Effects of Vitamin D Deficiency on Proliferation and Autophagy of Ovarian and Liver Tissues in a Rat Model of Polycystic Ovary Syndrome

 , , ,
, , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
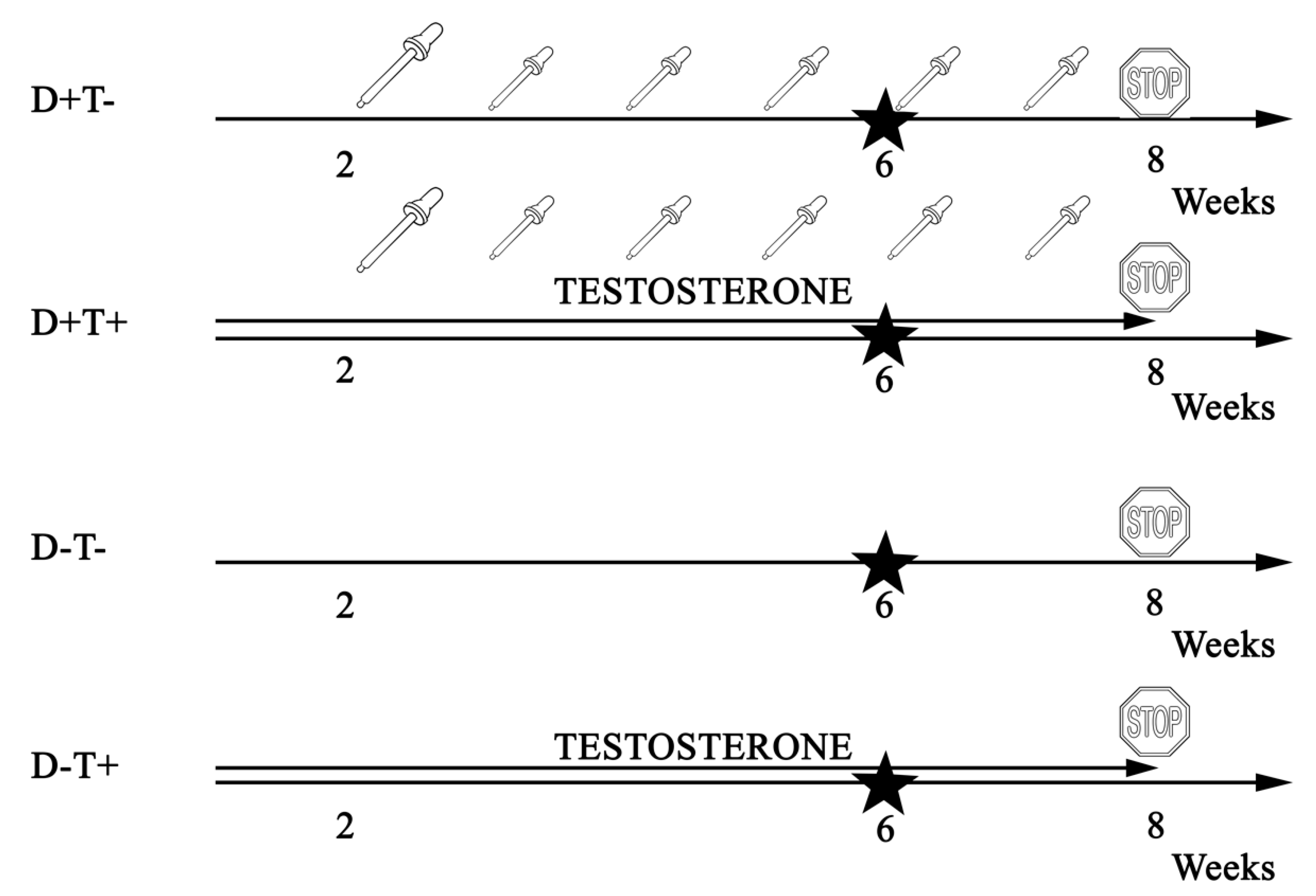
2.1. Animals
2.2. Immunohistochemistry
2.3. Western Blotting
2.4. Statistical Analysis
3. Results
3.1. Chracteristics of PCOS in Our Model
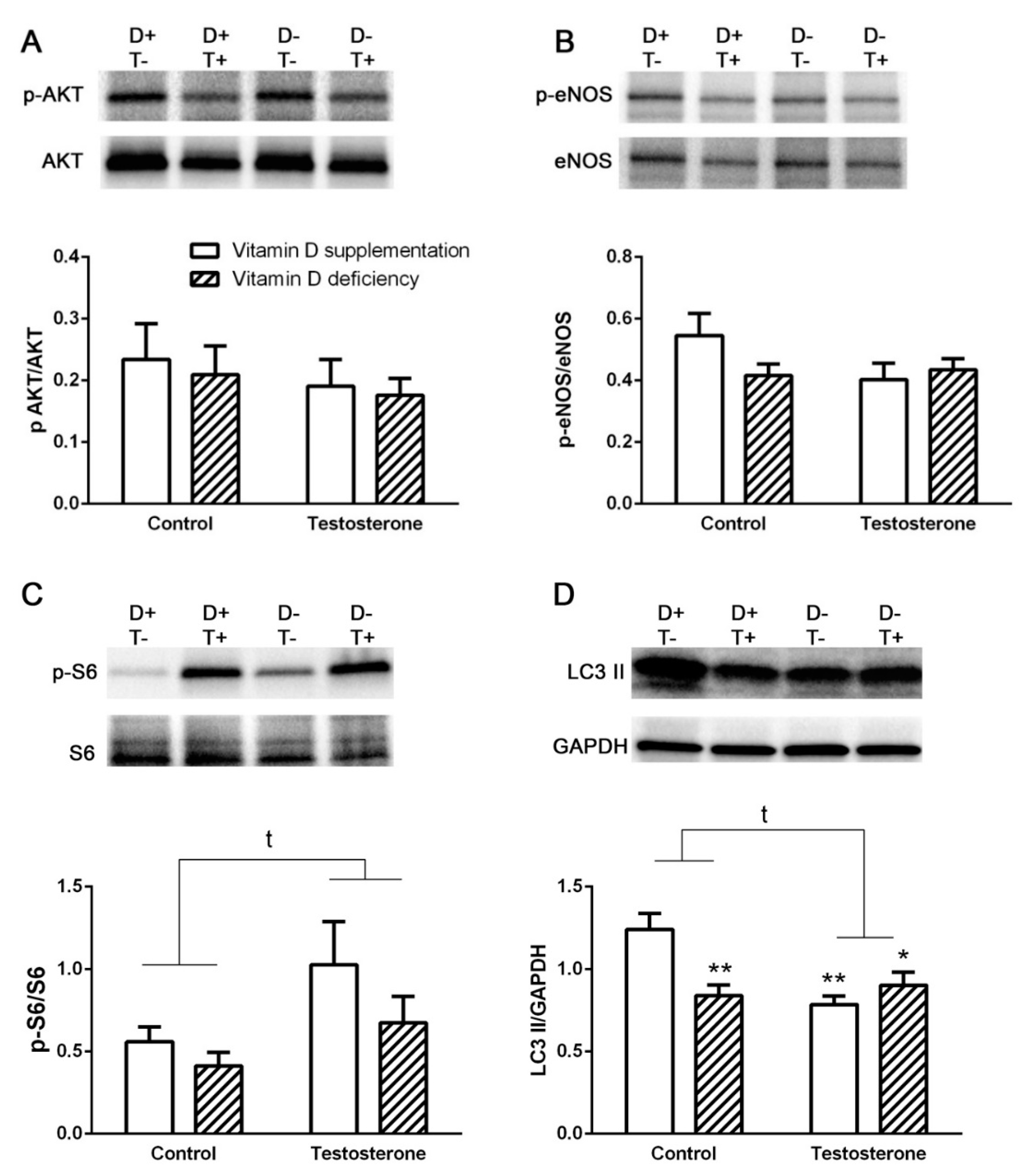
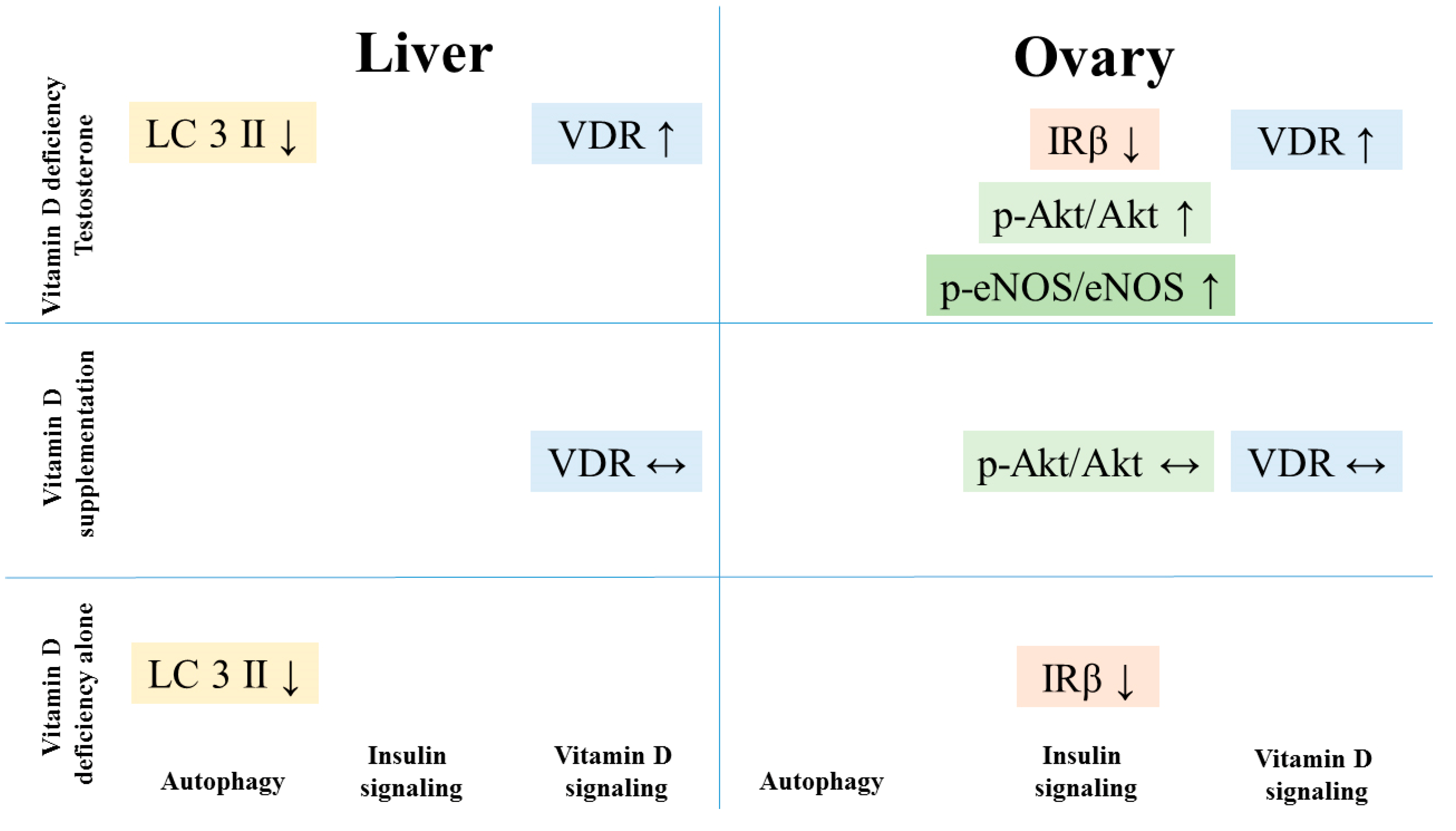
3.2. Alterations of Insulin Receptor Signaling and Autophagy in the Liver
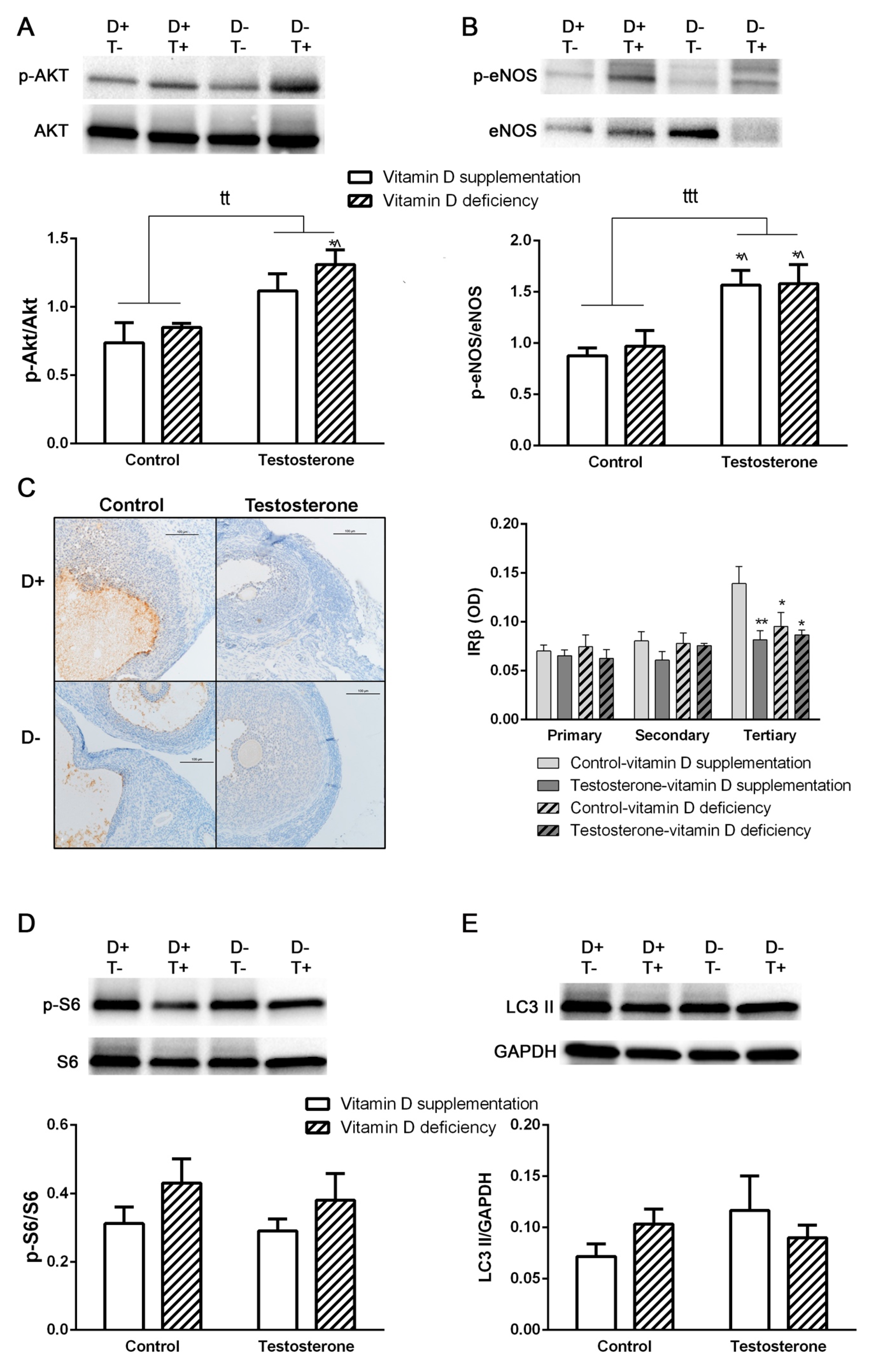
3.3. Alterations of Insulin Receptor Signaling and Autophagy in the Ovaries
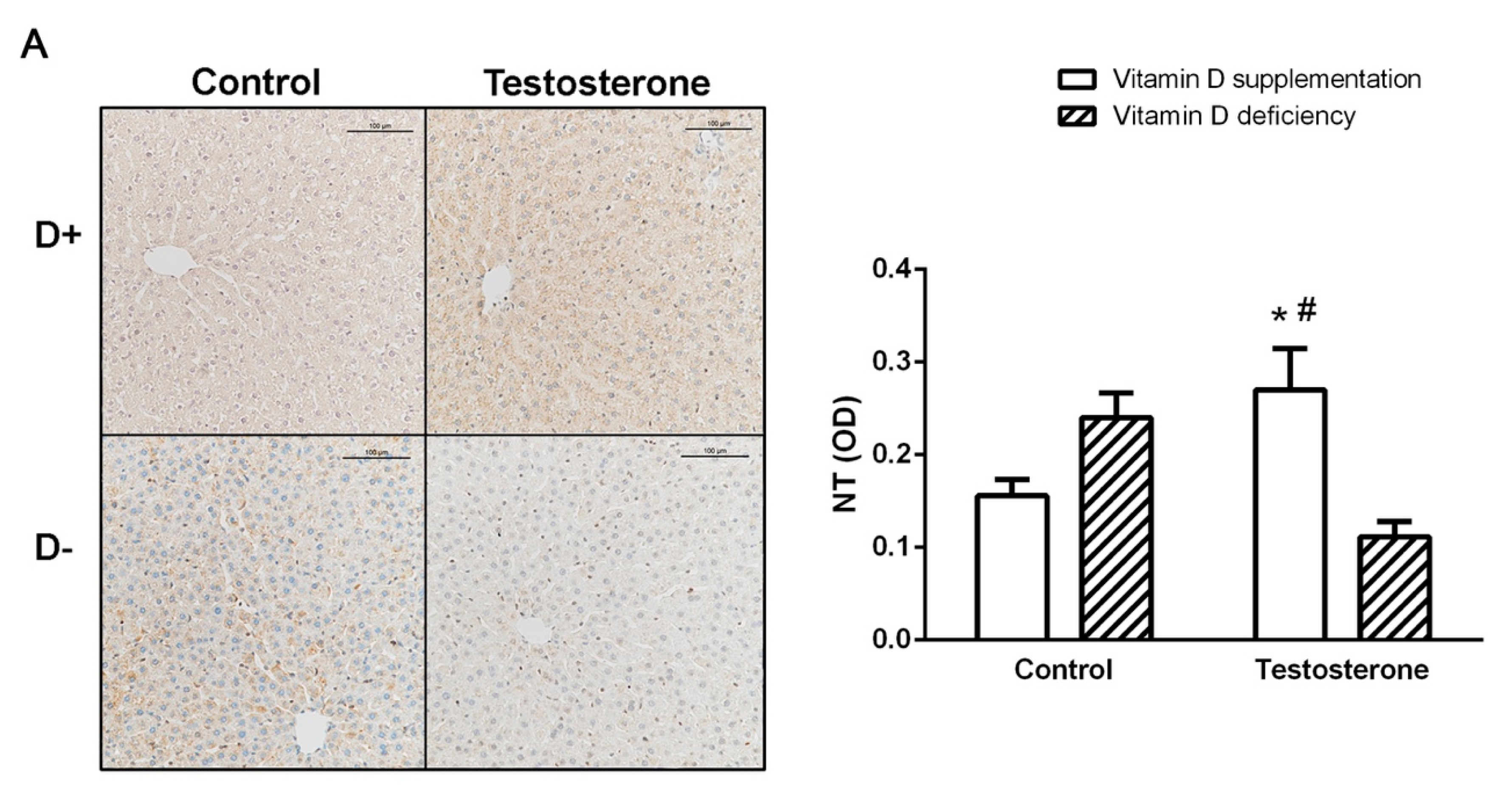
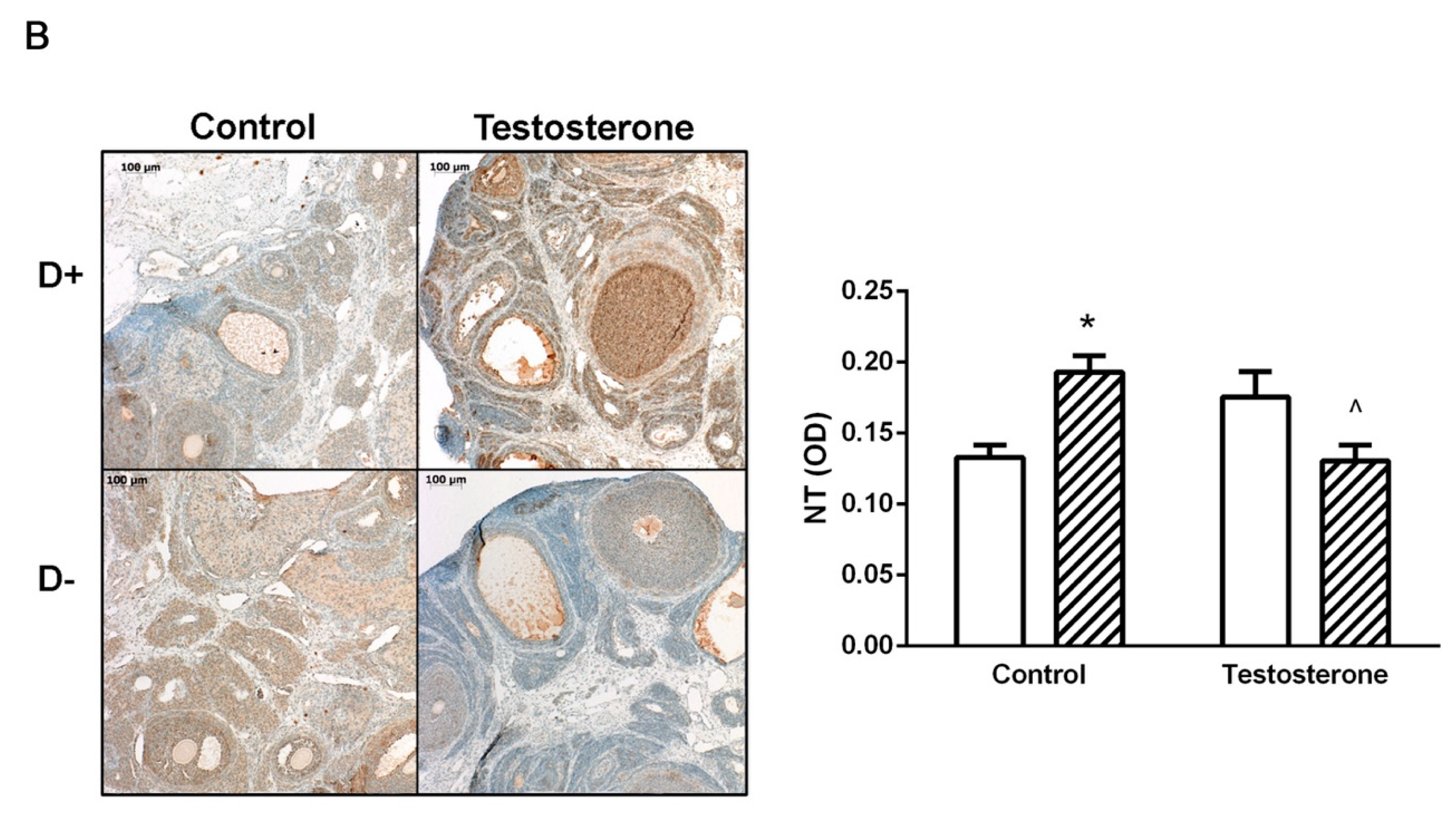
3.4. Nitrative Stress in the Liver and in the Ovaries
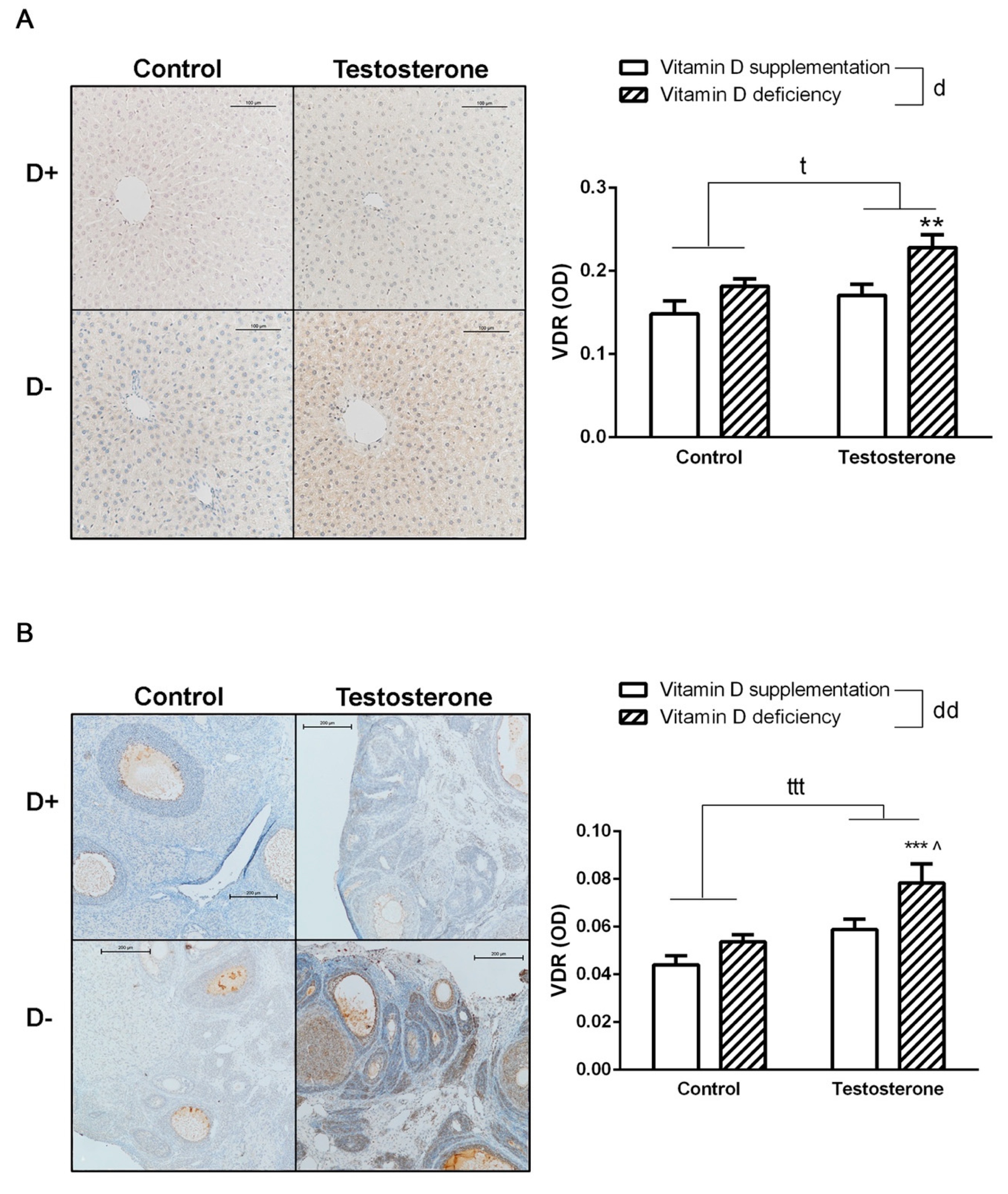
3.5. Changes of Vitamin D Receptor Immunostaining in the Liver and the Ovaries
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Palomba, S.; Santagni, S.; Falbo, A.; La Sala, G.B. Complications and challenges associated with polycystic ovary syndrome: current perspectives. Int. J. Women’s Heal. 2015, 7, 745–763. [Google Scholar]
- Mogili, K.D.; Karuppusami, R.; Thomas, S.; Chandy, A.; Kamath, M.S.; Tk, A. Prevalence of vitamin D deficiency in infertile women with polycystic ovarian syndrome and its association with metabolic syndrome—A prospective observational study. Eur. J. Obstet. Gynecol. Reprod. Boil. 2018, 229, 15–19. [Google Scholar]
- Davis, E.M.; Peck, J.D.; Hansen, K.R.; Neas, B.R.; Craig, L.B. Associations between vitamin D levels and polycystic ovary syndrome (PCOS) phenotypes. Minerva Endocrinol. 2018, 44, 176–184. [Google Scholar] [CrossRef]
