
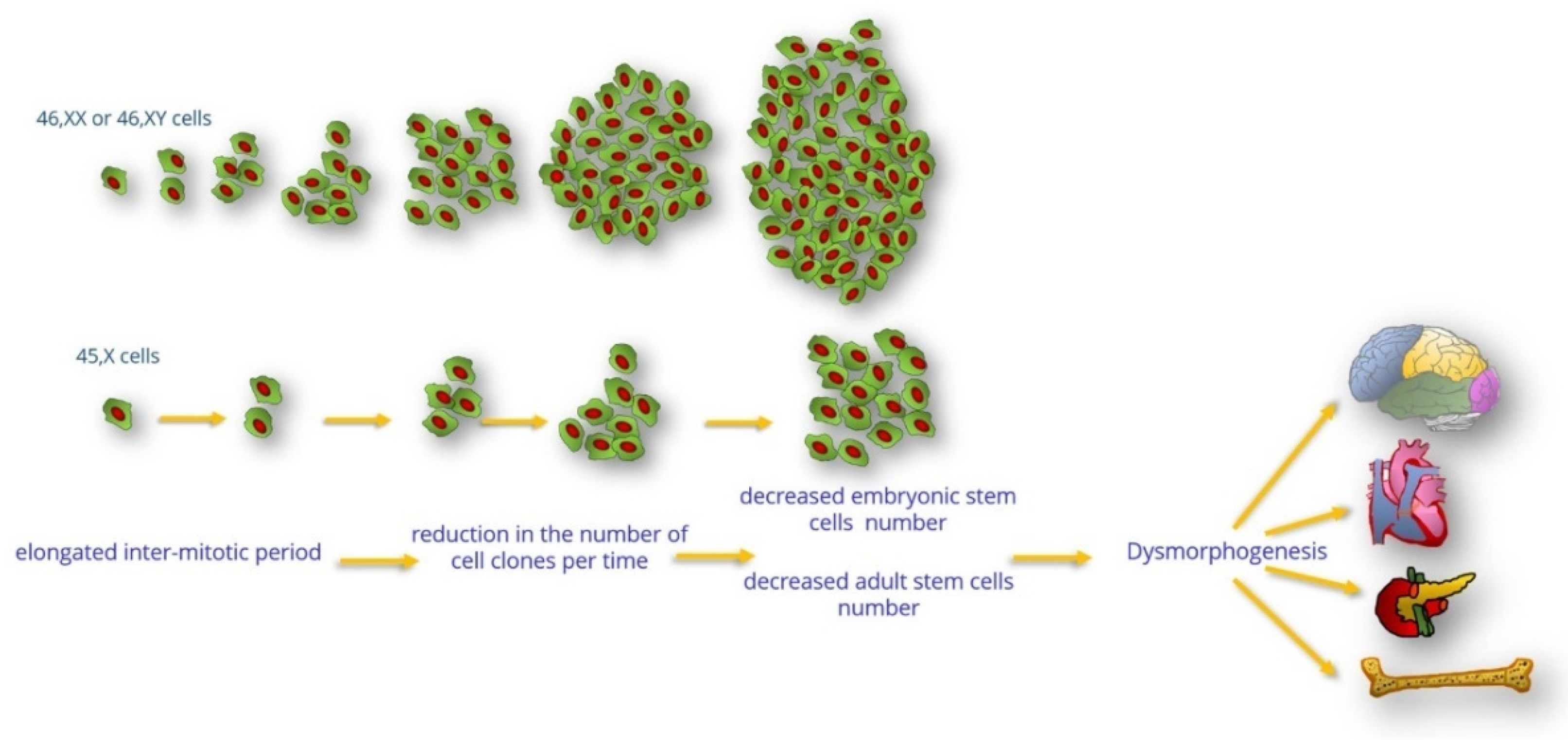
The Hypothesis of the Prolonged Cell Cycle in Turner Syndrome


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. XO Monosomy in Animals
3. Clinical Consequences of Reduced Cell Proliferation of 45,X Cells
3.1. Embryonic Lethality
3.2. Growth Retardation, Short Stature, and Osteopenia/Osteoporosis
3.2.1. Short Stature
3.2.2. Osteopenia/Osteoporosis
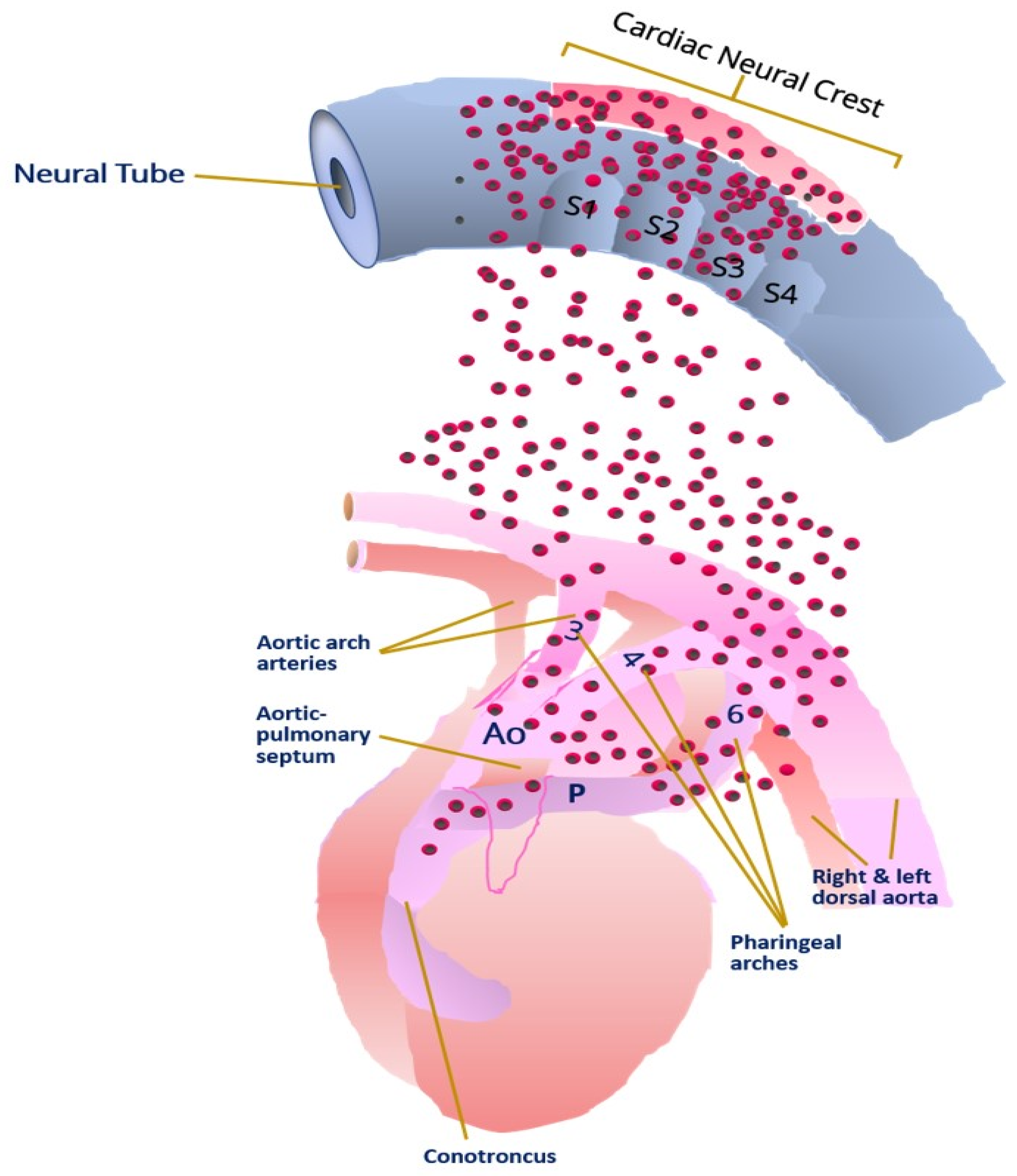
3.3. Congenital Heart Disease
3.4. Gonadal Dysgenesis
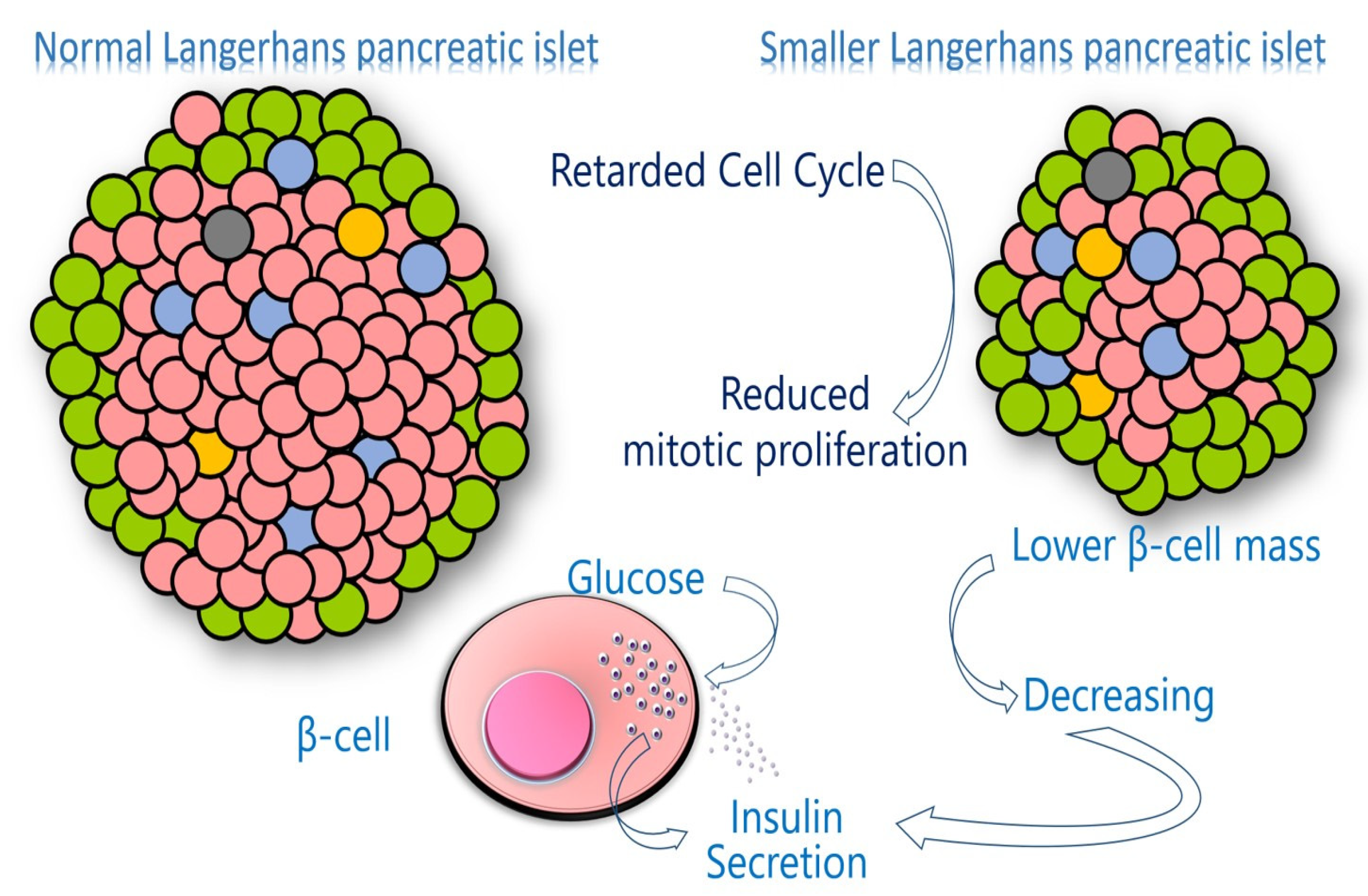
3.5. Impaired Pancreatic β-Cell Function
3.6. Neurologic Deficits
3.6.1. Neuropsychological Profile
3.6.2. Sensorineural Hearing Loss
3.7. Cancer in Turner Syndrome under the Prolonged Cell Cycle Hypothesis
4. Summary and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Homem, C.C.; Repic, M.; Knoblich, J.A. Proliferation control in neural stem and progenitor cells. Nat. Rev. Neurosci. 2015, 16, 647–659. [Google Scholar] [CrossRef] [PubMed]
- Simpson, J.L.; Le Beau, M.M. Gonadal and statural determinants on the X chromosome and their relationship to in vitro studies showing prolonged cell cycles in 45,X; 46,X,del(X)(p11); 46,X,del(X)(q13); and 46,X,del(X)(q22) fibroblasts. Am. J. Obstet. Gynecol. 1981, 141, 930–940. [Google Scholar] [CrossRef]
- Cure, S.; Boue, J.; Boue, A. Growth Characteristics of Human Embryonic Cell Lines with Chromosomal Anomalies. Biomedicine 1974, 21, 233–236. [Google Scholar] [PubMed]
- Verp, M.S.; Rosinsky, B.; Le Beau, M.M.; Martin, A.O.; Kaplan, R.; Wallemark, C.B.; Otano, L.; Simpson, J.L. Growth disadvantage of 45,X and 46,X,del(X)(p11) fibroblasts. Clin. Genet. 1988, 33, 277–285. [Google Scholar] [CrossRef]
- Opitz, J.M.; Gilbert, E.F. CNS anomalies and the midline as a ”developmental field”. Am. J. Med. Genet. 1982, 12, 443–455. [Google Scholar] [CrossRef]
- Paton, G.R.; Silver, M.F.; Allison, A.C. Comparison of cell cycle time in normal and trisomic cells. Humangenetik 1974, 23, 173–182. [Google Scholar] [CrossRef] [PubMed]
- Barlow, P.W. Differential cell division in human X chromosome mosaics. Humangenetik 1972, 14, 122–127. [Google Scholar] [CrossRef]
- Cloutier, J.M.; Mahadevaiah, S.K.; El Inati, E.; Nussenzweig, A.; Tóth, A.; Turner, J.M. Histone H2AFX links meiotic chromosome asynapsis to prophase I oocyte loss in mammals. PLoS Genet. 2015, 11, e1005462. [Google Scholar] [CrossRef]
- Jackson-Cook, C. A hypothesis: Could telomere length and/or epigenetic alterations contribute to infertility in females with Turner syndrome? Am. J. Med. Genet. C Semin. Med. Genet. 2019, 181, 108–116. [Google Scholar] [CrossRef]
- Julien, E.; Herr, W. Proteolytic processing is necessary to separate and ensure proper cell growth and cytokinesis functions of HCF-1. EMBO J. 2003, 22, 2360–2369. [Google Scholar] [CrossRef]
- Levine, M.S.; Holland, A.J. The impact of mitotic errors on cell proliferation and tumorigenesis. Genes Dev. 2018, 32, 620–638. [Google Scholar] [CrossRef] [PubMed]
- Pfau, S.J.; Silberman, R.E.; Knouse, K.A.; Amon, A. Aneuploidy impairs hematopoietic stem cell fitness and is selected against in regenerating tissues in vivo. Genes Dev. 2016, 30, 1395–1408. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, J.; Krag-Olsen, B. Cell selection in vivo. Follow-up of nine unselected mixoploid children. Hum. Genet. 1980, 55, 357–361. [Google Scholar] [CrossRef] [PubMed]
- Guttenbach, M.; Koschorz, B.; Bernthaler, U.; Grimm, T.; Schmid, M. Sex chromosome loss and aging: In situ hybridization studies on human interphase nuclei. Am. J. Hum. Genet. 1995, 57, 1143–1150. [Google Scholar] [PubMed]
- Russell, L.M.; Strike, P.; Browne, C.E.; Jacobs, P.A. X chromosome loss and ageing. Cytogenet. Genome Res. 2007, 116, 181–185. [Google Scholar] [CrossRef]
- Denes, A.M.; Landin-Wilhelmsen, K.; Wettergren, Y.; Bryman, I.; Hanson, C. The proportion of diploid 46,XX cells increases with time in women with Turner syndrome—A 10-year follow-up study. Genet. Test Mol. Biomarkers 2015, 19, 82–87. [Google Scholar] [CrossRef]
