A Multi-Biochemical and In Silico Study on Anti-Enzymatic Actions of Pyroglutamic Acid against PDE-5, ACE, and Urease Using Various Analytical Techniques: Unexplored Pharmacological Properties and Cytotoxicity Evaluation


 ,
,  ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Anti-PDE5A1 Inhibitory Properties
2.1.1. Protein Expression and Purification
2.1.2. Anti-PDE5A1 Activity
2.2. Anti-Angiotensin-Converting Enzyme (ACE) Properties
2.3. Anti-Urease Properties
2.3.1. Enzyme, Substrate, Inhibitors, and Chemicals
2.3.2. Instrumentation: ESI-MS
2.3.3. Determination of Anti-Urease Activity
2.4. Cytotoxicity Study
2.4.1. Cell Lines, Medium and Reagents
2.4.2. Assessment of Cytotoxicity
2.5. Molecular Docking Studies
Protein-Ligand Preparation and Performance of Docking Studies
3. Results and Discussion
3.1. Assessment of Anti-PDE5A1 Activity
3.2. Assessment of Anti-ACE Activity
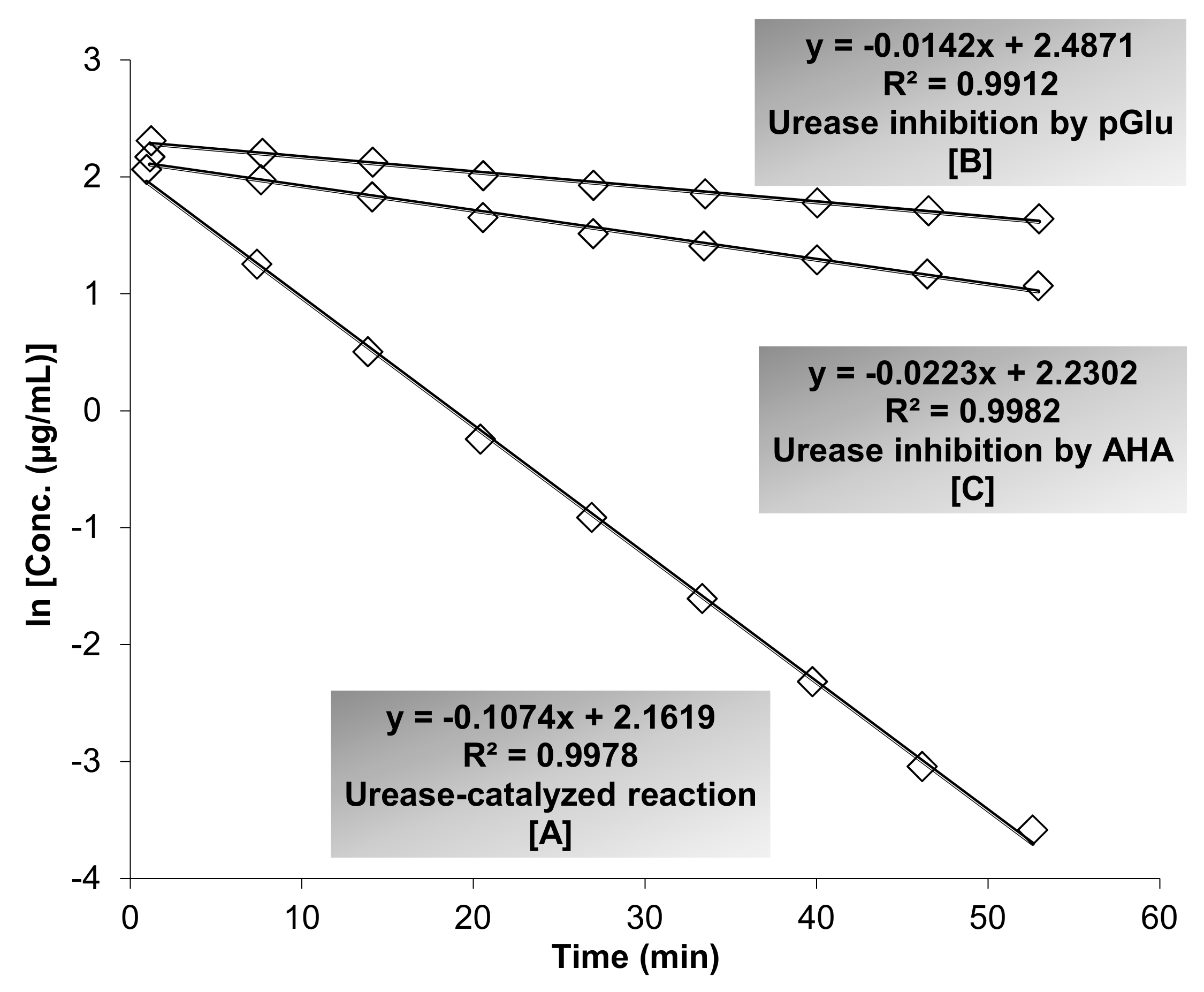
3.3. Evaluation of Anti-Urease Activity
3.4. Evaluation of Cytotoxicity Characteristics
3.5. Evaluation of Molecular Docking Analyses
3.5.1. Interaction of pGlu with Actice Site of PDE5A1
3.5.2. Interaction of pGlu with Actice Site of ACE
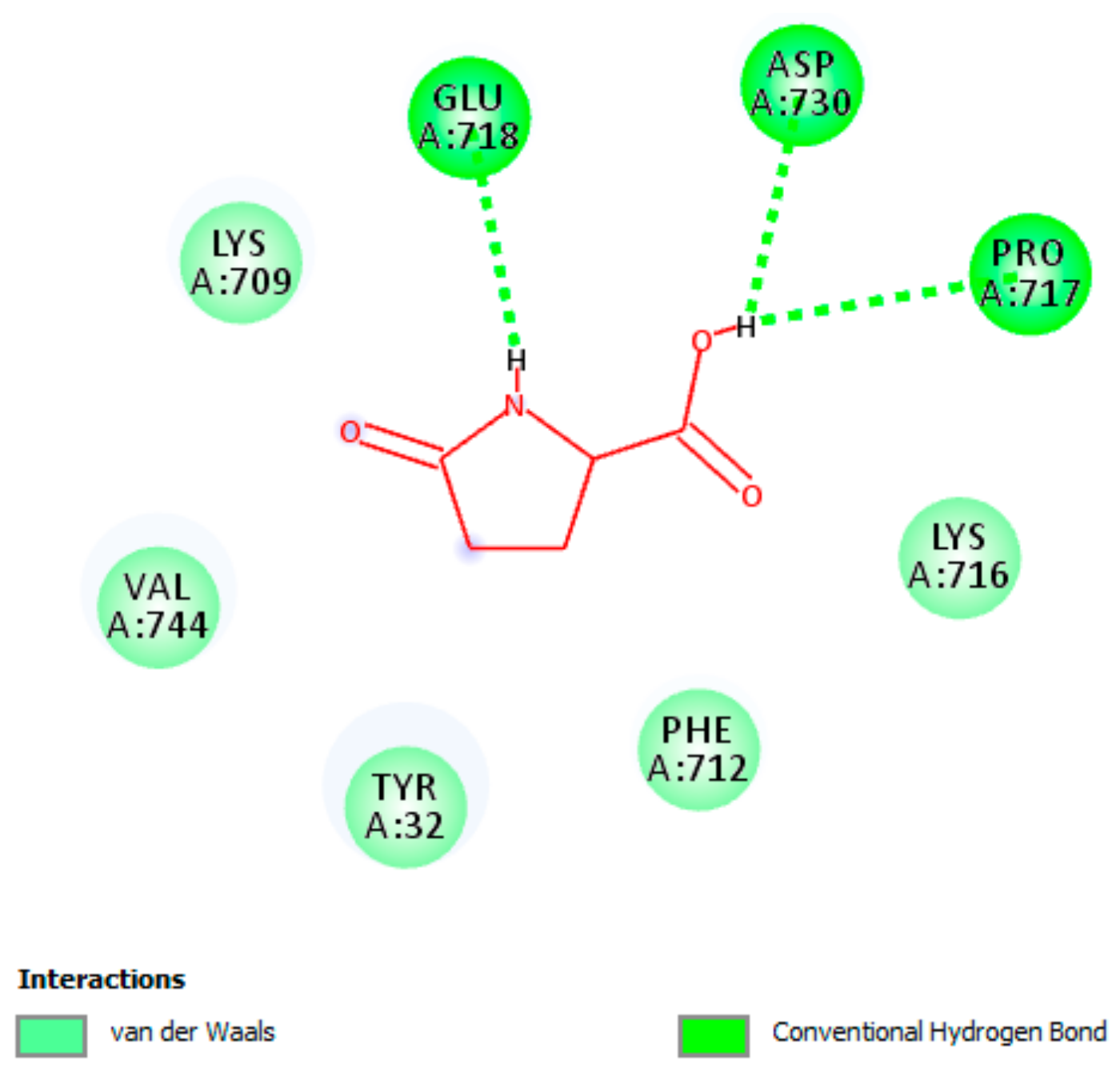
3.5.3. Interaction of pGlu with Actice Site of Urease
4. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Patel, K.; Ahmed, Z.S.; Huang, X.; Yang, Q.; Ekinci, E.; Neslund-Dudas, C.M.; Mitra, B.; Elnady, F.A.; Ahn, Y.H.; Yang, H.; et al. Discovering proteasomal deubiquitinating enzyme inhibitors for cancer therapy: lessons from rational design, nature and old drug reposition. Future Med Chem. 2018, 10, 2087–2108. [Google Scholar] [CrossRef] [PubMed]
- Eid, H.M.; Wright, M.L.; Anil Kumar, N.V.; Qawasmeh, A.; Hassan, S.T.S.; Mocan, A.; Nabavi, S.M.; Rastrelli, L.; Atanasov, A.G.; Haddad, P.S. Significance of Microbiota in Obesity and Metabolic Diseases and the Modulatory Potential by Medicinal Plant and Food Ingredients. Front. Pharmacol. 2017, 8, 387. [Google Scholar]