- Hahn, S.; Haselhorst, U.; Tan, S.; Quadbeck, B.; Schmidt, M.; Roesler, S.; Kimmig, R.; Mann, K.; Janssen, O.E. Low Serum 25-Hydroxyvitamin D Concentrations are Associated with Insulin Resistance and Obesity in Women with Polycystic Ovary Syndrome. Exp. Clin. Endocrinol. Diabetes 2006, 114, 577–583. [Google Scholar] [CrossRef]
- Hadjadj, L.; Várbíró, S.; Horváth, E.M.; Monori-Kiss, A.; Pál, É.; Karvaly, G.B.; Heinzlmann, A.; Magyar, A.; Szabo, I.; Sziva, R.E.; et al. Insulin resistance in an animal model of polycystic ovary disease is aggravated by vitamin D deficiency: Vascular consequences. Diabetes Vasc. Dis. Res. 2018, 15, 294–301. [Google Scholar] [CrossRef]
- Irani, M.; Merhi, Z. Role of vitamin D in ovarian physiology and its implication in reproduction: a systematic review. Fertil. Steril. 2014, 102, 460–468. [Google Scholar] [CrossRef]
- Menichini, D.; Facchinetti, F. Effects of vitamin D supplementation in women with polycystic ovary syndrome: a review. Gynecol. Endocrinol. 2019, 12, 1–5. [Google Scholar] [CrossRef]
- Javed, Z.; Papageorgiou, M.; Deshmukh, H.; Kilpatrick, E.S.; Mann, V.; Corless, L.; Abouda, G.; Rigby, A.S.; Atkin, S.L.; Sathyapalan, T. A Randomized, Controlled Trial of Vitamin D Supplementation on Cardiovascular Risk Factors, Hormones, and Liver Markers in Women with Polycystic Ovary Syndrome. Nutr. 2019, 11, 188. [Google Scholar]
- Basoli, V.; Santaniello, S.; Cruciani, S.; Ginesu, G.C.; Cossu, M.L.; Delitala, A.P.; Serra, P.A.; Ventura, C.; Maioli, M. Melatonin and Vitamin D Interfere with the Adipogenic Fate of Adipose-Derived Stem Cells. Int. J. Mol. Sci. 2017, 18, 981. [Google Scholar]
- Zhao, Y.; Zhang, C.; Huang, Y.; Yü, Y.; Li, R.; Li, M.; Liu, N.; Liu, P.; Qiao, J. Up-Regulated Expression of WNT5a Increases Inflammation and Oxidative Stress via PI3K/AKT/NF-κB Signaling in the Granulosa Cells of PCOS Patients. J. Clin. Endocrinol. Metab. 2015, 100, 201–211. [Google Scholar] [CrossRef]
