
Craniofacial Phenotypes and Genetics of DiGeorge Syndrome


Abstract
1. Introduction
2. Craniofacial Phenotypes of Patients with DGS/VCFS
3. Genetics of DGS/VCFS
3.1. TBX1 Gene
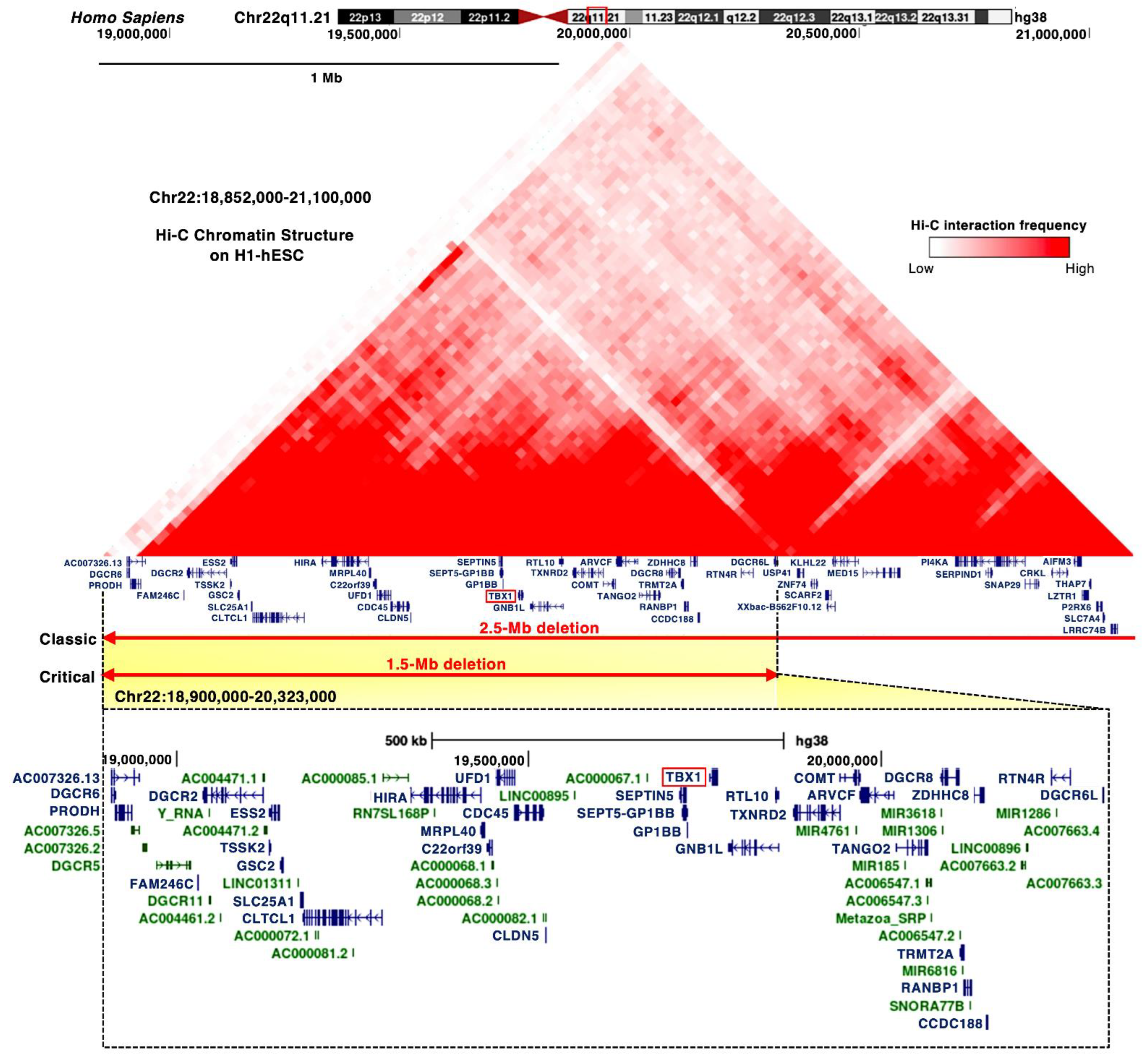
3.2. DiGeorge Syndrome Critical Region (DGCR)
3.3. MicroRNAs
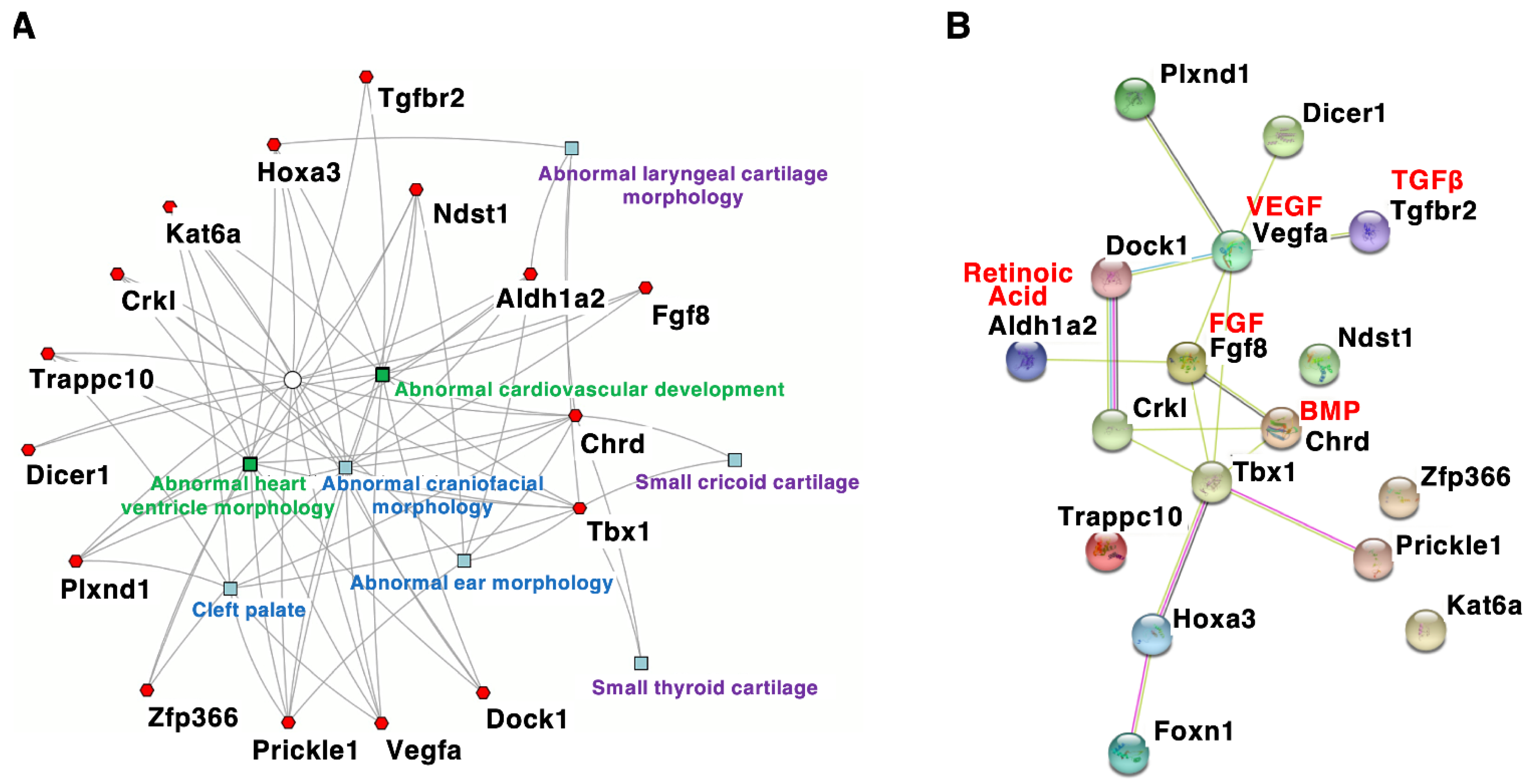
4. Craniofacial Phenotypes of DGS/VCFS Mouse Models
4.1. Tbx1
4.1.1. Cleft Palate
4.1.2. Abnormalities in Craniofacial Bones
4.1.3. Abnormalities in the Cranial Base and Cervical Spine
4.1.4. Dental Anomalies
4.1.5. Muscle Hypotonia
4.2. Chordin (Chrd) and Transforming Growth Factor, Beta Receptor II (Tgfbr2)
4.3. Vascular Endothelial Growth Factor A (Vegfa)
4.4. Fibroblast Growth Factor 8 (Fgf8) and FGF Receptor 2 (Fgfr2)
4.5. CRK like Proto-Oncogene, Adaptor Protein (Crkl)
4.6. Aldehyde Dehydrogenase Family 1, Subfamily A2 (Aldh1a2/Raldh2)
4.7. Homeobox A3 (Hoxa3)
4.8. Kat6a/Moz/Myst3 (Lysine Acetyltransferase 6A) and Epigenetic Modifiers
4.9. Sonic Hedgehog (Shh)
4.10. Paired-like Homeodomain Transcription Factor 2 (Pitx2)
5. Discussion
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Tézenas Du Montcel, S.; Mendizabai, H.; Aymé, S.; Lévy, A.; Philip, N. Prevalence of 22q11 microdeletion. J. Med. Genet. 1996, 33, 719. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Rivera, E.; Liu, Y.P.; Verbitsky, M.; Anderson, B.R.; Capone, V.P.; Otto, E.A.; Yan, Z.; Mitrotti, A.; Martino, J.; Steers, N.J.; et al. Genetic Drivers of Kidney Defects in the DiGeorge Syndrome. N. Engl. J. Med. 2017, 376, 742–754. [Google Scholar] [CrossRef] [PubMed]
- Du, Q.; de la Morena, M.T.; van Oers, N.S.C. The Genetics and Epigenetics of 22q11.2 Deletion Syndrome. Front. Genet. 2019, 10, 1365. [Google Scholar] [CrossRef] [PubMed]
- Gorlin, R.J.; Cohen, M.M., Jr.; Hennekam, R.C.M. Syndromes of the Head and the Neck; Oxford University Press: New York, NY, USA, 2001; pp. 850–853. [Google Scholar]
- Yagi, H.; Furutani, Y.; Hamada, H.; Sasaki, T.; Asakawa, S.; Minoshima, S.; Ichida, F.; Joo, K.; Kimura, M.; Imamura, S.; et al. Role of TBX1 in human del22q11.2 syndrome. Lancet 2003, 362, 1366–1373. [Google Scholar] [CrossRef]
- Jerome, L.A.; Papaioannou, V.E. DiGeorge syndrome phenotype in mice mutant for the T-box gene, Tbx1. Nat. Genet. 2001, 27, 286–291. [Google Scholar] [CrossRef]
- Lindsay, E.A.; Vitelli, F.; Su, H.; Morishima, M.; Huynh, T.; Pramparo, T.; Jurecic, V.; Ogunrinu, G.; Sutherland, H.F.; Scambler, P.J.; et al. Tbx1 haploinsufficieny in the DiGeorge syndrome region causes aortic arch defects in mice. Nature 2001, 410, 97–101. [Google Scholar] [CrossRef]
- Merscher, S.; Funke, B.; Epstein, J.A.; Heyer, J.; Puech, A.; Lu, M.M.; Xavier, R.J.; Demay, M.B.; Russell, R.G.; Factor, S.; et al. TBX1 is responsible for cardiovascular defects in velo-cardio-facial/DiGeorge syndrome. Cell 2001, 104, 619–629. [Google Scholar] [CrossRef]
- Hu, T.; Yamagishi, H.; Maeda, J.; McAnally, J.; Yamagishi, C.; Srivastava, D. Tbx1 regulates fibroblast growth factors in the anterior heart field through a reinforcing autoregulatory loop involving forkhead transcription factors. Development 2004, 131, 5491–5502. [Google Scholar] [CrossRef]
- Liao, J.; Kochilas, L.; Nowotschin, S.; Arnold, J.S.; Aggarwal, V.S.; Epstein, J.A.; Brown, M.C.; Adams, J.; Morrow, B.E. Full spectrum of malformations in velo-cardio-facial syndrome/DiGeorge syndrome mouse models by altering Tbx1 dosage. Hum. Mol. Genet. 2004, 13, 1577–1585. [Google Scholar] [CrossRef]
- Baldini, A. Dissecting contiguous gene defects: TBX1. Curr. Opin. Genet. Dev. 2005, 15, 279–284. [Google Scholar] [CrossRef]
- Aggarwal, V.S.; Morrow, B.E. Genetic modifiers of the physical malformations in velo-cardio-facial syndrome/DiGeorge syndrome. Dev. Disabil. Res. Rev. 2008, 14, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Papangeli, I.; Scambler, P. The 22q11 deletion: DiGeorge and velocardiofacial syndromes and the role of TBX1. Wiley Interdiscip. Rev. Dev. Biol. 2013, 2, 393–403. [Google Scholar] [CrossRef] [PubMed]
- Heliovaara, A.; Ranta, R.; Rautio, J. Pharyngeal morphology in children with submucous cleft palate with and without surgery. Eur. Arch. Oto-Rhino-Laryngol. Head Neck 2005, 262, 534–538. [Google Scholar] [CrossRef] [PubMed]
- Heliövaara, A.; Hurmerinta, K. Craniofacial cephalometric morphology in children with CATCH 22 syndrome. Orthod. Craniofac. Res. 2006, 9, 186–192. [Google Scholar] [CrossRef]
- Ryan, A.K.; Goodship, J.A.; Wilson, D.I.; Philip, N.; Levy, A.; Seidel, H.; Schuffenhauer, S.; Oechsler, H.; Belohradsky, B.; Prieur, M.; et al. Spectrum of clinical features associated with interstitial chromosome 22q11 deletions: A European collaborative study. J. Med. Genet. 1997, 34, 798–804. [Google Scholar] [CrossRef]
