β-Carotene: A Natural Compound Improves Cognitive Impairment and Oxidative Stress in a Mouse Model of Streptozotocin-Induced Alzheimer’s Disease

 , , and
, , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Experimental Animals
2.3. Study Design
2.4. Evaluation of Cognitive Performance
2.4.1. Elevated Plus-Maze
2.4.2. Passive Avoidance Apparatus
2.4.3. Open Field Apparatus
Behavior
- Latency (Initial time taken by the mice to leave the start area)
- Freezing (Time spent by the mice without any movement)
- Rearing
Location
- Field area visited (central and peripheral)
- Crossing
Autonomic Nervous System (ANS)
- Urination
- Defecation [26].
2.5. Neurochemical Studies
2.5.1. Estimation of Reduced Glutathione (GSH)
2.5.2. Estimation of Oxidized Glutathione (GSSG)
2.5.3. Determination of Superoxide Dismutase (SOD)
2.5.4. Estimation of CAT
2.5.5. Determination of Acetylcholinesterase Activity
2.6. Protein Analysis by ELISA
2.7. In Silico Modeling
2.8. Statistical Analysis
3. Results
3.1. Evaluation of Cognitive Performance
3.1.1. The Effect of β-Carotene on the Transfer Latency Using the Elevated Plus Maze Model in Mice that Received i.c.v STZ
3.1.2. Effect of β-Carotene on the Step Down Latency Using the Passive Avoidance Model in Mice that Received i.c.v STZ
3.1.3. The Effect of β-Carotene Following the Open Field Paradigm in Mice that Received i.c.v STZ
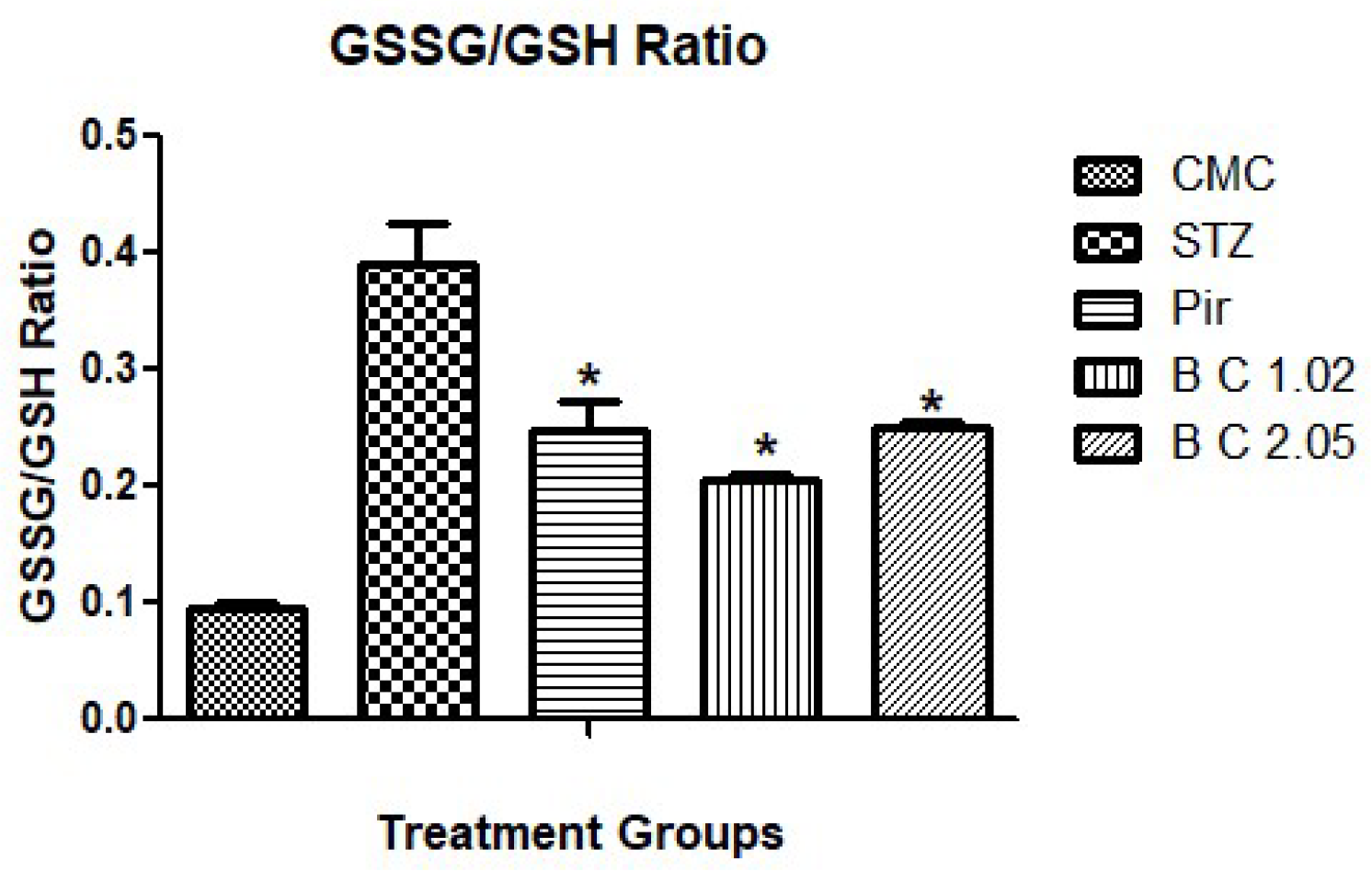
3.2. Determination of Biochemical Markers in Brain Tissues of Mice that Received I.STZ
3.3. Protein Analysis by ELISA
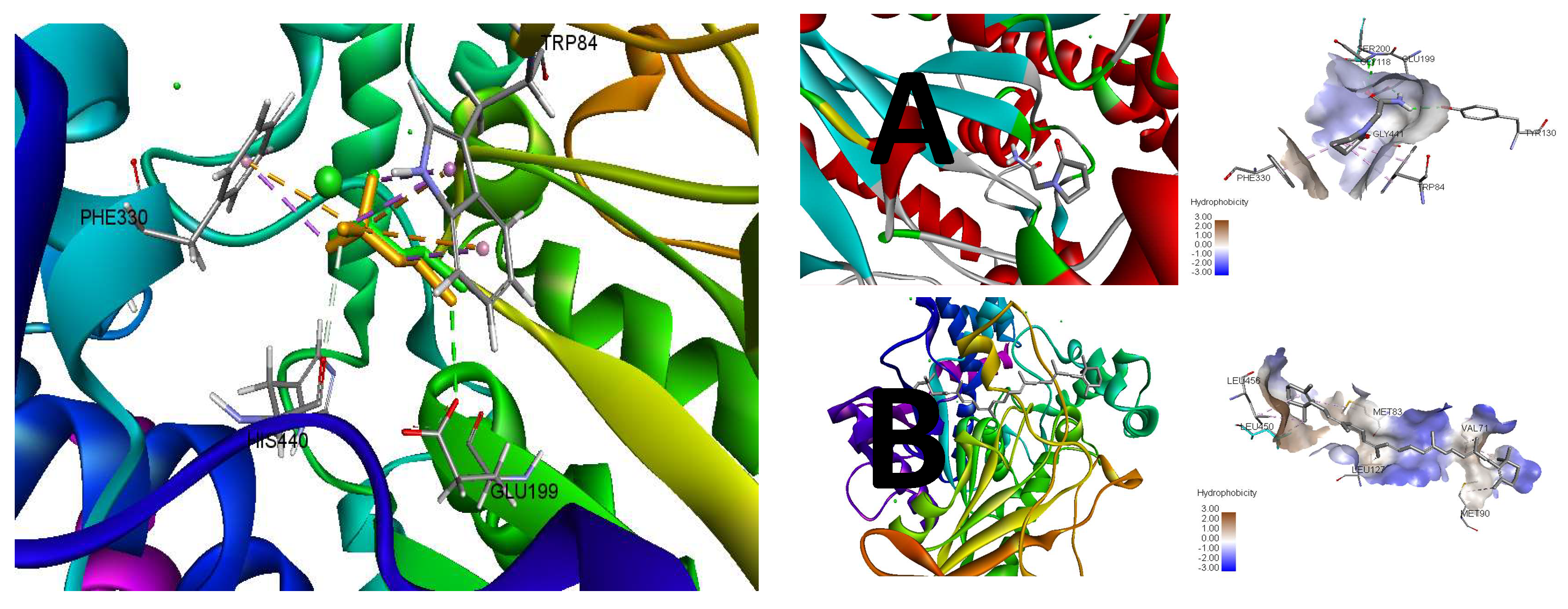
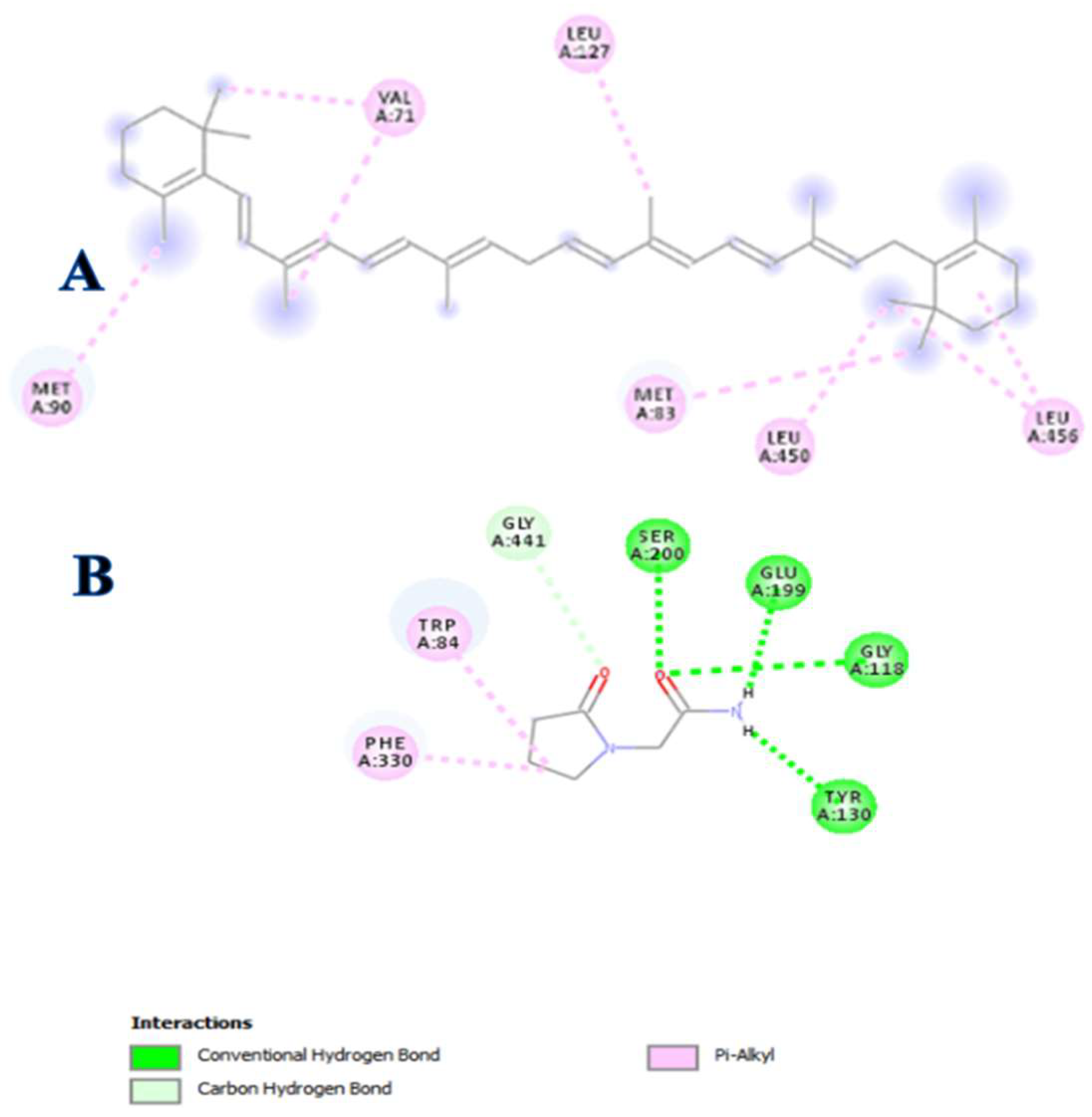
3.4. In Silico Modeling
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rajesh, V.; Riju, T.; Venkatesh, S.; Babu, G. Memory enhancing activity of Lawsonia inermis Linn. leaves against scopolamine induced memory impairment in Swiss albino mice. Orient. Pharm. Exp. Med. 2017, 17, 127–142. [Google Scholar] [CrossRef]