- Li, D.; You, Y.; Bi, F.-F.; Zhang, T.-N.; Jiao, J.; Wang, T.-R.; Zhou, Y.-M.; Shen, Z.-Q.; Wang, X.-X.; Yang, Q. Autophagy is activated in the ovarian tissue of polycystic ovary syndrome. Reproduction 2018, 155, 85–92. [Google Scholar] [CrossRef]
- Song, X.; Shen, Q.; Fan, L.; Yu, Q.; Jia, X.; Sun, Y.; Bai, W.; Kang, J. Dehydroepiandrosterone-induced activation of mTORC1 and inhibition of autophagy contribute to skeletal muscle insulin resistance in a mouse model of polycystic ovary syndrome. Oncotarget 2018, 9, 11905–11921. [Google Scholar] [CrossRef]
- Gao, L.; Cao, J.T.; Liang, Y.; Zhao, Y.C.; Lin, X.H.; Li, X.C.; Tan, Y.J.; Li, J.Y.; Zhou, C.L.; Xu, H.Y.; et al. Calcitriol attenuates cardiac remodeling and dysfunction in a murine model of polycystic ovary syndrome. Endocrine 2016, 52, 363–373. [Google Scholar] [CrossRef]
- Ainbinder, D.; Touitou, E. Testosterone Ethosomes for Enhanced Transdermal Delivery. Drug Deliv. 2005, 12, 297–303. [Google Scholar] [CrossRef]
- Basaria, S.; Lakshman, K.M. Safety and efficacy of testosterone gel in the treatment of male hypogonadism. Clin. Interv. Aging 2009, 4, 397–412. [Google Scholar] [CrossRef]
- Mannerås, L.; Cajander, S.; Holmäng, A.; Seleskovic, Z.; Lystig, T.; Lönn, M.; Stener-Victorin, E. A New Rat Model Exhibiting Both Ovarian and Metabolic Characteristics of Polycystic Ovary Syndrome. Endocrinology 2007, 148, 3781–3791. [Google Scholar] [CrossRef]
- Masszi, G.; Benkő, R.; Csibi, N.; Horvath, E.M.; Tokes, A.-M.; Novak, A.; Beres, N.J.; Tarszabo, R.; Buday, A.; Repas, C.; et al. Endothelial relaxation mechanisms and nitrative stress are partly restored by Vitamin D3 therapy in a rat model of polycystic ovary syndrome. Life Sci. 2013, 93, 133–138. [Google Scholar] [CrossRef][Green Version]
- Sara, L.; Antal, P.; Masszi, G.; Buday, A.; Horvath, E.M.; Hamar, P.; Monos, E.; Nadasy, G.L.; Varbiro, S. Arteriolar insulin resistance in a rat model of polycystic ovary syndrome. Fertil. Steril. 2012, 97, 462–468. [Google Scholar] [CrossRef]