- Raudsepp, T.; Das, P.J.; Avila, F.; Chowdhary, B.P. The pseudoautosomal region and sex chromosome aneuploidies in domestic species. Sex Dev. 2012, 6, 72–83. [Google Scholar] [CrossRef]
- Umeyama, K.; Nakano, K.; Matsunari, H.; Yamada, T.; Hasegawa, K.; Tang, K.; Tokuyama, Y.; Watanabe, M.; Nagaya, M.; Nagashima, H. The phenotype of a pig with monosomy X resembling Turner syndrome symptoms: A case report. J. Reprod. Dev. 2019, 65, 231–237. [Google Scholar] [CrossRef]
- Flaquer, A.; Rappold, G.A.; Wienker, T.F.; Fischer, C. The human pseudoautosomal regions: A review for genetic epidemiologists. Eur. J. Hum. Genet. 2008, 16, 771–779. [Google Scholar] [CrossRef]
- Lopes, A.M.; Burgoyne, P.S.; Ojarikre, A.; Bauer, J.; Sargent, C.A.; Amorim, A.; Affara, N.A. Transcriptional changes in response to X chromosome dosage in the mouse: Implications for X inactivation and the molecular basis of Turner Syndrome. BMC Genom. 2010, 11, 82. [Google Scholar] [CrossRef]
- Ashworth, A.; Rastan, S.; Lovell-Badge, R.; Kay, G. X-chromosome inactivation may explain the difference in viability of XO humans and mice. Nature 1991, 351, 406–408. [Google Scholar] [CrossRef] [PubMed]
- Rao, E.; Weiss, B.; Fukami, M.; Rump, A.; Niesler, B.; Mertz, A.; Muroya, K.; Binder, G.; Kirsch, S.; Winkelmann, M.; et al. Pseudoautosomal deletions encompassing a novel homeobox gene cause growth failure in idiopathic short stature and Turner syndrome. Nat. Genet. 1997, 16, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Probst, F.J.; Cooper, M.L.; Cheung, S.W.; Justice, M.J. Genotype, phenotype, and karyotype correlation in the XO mouse model of Turner Syndrome. J. Hered. 2008, 99, 512–517. [Google Scholar] [CrossRef] [PubMed]
- Urbach, A.; Benvenisty, N. Studying early lethality of 45, XO (Turner’s syndrome) embryos using human embryonic stem cells. PLoS ONE. 2009, 4, e4175. [Google Scholar] [CrossRef] [PubMed]
- Arnold, A.P. The mouse as a model of fundamental concepts related to Turner syndrome. Am. J. Med. Genet. C Semin. Med. Genet. 2019, 181, 76–85. [Google Scholar] [CrossRef]
- Burgoyne, P.S.; Biggers, J.D. The consequences of X-dosage deficiency in the germ line: Impaired development in vitro of preimplantation embryos from XO mice. Dev. Biol. 1976, 51, 109–117. [Google Scholar] [CrossRef]
- Lynn, P.M.; Davies, W. The 39,XO mouse as a model for the neurobiology of Turner syndrome and sex-biased neuropsychiatric disorders. Behav. Brain Res. 2007, 179, 173–182. [Google Scholar] [CrossRef]
- Nagaoka, S.I.; Hassold, T.J.; Hunt, P.A. Human aneuploidy: Mechanisms and new insights into an age-old problem. Nat. Rev. Genet. 2012, 13, 493–504. [Google Scholar] [CrossRef]
- Hook, E.B.; Topol, B.B.; Cross, P.K. The natural history of cytogenetically abnormal fetuses detected at midtrimester amniocentesis which are not terminated electively: New data and estimates of the excess and relative risk of late fetal death associated with 47, +21 and some other abnormal karyotypes. Am. J. Hum. Genet. 1989, 45, 855–856. [Google Scholar]
- Hook, E.B.; Warburton, D. Turner syndrome revisited: Review of new data supports the hypothesis that all viable 45, X cases are cryptic mosaics with a rescue cell line, implying an origin by mitotic loss. Hum. Genet. 2014, 133, 417–424. [Google Scholar] [CrossRef]
- Hook, E.B.; Warburton, D. The distribution of chromosomal genotypes associated with Turner’s syndrome: Livebirth prevalence rates and evidence for diminished fetal mortality and severity in genotypes associated with structural X abnormalities or mosaicism. Hum. Genet. 1983, 64, 24–27. [Google Scholar] [CrossRef] [PubMed]
- Hassold, T.; Hunt, P. To err (meiotically) is human: The genesis of human aneuploidy. Nat. Rev. Genet. 2001, 2, 280–291. [Google Scholar] [CrossRef] [PubMed]