- Mothet, J.P.; Rouaud, E.; Sinet, P.-M.; Potier, B.; Jouvenceau, A.; Dutar, P.; Videau, C.; Epelbaum, J.; Billard, J.M. A critical role for the glial-derived neuromodulator d-serine in the age-related deficits of cellular mechanisms of learning and memory. Aging Cell 2006, 5, 267–274. [Google Scholar] [CrossRef] [PubMed]
- Giovannini, M.; Casamenti, F.; Bartolini, L.; Pepeu, G. The brain cholinergic system as a target of cognition enhancers. Behav. Brain Res. 1997, 83, 1–5. [Google Scholar] [CrossRef]
- Francis, P.T.; Palmer, A.M.; Snape, M.; Wilcock, G.K. The cholinergic hypothesis of Alzheimer’s disease: A review of progress. J. Neurol. Neurosurg. Psychiatry 1999, 66, 137–147. [Google Scholar] [CrossRef] [PubMed]
- Başar, E.; Güntekin, B. A review of brain oscillations in cognitive disorders and the role of neurotransmitters. Brain Res. 2008, 1235, 172–193. [Google Scholar] [CrossRef] [PubMed]
- Maratha, S.R.; Mahadevan, N. Memory Enhancing Activity of Naringin in Unstressed and Stressed Mice: Possible Cholinergic and Nitriergic Modulation. Neurochem. Res. 2012, 37, 2206–2212. [Google Scholar] [CrossRef] [PubMed]
- Grober, E.; Hall, C.B.; Lipton, R.B.; Zonderman, A.B.; Resnick, S.M.; Kawas, C. Memory impairment, executive dysfunction, and intellectual decline in preclinical Alzheimer’s disease. J. Int. Neuropsychol. Soc. 2008, 14, 266–278. [Google Scholar] [CrossRef] [PubMed]
- Phelps, E.A.; Delgado, M.R.; Nearing, K.I.; LeDoux, J.E. Extinction learning in humans: Role of the amygdala and vmPFC. Neuron 2004, 43, 897–905. [Google Scholar] [CrossRef] [PubMed]
- Pickford, F.; Masliah, E.; Britschgi, M.; Lucin, K.; Narasimhan, R.; Jaeger, P.A.; Small, S.; Spencer, B.; Rockenstein, E.; Levine, B.; et al. The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid β accumulation in mice. J. Clin. Investig. 2008, 118, 2190–2199. [Google Scholar] [CrossRef]
- Goedert, M. Tau protein and the neurofibrillary pathology of Alzheimer’s disease. Trends Neurosci. 1993, 16, 460–465. [Google Scholar] [CrossRef]
- Mangialasche, F.; Solomon, A.; Winblad, B.; Mecocci, P.; Kivipelto, M. Alzheimer’s disease: Clinical trials and drug development. Lancet Neurol. 2010, 9, 702–716. [Google Scholar] [CrossRef]