- Pludowski, P.; Holick, M.F.; Grant, W.B.; Konstantynowicz, J.; Mascarenhas, M.R.; Haq, A.; Povoroznyuk, V.; Balatska, N.; Barbosa, A.P.; Karonova, T.; et al. Vitamin D supplementation guidelines. J. Steroid Biochem. Mol. Boil. 2018, 175, 125–135. [Google Scholar] [CrossRef]
- Pludowski, P.; Grant, W.B.; Bhattoa, H.P.; Bayer, M.; Povoroznyuk, V.; Rudenka, E.; Ramanau, H.; Varbiro, S.; Rudenka, A.; Karczmarewicz, E.; et al. Vitamin D Status in Central Europe. Int. J. Endocrinol. 2014, 2014, 1–12. [Google Scholar] [CrossRef]
- Samoto, T.; Maruo, T.; Matsuo, H.; Katayama, K.; Barnea, E.R.; Mochizuki, M. Altered expression of insulin and insulin-like growth factor-I receptors in follicular and stromal compartments of polycystic ovaries. Endocr. J. 1993, 40, 413–424. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Vassilatou, E. Nonalcoholic fatty liver disease and polycystic ovary syndrome. World J. Gastroenterol. 2014, 20, 8351–8363. [Google Scholar] [CrossRef] [PubMed]
- Lavallard, V.J.; Gual, P. Autophagy and Non-Alcoholic Fatty Liver Disease. BioMed Res. Int. 2014, 2014, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Guo, E.; Yang, J.; Li, A.; Yang, Y.; Liu, S.; Liu, A.; Jiang, X. 1,25(OH)2 D3 attenuates hepatic steatosis by inducing autophagy in mice. Obesity 2017, 25, 561–571. [Google Scholar] [CrossRef] [PubMed]
- Basualto-Alarcón, C.; Jorquera, G.; Altamirano, F.; Jaimovich, E.; Estrada, M. Testosterone Signals through mTOR and Androgen Receptor to Induce Muscle Hypertrophy. Med. Sci. Sports Exerc. 2013, 45, 1712–1720. [Google Scholar] [CrossRef]
- Bae, E.J.; Xu, J.; Oh, D.Y.; Bandyopadhyay, G.; Lagakos, W.S.; Keshwani, M.; Olefsky, J.M. Liver-specific p70 S6 Kinase Depletion Protects against Hepatic Steatosis and Systemic Insulin Resistance. J. Boil. Chem. 2012, 287, 18769–18780. [Google Scholar] [CrossRef] [PubMed]