- Tannus, S.; Cohen, Y.; Henderson, S.; Al Ma’mari, N.; Shavit, T.; Son, W.Y.; Dahan, M.H. Fresh transfer of Day 5 slow-growing embryos versus deferred transfer of vitrified, fully expanded Day 6 blastocysts: Which is the optimal approach? Hum. Reprod. 2019, 34, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Canki, N.; Warburton, D.; Byrne, J. Morphological characteristics of monosomy X in spontaneous abortions. Ann. Genet. 1988, 31, 4–13. [Google Scholar] [CrossRef]
- Vanneste, E.; Voet, T.; Le Caignec, C.; Ampe, M.; Konings, P.; Melotte, C.; Debrock, S.; Amyere, M.; Vikkula, M.; Schuit, F.; et al. Chromosome instability is common in human cleavage-stage embryos. Nat Med. 2009, 15, 577–583. [Google Scholar] [CrossRef]
- Hall, H.; Hunt, P.; Hassold, T. Meiosis and sex chromosome aneuploidy: How meiotic errors cause aneuploidy; how aneuploidy causes meiotic errors. Curr. Opin. Genet. Dev. 2006, 16, 323–329. [Google Scholar] [CrossRef]
- Rudd, M.K.; Schleede, J.B.; Williams, S.R.; Lee, K.; Laffin, J.; Pasion, R.; Papenhausen, P.R. Monosomy X rescue explains discordant NIPT results and leads to uniparental isodisomy. Prenat Diagn. 2018, 38, 920–923. [Google Scholar] [CrossRef]
- Álvarez-Nava, F.; Lanes, R. Epigenetics in Turner syndrome. Clin. Epigenet. 2018, 10, 45. [Google Scholar] [CrossRef]
- FitzSimmons, J.; Fantel, A.; Shepard, T. Growth parameters in mid-trimester fetal Turner syndrome. Early Hum. Dev. 1994, 38, 121–129. [Google Scholar] [CrossRef]
- Fiot, E.; Zenaty, D.; Boizeau, P.; Haigneré, J.; Dos Santos, S.; Léger, J.; French Turner Syndrome Study Group. X-chromosome gene dosage as a determinant of impaired pre and postnatal growth and adult height in Turner syndrome. Eur. J. Endocrinol. 2016, 174, 281–288. [Google Scholar] [CrossRef]
- Wisniewski, A.; Milde, K.; Stupnicki, R.; Szufladowicz-Wozniak, J. Weight deficit at birth and Turner’s syndrome. J. Pediatr. Endocrinol. Metab. 2007, 20, 607–613. [Google Scholar] [CrossRef] [PubMed]
- Davenport, M.L.; Punyasavatsut, N.; Stewart, P.W.; Gunther, D.F.; Sävendahl, L.; Sybert, V.P. Growth failure in early life: An important manifestation of Turner syndrome. Horm. Res. 2002, 57, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, H.; Banzai, M.; Yamauchi, T. Developmental retardation of XO mouse embryos at mid-gestation. J. Reprod. Fertil. 1999, 115, 263–267. [Google Scholar] [CrossRef] [PubMed]
- Burgoyne, P.S.; Tam, P.P.L.; Evans, E.P. Retarded development of XO conceptuses during early pregnancy in the mouse. J. Reprod. Fertil. 1983, 68, 387–393. [Google Scholar] [CrossRef] [PubMed]
- Jamieson, R.V.; Tan, S.S.; Tam, P.P.L. Retarded postimplantation development of X0 mouse embryos: Impact of the parental origin of the monosomic X chromosome. Dev. Biol. 1998, 201, 13–25. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zechner, U.; Reule, M.; Burgoyne, P.S.; Schubert, A.; Orth, A.; Hameister, H.; Fundele, R. Paternal transmission of X-linked dysplasia in mouse interspecific hybrids. Genetics 1997, 146, 1399–1405. [Google Scholar] [CrossRef]
- Ishikawa, H.; Rattigan, A.; Fundele, R.; Burgoyne, P.S. Effects of sex chromosome dosage on placental size in mice. Biol. Reprod. 2003, 69, 483–488. [Google Scholar] [CrossRef]
- Varmuza, S.; Prideaux, V.; Kothary, R.; Rossant, J. Polytene chromosomes in mouse trophoblast giant cells. Development 1988, 102, 127–134. [Google Scholar] [CrossRef]
- Clement-Jones, M.; Schiller, S.; Rao, E.; Blaschke, R.J.; Zuniga, A.; Zeller, R.; Robson, S.C.; Binder, G.; Glass, I.; Strachan, T.; et al. The short stature homeobox gene SHOX is involved in skeletal abnormalities in Turner syndrome. Hum. Mol. Genet. 2000, 9, 695–702. [Google Scholar] [CrossRef]
- Zinn, A.R.; Tonk, V.S.; Chen, Z.; Flejter, W.L.; Gardner, H.A.; Guerra, R.; Kushner, H.; Schwartz, S.; Sybert, V.P.; Van Dyke, D.L.; et al. Evidence for a Turner syndrome locus or loci at Xp11.2-p22.1. Am. J. Hum. Genet. 1998, 63, 1757–1766. [Google Scholar] [CrossRef]
- Therman, E.; Susman, B. The similarity of phenotypic effects caused by Xp and Xq deletions in the human female, a hypothesis. Hum. Genet. 1990, 85, 175–183. [Google Scholar] [CrossRef] [PubMed]