- Mecocci, P.; Polidori, M.C.; Praticò, D. Antioxidant Clinical Trials in Mild Cognitive Impairment and Alzheimer’s Disease. In Oxidative Stress in Applied Basic Research and Clinical Practice; Springer Science and Business Media LLC: Berlin, Germany, 2013; pp. 223–232. [Google Scholar]
- Silva, R.; Abílio, V.; Takatsu, A.; Kameda, S.; Grassl, C.; Chehin, A.; Medrano, W.; Calzavara, M.; Registro, S.; Andersen, M.; et al. Role of hippocampal oxidative stress in memory deficits induced by sleep deprivation in mice. Neuropharmacology 2004, 46, 895–903. [Google Scholar] [CrossRef] [PubMed]
- Rammal, H.; Bouayed, J.; Soulimani, R. A direct relationship between aggressive behavior in the resident/intruder test and cell oxidative status in adult male mice. Eur. J. Pharmacol. 2010, 627, 173–176. [Google Scholar] [CrossRef] [PubMed]
- Stadtman, E. Free radicals in the genesis of Alzheimer’s disease. Ann. N. Y. Acad. Sci. 1992, 695, 73–76. [Google Scholar]
- Tota, S.; Awasthi, H.; Kamat, P.K.; Nath, C.; Hanif, K.; Kamat, P.K. Protective effect of quercetin against intracerebral streptozotocin induced reduction in cerebral blood flow and impairment of memory in mice. Behav. Brain Res. 2010, 209, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Morris, M.C.; Evans, D.A.; Bienias, J.L.; Tangney, C.C.; Bennett, D.A.; Aggarwal, N.; Wilson, R.S.; Scherr, P.A. Dietary Intake of Antioxidant Nutrients and the Risk of Incident Alzheimer Disease in a Biracial Community Study. JAMA 2002, 287, 3230–3237. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.T.; Chang, W.N.; Tsai, N.W.; Huang, C.C.; Kung, C.T.; Su, Y.J.; Lin, W.C.; Cheng, B.C.; Su, C.M.; Chiang, Y.F.; et al. The Roles of Biomarkers of Oxidative Stress and Antioxidant in Alzheimer’s Disease: A Systematic Review. BioMed Res. Int. 2014, 2014, 1–14. [Google Scholar] [CrossRef]
- Praticò, D. Evidence of oxidative stress in Alzheimer’s disease brain and antioxidant therapy: Lights and shadows. Ann. N. Y. Acad. Sci. 2008, 1147, 70–78. [Google Scholar] [CrossRef]