- McDonald-McGinn, D.M.; Kirschner, R.; Goldmuntz, E.; Sullivan, K.; Eicher, P.; Gerdes, M.; Moss, E.; Solot, C.; Wang, P.; Jacobs, I.; et al. The Philadelphia story: The 22q11.2 deletion: Report on 250 patients. Genet. Couns. 1999, 10, 11–24. [Google Scholar]
- Klingberg, G.; Oskarsdóttir, S.; Johannesson, E.L.; Norén, J.G. Oral manifestations in 22q11 deletion syndrome. Int. J. Paediatr. Dent. 2002, 12, 14–23. [Google Scholar]
- Ricchetti, E.T.; States, L.; Hosalkar, H.S.; Tamai, J.; Maisenbacher, M.; McDonald-McGinn, D.M.; Zackai, E.H.; Drummond, D.S. Radiographic study of the upper cervical spine in the 22q11.2 deletion syndrome. J. Bone Joint Surg. Am. 2004, 86, 1751–1760. [Google Scholar] [CrossRef]
- Herman, S.B.; Guo, T.; McGinn, D.M.M.; Bolsa, A.; Shanske, A.L.; Bassett, A.S.; Chow, E.W.C.; Bowser, M.; Sheridan, M.; Beemer, F.; et al. Overt cleft palate phenotype and TBX1 genotype correlations in velo-cardio-facial/DiGeorge/22q11.2 deletion syndrome patients. Am. J. Med. Genet. A 2012, 158A, 2781–2787. [Google Scholar] [CrossRef]
- Hamidi, M.; Nabi, S.; Husein, M.; Mohamed, M.E.; Tay, K.Y.; McKillop, S. Cervical spine abnormalities in 22q11.2 deletion syndrome. Cleft Palate-Craniofacial J. 2014, 51, 230–233. [Google Scholar] [CrossRef]
- Jackson, O.; Crowley, T.B.; Sharkus, R.; Smith, R.; Jeong, S.; Solot, C.; McDonald-Mcginn, D. Palatal evaluation and treatment in 22q11.2 deletion syndrome. Am. J. Med. Genet. Part A 2019, 179, 1184–1195. [Google Scholar] [CrossRef] [PubMed]
- Bassett, A.S.; Chow, E.W.C.; Husted, J.; Weksberg, R.; Caluseriu, O.; Webb, G.D.; Gatzoulis, M.A. Clinical features of 78 adults with 22q11 Deletion Syndrome. Am. J. Med. Genet. A 2005, 138, 307–313. [Google Scholar] [CrossRef] [PubMed]
- Loos, E.; Verhaert, N.; Willaert, A.; Devriendt, K.; Swillen, A.; Hermans, R.; Op de Beeck, K.; Hens, G. Malformations of the middle and inner ear on CT imaging in 22q11 deletion syndrome. Am. J. Med. Genet. Part A 2016, 170, 2975–2983. [Google Scholar] [CrossRef] [PubMed]
- Verheij, E.; Kist, A.L.; Mink van der Molen, A.B.; Stegeman, I.; van Zanten, G.A.; Grolman, W.; Thomeer, H.G.X.M. Otologic and audiologic findings in 22q11.2 deletion syndrome. Eur. Arch. Oto-Rhino-Laryngol. Head Neck 2017, 274, 765–771. [Google Scholar] [CrossRef]
- Ford, L.C.; Sulprizio, S.L.; Rasgon, B.M. Otolaryngological manifestations of velocardiofacial syndrome: A retrospective review of 35 patients. Laryngoscope 2000, 110, 362–367. [Google Scholar] [CrossRef]
- Kobrynski, L.J.; Sullivan, K.E. Velocardiofacial syndrome, DiGeorge syndrome: The chromosome 22q11.2 deletion syndromes. Lancet 2007, 370, 1443–1452. [Google Scholar] [CrossRef]
- Oberoi, S.; Vargervik, K. Velocardiofacial syndrome with single central incisor. Am. J. Med. Genet. A 2005, 132A, 194–197. [Google Scholar] [CrossRef]
- Zhang, Z.; Huynh, T.; Baldini, A. Mesodermal expression of Tbx1 is necessary and sufficient for pharyngeal arch and cardiac outflow tract development. Development 2006, 133, 3587–3595. [Google Scholar] [CrossRef]
- Arnold, J.S.; Werling, U.; Braunstein, E.M.; Liao, J.; Nowotschin, S.; Edelmann, W.; Hebert, J.M.; Morrow, B.E. Inactivation of Tbx1 in the pharyngeal endoderm results in 22q11DS malformations. Development 2006, 133, 977–987. [Google Scholar] [CrossRef]
- Arnold, J.S.; Braunstein, E.M.; Ohyama, T.; Groves, A.K.; Adams, J.C.; Brown, M.C.; Morrow, B.E. Tissue-specific roles of Tbx1 in the development of the outer, middle and inner ear, defective in 22q11DS patients. Hum. Mol. Genet. 2006, 15, 1629–1639. [Google Scholar] [CrossRef]
- Choi, M.; Klingensmith, J. Chordin is a modifier of tbx1 for the craniofacial malformations of 22q11 deletion syndrome phenotypes in mouse. PLoS Genet. 2009, 5, e1000395. [Google Scholar] [CrossRef] [PubMed]
- Moraes, F.; Novoa, A.; Jerome-Majewska, L.A.; Papaioannou, V.E.; Mallo, M. Tbx1 is required for proper neural crest migration and to stabilize spatial patterns during middle and inner ear development. Mech. Dev. 2005, 122, 199–212. [Google Scholar] [CrossRef] [PubMed]
- Funato, N.; Nakamura, M.; Richardson, J.A.; Srivastava, D.; Yanagisawa, H. Tbx1 regulates oral epithelial adhesion and palatal development. Hum. Mol. Genet. 2012, 21, 2524–2537. [Google Scholar] [CrossRef] [PubMed]
- Funato, N.; Nakamura, M.; Richardson, J.A.; Srivastava, D.; Yanagisawa, H. Loss of Tbx1 induces bone phenotypes similar to cleidocranial dysplasia. Hum. Mol. Genet. 2015, 24, 424–435. [Google Scholar] [CrossRef] [PubMed]
- Funato, N.; Srivastava, D.; Shibata, S.; Yanagisawa, H. TBX1 Regulates Chondrocyte Maturation in the Spheno-occipital Synchondrosis. J. Dent. Res. 2020, 99, 1182–1191. [Google Scholar] [CrossRef]
