
Genetic Interaction of Thm2 and Thm1 Shapes Postnatal Craniofacial Bone

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Mouse Generation
2.2. Scanning, Reconstruction, and Landmark Placement
2.3. Measurements
2.4. Primary Osteoblast Generation and Differentiation Assay
2.5. SAG Treatment and qPCR
2.6. Statistics
3. Results
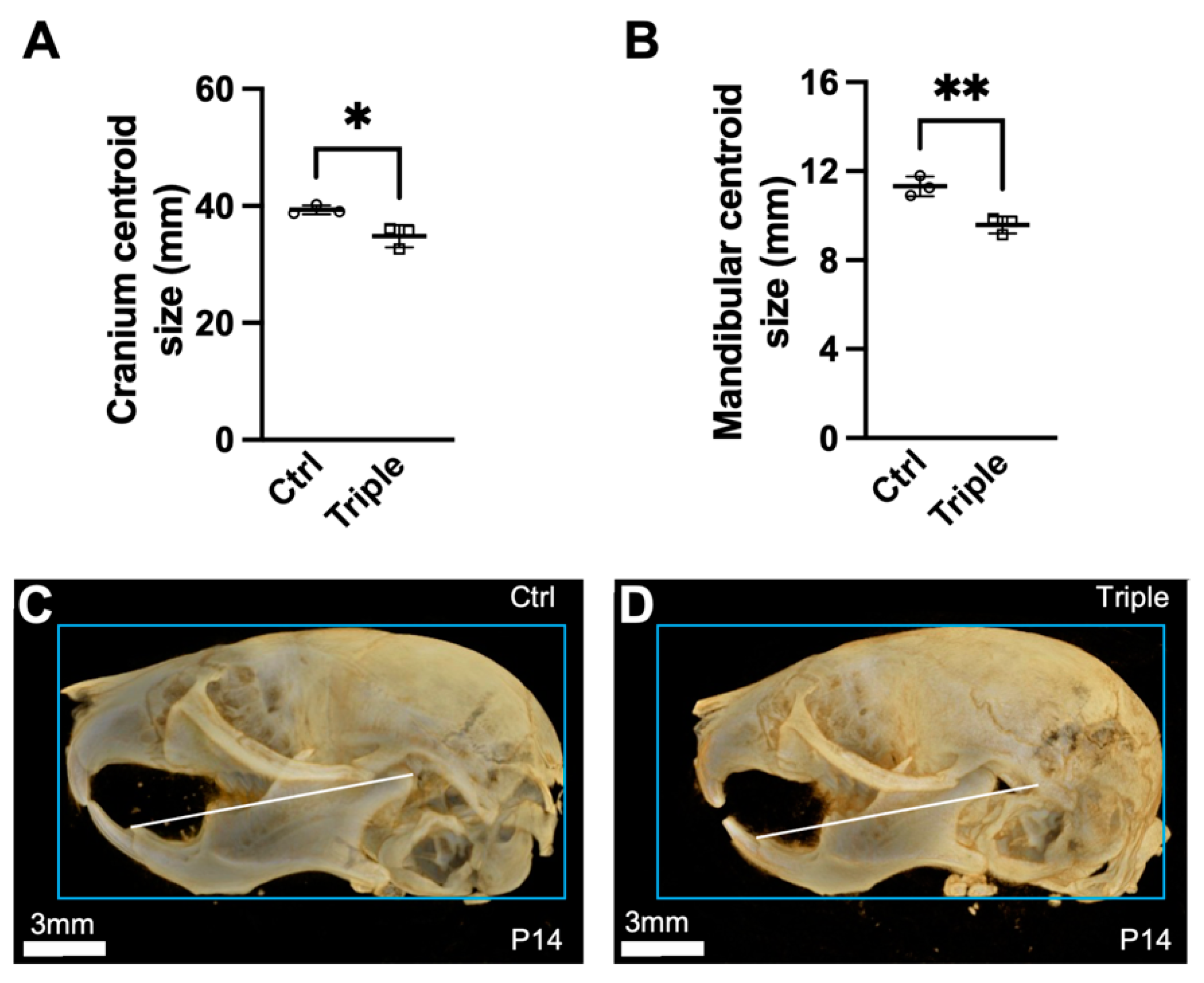
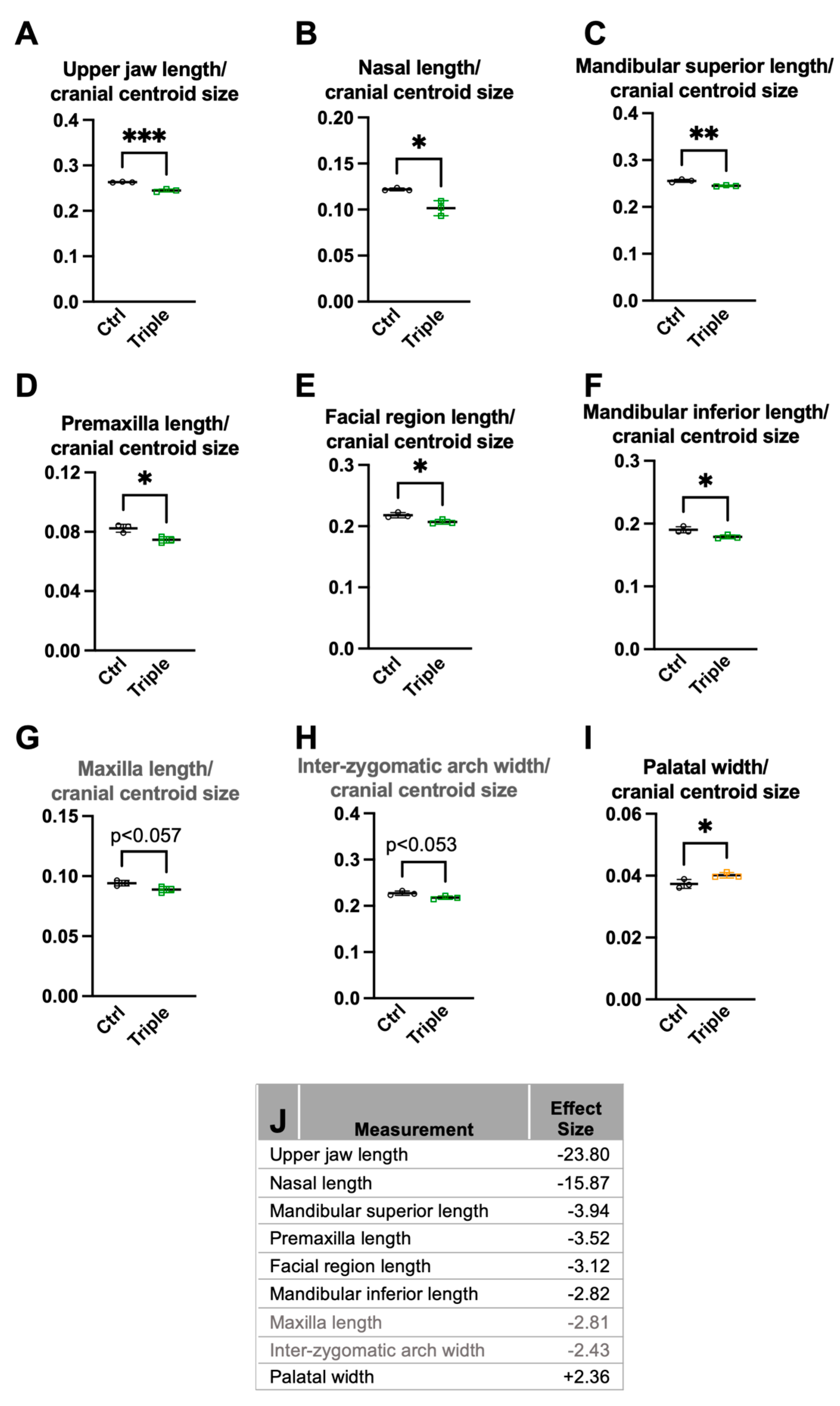
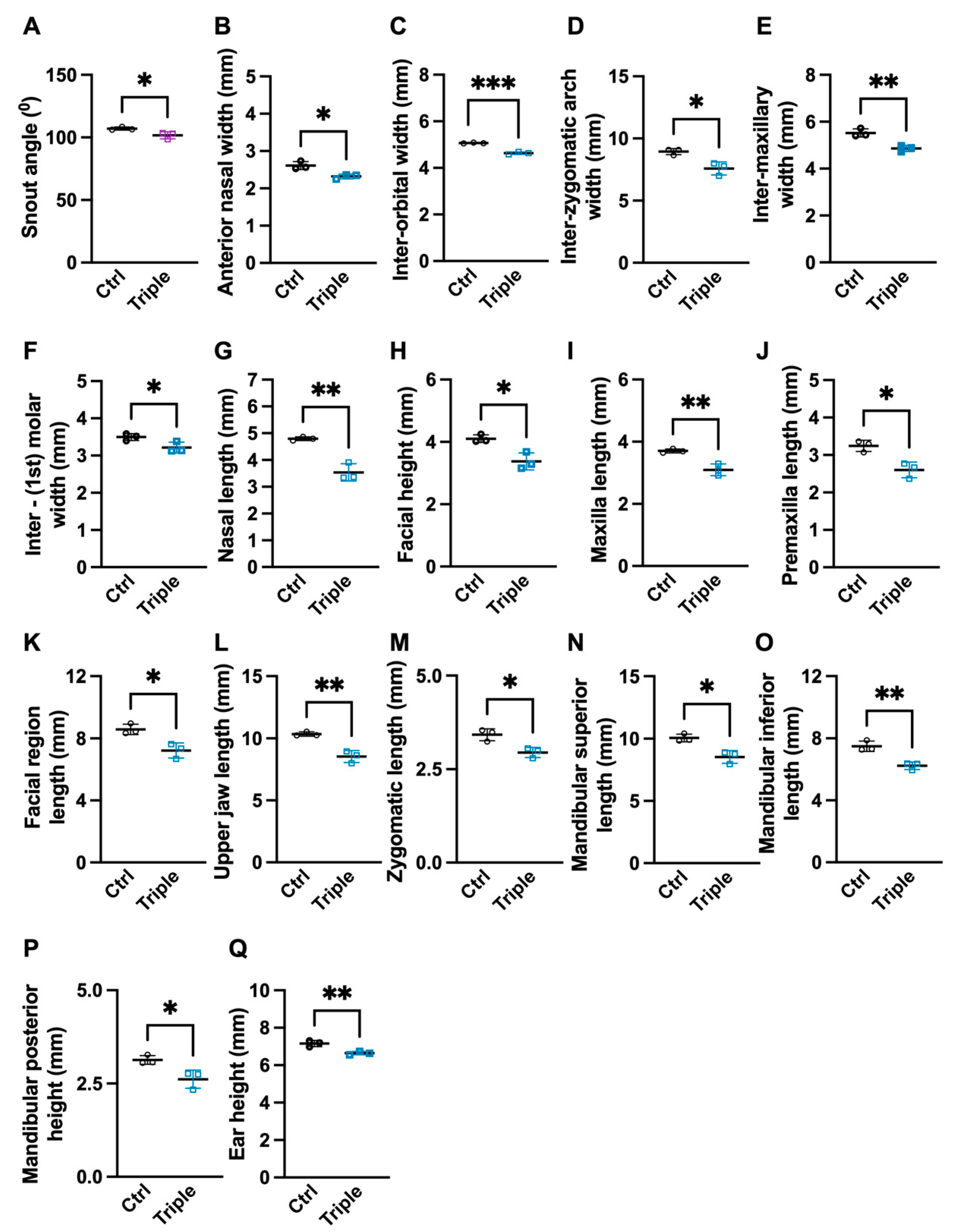
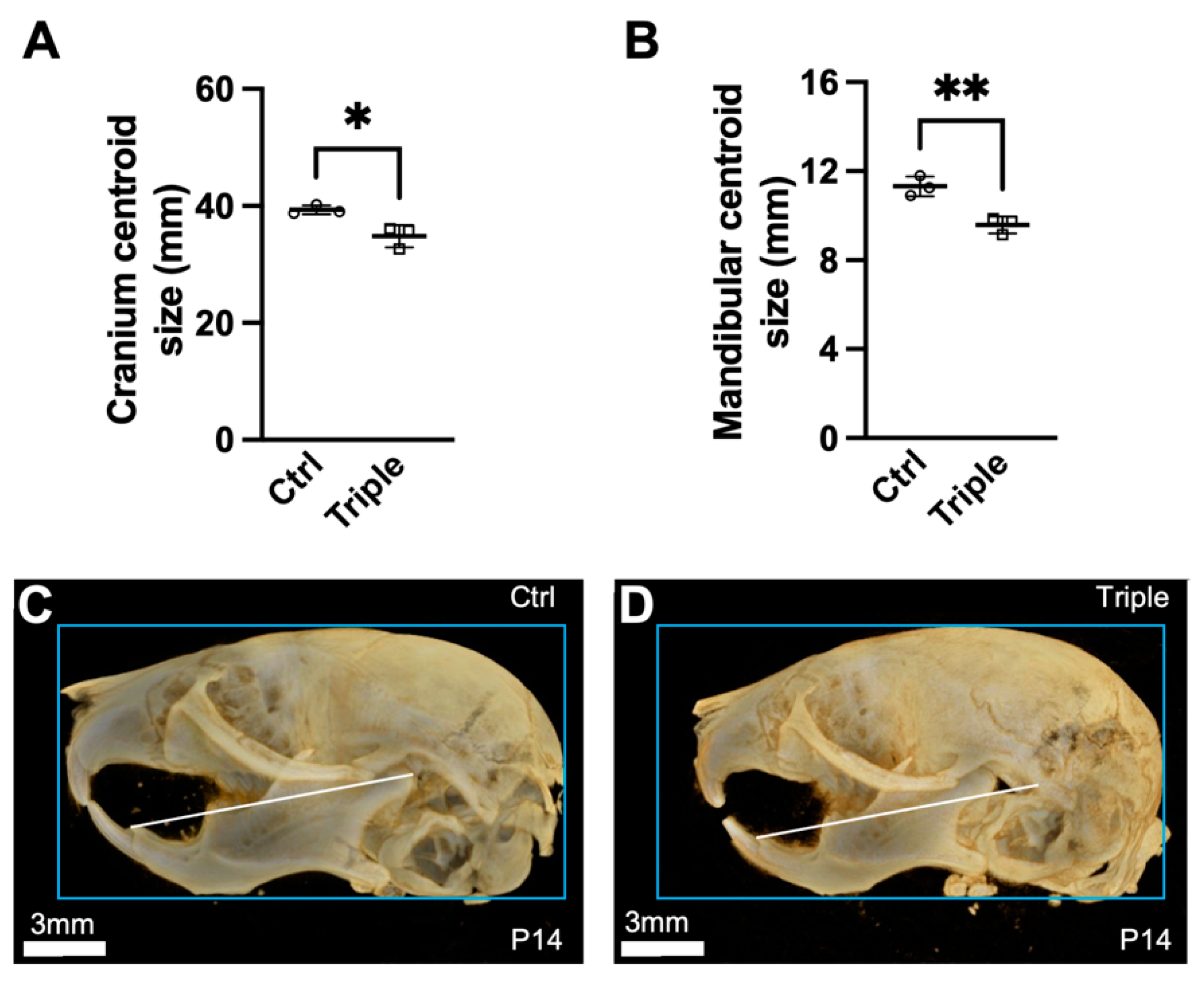
3.1. Triple Allele Mutants Have Reduced Skull and Mandibular Measurements
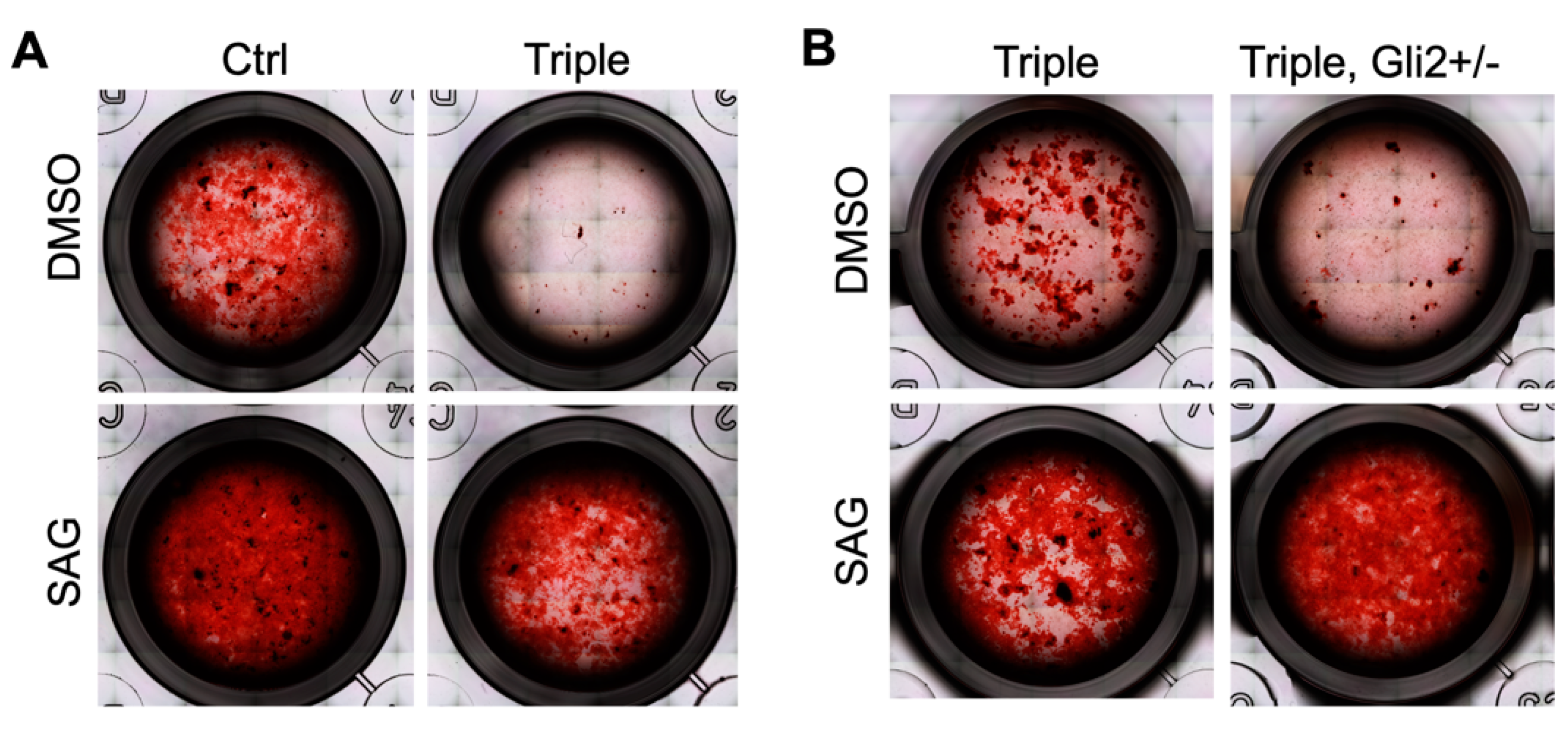
3.2. Hedgehog Agonist Enhances In Vitro Bone Formation of Calvarial-Derived Thm2−/−; Thm1aln/+ Osteoblasts
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Krakow, D.; Rimoin, D.L. The skeletal dysplasias. Genet. Med. 2010, 12, 327–341. [Google Scholar] [CrossRef] [Green Version]
- Poyner, S.E.; Bradshaw, W.T. Jeune Syndrome: Considerations for Management of Asphyxiating Thoracic Dystrophy. Neonatal Netw. 2013, 32, 342–352. [Google Scholar] [CrossRef]
- Xiao, Z.; Zhang, S.; Mahlios, J.; Zhou, G.; Magenheimer, B.S.; Guo, D.; Dallas, S.L.; Maser, R.; Calvet, J.P.; Bonewald, L.; et al. Cilia-like Structures and Polycystin-1 in Osteoblasts/Osteocytes and Associated Abnormalities in Skeletogenesis and Runx2 Expression. J. Biol. Chem. 2006, 281, 30884–30895. [Google Scholar] [CrossRef] [Green Version]
- Song, B.; Haycraft, C.J.; Seo, H.-S.; Yoder, B.K.; Serra, R. Development of the post-natal growth plate requires intraflagellar transport proteins. Dev. Biol. 2007, 305, 202–216. [Google Scholar] [CrossRef] [Green Version]