- Gilgun-Sherki, Y. Antioxidant Therapy in Acute Central Nervous System Injury: Current State. Pharmacol. Rev. 2002, 54, 271–284. [Google Scholar] [CrossRef]
- Sies, H.; Stahl, W.; Sundquist, A.R. Antioxidant Functions of Vitamins: Vitamins E and C, B-Carotene, and Other Carotenoids a. Ann. N. Y. Acad. Sci. 1992, 669, 7–20. [Google Scholar] [CrossRef]
- Nair, A.B.; Jacob, S. A simple practice guide for dose conversion between animals and human. J. Basic Clin. Pharm. 2016, 7, 27–31. [Google Scholar] [CrossRef] [PubMed]
- Pellow, S.; Chopin, P.; File, S.E.; Briley, M. Validation of open: Closed arm entries in an elevated plus-maze as a measure of anxiety in the rat. J. Neurosci. Methods 1985, 14, 149–167. [Google Scholar] [CrossRef]
- Vasudevan, M.; Parle, M. Memory enhancing activity of Anwala churna (Emblica officinalis Gaertn.): An Ayurvedic preparation. Physiol. Behav. 2007, 91, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Carola, V.; D’Olimpio, F.; Brunamonti, E.; Mangia, F.; Renzi, P. Evaluation of the elevated plus-maze and open-field tests for the assessment of anxiety-related behaviour in inbred mice. Behav. Brain Res. 2002, 134, 49–57. [Google Scholar] [CrossRef]
- Walsh, R.N.; Cummins, R.A. The open-field test: A critical review. Psychol. Bull. 1976, 83, 482–504. [Google Scholar] [CrossRef] [PubMed]
- Rahman, I.; Kode, A.; Biswas, S.K. Assay for quantitative determination of glutathione and glutathione disulfide levels using enzymatic recycling method. Nat. Protoc. 2006, 1, 3159–3165. [Google Scholar] [CrossRef] [PubMed]
- Suh, J.H.; Wang, H.; Liu, R.M.; Liu, J.; Hagen, T.M. (R)-α-Lipoic acid reverses the age-related loss in GSH redox status in post-mitotic tissues: Evidence for increased cysteine requirement for zGSH synthesis. Arch. Biochem. Biophys. 2004, 423, 126–135. [Google Scholar] [CrossRef] [PubMed]