- Harvey, A.L.; Edrada-Ebel, R.; Quinn, R.J. The re-emergence of natural products for drug discovery in the genomics era. Nat. Rev. Drug Discov. 2015, 14, 111–129. [Google Scholar] [PubMed] [Green Version]
- Kumar, A.; Bachhawat, A.K. Pyroglutamic acid: Throwing light on a lightly studied metabolite. Curr. Sci. 2012, 102, 288–297. [Google Scholar]
- Liss, D.B.; Paden, M.S.; Schwarz, E.S.; Mullins, M.E. What is the clinical significance of 5-oxoproline (pyroglutamic acid) in high anion gap metabolic acidosis following paracetamol (acetaminophen) exposure? Clin. Toxicol. 2013, 51, 817–827. [Google Scholar] [CrossRef]
- Sasaki, S.; Futagi, Y.; Kobayashi, M.; Ogura, J.; Iseki, K. Functional characterization of 5-oxoproline transport via SLC16A1/MCT1. J. Biol. Chem. 2015, 290, 2303–2311. [Google Scholar] [CrossRef] [PubMed]
- Yoshinari, O.; Igarashi, K. Anti-diabetic effect of pyroglutamic acid in type 2 diabetic Goto-Kakizaki rats and KK-Ay mice. Br. J. Nutr. 2011, 106, 995–1004. [Google Scholar] [CrossRef] [PubMed]
- Silva, A.R.; Silva, C.G.; Ruschel, C.; Helegda, C.; Wyse, A.T.; Wannmacher, C.M.; Wajner, M.; Dutra-Filho, C.S. L-pyroglutamic acid inhibits energy production and lipid synthesis in cerebral cortex of young rats in vitro. Neurochem. Res. 2001, 26, 1277–1283. [Google Scholar] [CrossRef] [PubMed]
- Corinaldesi, C.; Di Luigi, L.; Lenzi, A.; Crescioli, C. Phosphodiesterase type 5 inhibitors: back and forward from cardiac indications. J. Endocrinol. Invest. 2016, 39, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Hamet, P.; Tremblay, J. Platelet cGMP-binding Phosphodiesterase. Methods Enzymol. 1988, 159, 710–722. [Google Scholar] [PubMed]
- Francis, S.H.; Corbin, J.D. Purification of cGMP-Binding Protein Phosphodiesterase from Rat Lung. Methods Enzymol. 1988, 159, 722–729. [Google Scholar] [PubMed]
- Lin, C.S. Tissue Expression, Distribution, and Regulation of PDE5. Int. J. Impotence Res. 2004, 16, S8–S10. [Google Scholar] [CrossRef] [PubMed]