- Solot, C.B.; Sell, D.; Mayne, A.; Baylis, A.L.; Persson, C.; Jackson, O.; McDonald-McGinn, D.M. Speech-Language Disorders in 22q11.2 Deletion Syndrome: Best Practices for Diagnosis and Management. Am. J. Speech-Lang. Pathol. 2019, 28, 984–999. [Google Scholar] [CrossRef]
- Jaouadi, A.; Tabebi, M.; Abdelhedi, F.; Abid, D.; Kamoun, F.; Chabchoub, I.; Maatoug, S.; Doukali, H.; Belghuith, N.; Ksentini, M.A.; et al. A novel TBX1 missense mutation in patients with syndromic congenital heart defects. Biochem. Biophys. Res. Commun. 2018, 499, 563–569. [Google Scholar] [CrossRef]
- Zweier, C.; Sticht, H.; Aydin-Yaylagül, I.; Campbell, C.E.; Rauch, A. Human TBX1 Missense Mutations Cause Gain of Function Resulting in the Same Phenotype as 22q11.2 Deletions. Am. J. Hum. Genet. 2007, 80, 510–517. [Google Scholar] [CrossRef]
- Hasegawa, K.; Tanaka, H.; Higuchi, Y.; Hayashi, Y.; Kobayashi, K.; Tsukahara, H. Novel heterozygous mutation in TBX1 in an infant with hypocalcemic seizures. Clin. Pediatr. Endocrinol. 2018, 27, 159–164. [Google Scholar] [CrossRef]
- Alghamdi, M.; Al Khalifah, R.; Al Homyani, D.K.; Alkhamis, W.H.; Arold, S.T.; Ekhzaimy, A.; El-Wetidy, M.; Kashour, T.; Halwani, R. A novel TBX1 variant causing hypoparathyroidism and deafness. J. Endocr. Soc. 2020, 4, bvz028. [Google Scholar] [CrossRef]
- Ogata, T.; Niihori, T.; Tanaka, N.; Kawai, M.; Nagashima, T.; Funayama, R.; Nakayama, K.; Nakashima, S.; Kato, F.; Fukami, M.; et al. TBX1 mutation identified by exome sequencing in a Japanese family with 22q11.2 deletion syndrome-like craniofacial features and hypocalcemia. PLoS ONE 2014, 9, e91598. [Google Scholar] [CrossRef] [PubMed]
- Paylor, R.; Glaser, B.; Mupo, A.; Ataliotis, P.; Spencer, C.; Sobotka, A.; Sparks, C.; Choi, C.H.; Oghalai, J.; Curran, S.; et al. Tbx1 haploinsufficiency is linked to behavioral disorders in mice and humans: Implications for 22q11 deletion syndrome. Proc. Natl. Acad. Sci. USA 2006, 103, 7729–7734. [Google Scholar] [CrossRef] [PubMed]
- Rauch, R.; Hofbeck, M.; Zweier, C.; Koch, A.; Zink, S.; Trautmann, U.; Hoyer, J.; Kaulitz, R.; Singer, H.; Rauch, A. Comprehensive genotype-phenotype analysis in 230 patients with tetralogy of Fallot. J. Med. Genet. 2010, 47, 321–331. [Google Scholar] [CrossRef] [PubMed]
- Stark, K.L.; Xu, B.; Bagchi, A.; Lai, W.-S.; Liu, H.; Hsu, R.; Wan, X.; Pavlidis, P.; Mills, A.A.; Karayiorgou, M.; et al. Altered brain microRNA biogenesis contributes to phenotypic deficits in a 22q11-deletion mouse model. Nat. Genet. 2008, 40, 751–760. [Google Scholar] [CrossRef]
- Edelmann, L.; Stankiewicz, P.; Spiteri, E.; Pandita, R.K.; Shaffer, L.; Lupski, J.R.; Morrow, B.E.; Lupski, J. Two functional copies of the DGCR6 gene are present on human chromosome 22q11 due to a duplication of an ancestral locus. Genome Res. 2001, 11, 208–217. [Google Scholar] [CrossRef]
- Hierck, B.P.; Molin, D.G.M.; Boot, M.J.; Poelmann, R.E.; Gittenberger-De Groot, A.C. A chicken model for DGCR6 as a modifier gene in the DiGeorge critical region. Pediatr. Res. 2004, 56, 440–448. [Google Scholar] [CrossRef][Green Version]
- Gao, S.; Moreno, M.; Eliason, S.; Cao, H.; Li, X.; Yu, W.; Bidlack, F.B.; Margolis, H.C.; Baldini, A.; Amendt, B.A. TBX1 protein interactions and microRNA-96-5p regulation controls cell proliferation during craniofacial and dental development: Implications for 22q11.2 deletion syndrome. Hum. Mol. Genet. 2015, 24, 2330–2348. [Google Scholar] [CrossRef]
- Sun, H.; Jiang, P. MicroRNA-451a acts as tumor suppressor in cutaneous basal cell carcinoma. Mol. Genet. Genom. Med. 2018, 6, 1001–1009. [Google Scholar] [CrossRef]
- Wang, J.; Bai, Y.; Li, H.; Greene, S.B.; Klysik, E.; Yu, W.; Schwartz, R.J.; Williams, T.J.; Martin, J.F. MicroRNA-17-92, a direct Ap-2alpha transcriptional target, modulates T-box factor activity in orofacial clefting. PLoS Genet. 2013, 9, e1003785. [Google Scholar] [CrossRef]
- Kaimal, V.; Bardes, E.E.; Tabar, S.C.; Jegga, A.G.; Aronow, B.J. ToppCluster: A multiple gene list feature analyzer for comparative enrichment clustering and network-based dissection of biological systems. Nucleic Acids Res. 2010, 38, W96-102. [Google Scholar] [CrossRef]
- Bachiller, D.; Klingensmith, J.; Shneyder, N.; Tran, U.; Anderson, R.; Rossant, J.; De Robertis, E.M. The role of chordin/Bmp signals in mammalian pharyngeal development and DiGeorge syndrome. Development 2003, 130, 3567–3578. [Google Scholar] [CrossRef] [PubMed]
- Wurdak, H.; Ittner, L.M.; Lang, K.S.; Leveen, P.; Suter, U.; Fischer, J.A.; Karlsson, S.; Born, W.; Sommer, L. Inactivation of TGFβ signaling in neural crest stem cells leads to multiple defects reminiscent of DiGeorge syndrome. Genes Dev. 2005, 19, 530–535. [Google Scholar] [CrossRef] [PubMed]
