates of SARS-CoV-2 RNA-Dependent RNA Polymerase and their Implications on Protein Structure and Drug–Protein Binding. Molecules 2022, 27, 6475. [Google Scholar] [CrossRef]
- McCorvy, J.D.; Butler, K.V.; Kelly, B.; Rechsteiner, K.; Karpiak, J.; Betz, R.M.; Kormos, B.L.; Shoichet, B.K.; Dror, R.O.; Jin, J. Structure-inspired design of β-arrestin-biased ligands for aminergic GPCRs. Nat. Chem. Biol. 2018, 14, 126–134. [Google Scholar] [CrossRef]
- Roth, B.L.; Kroeze, W.K. Integrated approaches for genome-wide interrogation of the druggable non-olfactory G protein-coupled receptor superfamily. J. Biol. Chem. 2015, 290, 19471–19477. [Google Scholar] [CrossRef]
- Gallego-García, A.; Monera-Girona, A.J.; Pajares-Martínez, E.; Bastida-Martínez, E.; Pérez-Castaño, R.; Iniesta, A.A.; Fontes, M.; Padmanabhan, S.; Elías-Arnanz, M. A bacterial light response reveals an orphan desaturase for human plasmalogen synthesis. Science 2019, 366, 128–132. [Google Scholar] [CrossRef]
- Yan, P.; Eng, O.C.; Yu, C.J. A review on the expression and metabolic features of orphan human cytochrome P450 2S1 (CYP2S1). Curr. Drug Metab. 2018, 19, 917–929. [Google Scholar] [CrossRef]
- McCotter, S.W.; Horianopoulos, L.C.; Kronstad, J.W. Regulation of the fungal secretome. Curr. Genet. 2016, 62, 533–545. [Google Scholar] [CrossRef]
- Pellegrin, C.; Morin, E.; Martin, F.M.; Veneault-Fourrey, C. Comparative analysis of secretomes from ectomycorrhizal fungi with an emphasis on small-secreted proteins. Front. Microbiol. 2015, 6, 1278. [Google Scholar] [CrossRef]
- Alfaro, M.; Oguiza, J.A.; Ramírez, L.; Pisabarro, A.G. Comparative analysis of secretomes in basidiomycete fungi. J. Proteom. 2014, 102, 28–43. [Google Scholar] [CrossRef]
- Kim, K.-T.; Jeon, J.; Choi, J.; Cheong, K.; Song, H.; Choi, G.; Kang, S.; Lee, Y.-H. Kingdom-wide analysis of fungal small secreted proteins (SSPs) reveals their potential role in host association. Front. Plant Sci. 2016, 7, 186. [Google Scholar] [CrossRef]
- Voigt, C.A.; Schäfer, W.; Salomon, S. A secreted lipase of Fusarium graminearum is a virulence factor required for infection of cereals. Plant J. 2005, 42, 364–375. [Google Scholar] [CrossRef]
- Dong, L.; Wang, F.; Liu, T.; Dong, Z.; Li, A.; Jing, R.; Mao, L.; Li, Y.; Liu, X.; Zhang, K. Natural variation of TaGASR7-A1 affects grain length in common wheat under multiple cultivation conditions. Mol. Breed. 2014, 34, 937–947. [Google Scholar] [CrossRef]
- Wang, Z.; Li, J.; Chen, S.; Heng, Y.; Chen, Z.; Yang, J.; Zhou, K.; Pei, J.; He, H.; Deng, X.W. Poaceae-specific MS1 encodes a phospholipid-binding protein for male fertility in bread wheat. Proc. Natl. Acad. Sci. USA 2017, 114, 12614–12619. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.-S.; Li, Q.-F.; Zhang, C.-Q.; Zhang, C.; Yang, Q.-Q.; Pan, L.-X.; Ren, X.-Y.; Lu, J.; Gu, M.-H.; Liu, Q.-Q. GS9 acts as a transcriptional activator to regulate rice grain shape and appearance quality. Nat. Commun. 2018, 9, 1240. [Google Scholar] [CrossRef] [PubMed]
- Kasuga, T.; Mannhaupt, G.; Glass, N.L. Relationship between phylogenetic distribution and genomic features in Neurospora crassa. PLoS ONE 2009, 4, e5286. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, Y.; Kasuga, T.; Lopez-Giraldez, F.; Zhang, Y.; Zhang, Z.; Diaz, R.; Dong, C.; Sil, A.; Trail, F. Orphan genes are clustered with allorecognition loci and may be involved in incompatibility and speciation in Neurospora. bioRxiv 2022, 2022-06. [Google Scholar]
- Milde, S.; Hemmrich, G.; Anton-Erxleben, F.; Khalturin, K.; Wittlieb, J.; Bosch, T.C. Characterization of taxonomically restricted genes in a phylum-restricted cell type. Genome Biol. 2009, 10, 1–16. [Google Scholar] [CrossRef]
- Johnson, B.R. Taxonomically restricted genes are fundamental to biology and evolution. Front. Genet. 2018, 9, 407. [Google Scholar] [CrossRef]
- Kumar, A.; Gates, P.B.; Czarkwiani, A.; Brockes, J.P. An orphan gene is necessary for preaxial digit formation during salamander limb development. Nat. Commun. 2015, 6, 8684. [Google Scholar] [CrossRef]