- Tobin, J.L.; Beales, P.L. The nonmotile ciliopathies. Genet. Med. 2009, 11, 386–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McInerney-Leo, A.M.; Schmidts, M.; Cortés, C.R.; Leo, P.J.; Gener, B.; Courtney, A.D.; Gardiner, B.; Harris, J.A.; Lu, Y.; Marshall, M.; et al. Short-rib polydactyly and Jeune syndromes are caused by mutations in WDR60. Am. J. Hum. Genet. 2013, 93, 515–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cortes, C.; Metzis, V.; Wicking, C. Unmasking the ciliopathies: Craniofacial defects and the primary cilium. Wiley Interdiscip. Rev. Dev. Biol. 2015, 4, 637–653. [Google Scholar] [CrossRef]
- Huangfu, D.; Liu, A.; Rakeman, A.S.; Murcia, N.S.; Niswander, L.; Anderson, K.V. Hedgehog signalling in the mouse requires intraflagellar transport proteins. Nature 2003, 426, 83–87. [Google Scholar] [CrossRef] [PubMed]
- Eggenschwiler, J.T.; Anderson, K.V. Cilia and Developmental Signaling. Annu. Rev. Cell Dev. Biol. 2007, 23, 345–373. [Google Scholar] [CrossRef] [Green Version]
- Corbit, K.C.; Shyer, A.E.; Dowdle, W.E.; Gaulden, J.; Singla, V.; Reiter, J.F. Kif3a constrains beta-catenin-dependent Wnt signalling through dual ciliary and non-ciliary mechanisms. Nat. Cell Biol. 2008, 10, 70–76. [Google Scholar] [CrossRef]
- Schneider, L.; Clement, C.A.; Teilmann, S.C.; Pazour, G.J.; Hoffmann, E.K.; Satir, P.; Christensen, S.T. PDGFRalphaalpha signaling is regulated through the primary cilium in fibroblasts. Curr. Biol. 2005, 15, 1861–1866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clement, C.A.; Ajbro, K.D.; Koefoed, K.; Vestergaard, M.L.; Veland, I.R.; de Jesus, M.P.; Pedersen, L.B.; Benmerah, A.; Andersen, C.Y.; Larsen, L.A.; et al. TGF-beta signaling is associated with endocytosis at the pocket region of the primary cilium. Cell Rep. 2013, 3, 