- Stalmans, I.; Lambrechts, D.; De Smet, F.; Jansen, S.; Wang, J.; Maity, S.; Kneer, P.; von der Ohe, M.; Swillen, A.; Maes, C.; et al. VEGF: A modifier of the del22q11 (DiGeorge) syndrome? Nat. Med. 2003, 9, 173–182. [Google Scholar] [CrossRef] [PubMed]
- Abu-Issa, R.; Smyth, G.; Smoak, I.; Yamamura, K.; Meyers, E.N. Fgf8 is required for pharyngeal arch and cardiovascular development in the mouse. Development 2002, 129, 4613–4625. [Google Scholar] [CrossRef]
- Guris, D.L.; Fantes, J.; Tara, D.; Druker, B.J.; Imamoto, A. Mice lacking the homologue of the human 22q11.2 gene CRLK phenocopy neurocristopathies of DiGeorge syndrome. Nat. Genet. 2001, 27, 293–298. [Google Scholar] [CrossRef]
- Vermot, J.; Niederreither, K.; Garnier, J.M.; Chambon, P.; Dollé, P. Decreased embryonic retinoic acid synthesis results in a DiGeorge syndrome phenotype in newborn mice. Proc. Natl. Acad. Sci. USA 2003, 100, 1763–1768. [Google Scholar] [CrossRef]
- Chisaka, O.; Capecchi, M.R. Regionally restricted developmental defects resulting from targeted disruption of the mouse homeobox gene hox-1.5. Nature 1991, 350, 473–479. [Google Scholar] [CrossRef]
- Voss, A.K.; Vanyai, H.K.; Collin, C.; Dixon, M.P.; McLennan, T.J.; Sheikh, B.N.; Scambler, P.; Thomas, T. MOZ Regulates the Tbx1 Locus, and Moz Mutation Partially Phenocopies DiGeorge Syndrome. Dev. Cell 2012, 23, 652–663. [Google Scholar] [CrossRef]
- Sheehy, N.T.; Cordes, K.R.; White, M.P.; Ivey, K.N.; Srivastava, D. The neural crest-enriched microRNA miR-452 regulates epithelial-mesenchymal signaling in the first pharyngeal arch. Development 2010, 137, 4307–4316. [Google Scholar] [CrossRef]
- Gershwin, M.E. DiGeorge syndrome: Congenital thymic hypoplasia. Animal model: Congenitally athymic (nude) mouse. Am. J. Pathol. 1977, 89, 809–812. [Google Scholar]
- Yamagishi, H.; Maeda, J.; Hu, T.; McAnally, J.; Conway, S.J.; Kume, T.; Meyers, E.N.; Yamagishi, C.; Srivastava, D. Tbx1 is regulated by tissue-specific forkhead proteins through a common Sonic hedgehog-responsive enhancer. Genes Dev. 2003, 17, 269–281. [Google Scholar] [CrossRef] [PubMed]
- Chapman, D.L.; Garvey, N.; Hancock, S.; Alexiou, M.; Agulnik, S.I.; Gibson-Brown, J.J.; Cebra-Thomas, J.; Bollag, R.J.; Silver, L.M.; Papaioannou, V.E. Expression of the T-box family genes, Tbx1-Tbx5, during early mouse development. Dev. Dyn. 1996, 206, 379–390. [Google Scholar] [CrossRef]
- Zoupa, M.; Seppala, M.; Mitsiadis, T.; Cobourne, M.T. Tbx1 is expressed at multiple sites of epithelial-mesenchymal interaction during early development of the facial complex. Int. J. Dev. Biol. 2006, 50, 504–510. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Cerrato, F.; Xu, H.; Vitelli, F.; Morishima, M.; Vincentz, J.; Furuta, Y.; Ma, L.; Martin, J.F.; Baldini, A.; et al. Tbx1 expression in pharyngeal epithelia is necessary for pharyngeal arch artery development. Development 2005, 132, 5307–5315. [Google Scholar] [CrossRef]
- Aggarwal, V.S.; Carpenter, C.; Freyer, L.; Liao, J.; Petti, M.; Morrow, B.E. Mesodermal Tbx1 is required for patterning the proximal mandible in mice. Dev. Biol. 2010, 344, 669–681. [Google Scholar] [CrossRef]
- Bush, J.O.; Jiang, R. Palatogenesis: Morphogenetic and molecular mechanisms of secondary palate development. Development 2012, 139, 828. [Google Scholar] [CrossRef]
- Funato, N. Molecular basis of cleft palates in mice. World J. Biol. Chem. 2015, 6, 121. [Google Scholar] [CrossRef]
- Goudy, S.; Law, A.; Sanchez, G.; Baldwin, H.S.; Brown, C. Tbx1 is necessary for palatal elongation and elevation. Mech. Dev. 2010, 127, 292–300. [Google Scholar] [CrossRef]
- Peters, H.; Neubuser, A.; Kratochwil, K.; Balling, R. Pax9-deficient mice lack pharyngeal pouch derivatives and teeth and exhibit craniofacial and limb abnormalities. Genes Dev. 1998, 12, 2735–2747. [Google Scholar] [CrossRef]
- Ivins, S.; Van Beuren, K.L.; Roberts, C.; James, C.; Lindsay, E.; Baldini, A.; Ataliotis, P.; Scambler, P.J. Microarray analysis detects differentially expressed genes in the pharyngeal region of mice lacking Tbx1. Dev. Biol. 2005, 285, 554–569. [Google Scholar] [CrossRef]
- Funato, N.; Yanagisawa, H. Deletion of the T-box transcription factor gene, Tbx1, in mice induces differential expression of genes associated with cleft palate in humans. Arch. Oral Biol. 2018, 95, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Komori, T.; Yagi, H.; Nomura, S.; Yamaguchi, A.; Sasaki, K.; Deguchi, K.; Shimizu, Y.; Bronson, R.T.; Gao, Y.H.; Inada, M.; et al. Targeted disruption of Cbfa1 results in a complete lack of bone formation owing to maturational arrest of osteoblasts. Cell 1997, 89, 755–764. [Google Scholar] [CrossRef]
- Otto, F.; Thornell, A.P.; Crompton, T.; Denzel, A.; Gilmour, K.C.; Rosewell, I.R.; Stamp, G.W.; Beddington, R.S.; Mundlos, S.; Olsen, B.R.; et al. Cbfa1, a candidate gene for cleidocranial dysplasia syndrome, is essential for osteoblast differentiation and bone development. Cell 1997, 89, 765–771. [Google Scholar] [CrossRef]