- Kouvelas, D.; Goulas, A.; Papazisis, G.; Sardeli, C.; Pourzitaki, C. PDE5 Inhibitors: In Vitro and In Vivo Pharmacological Profile. Curr. Pharm. Des. 2009, 15, 3464–3475. [Google Scholar] [CrossRef] [PubMed]
- Giordano, D.; De Stefano, M.E.; Citro, G.; Modica, A.; Giorgi, M. Expression of cGMP-Binding cGMP-Specific Phosphodiesterase (PDE5) in Mouse Tissues and Cell Lines Using an Antibody Against the Eenzyme Aamino-Terminal Domain. Biochim. Biophys. Acta Mol. Cell Res. 2001, 1539, 16–27. [Google Scholar]
- Regulska, K.; Stanisz, B.; Regulski, M.; Murias, M. How to design a potent, specific, and stable angiotensin-converting enzyme inhibitor. Drug Discov. Today. 2014, 19, 1731–1743. [Google Scholar] [CrossRef] [PubMed]
- Lever, A.F.; Hole, D.J.; Gillis, C.R.; McCallum, I.R.; McInnes, G.T.; MacKinnon, P.L.; Meredith, P.A.; Murray, L.S.; Reid, J.L.; Robertson, J.W. Do inhibitors of angiotensin-I-converting enzyme protect against risk of cancer? Lancet. 1998, 352, 179–184. [Google Scholar] [CrossRef]
- Regulski, M.; Regulska, K.; Stanisz, B.J.; Murias, M.; Gieremek, P.; Wzgarda, A.; Niznik, B. Chemistry and pharmacology of Angiotensin-converting enzyme inhibitors. Curr. Pharm. Des. 2015, 21, 1764–1775. [Google Scholar] [CrossRef]
- Hassan, S.T.S.; Švajdlenka, E.; Rengasamy, K.R.R.; Melichárková, R.; Pandian, S.K. S. Afr. J. Bot. 2019, 120, 175–178. [CrossRef]
- Kappaun, K.; Piovesan, A.R.; Carlini, C.R.; Ligabue-Braun, R. Ureases: Historical aspects, catalytic, and non-catalytic properties - A review. J. Adv. Res. 2018, 13, 3–17. [Google Scholar] [CrossRef]
- Hassan, S.T.; Šudomová, M. The Development of Urease Inhibitors: What Opportunities Exist for Better Treatment of Helicobacter pylori Infection in Children? Children 2017, 4, 2. [Google Scholar] [CrossRef]
- Amtul, Z.; Rahman, A.U.; Siddiqui, R.A.; Choudhary, M.I. Chemistry and mechanism of urease inhibition. Curr. Med. Chem. 2002, 9, 1323–1348. [Google Scholar] [CrossRef]