1806–1814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ezratty, E.J.; Stokes, N.; Chai, S.; Shah, A.S.; Williams, S.; Fuchs, E. A Role for the Primary Cilium in Notch Signaling and Epidermal Differentiation during Skin Development. Cell 2011, 145, 1129–1141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miceli, C.; Roccio, F.; Penalva-Mousset, L.; Burtin, M.; Leroy, C.; Nemazanyy, I.; Kuperwasser, N.; Pontoglio, M.; Friedlander, G.; Morel, E.; et al. The primary cilium and lipophagy translate mechanical forces to direct metabolic adaptation of kidney epithelial cells. Nat. Cell Biol. 2020, 22, 1091–1102. [Google Scholar] [CrossRef]
- Wheway, G.; Nazlamova, L.; Hancock, J.T. Signaling through the Primary Cilium. Front. Cell Dev. Biol. 2018, 6, 8. [Google Scholar] [CrossRef]
- Goetz, S.C.; Ocbina, P.J.; Anderson, K.V. The Primary Cilium as a Hedgehog Signal Transduction Machine. Methods Cell Biol. 2009, 94, 199–222. [Google Scholar] [CrossRef] [Green Version]
- Veland, I.R.; Awan, A.; Pedersen, L.; Yoder, B.K.; Christensen, S.T. Primary Cilia and Signaling Pathways in Mammalian Development, Health and Disease. Nephron Physiol. 2009, 111, 39–53. [Google Scholar] [CrossRef] [Green Version]
- Valente, E.M.; Rosti, R.O.; Gibbs, E.; Gleeson, J.G. Primary cilia in neurodevelopmental disorders. Nat. Rev. Neurol. 2014, 10, 27–36. [Google Scholar] [CrossRef] [Green Version]
- Christensen, S.T.; Ott, C.M. A Ciliary Signaling Switch. Science 2007, 317, 330–331. [Google Scholar] [CrossRef]
- Herron, B.J.; Lu, W.; Rao, C.; Liu, S.; Peters, H.; Bronson, R.T.; Justice, M.J.; McDonald, J.D.; Beier, D.R. Efficient generation and mapping of recessive developmental mutations using ENU mutagenesis. Nat. Genet. 2002, 30, 185–189. [Google Scholar] [CrossRef]