- Mundlos, S.; Otto, F.; Mundlos, C.; Mulliken, J.B.; Aylsworth, A.S.; Albright, S.; Lindhout, D.; Cole, W.G.; Henn, W.; Knoll, J.H.; et al. Mutations involving the transcription factor CBFA1 cause cleidocranial dysplasia. Cell 1997, 89, 773–779. [Google Scholar] [CrossRef]
- Funato, N. New Insights Into Cranial Synchondrosis Development: A Mini Review. Front. Cell Dev. Biol. 2020, 8, 706. [Google Scholar] [CrossRef]
- Catón, J.; Luder, H.U.; Zoupa, M.; Bradman, M.; Bluteau, G.; Tucker, A.S.; Klein, O.; Mitsiadis, T.A. Enamel-free teeth: Tbx1 deletion affects amelogenesis in rodent incisors. Dev. Biol. 2009, 328, 493–505. [Google Scholar] [CrossRef]
- Kelly, R.G.; Jerome-Majewska, L.A.; Papaioannou, V.E. The del22q11.2 candidate gene Tbx1 regulates branchiomeric myogenesis. Hum. Mol. Genet. 2004, 13, 2829–2840. [Google Scholar] [CrossRef]
- Kong, P.; Racedo, S.E.; Macchiarulo, S.; Hu, Z.; Carpenter, C.; Guo, T.; Wang, T.; Zheng, D.; Morrow, B.E. Tbx1 is required autonomously for cell survival and fate in the pharyngeal core mesoderm to form the muscles of mastication. Hum. Mol. Genet. 2014, 23, 4215–4231. [Google Scholar] [CrossRef]
- Harel, I.; Maezawa, Y.; Avraham, R.; Rinon, A.; Ma, H.Y.; Cross, J.W.; Leviatan, N.; Hegesh, J.; Roy, A.; Jacob-Hirsch, J.; et al. Pharyngeal mesoderm regulatory network controls cardiac and head muscle morphogenesis. Proc. Natl. Acad. Sci. USA. 2012, 109, 18839–18844. [Google Scholar] [CrossRef]
- Grifone, R.; Jarry, T.; Dandonneau, M.; Grenier, J.; Duprez, D.; Kelly, R.G. Properties of branchiomeric and somite-derived muscle development in Tbx1 mutant embryos. Dev. Dyn. 2008, 237, 3071–3078. [Google Scholar] [CrossRef]
- Pane, L.S.; Zhang, Z.; Ferrentino, R.; Huynh, T.; Cutillo, L.; Baldini, A. Tbx1 is a negative modulator of Mef2c. Hum. Mol. Genet. 2012, 21, 2485–2496. [Google Scholar] [CrossRef] [PubMed]
- Dong, F.; Sun, X.; Liu, W.; Ai, D.; Klysik, E.; Lu, M.F.; Hadley, J.; Antoni, L.; Chen, L.; Baldini, A.; et al. Pitx2 promotes development of splanchnic mesoderm-derived branchiomeric muscle. Development 2006, 133, 4891–4899. [Google Scholar] [CrossRef] [PubMed]
- Shih, H.P.; Gross, M.K.; Kioussi, C. Cranial muscle defects of Pitx2 mutants result from specification defects in the first branchial arch. Proc. Natl. Acad. Sci. USA 2007, 104, 5907–5912. [Google Scholar] [CrossRef] [PubMed]
- Papangeli, I.; Scambler, P.J. Tbx1 genetically interacts with the transforming growth factor-β/bone morphogenetic protein inhibitor Smad7 during great vessel remodeling. Circ. Res. 2013, 112, 90–102. [Google Scholar] [CrossRef]
- Vieira, J.M.; Ruhrberg, C.; Schwarz, Q. VEGF receptor signaling in vertebrate development. Organogenesis 2010, 6, 97–106. [Google Scholar] [CrossRef]
- Lambrechts, D.; Devriendt, K.; Driscoll, D.A.; Goldmuntz, E.; Gewillig, M.; Vlietinck, R.; Collen, D.; Carmeliet, P. Low expression VEGF haplotype increases the risk for tetralogy of Fallot: A family based association study. J. Med. Genet. 2005, 42, 519–522. [Google Scholar] [CrossRef]
- Frank, D.U.; Fotheringham, L.K.; Brewer, J.A.; Muglia, L.J.; Tristani-Firouzi, M.; Capecchi, M.R.; Moon, A.M. An Fgf8 mouse mutant phenocopies human 22q11 deletion syndrome. Development 2002, 129, 4591–4603. [Google Scholar] [CrossRef]
- Vitelli, F.; Taddei, I.; Morishima, M.; Meyers, E.N.; Lindsay, E.A.; Baldini, A. A genetic link between Tbx1 and fibroblast growth factor signaling. Development 2002, 129, 4605–4611. [Google Scholar] [CrossRef]
- Brown, C.B.; Wenning, J.M.; Lu, M.M.; Epstein, D.J.; Meyers, E.N.; Epstein, J.A. Cre-mediated excision of Fgf8 in the Tbx1 expression domain reveals a critical role for Fgf8 in cardiovascular development in the mouse. Dev. Biol. 2004, 267, 190–202. [Google Scholar] [CrossRef]
- Mitsiadis, T.A.; Tucker, A.S.; De Bari, C.; Cobourne, M.T.; Rice, D.P.C. A regulatory relationship between Tbx1 and FGF signaling during tooth morphogenesis and ameloblast lineage determination. Dev. Biol. 2008, 320, 39–48. [Google Scholar] [CrossRef]
- Lin, C.-Y.; Chen, W.-T.; Lee, H.-C.; Yang, P.-H.; Yang, H.-J.; Tsai, H.-J. The transcription factor Six1a plays an essential role in the craniofacial myogenesis of zebrafish. Dev. Biol. 2009, 331, 152–166. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Sun, Y.; Zhou, B.; Adam, R.M.; Li, X.; Pu, W.T.; Morrow, B.E.; Moon, A.; Li, X. A Tbx1-Six1/Eya1-Fgf8 genetic pathway controls mammalian cardiovascular and craniofacial morphogenesis. J. Clin. Investig. 2011, 121, 1585–1595. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Diacou, A.; Johnston, H.R.; Musfee, F.I.; McDonald-McGinn, D.M.; McGinn, D.; Crowley, T.B.; Repetto, G.M.; Swillen, A.; Breckpot, J.; et al. Complete Sequence of the 22q11.2 Allele in 1,053 Subjects with 22q11.2 Deletion Syndrome Reveals Modifiers of Conotruncal Heart Defects. Am. J. Hum. Genet. 2020, 106, 26–40. [Google Scholar] [CrossRef] [PubMed]