- Awllia, J.A.J.; Sara, A.; Wahab, A.-T.; Al-Ghamdi, M.; Rasheed, S.; Huwait, E.; Iqbal Choudhary, M. Discovery of new inhibitors of urease enzyme: A study using STD-NMR spectroscopy. Lett. Drug Des. Discov. 2015, 12, 819–827. [Google Scholar] [CrossRef]
- Wang, H.; Liu, Y.; Huai, Q.; Cai, J.; Zoraghi, R.; Francis, S.H.; Corbin, J.D.; Robinson, H.; Xin, Z.; Lin, G.; et al. Multiple conformations of phosphodiesterase-5: implications for enzyme function and drug development. J. Biol. Chem. 2006, 281, 469–479. [Google Scholar] [CrossRef] [PubMed]
- Shang, N.N.; Shao, Y.X.; Cai, Y.H.; Guan, M.; Huang, M.; Cui, W.; He, L.; Yu, Y.J.; Huang, L.; Li, Z.; et al. Discovery of 3-(4-Hydroxybenzyl)-1-(thiophen-2-yl)chromeno[2,3-c]pyrrol-9(2H)-one as a Phosphodiesterase-5 Inhibitor and Its Complex Crystal Structure. Biochem. Pharmacol. 2014, 89, 86–98. [Google Scholar] [CrossRef] [PubMed]
- Cushman, D.W.; Cheung, H.S. Spectrophotometric assay and properties of the angiotensin-converting enzyme of rabbit lung. Biochem. Pharmacol. 1971, 20, 1637–1648. [Google Scholar] [CrossRef]
- Hassan, S.T.S.; Švajdlenka, E.; Berchová-Bímová, K. Hibiscus sabdariffa L. and Its Bioactive Constituents Exhibit Antiviral Activity against HSV-2 and Anti-enzymatic Properties against Urease by an ESI-MS Based Assay. Molecules. 2017, 22, 722. [Google Scholar] [CrossRef]
- Supino, R. Methods in molecular biology. In In Vitro Toxicity Testing Protocols; Hare, S., Atterwill, C.K., Eds.; Humana Press: Totowa, NJ, USA, 1995; pp. 137–149. [Google Scholar]
- Hassan, S.T.S.; Švajdlenka, E. Biological evaluation and molecular docking of protocatechuic acid from Hibiscus sabdariffa L. as a potent urease inhibitor by an ESI-MS based method. Molecules 2017, 22, 1696. [Google Scholar] [CrossRef] [PubMed]