- Andrisse, S.; Billings, K.; Xue, P.; Wu, S. Insulin signaling displayed a differential tissue-specific response to low-dose dihydrotestosterone in female mice. Am. J. Physiol. Metab. 2017, 314, E353–E365. [Google Scholar] [CrossRef]
- E Ryan, K.; Casey, S.M.; Canty, M.J.; A Crowe, M.; Martin, F.; O Evans, A.C. Akt and Erk signal transduction pathways are early markers of differentiation in dominant and subordinate ovarian follicles in cattle. Reproduction 2007, 133, 617–626. [Google Scholar] [CrossRef]
- Li, T.; Mo, H.; Chen, W.; Li, L.; Xiao, Y.; Zhang, J.; Li, X.; Lu, Y. Role of the PI3K-Akt Signaling Pathway in the Pathogenesis of Polycystic Ovary Syndrome. Reprod. Sci. 2017, 24, 646–655. [Google Scholar] [CrossRef]
- Yu, J.; Akishita, M.; Eto, M.; Ogawa, S.; Son, B.-K.; Kato, S.; Ouchi, Y.; Okabe, T. Androgen Receptor-Dependent Activation of Endothelial Nitric Oxide Synthase in Vascular Endothelial Cells: Role of Phosphatidylinositol 3-Kinase/Akt Pathway. Endocrinology 2010, 151, 1822–1828. [Google Scholar] [CrossRef]
- Liu, W.-J.; Xin, Z.-C.; Xin, H.; Yuan, Y.-M.; Tian, L.; Guo, Y.-L. Effects of icariin on erectile function and expression of nitric oxide synthase isoforms in castrated rats. Asian J. Androl. 2005, 7, 381–388. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.; Tang, Y.; Tang, Z.; Zu, X.; Qi, L.; Zhang, X.; Wang, G. Icariin improves the sexual function of male mice through the PI3K/AKT/eNOS/NO signalling pathway. Andrologia 2018, 50. [Google Scholar] [CrossRef] [PubMed]
- Dupont, J.; Scaramuzzi, R.J. Insulin signalling and glucose transport in the ovary and ovarian function during the ovarian cycle. Biochem. J. 2016, 473, 1483–1501. [Google Scholar] [CrossRef] [PubMed]