- Moon, A.M.; Guris, D.L.; Seo, J.H.; Li, L.; Hammond, J.; Talbot, A.; Imamoto, A. Crkl deficiency disrupts Fgf8 signaling in a mouse model of 22q11 deletion syndromes. Dev. Cell 2006, 10, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Rosa, F.W.; Wilk, A.L.; Kelsey, F.O. Teratogen update: Vitamin A congeners. Teratology 1986, 33, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Roberts, C.; Ivins, S.M.; James, C.T.; Scambler, P.J. Retinoic acid down-regulatesTbx1 expression in vivo and in vitro. Dev. Dyn. 2005, 232, 928–938. [Google Scholar] [CrossRef] [PubMed]
- Wendling, O.; Dennefeld, C.; Chambon, P.; Mark, M. Retinoid signaling is essential for patterning the endoderm of the third and fourth pharyngeal arches. Development 2000, 127, 1553–1562. [Google Scholar] [CrossRef]
- Niederreither, K.; Vermot, J.; Le Roux, I.; Schuhbaur, B.; Chambon, P.; Dollé, P. The regional pattern of retinoic acid synthesis by RALDH2 is essential for the development of posterior pharyngeal arches and the enteric nervous system. Development 2003, 130, 2525–2534. [Google Scholar] [CrossRef]
- Ryckebüsch, L.; Bertrand, N.; Mesbah, K.; Bajolle, F.; Niederreither, K.; Kelly, R.G.; Zaffran, S. Decreased levels of embryonic retinoic acid synthesis accelerate recovery from arterial growth delay in a mouse model of DiGeorge syndrome. Circ. Res. 2010, 106, 686–694. [Google Scholar] [CrossRef]
- Roberts, C.; Ivins, S.; Cook, A.C.; Baldini, A.; Scambler, P.J. Cyp26 genes a1, b1 and c1 are down-regulated in Tbx1 null mice and inhibition of Cyp26 enzyme function produces a phenocopy of DiGeorge Syndrome in the chick. Hum. Mol. Genet. 2006, 15, 3394–3410. [Google Scholar] [CrossRef]
- Guris, D.L.; Duester, G.; Papaioannou, V.E.; Imamoto, A. Dose-dependent interaction of Tbx1 and Crkl and locally aberrant RA signaling in a model of del22q11 syndrome. Dev. Cell 2006, 10, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Mulder, G.B.; Manley, N.; Maggio-Price, L. Retinoic acid-induced thymic abnormalities in the mouse are associated with altered pharyngeal morphology, thymocyte maturation defects, and altered expression of Hoxa3 and Pax1. Teratology 1998, 58, 263–275. [Google Scholar] [CrossRef]
- Chen, L.; Zhao, P.; Wells, L.; Amemiya, C.T.; Condie, B.G.; Manley, N.R. Mouse and zebrafish Hoxa3 orthologues have nonequivalent in vivo protein function. Proc. Natl. Acad. Sci. USA 2010, 107, 10555–10560. [Google Scholar] [CrossRef] [PubMed]
- Fulcoli, F.G.; Franzese, M.; Liu, X.; Zhang, Z.; Angelini, C.; Baldini, A. Rebalancing gene haploinsufficiency in vivo by targeting chromatin. Nat. Commun. 2016, 7, 11688. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Fulcoli, F.G.; Ferrentino, R.; Martucciello, S.; Illingworth, E.A.; Baldini, A. Transcriptional control in cardiac progenitors: Tbx1 interacts with the BAF chromatin remodeling complex and regulates Wnt5a. PLoS Genet. 2012, 8, e1002571. [Google Scholar] [CrossRef]
- León, L.E.; Benavides, F.; Espinoza, K.; Vial, C.; Alvarez, P.; Palomares, M.; Lay-Son, G.; Miranda, M.; Repetto, G.M. Partial microduplication in the histone acetyltransferase complex member KANSL1 is associated with congenital heart defects in 22q11.2 microdeletion syndrome patients. Sci. Rep. 2017, 7, 1795. [Google Scholar] [CrossRef]
- Zhang, Z.; Shi, L.; Song, L.; Maurer, K.; Zhao, X.; Zackai, E.H.; McGinn, D.E.; Crowley, T.B.; McGinn, D.M.M.; Sullivan, K.E. Chromatin Modifications in 22q11.2 Deletion Syndrome. J. Clin. Immunol. 2021, 41, 1853–1864. [Google Scholar] [CrossRef]
- Washington Smoak, I.; Byrd, N.A.; Abu-Issa, R.; Goddeeris, M.M.; Anderson, R.; Morris, J.; Yamamura, K.; Klingensmith, J.; Meyers, E.N. Sonic hedgehog is required for cardiac outflow tract and neural crest cell development. Dev. Biol. 2005, 283, 357–372. [Google Scholar] [CrossRef]
- Garg, V.; Yamagishi, C.; Hu, T.; Kathiriya, I.S.; Yamagishi, H.; Srivastava, D. Tbx1, a DiGeorge syndrome candidate gene, is regulated by sonic hedgehog during pharyngeal arch development. Dev. Biol. 2001, 235, 62–73. [Google Scholar] [CrossRef]
- Kou, I.; Otomo, N.; Takeda, K.; Momozawa, Y.; Lu, H.-F.; Kubo, M.; Kamatani, Y.; Ogura, Y.; Takahashi, Y.; Nakajima, M.; et al. Genome-wide association study identifies 14 previously unreported susceptibility loci for adolescent idiopathic scoliosis in Japanese. Nat. Commun. 2019, 10, 3685. [Google Scholar] [CrossRef]
- Brooks, J.K.; Coccaro, P.J.; Zarbin, M.A. The Rieger anomaly concomitant with multiple dental, craniofacial, and somatic midline anomalies and short stature. Oral Surg. Oral Med. Oral Pathol. 1989, 68, 717–724. [Google Scholar] [CrossRef]