- Bin Choi, Y.; Kim, Y.I.; Lee, K.S.; Kim, B.S.; Kim, D.J. Protective effect of epigallocatechin gallate on brain damage after transient middle cerebral artery occlusion in rats. Brain Res. 2004, 1019, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Okado-Matsumoto, A.; Fridovich, I. Subcellular Distribution of Superoxide Dismutases (SOD) in Rat Liver: Cu, Zn-SOD in mitochondria. J. Biol. Chem. 2001, 276, 38388–38393. [Google Scholar] [CrossRef] [PubMed]
- Saleem, U.; Ahmad, B.; Ahmad, M.; Hussain, K.; Bukhari, N.I. Investigation of in vivo antioxidant activity of Euphorbia helioscopia latex and leaves methanol extract: A target against oxidative stress induced toxicity. Asian Pac. J. Trop. Med. 2014, 7, S369–S375. [Google Scholar] [CrossRef]
- Waterborg, J.H. The Lowry method for protein quantitation. In The Protein Protocols Handbook; Springer: Berlin/Heidelberg, Germany, 2002; pp. 7–9. [Google Scholar]
- Ellman, G.L.; Courtney, K.D.; Andres, V., Jr.; Featherstone, R.M. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 1961, 7, 88–95. [Google Scholar] [CrossRef]
- DeMattos, R.B.; Bales, K.R.; Cummins, D.J.; Paul, S.M.; Holtzman, D.M. Brain to plasma amyloid-β efflux: A measure of brain amyloid burden in a mouse model of Alzheimer’s disease. Science 2002, 295, 2264–2267. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Chemin. Form. 2011, 3, 33. [Google Scholar]
- Biovia, D.S. Discovery Studio Modeling Environment; Dassault Systèmes: San Diego, CA, USA, 2017. [Google Scholar]
- Esch, T.; Stefano, G.B.; Fricchione, G.L.; Benson, H. The role of stress in neurodegenerative diseases and mental disorders. Neuro Endocrinol. Lett. 2002, 23, 199–208. [Google Scholar] [PubMed]
- Hira, S.; Saleem, U.; Anwar, F.; Ahmad, B. Antioxidants Attenuate Isolation- and L-DOPA-Induced Aggression in Mice. Front. Pharmacol. 2018, 8, 945. [Google Scholar] [CrossRef] [PubMed]
- Farr, S.A.; Poon, H.F.; Dogrukol-Ak, D.; Drake, J.; Banks, W.A.; Eyerman, E.; Butterfield, D.A.; Morley, J.E. The antioxidants α-lipoic acid and N-acetylcysteine reverse memory impairment and brain oxidative stress in aged SAMP8 mice. J. Neurochem. 2003, 84, 1173–1183. [Google Scholar] [CrossRef]