- Samoto, T.; Maruo, T.; Ladines-Llave, C.A.; Matsuo, H.; Deguchi, J.; Barnea, E.R.; Mochizuki, M. Insulin Receptor Expression in Follicular and Stromal Compartments of the Human Ovary over the Course of Follicular Growth, Regression and Atresia. Endocr. J. 1993, 40, 715–726. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, Z.; Smith, B.; Pillay, T. The APS adapter protein couples the insulin receptor to the phosphorylation of c-Cbl and facilitates ligand-stimulated ubiquitination of the insulin receptor. FEBS Lett. 2000, 475, 31–34. [Google Scholar] [CrossRef]
- Katsanakis, K.D.; Pillay, T.S. Cross-talk between the Two Divergent Insulin Signaling Pathways Is Revealed by the Protein Kinase B (Akt)-mediated Phosphorylation of Adapter Protein APS on Serine 588. J. Boil. Chem. 2005, 280, 37827–37832. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Wang, Z.; Cao, J.; Chen, Y.; Dong, Y. A novel and compact review on the role of oxidative stress in female reproduction. Reprod. Boil. Endocrinol. 2018, 16, 80. [Google Scholar] [CrossRef] [PubMed]
- Temiz-Resitoglu, M.; Kucukkavruk, S.P.; Guden, D.S.; Cecen, P.; Sari, A.N.; Tunctan, B.; Gorur, A.; Tamer-Gumus, L.; Buharalioglu, C.K.; Malik, K.U.; et al. Activation of mTOR/IκB-α/NF-κB pathway contributes to LPS-induced hypotension and inflammation in rats. Eur. J. Pharmacol. 2017, 802, 7–19. [Google Scholar] [CrossRef]
- Keeney, J.T.R.; Forster, S.; Sultana, R.; Brewer, L.D.; Latimer, C.S.; Cai, J.; Klein, J.B.; Porter, N.M.; Butterfield, D.A. Dietary Vitamin D Deficiency in Rats from Middle- to Old-age Leads to Elevated Tyrosine Nitration and Proteomics Changes in Levels of Key Proteins in Brain: Implications for Low Vitamin D-dependent Age-Related Cognitive Decline. Free. Radic. Boil. Med. 2013, 65, 324–334. [Google Scholar] [CrossRef]
- Atiomo, W.; Daykin, C.A. Metabolomic biomarkers in women with polycystic ovary syndrome: a pilot study. Mol. Hum. Reprod. 2012, 18, 546–553. [Google Scholar] [CrossRef]