- Mucchielli, M.L.; Mitsiadis, T.A.; Raffo, S.; Brunet, J.F.; Proust, J.P.; Goridis, C. Mouse Otlx2/RIEG expression in the odontogenic epithelium precedes tooth initiation and requires mesenchyme-derived signals for its maintenance. Dev. Biol. 1997, 189, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Nowotschin, S.; Liao, J.; Gage, P.J.; Epstein, J.A.; Campione, M.; Morrow, B.E. Tbx1 affects asymmetric cardiac morphogenesis by regulating Pitx2 in the secondary heart field. Development 2006, 133, 1565–1573. [Google Scholar] [CrossRef] [PubMed]
- Cao, H.; Florez, S.; Amen, M.; Huynh, T.; Skobe, Z.; Baldini, A.; Amendt, B.A. Tbx1 regulates progenitor cell proliferation in the dental epithelium by modulating Pitx2 activation of p21. Dev. Biol. 2010, 347, 289–300. [Google Scholar] [CrossRef]
- Lindsay, E.A.; Botta, A.; Jurecic, V.; Carattini-Rivera, S.; Cheah, Y.C.; Rosenblatt, H.M.; Bradley, A.; Baldini, A. Congenital heart disease in mice deficient for the DiGeorge syndrome region. Nature 1999, 401, 379–383. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Phenotypes | Features | Frequency |
|---|---|---|
| Palatal anomalies | Overt cleft palate | 7–11% |
| Submucous cleft palate | 5–23% | |
| Bifid uvula | 5–10% | |
| Velopharyngeal insufficiency | 27–92% | |
| Dental anomalies | Tooth agenesis | 15% |
| Hypoplasia of primary teeth | 32% | |
| Hypoplasia of permanent teeth | 10% | |
| Enamel hypomineralization of primary teeth | 39% | |
| Enamel hypomineralization of permanent teeth | 41% | |
| Ear-nose-throat abnormalities | Hearing loss | 33–39% |
| Otitis media with effusion | 2% | |
| Tracheomalacia/laryngomalacia | 2% | |
| Laryngeal web | 1% | |
| Ocular abnormalities | Hooding of the upper lid | 41% |
| Ptosis | 9% | |
| Hooding of the lower lid | 6% | |
| Epicanthal folds | 3% | |
| Distichiasis | 3% | |
| Cranial base anomalies | Platybasia | 50–91% |
| Basilar impression | 3% | |
| Cervical spine anomalies | Atlas (C1) anomalies | 75% |
| Axis (C2) anomalies | 59% | |
| Fusion of C2–C3 | 34% |
| DGS/VCFS | Tbx1-Null Mice | |
|---|---|---|
| Cranium | Dolichocephaly | Small cranium |
| Abnormal skull morphology | Hypoplastic parietal bone | |
| Malar flattening | Hypoplastic interparietal bone | |
| Long face | Unfused cranial sutures between frontal and parietal bones | |
| Temporal bone hypoplasia | ||
| Absent zygomatic arch | ||
| Abnormal zygomatic arch morphology | ||
| Cranial Base | Platybasia | Abnormal fusion of the basioccipital and basisphenoid bones |
| Basilar impression | Abnormal presphenoid bone morphology | |
| Abnormal basioccipital bone morphology | ||
| Palate | Cleft palate | Cleft palate |
| Submucous cleft palate | Submucous cleft palate | |
| Bifid uvula | Bifid uvula | |
| Highly arched palate | ||
| Velopharyngeal insufficiency | ||
| Mandible | Retrognathia | Absent mandibular coronoid process |
| Short mandible | Short mandible | |
| Micrognathia | Micrognathia | |
| Teeth | Enamel hypoplasia | Abnormal upper incisor morphology |
| Single central incisor | Absent upper incisors | |
| Small teeth | ||
| Abnormality of the dentition | ||
| Carious teeth | ||
| Muscles | Pharyngeal hypotonia | Absent masseter muscle |
| Absent pterygoid muscle | ||
| Absent temporalis muscle | ||
| Eyes | Hypertelorism/telecanthus | Hypertelorism |
| Downslanted palpebral fissures | ||
| Proptosis | ||
| Strabismus | ||
| Abnormal eyelid morphology | ||
| Epicanthus | ||
| Microphthalmia | ||
| External Ears | Small earlobe | Ear lobe hypoplasia |
| Low-set ears | Lowered ear position | |
| Abnormally folded pinna | Abnormal ear shape | |
| Preauricular pit | Absent outer ear | |
| Anotia | ||
| Middle and Inner Ears | Chronic otitis media | Abnormal middle ear ossicle morphology |
| Conductive hearing loss | Absent middle ear ossicles | |
| Sensorineural hearing loss | Abnormal stapes morphology | |
| Auditory canal stenosis | Abnormal incus morphology | |
| Pulsatile tympanic membrane | Abnormal malleus morphology | |
| Thickened tympanic membrane | Absent stapes | |
| Tympanic membrane retraction | Abnormal external auditory canal morphology | |
| Decreased tympanic ring size | ||
| Nose | Prominent nasal bridge | Short snout |
| Abnormal nasal morphology | ||
| Underdeveloped nasal alae | ||
| Choanal atresia | ||
| Throat | Abnormal thorax morphology | Small thyroid cartilage |
| Abnormality of the pharynx | Small cricoid cartilage | |
| Abnormal thyroid cartilage morphology | ||
| Pharynx hypoplasia | ||
| Hyoid bones | Delayed development of the hyoid bone | Hyoid bone hypoplasia |
| Invisible hyoid ossification center | Abnormal hyoid bone morphology | |