- Fernández-Checa, J.C.; Kaplowitz, N.; Garcia-Ruiz, C.; Colell, A.; Miranda, M.; Mari, M.; Ardite, E.; Morales, A. GSH transport in mitochondria: Defense against TNF-induced oxidative stress and alcohol-induced defect. Am. J. Physiol. Liver Physiol. 1997, 273, G7–G17. [Google Scholar] [CrossRef]
- Kim, J.; Keum, Y.S. NRF2, a Key Regulator of Antioxidants with Two Faces towards Cancer. Oxidative Med. Cell. Longev. 2016, 2016, 1–7. [Google Scholar] [CrossRef]
- Doroshow, J.H. Glutathione peroxidase and oxidative stress. Toxicol. Lett. 1995, 82, 395–398. [Google Scholar] [CrossRef]
- Shahripour, R.B.; Harrigan, M.R.; Alexandrov, A.V. N-acetylcysteine (NAC) in neurological disorders: Mechanisms of action and therapeutic opportunities. Brain Behav. 2014, 4, 108–122. [Google Scholar] [CrossRef] [PubMed]
- Finkel, T.; Holbrook, N.J. Oxidants, oxidative stress and the biology of ageing. Nature 2000, 408, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, V.; Kuhad, A.; Bishnoi, M.; Chopra, K. Chronic treatment with tocotrienol, an isoform of vitamin E, prevents intracerebroventricular streptozotocin-induced cognitive impairment and oxidative–nitrosative stress in rats. Pharmacol. Biochem. Behav. 2009, 93, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Ansari, M.A.; Ahmad, A.S.; Salim, S.; Yousuf, S.; Ishrat, T.; Islam, F. Selenium Protects Cerebral Ischemia in Rat Brain Mitochondria. Biol. Trace Element Res. 2004, 101, 73–86. [Google Scholar] [CrossRef]
- Chang, Q.; Gold, E.P. Switching memory systems during learning: Changes in patterns of brain acetylcholine release in the hippocampus and striatum in rats. F1000 Post Publ. Peer Rev. Biomed. Lit. 2003, 23, 3001–3005. [Google Scholar] [CrossRef]
- Gold, P.E. Acetylcholine modulation of neural systems involved in learning and memory. Neurobiol. Learn. Mem. 2003, 80, 194–210. [Google Scholar] [CrossRef]
- Jarrett, J.T.; Lansbury, P.T., Jr. Seeding “one-dimensional crystallization” of amyloid: A pathogenic mechanism in Alzheimer’s disease and scrapie? Cell 1993, 73, 1055–1058. [Google Scholar] [CrossRef]