- Hassan, S.T.S.; Šudomová, M.; Berchová-Bímová, K.; Gowrishankar, S.; Rengasamy, K.R.R. Antimycobacterial, Enzyme Inhibition, and Molecular Interaction Studies of Psoromic Acid in Mycobacterium tuberculosis: Efficacy and Safety Investigations. J. Clin. Med. 2018, 7, 226. [Google Scholar] [CrossRef] [PubMed]
- Dassault Systèmes BIOVIA. Discovery Studio Modeling Environment, Release 2017; Dassault Systèmes: San Diego, CA, USA, 2017. [Google Scholar]
- Oh, T.Y.; Kang, K.K.; Ahn, B.O.; Yoo, M.; Kim, W.B. Erectogenic Effect of the Selective Phosphodiesterase Type 5 Inhibitor, DA-8159. Arch. Pharmacal. Res. 2000, 23, 471–476. [Google Scholar] [CrossRef]
- Keating, G.M.; Scott, L.J. Vardenafil. Drugs 2003, 63, 2673–2702. [Google Scholar] [CrossRef]
- Jung, J.Y.; Kim, S.K.; Kim, B.S.; Lee, S.H.; Park, Y.S.; Kim, S.J.; Choi, C.; Yoon, S.I.; Kim, J.S.; Cho, S.D.; et al. The Penile Erection Efficacy of a New Phosphodiesterase Type 5 Inhibitor, Mirodenafil (SK3530), in Rabbits with Acute Spinal Cord Injury. J. Vet. Med. Sci. 2008, 70, 1199–1204. [Google Scholar] [CrossRef] [Green Version]
- Galieè, N.; Ghofrani, H.A.; Torbicki, A.; Barst, R.J.; Rubin, L.J.; Badesch, D.; Fleming, T.; Parpia, T.; Burgess, G.; Branzi, A.; et al. Sildenafil Citrate Therapy for Pulmonary Arterial Hypertension. N. Engl. J. Med. 2005, 353, 2148–2157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azzouni, F.; Abu samra, K. Are Phosphodiesterase Type 5 Inhibitors Associated with Vision-Threatening Adverse Events? A Critical Analysis and Review of the Literature. J. Sex. Med. 2011, 8, 2894–2903. [Google Scholar] [CrossRef]
- Khan, A.S.; Sheikh, Z.; Khan, S.; Dwivedi, R.; Benjamin, E. Viagra Deafness—Sensorineural Hearing Loss and Phosphodiesterase-5 Inhibitors. Laryngoscope 2011, 121, 1049–1054. [Google Scholar] [CrossRef]
- Wu, D.; Zhang, T.; Chen, Y.; Huang, Y.; Geng, H.; Yu, Y.; Zhang, C.; Lai, Z.; Wu, Y.; Guo, X.; et al. Discovery and Optimization of Chromeno[2,3-c]pyrrol-9(2H)-ones as Novel Selective and Orally Bioavailable Phosphodiesterase 5 Inhibitors for the Treatment of Pulmonary Arterial Hypertension. J Med Chem. 2017, 60, 6622–6637. [Google Scholar] [CrossRef]