- Miller, K.A.; Ah-Cann, C.J.; Welfare, M.F.; Tan, T.Y.; Pope, K.; Caruana, G.; Freckmann, M.-L.; Savarirayan, R.; Bertram, J.F.; Dobbie, M.S.; et al. Cauli: A Mouse Strain with an Ift140 Mutation That Results in a Skeletal Ciliopathy Modelling Jeune Syndrome. PLoS Genet. 2013, 9, e1003746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashe, A.; Butterfield, N.C.; Town, L.; Courtney, A.D.; Cooper, A.N.; Ferguson, C.; Barry, R.; Olsson, F.; Liem, K.F.; Parton, R.G.; et al. Mutations in mouse Ift144 model the craniofacial, limb and rib defects in skeletal ciliopathies. Hum. Mol. Genet. 2012, 21, 1808–1823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rix, S.; Calmont, A.; Scambler, P.J.; Beales, P.L. An Ift80 mouse model of short rib polydactyly syndromes shows defects in hedgehog signalling without loss or malformation of cilia. Hum. Mol. Genet. 2011, 20, 1306–1314. [Google Scholar] [CrossRef] [Green Version]
- Haycraft, C.J.; Zhang, Q.; Song, B.; Jackson, W.; Detloff, P.J.; Serra, R.; Yoder, B.K. Intraflagellar transport is essential for endochondral bone formation. Development 2007, 134, 307–316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murcia, N.S.; Richards, W.G.; Yoder, B.K.; Mucenski, M.L.; Dunlap, J.R.; Woychik, R.P. The Oak Ridge Polycystic Kidney (orpk) disease gene is required for left-right axis determination. Development 2000, 127, 2347–2355. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Murcia, N.S.; Chittenden, L.R.; Richards, W.G.; Michaud, E.J.; Woychik, R.P.; Yoder, B.K. Loss of theTg737 protein results in skeletal patterning defects. Dev. Dyn. 2003, 227, 78–90. [Google Scholar] [CrossRef]