- Yao, X.; Zhang, G.; Guo, Y.; Ei-Samahy, M.; Wang, S.; Wan, Y.; Han, L.; Liu, Z.; Wang, F.; Zhang, Y. Vitamin D receptor expression and potential role of vitamin D on cell proliferation and steroidogenesis in goat ovarian granulosa cells. Theriogenology 2017, 102, 162–173. [Google Scholar] [CrossRef] [PubMed]
- Nizami, H.L.; Katare, P.; Prabhakar, P.; Kumar, Y.; Arava, S.K.; Chakraborty, P.; Maulik, S.K.; Banerjee, S.K. Vitamin D deficiency in rats causes cardiac dysfunction by inducing myocardial insulin resistance. Mol. Nutr. Food Res. 2019, 16, e1900109. [Google Scholar] [CrossRef] [PubMed]
- Shirvani-Farsani, Z.; Kakhki, M.P.; Gargari, B.N.; Doosti, R.; Moghadasi, A.N.; Azimi, A.R.; Behmanesh, M. The expression of VDR mRNA but not NF-κB surprisingly decreased after vitamin D treatment in multiple sclerosis patients. Neurosci. Lett. 2017, 653, 258–263. [Google Scholar] [CrossRef] [PubMed]
- Herian, M.; Luck, M.R.; Grzesiak, M. The Influence of Testosterone on the Expression and Function of Vitamin D3 Receptor (VDR) Protein in the Porcine Ovarian Follicle. Physiol. Res. 2018, 67, 515–519. [Google Scholar] [CrossRef] [PubMed]
- Saki, F.; Kasaee, S.R.; Sadeghian, F.; Koohpeyma, F.; Omrani, G.H.R. Investigating the effect of testosterone by itself and in combination with letrozole on 1,25-dihydroxy vitamin D and FGF23 in male rats. J. Endocrinol. Invest. 2019, 42, 19–25. [Google Scholar] [CrossRef]
- Shaw, P.A.; Rittenberg, P.V.; Brown, T.J. Activation of Androgen Receptor-Associated Protein 70 (ARA70) mRNA Expression in Ovarian Cancer. Gynecol. Oncol. 2001, 80, 132–138. [Google Scholar] [CrossRef] [PubMed]