- Saleem, U.; Raza, Z.; Anwar, F.; Ahmad, B.; Hira, S.; Ali, T. Experimental and Computational Studies to Characterize and Evaluate the Therapeutic Effect of Albizia lebbeck (L.) Seeds in Alzheimer’s Disease. Medicina 2019, 55, 184. [Google Scholar] [CrossRef]
- Herholz, K. Acetylcholine esterase activity in mild cognitive impairment and Alzheimer’s disease. Eur. J. Nucl. Med. Mol. Imaging 2008, 35, 25–29. [Google Scholar] [CrossRef]







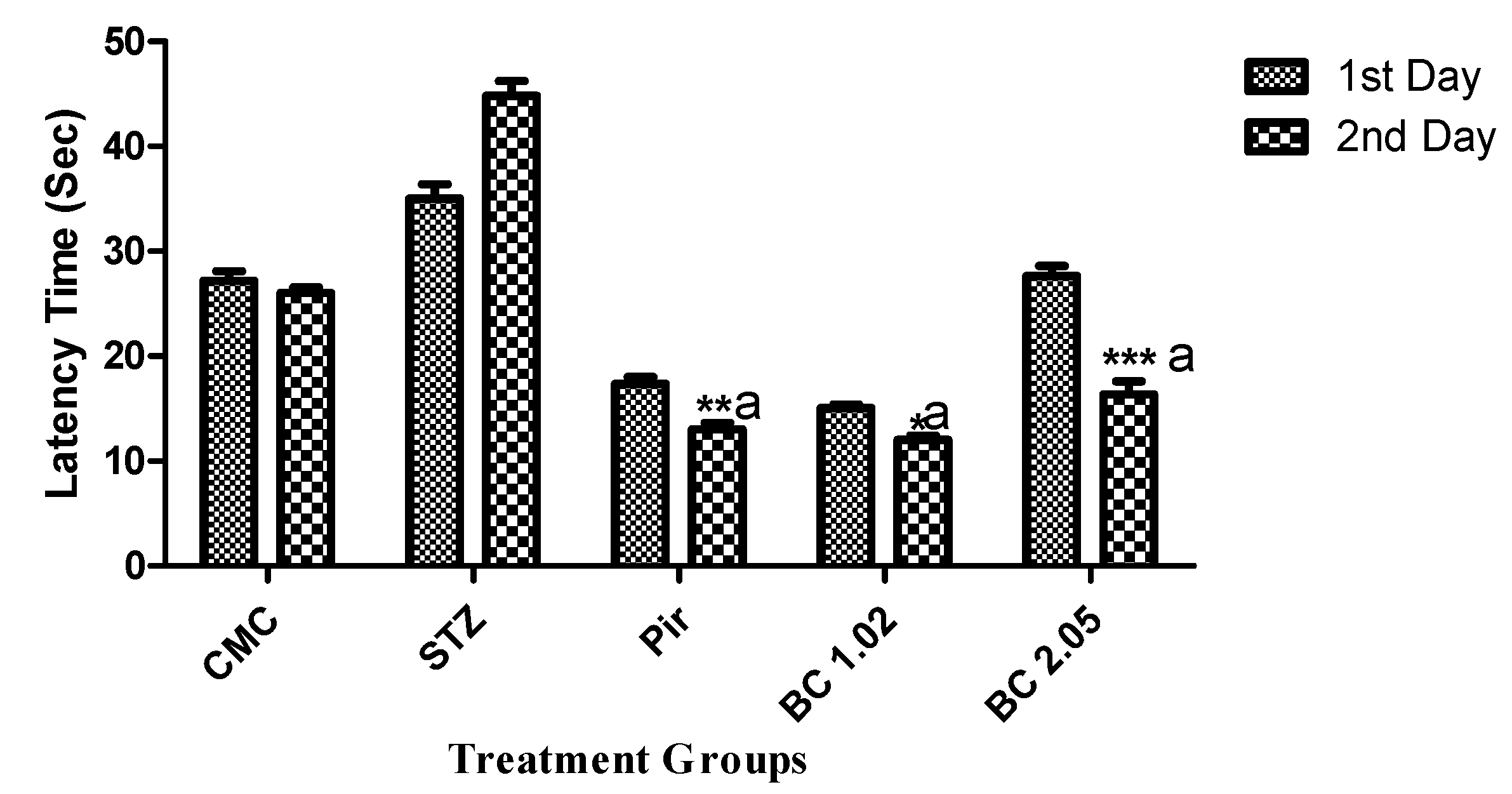{kind=link}
{kind=link}
{kind=link}
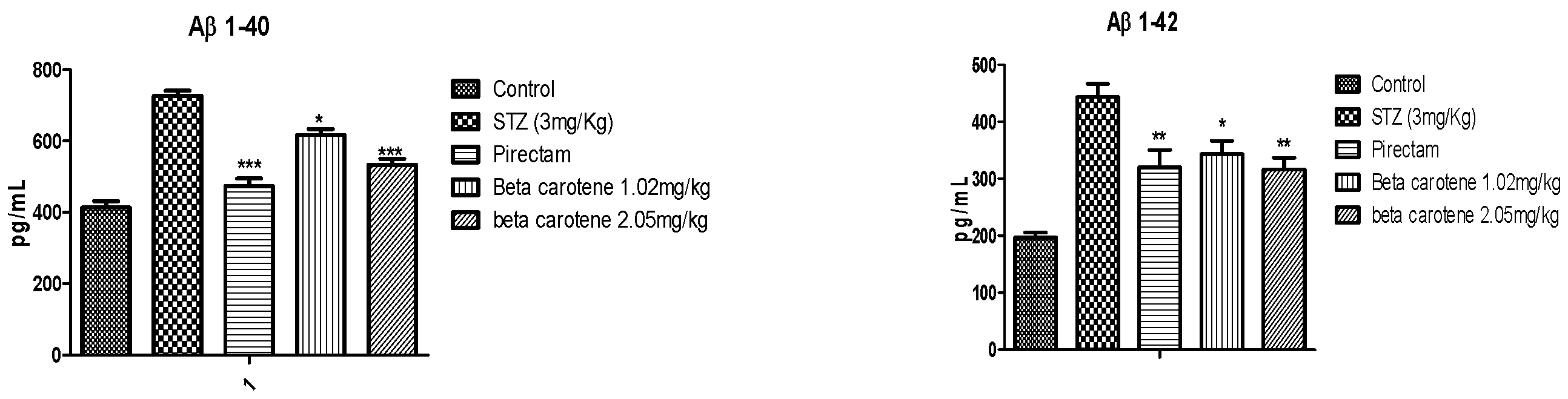{kind=link}
{kind=link}
{kind=link}
| Parameters | Control | Disease (STZ) | β-Carotene | |||
|---|---|---|---|---|---|---|
| Dose mg/kg | CMC 1 mL/kg | 3.0 | 1.02 | 2.05 | ||
| Whole body movement | Latency (s.) | 2.80 ± 0.374 | 5.6 ± 0.510 | 3.20 ± 0.200 | 2.00 ± 0.316 | |
| Rearing (no.) | 27.40 ± 1.030 | 16.60 ± 0.245 | 39.60 ± 1.435 ** | 29.80 ± 9.749 | ||
| Freezing (s.) | 32.00 ± 1.225 *** | 89.00 ± 3.317 | 29.00 ± 1.789 *** | 16.00 ± 2.258 *** | ||
| Part body Movement | Scratching (s.) | 3.80 ± 0.97 *** | 5.40 ± 0.400 | 2.60 ± 0.400 *** | 1.20 ± 0.490 | |
| Teeth Chattering | No | Yes | No | No | ||
| Digging | No | Yes | No | No | ||
| Location | Field area Visited | Central (s.) | 52.40 ± 7.019 *** | 112.00 ± 16.778 | 30.20 ± 1.985 *** | 30.00 ± 3.146 *** |
| Peripheral (s.) | 209 ± 9.066 *** | 88.00 ± 3.742 | 235.0 ± 2.236 *** | 250.080 ± 15.203 *** | ||
| Crossing (no.) | 31.6 0 ± 0.927 *** | 16.30 ± 0.200 | 49.40 ± 631 *** | 16.00 ± 1.342 *** | ||
| ANS | Defecation (no.) | 2.60 ± 0.670 *** | 17.00 ± 0.447 | 0.60 ± 0.400 *** | 0.400 ± 0.245 *** | |
| Urination (no.) | 4.6 ± 0.400 *** | 10.00 ± 0.707 | 0.80 ± 0.374 *** | 0.200 ± 0.200 *** | ||
| Sr. # | Treatment Groups | Dose (mg/kg) | GSH (µg/mg of Brain Tissue) | SOD (µg/mg of Brain Tissue) | CAT (µg/mg of Brain Tissue) | Acetylcholine Esterase (µmol/mg of Brain Tissue) |
|---|---|---|---|---|---|---|