- Schock, E.N.; Struve, J.N.; Chang, C.-F.; Williams, T.J.; Snedeker, J.; Attia, A.C.; Stottmann, R.W.; Brugmann, S.A. A tissue-specific role for intraflagellar transport genes during craniofacial development. PLoS ONE 2017, 12, e0174206. [Google Scholar] [CrossRef]
- Tran, P.V.; Haycraft, C.J.; Besschetnova, T.Y.; Turbe-Doan, A.; Stottmann, R.W.; Herron, B.J.; Chesebro, A.L.; Qiu, H.; Scherz, P.J.; Shah, J.V.; et al. THM1 negatively modulates mouse sonic hedgehog signal transduction and affects retrograde intraflagellar transport in cilia. Nat. Genet. 2008, 40, 403–410. [Google Scholar] [CrossRef] [Green Version]
- Davis, E.E.; Zhang, Q.; Liu, Q.; Diplas, B.H.; Davey, L.M.; Hartley, J.; Stoetzel, C.; Szymanska, K.; Ramaswami, G.; Logan, C.V. TTC21B contributes both causal and modifying alleles across the ciliopathy spectrum. Nat. Genet. 2011, 43, 189–196. [Google Scholar] [CrossRef]
- Allard, B.A.; Wang, W.; Pottorf, T.S.; Mumtaz, H.; Jack, B.M.; Wang, H.H.; Silva, L.M.; Jacobs, D.T.; Wang, J.; Bumann, E.E.; et al. Thm2 interacts with paralog, Thm1, and sensitizes to Hedgehog signaling in postnatal skeletogenesis. Cell Mol. Life Sci. 2021, 78, 3743–3762. [Google Scholar] [CrossRef]
- Vora, S.R.; Camci, E.D.; Cox, T.C. Postnatal Ontogeny of the Cranial Base and Craniofacial Skeleton in Male C57BL/6J Mice: A Reference Standard for Quantitative Analysis. Front. Physiol. 2015, 6, 417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klingenberg, C.P. Size, shape, and form: Concepts of allometry in geometric morphometrics. Dev. Genes Evol. 2016, 226, 113–137. [Google Scholar] [CrossRef] [PubMed]
- Dallas, S.L.; Miyazono, K.; Skerry, T.; Mundy, G.R.; Bonewald, L.F. Dual role for the latent transforming growth factor-beta binding protein in storage of latent TGF-beta in the extracellular matrix and as a structural matrix protein. J. Cell Biol. 1995, 131, 539–549. [Google Scholar] [CrossRef]