- Prueih, R.L.; Ross, J.L.; Zinn, A.R. Physical mapping of nine Xq translocation breakpoints and identification of XPNPEP2 as a premature ovarian failure candidate gene. Cytogenet. Cell Genet. 2000, 89, 44–50. [Google Scholar]
- Burgoyne, P.S.; Ojarikre, O.A.; Turner, J.M. Evidence that postnatal growth retardation in XO mice is due to haploinsufficiency for a non-PAR X gene. Cytogenet. Genome Res. 2002, 99, 252–256. [Google Scholar] [CrossRef]
- Haverkamp, F.; Wölfle, J.; Zerres, K.; Butenandt, O.; Amendt, P.; Hauffa, B.P.; Weimann, E.; Bettendorf, M.; Keller, E.; Mühlenberg, R.; et al. Growth retardation in Turner syndrome: Aneuploidy, rather than specific gene loss, may explain growth failure. J. Clin. Endocrinol. Metab. 1999, 84, 4578–4582. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Pritchard, M.A.; Kola, I. The “gene dosage effect” hypothesis versus the “amplified developmental instability” hypothesis in Down syndrome. J. Neural. Transm. Suppl. 1999, 57, 293–303. [Google Scholar] [PubMed]
- Trolle, C.; Nielsen, M.M.; Skakkebæk, A.; Lamy, P.; Vang, S.; Hedegaard, J.; Nordentoft, I.; Ørntoft, T.F.; Pedersen, J.S.; Gravholt, C.H. Widespread DNA hypomethylation and differential gene expression in Turner syndrome. Sci. Rep. 2016, 6, 34220. [Google Scholar]
- Kelkar, A.; Deobagkar, D. Methylation profile of genes on the human X chromosome. Epigenetics 2010, 5, 612–618. [Google Scholar] [CrossRef][Green Version]
- Rajpathak, S.N.; Vellarikkal, S.K.; Patowary, A.; Scaria, V.; Sivasubbu, S.; Deobagkar, D.D. Human 45,X fibroblast transcriptome reveals distinct differentially expressed genes including long noncoding RNAs potentially associated with the pathophysiology of Turner syndrome. PLoS ONE. 2014, 9, e100076. [Google Scholar] [CrossRef]
- Zhang, R.; Hao, L.; Wang, L.; Chen, M.; Li, W.; Li, R.; Yu, J.; Xiao, J.; Wu, J. Gene expression analysis of induced pluripotent stem cells from aneuploid chromosomal syndromes. BMC Genom. 2013, 14 (Suppl S5), S8. [Google Scholar] [CrossRef]
- Sharma, A.; Jamil, M.A.; Nuesgen, N.; Schreiner, F.; Priebe, L.; Hoffmann, P.; Herns, S.; Nöthen, M.M.; Fröhlich, H.; Oldenburg, J.; et al. DNA methylation signature in peripheral blood reveals distinct characteristics of human X chromosome numerical aberrations. Clin. Epigenet. 2015, 7, 76. [Google Scholar] [CrossRef]
- Li, W.; Wang, X.; Fan, W.; Zhao, P.; Chan, Y.C.; Chen, S.; Zhang, S.; Guo, X.; Zhang, Y.; Li, Y.; et al. Modeling abnormal early development with induced pluripotent stem cells from aneuploid syndromes. Hum. Mol. Genet. 2012, 21, 32–45. [Google Scholar] [CrossRef] [PubMed]
- Dória, S.; Sousa, M.; Fernandes, S.; Ramalho, C.; Brandão, O.; Matias, A.; Barros, A.; Carvalho, F. Gene expression pattern of IGF2, PHLDA2, PEG10 and CDKN1C imprinted genes in spontaneous miscarriages or fetal deaths. Epigenetics 2010, 5, 444–450. [Google Scholar] [CrossRef]
- Jilka, R.L.; Weinstein, R.S.; Bellido, T.; Roberson, P.; Parfitt, A.M.; Manolagas, S.C. Increased bone formation by prevention of osteoblast apoptosis with parathyroid hormone. J. Clin. Investig. 1999, 104, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Gravholt, C.H.; Andersen, N.H.; Conway, G.S.; Dekkers, O.M.; Geffner, M.E.; Klein, K.O.; Lin, A.E.; Mauras, N.; Quigley, C.A.; Rubin, K.; et al. Clinical practice guidelines for the care of girls and women with Turner syndrome: Proceedings from the 2016 Cincinnati International Turner Syndrome Meeting. Eur. J. Endocrinol. 2017, 177, G1–G70. [Google Scholar] [CrossRef] [PubMed]
- Rochiccioli, P.; David, M.; Malpuech, G.; Colle, M.; Limal, J.M.; Battin, J.; Mariani, R.; Sultan, C.; Nivelon, J.L.; Simonin, G.; et al. Study of final height in Turner’s syndrome: Ethnic and genetic influences. Acta Paediatr. 1994, 83, 305–308. [Google Scholar] [CrossRef] [PubMed]
- Cooke, D.W.; Divall, S.A.; Radovick, S. Normal and Aberrant Growth in Children. In Williams Textbook of Endocrinolog, 14th ed.; Rosen, C.J., Melmed, S., Auchus, R.J., Goldfine, A.B., Koenig, R.J., Eds.; Elsevier: Philadelphia, PA, USA; pp. 964–1073.