- Maguire, O.; Pollock, C.; Martin, P.; Owen, A.; Smyth, T.; Doherty, D.; Campbell, M.J.; McClean, S.; Thompson, P. Regulation of CYP3A4 and CYP3A5 expression and modulation of “intracrine” metabolism of androgens in prostate cells by liganded vitamin D receptor. Mol. Cell. Endocrinol. 2012, 364, 54–64. [Google Scholar] [CrossRef]
- Chatterjee, B.; Echchgadda, I.; Song, C.S. Vitamin D Receptor Regulation of the Steroid/Bile Acid Sulfotransferase SULT2A1. Methods Enzymol. 2005, 400, 165–191. [Google Scholar]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lajtai, K.; Nagy, C.T.; Tarszabó, R.; Benkő, R.; Hadjadj, L.; Sziva, R.E.; Gerszi, D.; Bányai, B.; Ferdinandy, P.; Nádasy, G.L.; et al. Effects of Vitamin D Deficiency on Proliferation and Autophagy of Ovarian and Liver Tissues in a Rat Model of Polycystic Ovary Syndrome. Biomolecules 2019, 9, 471. https://doi.org/10.3390/biom9090471
Lajtai K, Nagy CT, Tarszabó R, Benkő R, Hadjadj L, Sziva RE, Gerszi D, Bányai B, Ferdinandy P, Nádasy GL, et al. Effects of Vitamin D Deficiency on Proliferation and Autophagy of Ovarian and Liver Tissues in a Rat Model of Polycystic Ovary Syndrome. Biomolecules. 2019; 9(9):471. https://doi.org/10.3390/biom9090471
Chicago/Turabian StyleLajtai, Krisztina, Csilla Terézia Nagy, Róbert Tarszabó, Rita Benkő, Leila Hadjadj, Réka Eszter Sziva, Dóra Gerszi, Bálint Bányai, Péter Ferdinandy, György László Nádasy, and et al. 2019. "Effects of Vitamin D Deficiency on Proliferation and Autophagy of Ovarian and Liver Tissues in a Rat Model of Polycystic Ovary Syndrome" Biomolecules 9, no. 9: 471. https://doi.org/10.3390/biom9090471
APA StyleLajtai, K., Nagy, C. T., Tarszabó, R., Benkő, R., Hadjadj, L., Sziva, R. E., Gerszi, D., Bányai, B., Ferdinandy, P., Nádasy, G. L., Giricz, Z., Horváth, E. M., & Várbíró, S. (2019). Effects of Vitamin D Deficiency on Proliferation and Autophagy of Ovarian and Liver Tissues in a Rat Model of Polycystic Ovary Syndrome. Biomolecules, 9(9), 471. https://doi.org/10.3390/biom9090471