- Andersson, D.I.; Jerlström-Hultqvist, J.; Näsvall, J. Evolution of new functions de novo and from preexisting genes. Cold Spring Harb. Perspect. Biol. 2015, 7, a017996. [Google Scholar] [CrossRef]
- Wissler, L.; Gadau, J.; Simola, D.F.; Helmkampf, M.; Bornberg-Bauer, E. Mechanisms and dynamics of orphan gene emergence in insect genomes. Genome Biol. Evol. 2013, 5, 439–455. [Google Scholar] [CrossRef]
- Hsiao, W.W.L.; Ung, K.; Aeschliman, D.; Bryan, J.; Finlay, B.B.; Brinkman, F.S.L. Evidence of a large novel gene pool associated with prokaryotic genomic islands. PLoS Genet. 2005, 1, e62. [Google Scholar] [CrossRef]
- Ho Sui, S.J.; Fedynak, A.; Hsiao, W.W.; Langille, M.G.; Brinkman, F.S. The association of virulence factors with genomic islands. PLoS ONE 2009, 4, e8094. [Google Scholar] [CrossRef] [PubMed]
- Beres, S.B.; Carroll, R.K.; Shea, P.R.; Sitkiewicz, I.; Martinez-Gutierrez, J.C.; Low, D.E.; McGeer, A.; Willey, B.M.; Green, K.; Tyrrell, G.J. Molecular complexity of successive bacterial epidemics deconvoluted by comparative pathogenomics. Proc. Natl. Acad. Sci. USA 2010, 107, 4371–4376. [Google Scholar] [CrossRef]
- Brüssow, H.; Hendrix, R.W. Phage genomics: Small is beautiful. Cell 2002, 108, 13–16. [Google Scholar] [CrossRef]
- Yin, Y.; Fischer, D. On the origin of microbial ORFans: Quantifying the strength of the evidence for viral lateral transfer. BMC Evol. Biol. 2006, 6, 1–7. [Google Scholar] [CrossRef]
- Koonin, E.V.; Dolja, V.V. Evolution of complexity in the viral world: The dawn of a new vision. Virus Res. 2006, 117, 1–4. [Google Scholar] [CrossRef]
- Kaur, N.; Chen, W.; Zheng, Y.; Hasegawa, D.K.; Ling, K.-S.; Fei, Z.; Wintermantel, W.M. Transcriptome analysis of the whitefly, Bemisia tabaci MEAM1 during feeding on tomato infected with the crinivirus, Tomato chlorosis virus, identifies a temporal shift in gene expression and differential regulation of novel orphan genes. BMC Genom. 2017, 18, 1–20. [Google Scholar] [CrossRef]
- Yin, Y.; Fischer, D. Identification and investigation of ORFans in the viral world. BMC Genom. 2008, 9, 1–10. [Google Scholar] [CrossRef]
- Siew, N.; Fischer, D. Twenty thousand ORFan microbial protein families for the biologist? Structure 2003, 11, 7–9. [Google Scholar] [CrossRef]
- Kuchibhatla, D.B.; Sherman, W.A.; Chung, B.Y.; Cook, S.; Schneider, G.; Eisenhaber, B.; Karlin, D.G. Powerful sequence similarity search methods and in-depth manual analyses can identify remote homologs in many apparently “orphan” viral proteins. J. Virol. 2014, 88, 10–20. [Google Scholar] [CrossRef]
- Kallies, R.; Kopp, A.; Zirkel, F.; Estrada, A.; Gillespie, T.R.; Drosten, C.; Junglen, S. Genetic characterization of goutanap virus, a novel virus related to negeviruses, cileviruses and higreviruses. Viruses 2014, 6, 4346–4357. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Zheng, W.; Zhu, Y.; Ye, H.; Tang, B.; Arendsee, Z.W.; Jones, D.; Li, R.; Ortiz, D.; Zhao, X. QQS orphan gene regulates carbon and nitrogen partitioning across species via NF-YC interactions. Proc. Natl. Acad. Sci. USA 2015, 112, 14734–14739. [Google Scholar] [CrossRef]
- Wu, Z.; Gao, S.; Diamond, C.; Kajigaya, S.; Chen, J.; Shi, R.; Palmer, C.; Hsu, A.P.; Calvo, K.R.; Hickstein, D.D. Sequencing of RNA in single cells reveals a distinct transcriptome signature of hematopoiesis in GATA2 deficiency. Blood Adv. 2020, 4, 2702–2716. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.; Mishra, B.; Mehmood, A.; Athar, M.; Mukhtar, M.S. Integrative network biology framework elucidates molecular mechanisms of SARS-CoV-2 pathogenesis. Iscience 2020, 23, 101526. [Google Scholar] [CrossRef] [PubMed]
- Mishra, B.; Kumar, N.; Mukhtar, M.S. Network biology to uncover functional and structural properties of the plant immune system. Curr. Opin. Plant Biol. 2021, 62, 102057. [Google Scholar] [CrossRef]
- Gao, R.; Bai, S.; Henderson, Y.C.; Lin, Y.; Schalck, A.; Yan, Y.; Kumar, T.; Hu, M.; Sei, E.; Davis, A. Delineating copy number and clonal substructure in human tumors from single-cell transcriptomes. Nat. Biotechnol. 2021, 39, 599–608. [Google Scholar] [CrossRef]