- Barrenäs, M.L.; Landin-Wilhelmsen, K.; Hanson, C. Ear and hearing in relation to genotype and growth in Turner syndrome. Hear. Res. 2000, 144, 21–28. [Google Scholar] [CrossRef]
- Brook, C.G.; Gasser, T.; Werder, E.A.; Prader, A.; Vanderschueren-Lodewykx, M.A. Height correlations between parents and mature offspring in normal subjects and in subjects with Turner’s and Klinefelter’s and other syndromes. Ann. Hum. Biol. 1977, 4, 17–22. [Google Scholar] [CrossRef]
- Álvarez-Nava, F.; Lanes, R.; Quintero, J.M.; Miras, M.; Fideleff, H.; Mericq, V.; Marcano, H.; Zabala, W.; Soto, M.; Pardo, T.; et al. Effect of the Parental Origin of the X-Chromosome on the Clinical Features, Associated Complications, the Two-Year-Response to Growth Hormone (RhGH) and the Biochemical Profile in Patients with Turner Syndrome. Int. J. Pediatr. Endocrinol. 2013, 10, 2013. [Google Scholar] [CrossRef]
- Binder, G. Short stature due to SHOX deficiency: Genotype, phenotype, and therapy. Horm. Res. Paediatr. 2011, 75, 81–89. [Google Scholar] [CrossRef]
- Ross, J.L.; Long, L.M.; Loriaux, D.L.; Cutler, G.B., Jr. Growth hormone secretory dynamics in Turner syndrome. J. Pediatr. 1985, 106, 202–206. [Google Scholar] [CrossRef]
- Cuttler, L.; Van Vliet, G.; Conte, F.A.; Kaplan, S.L.; Grumbach, M.M. Somatomedin-C levels in children and adolescents with gonadal dysgenesis: Differences from age-matched normal females and effect of chronic estrogen replacement therapy. J. Clin. Endocrinol. Metab. 1985, 60, 1087–1092. [Google Scholar] [CrossRef] [PubMed]
- Gravholt, C.H.; Frystyk, J.; Flyvbjerg, A.; Orskov, H.; Christiansen, J.S. Reduced free IGF-I and increased IGFBP-3 proteolysis in Turner syndrome: Modulation by female sex steroids. Am. J. Physiol. Endocrinol. Metab. 2001, 280, E308–E314. [Google Scholar] [CrossRef] [PubMed]
- Gravholt, C.H.; Svenstrup, B.; Bennett, P.; Christiansen, J.S. Reduced androgen levels in adult Turner syndrome: Influence of female sex steroids and growth hormone status. Clin. Endocrinol. 1999, 50, 791–800. [Google Scholar] [CrossRef] [PubMed]
- Rubin, K. Turner Syndrome and Osteoporosis: Mechanisms and Prognosis. Pediatrics 1998, 102, 481–485. [Google Scholar] [CrossRef] [PubMed]
- Bercu, B.B.; Kramer, S.S.; Bode, H.H. A useful radiologic sign for the diagnosis of Turner’s syndrome. Pediatrics 1976, 58, 737–739. [Google Scholar] [CrossRef]
- Frisancho, A.R.; Garn, S.M.; Ascoli, W. Subperiosteal and endosteal bone apposition during adolescence. Hum. Biol. 1970, 42, 639–664. [Google Scholar]
- Bakalov, V.K.; Bondy, C.A. Fracture risk and bone mineral density in Turner syndrome. Rev. Endocr. Metab. Disord. 2008, 9, 145–151. [Google Scholar] [CrossRef]
- Landin-Wilhelmsen, K.; Bryman, I.; Windh, M.; Wilhelmsen, L. Osteoporosis and fractures in Turner syndrome-importance of growth promoting and oestrogen therapy. Clin. Endocrinol. 1999, 51, 497–502. [Google Scholar] [CrossRef]
- El-Mansoury, M.; Barrenäs, M.L.; Bryman, I.; Hanson, C.; Larsson, C.; Wilhelmsen, L.; Landin-Wilhelmsen, K. Chromosomal mosaicism mitigates stigmata and cardiovascular risk factors in Turner syndrome. Clin. Endocrinol. 2007, 66, 744–751. [Google Scholar] [CrossRef]
- Gravholt, C.H.; Vestergaard, P.; Hermann, A.P.; Mosekilde, L.; Brixen, K.; Christiansen, J.S. Increased fracture rates in Turner’s syndrome: A nationwide questionnaire survey. Clin. Endocrinol. 2003, 59, 89–96. [Google Scholar] [CrossRef]
- Ensrud, K.E.; Lipschutz, R.C.; Cauley, J.A.; Seeley, D.; Nevitt, M.C.; Scott, J.; Orwoll, E.S.; Genant, H.K.; Cummings, S.R. Body size and hip fracture risk in older women: A prospective study. Study of Osteoporotic Fractures Research Group. Am. J. Med. 1997, 103, 274–280. [Google Scholar] [CrossRef]
- Jilka, R.L.; Weinstein, R.S.; Bellido, T.; Parfitt, A.M.; Manolagas, S.C. Osteoblast programmed cell death (apoptosis): Modulation by growth factors and cytokines. J. Bone Miner. Res. 1998, 13, 793–802. [Google Scholar] [CrossRef] [PubMed]
- Gravholt, C.H.; Lauridsen, A.L.; Brixen, K.; Mosekilde, L.; Heickendorff, L.; Christiansen, J.S. Marked disproportionality in bone size and mineral, and distinct abnormalities in bone markers and calcitropic hormones in adult turner syndrome: A cross-sectional study. J. Clin. Endocrinol. Metab. 2002, 87, 2798–2808. [Google Scholar] [CrossRef] [PubMed]
- Nissen, N.; Gravholt, C.H.; Abrahamsen, B.; Hauge, E.M.; Jensen, J.E.; Mosekilde, L.; Brixen, K. Disproportional geometry of the proximal femur in patients with Turner syndrome: A cross-sectional study. Clin. Endocrinol. 2007, 67, 897–903. [Google Scholar] [CrossRef]
- Mortensen, K.H.; Andersen, N.H.; Gravholt, C.H. Cardiovascular phenotype in Turner syndrome—Integrating cardiology, genetics, and endocrinology. Endocr. Rev. 2012, 33, 677–714. [Google Scholar] [CrossRef]
- Surerus, E.; Huggon, I.C.; Allan, L.D. Turner’s syndrome in fetal life. Ultrasound Obstet. Gynecol. 2003, 22, 264–267. [Google Scholar] [CrossRef] [PubMed]
- Clark, E.B. Neck web and congenital heart defects: A pathogenic association in 45 X-O Turner syndrome? Teratology 1984, 29, 355–361. [Google Scholar] [CrossRef]
- Mazzanti, L.; Cacciari, E. Congenital heart disease in patients with Turner’s syndrome. Italian Study Group for Turner Syndrome (ISGTS). J. Pediatr. 1998, 133, 688–692. [Google Scholar] [CrossRef]
- Loscalzo, M.L.; Van, P.L.; Ho, V.B.; Bakalov, V.K.; Rosing, D.R.; Malone, C.A.; Dietz, H.C.; Bondy, C.A. Association between fetal lymphedema and congenital cardiovascular defects in Turner syndrome. Pediatrics 2005, 115, 732–735. [Google Scholar] [CrossRef]
- Smith, D.W.; Jones, K.L. Recognizable Patterns of Human Malformation: Genetic, Embryologic and Clinical Aspects, 6th ed.; Elsevier Saunders: Philadelphia, PA, USA, 2006; 738p. [Google Scholar]
- Lacro, R.V.; Jones, K.L.; Benirschke, K. Coarctation of the Aorta in Turner Syndrome: A Pathologic Study of Fetuses with Nuchal Cystic Hygromas, Hydrops Fetalis, and Female Genitalia. Pediatrics 1988, 81, 445–451. [Google Scholar]