| 1 | Control | CMC (1 mL/kg) | 14.4 ± 1.03 *** | 10.320 ± 0.185 *** | 4.96 ± 0.163 *** | 2.8 ± 0.170 *** |
| 2 | Diseased (STZ) | 300 | 3.54 ± 0.383 | 0.043 ± 0.002 | 1.11 ± 0.206 | 7.54 ± 0.220 *** |
| 3 | Standard (Piracetam) | 200 | 9.40 ± 0.678 *** | 0.262 ± 0.020 | 4.93 ± 1.00 *** | 4.620 ± 0.206 *** |
| 4 | β-carotene | 1.02 | 10.30 ± 0.200 *** | 0.284 ± 0.012 | 6.15 ± 0.01 *** | 3.430 ± 0.187 *** |
| 2.05 | 10.320 ± 0.185 *** | 0.492 ± 0.012 | 6.84 ± 0.01 *** | 3.380 ± 0.080 *** |
| Compound | Binding Energy (ΔG) kcal/mol | Inhibition Constant (Ki) μM | Interacting Residues | Interaction Type |
|---|---|---|---|---|
| β-carotene | −7.7 | 2.27 | LEU456, LEU450, LEU127, MET83, VAL71, MET90 | Alkyl |
| Piracetam | −5.8 | 56.05 | TYR130, GLY118, GLU199, SER200, GLY441, TRP84, PHE330 | H-Bonding, Alkyl |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hira, S.; Saleem, U.; Anwar, F.; Sohail, M.F.; Raza, Z.; Ahmad, B. β-Carotene: A Natural Compound Improves Cognitive Impairment and Oxidative Stress in a Mouse Model of Streptozotocin-Induced Alzheimer’s Disease. Biomolecules 2019, 9, 441. https://doi.org/10.3390/biom9090441
Hira S, Saleem U, Anwar F, Sohail MF, Raza Z, Ahmad B. β-Carotene: A Natural Compound Improves Cognitive Impairment and Oxidative Stress in a Mouse Model of Streptozotocin-Induced Alzheimer’s Disease. Biomolecules. 2019; 9(9):441. https://doi.org/10.3390/biom9090441
Chicago/Turabian StyleHira, Sundas, Uzma Saleem, Fareeha Anwar, Muhammad Farhan Sohail, Zohaib Raza, and Bashir Ahmad. 2019. "β-Carotene: A Natural Compound Improves Cognitive Impairment and Oxidative Stress in a Mouse Model of Streptozotocin-Induced Alzheimer’s Disease" Biomolecules 9, no. 9: 441. https://doi.org/10.3390/biom9090441
APA StyleHira, S., Saleem, U., Anwar, F., Sohail, M. F., Raza, Z., & Ahmad, B. (2019). β-Carotene: A Natural Compound Improves Cognitive Impairment and Oxidative Stress in a Mouse Model of Streptozotocin-Induced Alzheimer’s Disease. Biomolecules, 9(9), 441. https://doi.org/10.3390/biom9090441