- Raza, A.; Tabassum, J.; Fakhar, A.Z.; Sharif, R.; Chen, H.; Zhang, C.; Ju, L.; Fotopoulos, V.; Siddique, K.H.; Singh, R.K. Smart reprograming of plants against salinity stress using modern biotechnological tools. Crit. Rev. Biotechnol. 2022, 1–28. [Google Scholar] [CrossRef]
- Gleba, Y.Y.; Tusé, D.; Giritch, A. Plant viral vectors for delivery by Agrobacterium. Plant Viral Vectors 2013, 155–192. [Google Scholar]
- Buckholz, R.G.; Gleeson, M.A. Yeast systems for the commercial production of heterologous proteins. Bio/Technology 1991, 9, 1067–1072. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Orphan Gene | Corresponding Host | Function/s | Reference |
|---|---|---|---|
| Arabidopsis thaliana | Reduces susceptibility to pathogens and pests | [20] |
| Nematodes | Inhibitor of Dauer development | [3] |
| Vertebrates | Antiviral activity | [21] |
| Brassica rapa | Primary metabolism | [22] |
| Physcomitrium patens | Drought tolerance | [23] |
| Soybean | Modulates carbon and nitrogen allocation | [24] |
| Oryza sativa | Executor resistance gene against Xanthomonas oryzae pv. oryzae (Xoo) | [25] |
| Wheat | Biotic stress resistance | [26] |
| C. canephora | Protects plants against drought | [27] |
| Oryza | Triggers enhance resistance to Xanthomonas oryzae pv. oryzae in rice | [28] |
| Plutella xylostella | Role in male fertility in P. xylostella | [29] |
| Magnaporthe oryzae | Species-specific adaptive processes | [30] |
| Coffee | Involved in abiotic and biotic stress responses | [31] |
| Wheat | For recurrent selection and hybrid seed production in wheat | [32] |
| Coffee | Drought tolerance in coffee | [33] |
| Wheat | Contributes positively to wheat tolerance of DON | [34] |
| C. elegans | Involved as RNA intermediate | [35] |
| Pseudomonas fluorescens | Produces six secondary metabolites | [36] |
| Escherichia coli | Involved in E. coli’s central metabolism | [14] |
| Escherichia coli | Regulating bacterial multicellular behavior and metabolism | [37] |
| Escherichia coli, Shigella spp. | Responsible for lineage-specific adaptations | [38] |
| Saccharomyces cerevisiae | Compromises cell growth | [39] |
| Escherichia coli | Auto-regulated and TBT-inducible repressor | [40] |
| Myxococcus | Negative regulatory role in M. xanthus | [41] |
| Aspergillus terreus | Monitoring conditions for secondary metabolite production | [42] |
| Human | New therapeutic target in the treatment of HCC | [43] |
| Human | Contributes to the diversity of KIR haplotypes | [44] |
| Human | Causes a distinct clinical subtype of neurodegeneration with brain iron accumulation | [45] |
| Escherichia coli | Key role in the virulence of ExPEC in zebrafish embryos | [46] |
| Arabidopsis thaliana | Unknown function, showing tissue-specific expression | [47] |
| Arabidopsis thaliana | Codes for a novel protein family unique to plants | [48] |
| Atlantis II Red Sea brine pool | Confers antibiotic and anticancer effects | [16] |
| Streptococcus pneumoniae | Maintains iron homeostasis in S. pneumonia | [49] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fakhar, A.Z.; Liu, J.; Pajerowska-Mukhtar, K.M.; Mukhtar, M.S. The Lost and Found: Unraveling the Functions of Orphan Genes. J. Dev. Biol. 2023, 11, 27. https://doi.org/10.3390/jdb11020027
Fakhar AZ, Liu J, Pajerowska-Mukhtar KM, Mukhtar MS. The Lost and Found: Unraveling the Functions of Orphan Genes. Journal of Developmental Biology. 2023; 11(2):27. https://doi.org/10.3390/jdb11020027
Chicago/Turabian StyleFakhar, Ali Zeeshan, Jinbao Liu, Karolina M. Pajerowska-Mukhtar, and M. Shahid Mukhtar. 2023. "The Lost and Found: Unraveling the Functions of Orphan Genes" Journal of Developmental Biology 11, no. 2: 27. https://doi.org/10.3390/jdb11020027
APA StyleFakhar, A. Z., Liu, J., Pajerowska-Mukhtar, K. M., & Mukhtar, M. S. (2023). The Lost and Found: Unraveling the Functions of Orphan Genes. Journal of Developmental Biology, 11(2), 27. https://doi.org/10.3390/jdb11020027