- Berdahl, L.D.; Wenstrom, K.D.; Hanson, J. Web neck anomaly and its association with congenital heart disease. Am. J. Med. Genet. 1995, 56, 304–307. [Google Scholar] [CrossRef] [PubMed]
- Miyabara, S.; Nakayama, M.; Suzumori, K.; Yonemitsu, N.; Sugihara, H. Developmental analysis of cardiovascular system of 45, X fetuses with cystic hygroma. Am. J. Med. Genet. 1997, 68, 135–141. [Google Scholar] [CrossRef]
- Van Der Putte, S.C. Lymphatic malformation in human fetuses. A study of fetuses with Turner’s syndrome or status Bonnevie-Ullrich. Virchows Arch. A Pathol. Anat. Histol. 1977, 376, 233–246. [Google Scholar]
- Sachdev, V.; Matura, L.A.; Sidenko, S.; Ho, V.B.; Arai, A.E.; Rosing, D.R.; Bondy, C.A. Aortic valve disease in Turner syndrome. J. Am. Coll. Cardiol. 2008, 51, 1904–1909. [Google Scholar] [CrossRef] [PubMed]
- Kappetein, A.P.; Gittenberger-De Groot, A.C.; Zwinderman, A.H.; Rohmer, J.; Poelmann, R.E.; Huysmans, H.A. The neural crest as a possible pathogenetic factor in coarctation of the aorta and bicuspid aortic valve. J. Thorac. Cardiovasc. Surg. 1991, 102, 830–836. [Google Scholar] [CrossRef]
- Prakash, S.K. The impact of somatic mosaicism on bicuspid aortic valve and aortic dissection in Turner Syndrome. Am. J. Med. Genet. C Semin. Med. Genet. 2019, 181, 7–12. [Google Scholar] [CrossRef]
- Wu, B.; Wang, Y.; Lui, W.; Langworthy, M.; Tompkins, K.L.; Hatzopoulos, A.K.; Baldwin, H.S.; Zhou, B. Nfatc1 coordinates valve endocardial cell lineage development required for heart valve formation. Circ. Res. 2011, 109, 183–192. [Google Scholar] [CrossRef]
- El-Hamamsy, I.; Yacoub, M.H. Cellular and molecular mechanisms of thoracic aortic aneurysms. Nat. Rev. Cardiol. 2009, 6, 771–786. [Google Scholar] [CrossRef]
- Risau, W.; Lemmon, V. Changes in the vascular extracellular matrix during embryonic vasculogenesis and angiogenesis. Dev. Biol. 1988, 125, 441–450. [Google Scholar] [CrossRef]
- Corbitt, H.; Gutierrez, J.; Silberbach, M.; Maslen, C.L. The genetic basis of Turner syndrome aortopathy. Am. J. Med. Genet. C Semin. Med. Genet. 2019, 181, 117–125. [Google Scholar] [CrossRef]
- Gittenberger-De Groot, A.C.; Azhar, M.; Molin, D.G. Transforming growth factor beta-SMAD2 signaling and aortic arch development. Trends Cardiovasc. Med. 2006, 16, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Gomez, D.; Al Haj Zen, A.; Borges, L.F.; Philippe, M.; Gutierrez, P.S.; Jondeau, G.; Michel, J.B.; Vranckx, R. Syndromic and non-syndromic aneurysms of the human ascending aorta share activation of the Smad2 pathway. J. Pathol. 2009, 218, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Arepalli, S.; Cheng, C.M.; Bakalov, V.K.; Bondy, C.A. Perturbation of the transforming growth factor β system in Turner syndrome. Beijing Da Xue Xue Bao Yi Xue Ban 2012, 44, 720–724. [Google Scholar] [PubMed]
- Hankus, M.; Soltysik, K.; Szeliga, K.; Antosz, A.; Drosdzol-Cop, A.; Wilk, K.; Zachurzok, A.; Malecka-Tendera, E.; Gawlik, A.M. Prediction of Spontaneous Puberty in Turner Syndrome Based on Mid-Childhood Gonadotropin Concentrations, Karyotype, and Ovary Visualization: A Longitudinal Study. Horm. Res. Paediatr. 2018, 89, 90–97. [Google Scholar] [CrossRef]
- Pasquino, A.M.; Passeri, F.; Pucarelli, I.; Segni, M.; Municchi, G. Spontaneous pubertal development in Turner’s syndrome. Italian Study Group for Turner’s Syndrome. J. Clin. Endocrinol. Metab. 1997, 82, 1810–1813. [Google Scholar] [PubMed]
- Hadnott, T.N.; Gould, H.N.; Gharib, A.M.; Bondy, C.A. Outcomes of spontaneous and assisted pregnancies in Turner syndrome: The U.S. National Institutes of Health experience. Fertil. Steril. 2011, 95, 2251–2256. [Google Scholar] [CrossRef]
- Bryman, I.; Sylvén, L.; Berntorp, K.; Innala, E.; Bergström, I.; Hanson, C.; Oxholm, M.; Landin-Wilhelmsen, K. Pregnancy Rate and Outcome in Swedish Women with Turner Syndrome. Fertil. Steril. 2011, 95, 2507–2510. [Google Scholar] [CrossRef]
- Bernard, V.; Donadille, B.; Zenaty, D.; Courtillot, C.; Salenave, S.; Brac de la Perrière, A.; Albarel, F.; Fèvre, A.; Kerlan, V.; Brue, T.; et al. Spontaneous fertility and pregnancy outcomes amongst 480 women with Turner syndrome. Hum. Reprod. 2016, 31, 782–788. [Google Scholar] [CrossRef]
- Modi, D.N.; Sane, S.; Bhartiya, D. Accelerated germ cell apoptosis in sex chromosome aneuploid fetal human gonads. Mol. Hum. Reprod. 2003, 9, 219–225. [Google Scholar] [CrossRef]
- Balen, A.H.; Harris, S.E.; Chambers, E.L.; Picton, H.M. Conservation of fertility and oocyte genetics in a young woman with mosaic Turner syndrome. BJOG 2010, 117, 238–242. [Google Scholar] [CrossRef]
- Grynberg, M.; Bidet, M.; Benard, J.; Poulain, M.; Sonigo, C.; Cédrin-Durnerin, I.; Polak, M. Fertility preservation in Turner syndrome. Fertil. Steril. 2016, 105, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Rivelis, C.F.; Coco, R.; Bergada, C. Ovarian differentiation in Turner’s syndrome. J. Genet. Hum. 1978, 26, 69–83. [Google Scholar] [PubMed]
- Mamsen, L.S.; Lutterodt, M.C.; Andersen, E.W.; Byskov, A.G.; Andersen, C.Y. Germ cell numbers in human embryonic and fetal gonads during the first two trimesters of pregnancy: Analysis of six published studies. Hum. Reprod. 2011, 26, 2140–2145. [Google Scholar] [CrossRef]
- Jacobs, P.; Dalton, P.; James, R.; Mosse, K.; Power, M.; Robinson, D.; Skuse, D. Turner syndrome: A cytogenetic and molecular study. Ann. Hum. Genet. 1997, 61, 471–483. [Google Scholar] [CrossRef]
- Hanson, L.; Bryman, I.; Barrenäs, M.L.; Janson, P.O.; Wahlström, J.; Albertsson-Wikland, K.; Hanson, C. Genetic analysis of mosaicism in 53 women with Turner syndrome. Hereditas 2001, 134, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Hreinsson, J.G.; Otala, M.; Fridström, M.; Borgström, B.; Rasmussen, C.; Lundqvist, M.; Tuuri, T.; Simberg, N.; Mikkola, M.; Dunkel, L.; et al. Follicles are found in the ovaries of adolescent girls with Turner’s syndrome. J. Clin. Endocrinol. Metab. 2002, 87, 3618–3623. [Google Scholar]