- Kim, J.H.; Seong, S.; Kim, K.; Kim, I.; Jeong, B.C.; Kim, N. Downregulation of Runx2 by 1,25-Dihydroxyvitamin D(3) Induces the Transdifferentiation of Osteoblasts to Adipocytes. Int. J. Mol. Sci. 2016, 17, 770. [Google Scholar] [CrossRef] [Green Version]
- Berthet, E.; Chen, C.; Butcher, K.; Schneider, R.; Alliston, T.; Amirtharajah, M. Smad3 binds scleraxis and mohawk and regulates tendon matrix organization. J. Orthop. Res. 2013, 31, 1475–1483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palazuelos, J.; Klingener, M.; Aguirre, A. TGFbeta signaling regulates the timing of CNS myelination by modulating oligodendrocyte progenitor cell cycle exit through SMAD3/4/FoxO1/Sp1. J. Neurosci. 2014, 34, 7917–7930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Estrada, K.D.; Retting, K.N.; Chin, A.M.; Lyons, K.M. Smad6 is essential to limit BMP signaling during cartilage development. J. Bone Miner. Res. 2011, 26, 2498–2510. [Google Scholar] [CrossRef] [Green Version]
- de Jonge, H.J.; Fehrmann, R.S.N.; de Bont, E.S.J.M.; Hofstra, R.M.W.; Gerbens, F.; Kamps, W.A.; de Vries, E.G.E.; van der Zee, A.G.J.; te Meerman, G.J.; ter Elst, A. Evidence based selection of housekeeping genes. PLoS ONE 2007, 2, e898. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Andre, P.; Ye, L.; Yang, Y.-Z. The Hedgehog signalling pathway in bone formation. Int. J. Oral Sci. 2015, 7, 73–79. [Google Scholar] [CrossRef]
- Biosse Duplan, M.; Komla-Ebri, D.; Heuzé, Y.; Estibals, V.; Gaudas, E.; Kaci, N.; Benoist-Lasselin, C.; Zerah, M.; Kramer, I.; Kneissel, M.; et al. Meckel’s and condylar cartilages anomalies in achondroplasia result in defective development and growth of the mandible. Hum. Mol. Genet. 2016, 25, 2997–3010. [Google Scholar] [CrossRef] [Green Version]