| Cervical spine | Dysmorphic C1 | Abnormal cervical atlas (C1) morphology |
| Anterior arch cleft of C1 | Absent arcus anterior of C1 | |
| Open posterior arch C1 | ||
| Fusion of C1–C2 | ||
| Fusion of C2–C3 | ||
| Upswept C2 lamina | ||
| Platyspondyly | ||
| Others | Short clavicle | |
| References | [14,15,16,17,18,19,20,21,22,23,24,25,26,27,28] | [6,7,8,9,10,29,30,31,32,33,34,35,36] |
| Mutation | Domain | Condition | Craniofacial Anomalies | References |
|---|---|---|---|---|
| c.89_284del | N-terminal | DiGeorge syndrome | Yes | ClinVar Variant: 971780 |
| c.199_224del | N-terminal | DiGeorge syndrome | Yes | ClinVar Variant: 949172 |
| c.292A>T | N-terminal | DiGeorge syndrome | Yes | ClinVar Variant: 526036 |
| c.385G>A | T-box | Tetralogy of Fallot | No | ClinVar Variant: 488618 |
| c.443T>A (F148Y) | T-box | Conotruncal anomaly face syndrome | Yes | [5] |
| c.503T>C | T-box | DiGeorge syndrome Velocardiofacial syndrome (Shprintzen syndrome) Tetralogy of Fallot | Yes | ClinVar Variant: 973222 |
| c.569C > A (P190Q) | T-box | Congenital heart defects | No | [38] |
| c.582C>G (H194Q) | T-box | Velocardiofacial syndrome | Yes | [39] |
| c.928G>A (G310S) | C-terminal | DiGeorge syndrome | Yes | [5] |
| c.967_977dup AACCCCGTGGC | C-terminal | Thymic hypoplasia Postaxial polydactyly of the right fifth toe | No | [40] |
| c.1158_1159delinsT | C-terminal | Hypoparathyroidism and hypocalcemia Facial asymmetry Deafness | Yes | [41] |
| c.1223delC | C-terminal | Conotruncal anomaly face syndrome Velocardiofacial syndrome | Yes | [5] |
| c.1253delA | C-terminal | DiGeorge syndrome | Yes | [42] |
| c.1320-1342del23bp | C-terminal | Velocardiofacial syndrome | Yes/No | [43] |
| c.1399-1428dup30 | C-terminal | Tetralogy of Fallot Scoliosis Facial asymmetry Upslanting palpebral fissures Absent pulmonary valve Isolated left pulmonary artery | Yes | [44] |
| Gene Symbol | Induced Mutation Type | Cranium | Palate | Teeth | Muscles | Ear-Nose-Throat | Hyoid Bones | Cardio-Vascular |
|---|---|---|---|---|---|---|---|---|
| Tbx1 | Null | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| Chrd | Null | Yes | Yes | nr | nr | Yes | Yes | Yes |
| Tgfbr2 | Deletion (Wnt1-Cre) | Yes | Yes | nr | nr | nr | nr | Yes |
| Vegfa | Null | Yes | Yes | Yes | nr | nr | nr | Yes |
| Fgf8 | Hypomorphic allele | Yes | Yes | Yes | nr | Yes | Yes | Yes |
| Crkl | Null | Yes | nr | nr | nr | Yes | nr | Yes |
| Aldh1a2 | Hypomorphic allele | nr | nr | nr | nr | Yes | Yes | Yes |
| Hoxa3 | Null | nr | Yes | nr | Yes | Yes | Yes | Yes |
| Kat6a | Null | nr | Yes | nr | nr | Yes | nr | Yes |
| Dicer1 | Deletion (Wnt1-Cre) | Yes | nr | nr | nr | nr | nr | Yes |
| Plxnd1 | Single point mutation | nr | Yes | nr | nr | Yes | nr | Yes |
| Dock1 | Undefined | nr | nr | nr | nr | Yes | nr | Yes |
| Ndst1 | Single point mutation | nr | nr | nr | nr | Yes | nr | Yes |
| Prickle1 | Single point mutation | Yes | Yes | nr | nr | Yes | nr | Yes |
| Trappc10 | Undefined | Yes | Yes | nr | nr | nr | nr | Yes |
| Zfp366 | Single point mutation | nr | nr | nr | nr | Yes | nr | Yes |
| Foxn1 | Intragenic deletion | nr | nr | nr | nr | Yes | nr | Yes |
| Tbx1-Mutant Mice | Craniofacial Phenotypes | ||||||
|---|---|---|---|---|---|---|---|
| Mutation Type | Tissue/Cell | Cranium | Cranial Base | Palate | Mandible | Hyoid Bone | Cervical Spine |
| Tbx1+/− | Entire body | Normal | Normal | Normal | Normal | Normal | Normal |
| Tbx1-null | Entire body | Abnormal | Abnormal | CP | Hypoplastic | Hypoplastic | Abnormal |
| Deletion (Foxg1-Cre) | Pharyngeal tissues * | Abnormal | Abnormal | CP | Hypoplastic | Hypoplastic | NA |
| Deletion (KRT14-Cre) | Epithelium | Normal | Normal | Anterior CP | Normal | Normal | Normal |
| Deletion (Mesp1-Cre) | Mesoderm | Abnormal | Abnormal | NA | Hypoplastic | Hypoplastic | Abnormal |
| Deletion (Twist2-Cre) | Osteochondral progenitors | Abnormal | Abnormal | Normal | Normal | Hypoplastic | Abnormal |
| Deletion (Wnt1-Cre) | Neural crest | Normal | Normal | Normal | Normal | Hypoplastic | Normal |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Funato, N. Craniofacial Phenotypes and Genetics of DiGeorge Syndrome. J. Dev. Biol. 2022, 10, 18. https://doi.org/10.3390/jdb10020018
Funato N. Craniofacial Phenotypes and Genetics of DiGeorge Syndrome. Journal of Developmental Biology. 2022; 10(2):18. https://doi.org/10.3390/jdb10020018
Chicago/Turabian StyleFunato, Noriko. 2022. "Craniofacial Phenotypes and Genetics of DiGeorge Syndrome" Journal of Developmental Biology 10, no. 2: 18. https://doi.org/10.3390/jdb10020018
APA StyleFunato, N. (2022). Craniofacial Phenotypes and Genetics of DiGeorge Syndrome. Journal of Developmental Biology, 10(2), 18. https://doi.org/10.3390/jdb10020018