- Fechner, P.Y.; Davenport, M.L.; Qualy, R.L.; Ross, J.L.; Gunther, D.F.; Eugster, E.A.; Huseman, C.; Zagar, A.J.; Quigley, C.A.; Toddler Turner Study Group. Differences in follicle-stimulating hormone secretion between 45, X monosomy Turner syndrome and 45, X/46, XX mosaicism are evident at an early age. J. Clin. Endocrinol. Metab. 2006, 91, 4896–4902. [Google Scholar] [CrossRef]
- Burgoyne, P.S.; Baker, T.G. Perinatal oocyte loss in XO mice and its implications for the aetiology of gonadal dysgenesis in XO women. J. Reprod. Fertil. 1985, 75, 633–645. [Google Scholar] [CrossRef]
- Reynaud, K.; Cortvrindt, R.; Verlinde, F.; De Schepper, J.; Bourgain, C.; Smitz, J. Number of ovarian follicles in human fetuses with the 45, X karyotype. Fertil. Steril. 2004, 81, 1112–1119. [Google Scholar] [CrossRef]
- Speed, R.M. Oocyte Development in XO Foetuses of Man and Mouse: The Possible Role of Heterologous X-Chromosome Pairing in Germ Cell Survival. Chromosoma 1986, 94, 115–124. [Google Scholar] [CrossRef]
- Kidder, G.M.; Mhawi, A.A. Gap junctions and ovarian folliculogenesis. Reproduction 2002, 123, 613–620. [Google Scholar] [CrossRef] [PubMed]
- Peek, R.; Schleedoorn, M.; Smeets, D.; van de Zande, G.; Groenman, F.; Braat, D.; van der Velden, J.; Fleischer, K. Ovarian follicles of young patients with Turner’s syndrome contain normal oocytes but monosomic 45, X granulosa cells. Hum. Reprod. 2019, 34, 1686–1696. [Google Scholar] [CrossRef] [PubMed]
- Hirshfield, A.N. Development of Follicles in the Mammalian Ovary. Int. Rev. Cytol. 1991, 124, 43–101. [Google Scholar] [PubMed]
- Kulkarni, R.N. New insights into the roles of insulin/IGF-I in the development and maintenance of beta-cell mass. Rev. Endocr. Metab. Disord. 2005, 6, 199–210. [Google Scholar] [CrossRef]
- Singh, S.P.; Ninov, N. The triumvirate of beta-cell regeneration: Solutions and bottlenecks to curing diabetes. Int. J. Dev. Biol. 2018, 62, 453–464. [Google Scholar] [CrossRef]
- Van Haeften, T.W.; Twickler, T.B. Insulin-like growth factors and pancreas beta cells. Eur. J. Clin. Investig. 2004, 34, 249–255. [Google Scholar] [CrossRef]
- DeFronzo, R.A.; Ferrannini, E.; Groop, L.; Henry, R.R.; Herman, W.H.; Holst, J.J.; Hu, F.B.; Kahn, C.R.; Raz, I.; Shulman, G.I.; et al. Type 2 Diabetes Mellitus. Nat. Rev. Dis. Primers 2015, 1, 15019. [Google Scholar] [CrossRef]
- DeFronzo, R.A.; Eldor, R.; Abdul-Ghani, M. Pathophysiologic approach to therapy in patients with newly diagnosed type 2 diabetes. Diabetes Care 2013, 36, S127–S138. [Google Scholar] [CrossRef]
- Davis, S.M.; Geffner, M.E. Cardiometabolic health in Turner syndrome. Am. J. Med. Genet. C Semin. Med. Genet. 2019, 181, 52–58. [Google Scholar] [CrossRef]
- Agudo, J.; Ayuso, E.; Jimenez, V.; Salavert, A.; Casellas, A.; Tafuro, S.; Haurigot, V.; Ruberte, J.; Segovia, J.C.; Bueren, J.; et al. IGF-I Mediates Regeneration of Endocrine Pancreas by Increasing Beta Cell Replication through Cell Cycle Protein Modulation in Mice. Diabetologia 2008, 51, 1862–1872. [Google Scholar] [CrossRef]
- Álvarez-Nava, F.; Bastidas, D.; Racines-Orbe, M.; Guarderas, J. Insulin Sensitivity and Pancreatic β-Cell Function in Ecuadorian Women with Turner Syndrome. Front. Endocrinol. 2020, 11, 482. [Google Scholar] [CrossRef] [PubMed]
- Gershengorn, M.C.; Hardikar, A.A.; Wei, C.; Geras-Raaka, E.; Marcus-Samuels, B.; Raaka, B.M. Epithelial-to-mesenchymal transition generates proliferative human islet precursor cells. Science 2004, 306, 2261–2264. [Google Scholar] [CrossRef] [PubMed]
- Dor, Y.; Brown, J.; Martinez, O.L.; Melton, D.A. Adult pancreatic beta-cells are formed by self-duplication rather than stem-cell differentiation. Nature 2004, 429, 41–46. [Google Scholar] [CrossRef] [PubMed]
- Hutaff-Lee, C.; Bennett, E.; Howell, S.; Tartaglia, N. Clinical developmental, neuropsychological, and social-emotional features of Turner syndrome. Am. J. Med. Genet. C Semin. Med. Genet. 2019, 181, 126–134. [Google Scholar] [CrossRef]
- Ross, J.; Roeltgen, D.; Zinn, A. Cognition and the sex chromosomes: Studies in Turner syndrome. Horm. Res. 2006, 65, 47–56. [Google Scholar] [CrossRef]
- McCauley, E.; Kay, T.; Ito, J.; Treder, R. The Turner Syndrome: Cognitive Deficits, Affective Discrimination, and Behavior Problems. Child. Dev. 1987, 58, 464–473. [Google Scholar] [CrossRef]
- Murphy, D.G.; Allen, G.; Haxby, J.V.; Largay, K.A.; Daly, E.; White, B.J.; Powell, C.M.; Schapiro, M.B. The effects of sex steroids, and the X chromosome, on female brain function: A study of the neuropsychology of adult Turner syndrome. Neuropsychologia 1994, 32, 1309–1323. [Google Scholar] [CrossRef]
- Hart, S.J.; Davenport, M.L.; Hooper, S.R.; Belger, A. Visuospatial executive function in Turner syndrome: Functional MRI and neurocognitive findings. Brain 2006, 129, 1125–1136. [Google Scholar] [CrossRef]
- Knickmeyer, R.C.; Hooper, S.R. The deep biology of cognition: Moving toward a comprehensive neurodevelopmental model of Turner syndrome. Am. J. Med. Genet. C Semin. Med. Genet. 2019, 181, 91–99. [Google Scholar] [CrossRef]
- Ross, J.; Zinn, A.; McCauley, E. Neurodevelopmental and psychosocial aspects of Turner syndrome. Ment. Retard. Dev. Disabil. Res. Rev. 2000, 6, 135–141. [Google Scholar] [CrossRef]
- Reiss, A.L.; Freund, L.; Plotnick, L.; Baumgardner, T.; Green, K.; Sozer, A.C.; Reader, M.; Boehm, C.; Denckla, M.B. The effects of X monosomy on brain development: Monozygotic twins discordant for Turner’s syndrome. Ann. Neurol. 1993, 34, 95–107. [Google Scholar] [CrossRef] [PubMed]
- Brown, W.E.; Kesler, S.R.; Eliez, S.; Warsofsky, I.S.; Haberecht, M.; Reiss, A.L. A volumetric study of parietal lobe subregions in Turner syndrome. Dev. Med. Child. Neurol. 2004, 46, 607–609. [Google Scholar] [CrossRef] [PubMed]