- Long, F.; Chung, U.-I.; Ohba, S.; McMahon, J.; Kronenberg, H.M.; McMahon, A.P. Ihh signaling is directly required for the osteoblast lineage in the endochondral skeleton. Development 2004, 131, 1309–1318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, X.; Cao, J.; He, X.; Serra, R.; Qu, J.; Cao, X.; Yang, S. Ciliary IFT80 balances canonical versus non-canonical hedgehog signalling for osteoblast differentiation. Nat. Commun. 2016, 7, 11024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimoyama, A.; Wada, M.; Ikeda, F.; Hata, K.; Matsubara, T.; Nifuji, A.; Noda, M.; Amano, K.; Yamaguchi, A.; Nishimura, R.; et al. Ihh/Gli2 Signaling Promotes Osteoblast Differentiation by Regulating Runx2 Expression and Function. Mol. Biol. Cell 2007, 18, 2411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paese, C.L.B.; Brooks, E.C.; Aarnio-Peterson, M.; Brugmann, S.A. Ciliopathic micrognathia is caused by aberrant skeletal differentiation and remodeling. Development 2021, 148, dev194175. [Google Scholar] [CrossRef] [PubMed]
- Ealba, E.L.; Jheon, A.H.; Hall, J.; Curantz, C.; Butcher, K.D.; Schneider, R.A. Neural crest-mediated bone resorption is a determinant of species-specific jaw length. Dev. Biol. 2015, 408, 151–163. [Google Scholar] [CrossRef]
- Esenlik, E.; Şener, E.H.; Yılmaz, H.H.; Malas, M.A. Cephalometric investigation of craniomaxillofacial structures during the prenatal period: A cadaver study. Am. J. Orthod. Dentofac. Orthop. 2014, 145, 217–227. [Google Scholar] [CrossRef]