- Mullaney, R.; Murphy, D. Turner syndrome: Neuroimaging findings: Structural and functional. Dev. Disabil. Res. Rev. 2009, 15, 279–283. [Google Scholar] [CrossRef] [PubMed]
- O’Donoghue, S.; Green, T.; Ross, J.L.; Hallmayer, J.; Lin, X.; Jo, B.; Huffman, L.C.; Hong, D.S.; Reiss, A.L. Brain Development in School-Age and Adolescent Girls: Effects of Turner Syndrome, Estrogen Therapy, and Genomic Imprinting. Biol. Psychiatry 2020, 87, 113–122. [Google Scholar] [CrossRef]
- Green, T.; Hosseini, H.; Piccirilli, A.; Ishak, A.; Grill-Spector, K.; Reiss, A.L. X-Chromosome Insufficiency Alters Receptive Fields across the Human Early Visual Cortex. J. Neurosci. 2019, 39, 8079–8088. [Google Scholar] [CrossRef]
- Murphy, D.G.; DeCarli, C.; Daly, E.; Haxby, J.V.; Allen, G.; White, B.J.; McIntosh, A.R.; Powell, C.M.; Horwitz, B.; Rapoport, S.I.; et al. X-chromosome effects on female brain: A magnetic resonance imaging study of Turner’s syndrome. Lancet 1993, 342, 1197–1200. [Google Scholar] [CrossRef]
- Clark, C.; Klonoff, H.; Hayden, M. Regional cerebral glucose metabolism in Turner syndrome. Can. J. Neurol. Sci. 1990, 17, 140–144. [Google Scholar] [CrossRef]
- Xie, S.; Yang, J.; Zhang, Z.; Zhao, C.; Bi, Y.; Zhao, Q.; Pan, H.; Gong, G. The Effects of the X Chromosome on Intrinsic Functional Connectivity in the Human Brain: Evidence from Turner Syndrome Patients. Cereb. Cortex 2017, 27, 474–484. [Google Scholar] [CrossRef]
- Green, T.; Saggar, M.; Ishak, A.; Hong, D.S.; Reiss, A.L. X-Chromosome Effects on Attention Networks: Insights from Imaging Resting-State Networks in Turner Syndrome. Cereb. Cortex 2018, 28, 3176–3183. [Google Scholar] [CrossRef]
- Ross, J.L.; Roeltgen, D.; Kushner, H.; Wei, F.; Zinn, A.R. The Turner syndrome-associated neurocognitive phenotype maps to distal Xp. Am. J. Hum. Genet. 2000, 67, 672–681. [Google Scholar] [CrossRef]
- Ross, J.L.; Stefanatos, G.A.; Kushner, H.; Zinn, A.R.; Bondy, C.; Roeltgen, D. Persistent cognitive deficits in adult women with Turner syndrome. Neurology 2002, 58, 218–225. [Google Scholar] [CrossRef] [PubMed]
- Temple, C.M.; Carney, R.A. Intellectual functioning of children with Turner syndrome: A comparison of behavioural phenotypes. Dev. Med. Child. Neurol. 1993, 35, 691–698. [Google Scholar] [CrossRef] [PubMed]
- Barrenäs, M.L.; Bratthall, A.; Dahlgren, J. The association between short stature and sensorineural hearing loss. Hear. Res. 2005, 205, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Bonnard, Å.; Bark, R.; Hederstierna, C. Clinical update on sensorineural hearing loss in Turner syndrome and the X-chromosome. Am. J. Med. Genet. C Semin. Med. Genet. 2019, 181, 18–24. [Google Scholar] [CrossRef]
- Camarero, G.; Avendano, C.; Fernandez-Moreno, C.; Villar, A.; Contreras, J.; de Pablo, F.; Pichel, J.G.; Varela-Nieto, I. Delayed inner ear maturation and neuronal loss in postnatal Igf-1-deficient mice. J. Neurosci. 2001, 21, 7630–7641. [Google Scholar] [CrossRef]
- Álvarez-Nava, F.; Racines-Orbe, M.; Witt, J.; Guarderas, J.; Vicuña, Y.; Estévez, M.; Lanes, R. Metabolic Syndrome as a Risk Factor for Sensorineural Hearing Loss in Adult Patients with Turner Syndrome. Appl. Clin. Genet. 2020, 13, 25–35. [Google Scholar] [CrossRef]
- Schoemaker, M.J.; Swerdlow, A.J.; Higgins, C.D.; Wright, A.F.; Jacobs, P.A.; UK Clinical Cytogenetics Group. Cancer incidence in women with Turner syndrome in Great Britain: A national cohort study. Lancet Oncol. 2008, 9, 239–246. [Google Scholar] [CrossRef]
- Claus, E.B.; Black, P.M.; Bondy, M.L.; Calvocoressi, L.; Schildkraut, J.M.; Wiemels, J.L.; Wrensch, M. Exogenous hormone use and meningioma risk: What do we tell our patients? Cancer 2007, 110, 471–476. [Google Scholar] [CrossRef]
- Jostel, A.; Mukherjee, A.; Hulse, P.A.; Shalet, S.M. Adult growth hormone replacement therapy and neuroimaging surveillance in brain tumour survivors. Clin. Endocrinol. 2005, 62, 698–705. [Google Scholar] [CrossRef]
- Roh, M.R.; Eliades, P.; Gupta, S.; Tsao, H. Genetics of melanocytic nevi. Pigment Cell Melanoma Res. 2015, 28, 661–672. [Google Scholar] [CrossRef]
- Patruno, C.; Scalvenzi, M.; Megna, M.; Russo, I.; Gaudiello, F.; Balato, N. Melanocytic nevi in children of southern Italy: Dermoscopic, constitutional, and environmental factors. Pediatr. Dermatol. 2014, 31, 38–42. [Google Scholar] [CrossRef] [PubMed]
- Lekanne Deprez, R.H.; Riegman, P.H.; van Drunen, E.; Warringa, U.L.; Groen, N.A.; Stefanko, S.Z.; Koper, J.W.; Avezaat, C.J.; Mulder, P.G.; Zwarthoff, E.C.; et al. Cytogenetic, molecular genetic and pathological analyses in 126 meningiomas. J. Neuropathol. Exp. Neurol. 1995, 54, 224–235. [Google Scholar] [CrossRef] [PubMed]
- Indsto, J.O.; Nassif, N.T.; Kefford, R.F.; Mann, G.J. Frequent loss of heterozygosity targeting the inactive X chromosome in melanoma. Clin. Cancer Res. 2003, 9, 6476–6482. [Google Scholar]
- Cheng, L.; MacLennan, G.T.; Pan, C.X.; Jones, T.D.; Moore, C.R.; Zhang, S.; Gu, J.; Patel, N.B.; Kao, C.; Gardner, T.A. Allelic loss of the active X chromosome during bladder carcinogenesis. Arch. Pathol. Lab. Med. 2004, 128, 187–190. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Álvarez-Nava, F.; Soto-Quintana, M. The Hypothesis of the Prolonged Cell Cycle in Turner Syndrome. J. Dev. Biol. 2022, 10, 16. https://doi.org/10.3390/jdb10020016
Álvarez-Nava F, Soto-Quintana M. The Hypothesis of the Prolonged Cell Cycle in Turner Syndrome. Journal of Developmental Biology. 2022; 10(2):16. https://doi.org/10.3390/jdb10020016
Chicago/Turabian StyleÁlvarez-Nava, Francisco, and Marisol Soto-Quintana. 2022. "The Hypothesis of the Prolonged Cell Cycle in Turner Syndrome" Journal of Developmental Biology 10, no. 2: 16. https://doi.org/10.3390/jdb10020016
APA StyleÁlvarez-Nava, F., & Soto-Quintana, M. (2022). The Hypothesis of the Prolonged Cell Cycle in Turner Syndrome. Journal of Developmental Biology, 10(2), 16. https://doi.org/10.3390/jdb10020016