- Yoon, Y.-J.; Perkiomaki, M.R.; Tallents, R.H.; Barillas, I.; Herrera-Guido, R.; Fong, C.-T.; Kyrkanides, S. Transverse Craniofacial Features and Their Genetic Predisposition in Families with Nonsyndromic Unilateral Cleft Lip and Palate. Cleft Palate Craniofac. J. 2004, 41, 256–261. [Google Scholar] [CrossRef]
- Vergato, L.A.; Doerfler, R.J.; Mooney, M.P.; Siegel, M.I. Mouse palatal width growth rates as an "at risk" factor in the development of cleft palate induced by hypervitaminosis A. J. Craniofac. Genet. Dev. Biol. 1997, 17, 204–210. [Google Scholar]
- Siegel, M.I.; Mooney, M.P. Palatal width growth rates as the genetic determinant of cleft palate induced by vitamin A. J. Craniofac. Genet. Dev. Biol. Suppl. 1986, 2, 187–191. [Google Scholar]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bumann, E.E.; Hahn Leat, P.; Wang, H.H.; Hufft-Martinez, B.M.; Wang, W.; Tran, P.V. Genetic Interaction of Thm2 and Thm1 Shapes Postnatal Craniofacial Bone. J. Dev. Biol. 2022, 10, 17. https://doi.org/10.3390/jdb10020017
Bumann EE, Hahn Leat P, Wang HH, Hufft-Martinez BM, Wang W, Tran PV. Genetic Interaction of Thm2 and Thm1 Shapes Postnatal Craniofacial Bone. Journal of Developmental Biology. 2022; 10(2):17. https://doi.org/10.3390/jdb10020017
Chicago/Turabian StyleBumann, Erin E., Portia Hahn Leat, Henry H. Wang, Brittany M. Hufft-Martinez, Wei Wang, and Pamela V. Tran. 2022. "Genetic Interaction of Thm2 and Thm1 Shapes Postnatal Craniofacial Bone" Journal of Developmental Biology 10, no. 2: 17. https://doi.org/10.3390/jdb10020017
APA StyleBumann, E. E., Hahn Leat, P., Wang, H. H., Hufft-Martinez, B. M., Wang, W., & Tran, P. V. (2022). Genetic Interaction of Thm2 and Thm1 Shapes Postnatal Craniofacial Bone. Journal of Developmental Biology, 10(2), 17. https://doi.org/10.3390/jdb10020017