- Patten, G.S.; Abeywardena, M.Y.; Bennett, L.E. Inhibition of Angiotensin Converting Enzyme, Angiotensin II Receptor Blocking, and Blood Pressure Lowering Bioactivity across Plant Families. Crit. Rev. Food Sci. Nutr. 2016, 56, 181–214. [Google Scholar] [CrossRef] [PubMed]
- Bullo, M.; Tschumi, S.; Bucher, B.S.; Bianchetti, M.G.; Simonetti, G.D. Pregnancy outcome following exposure to angiotensin-converting enzyme inhibitors or angiotensin receptor antagonists: A systematic review. Hypertension 2012, 60, 444–450. [Google Scholar] [CrossRef]
- Dicpinigaitis, P.V. Angiotensin-converting enzyme inhibitor-induced cough: ACCP evidence-based clinical practice guidelines. Chest 2006, 129, 169S–173S. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Xu, X.; Liang, Y.; Head, R.; Bennett, L. Inhibition of angiotensin converting enzyme (ACE) activity by polyphenols from tea (Camellia sinensis) and links to processing method. Food Funct. 2011, 2, 310–319. [Google Scholar] [CrossRef]
- Balasuriya, B.N.; Rupasinghe, H.V. Plant flavonoids as angiotensin converting enzyme inhibitors in regulation of hypertension. Funct. Foods Health Dis. 2011, 1, 172–188. [Google Scholar]
- Liu, J.-C.; Hsu, F.-L.; Tsai, J.-C.; Chan, P.; Liu, J.Y.-H.; Thomas, G.N.; Tomlinson, B.; Lo, M.-Y.; Lin, J.-Y. Antihypertensive effects of tannins isolated from traditional chinese herbs as non-specific inhibitors of angiontensin converting enzyme. Life Sci. 2003, 73, 1543–1555. [Google Scholar] [CrossRef]
- Daskaya-Dikmen, C.; Yucetepe, A.; Karbancioglu-Guler, F.; Daskaya, H.; Ozcelik, B. Angiotensin-I-Converting Enzyme (ACE)-Inhibitory Peptides from Plants. Nutrients 2017, 9, 316. [Google Scholar] [CrossRef] [PubMed]
- Follmer, C.J. Ureases as a target for the treatment of gastric and urinary infections. Clin. Pathol. 2010, 63, 424–430. [Google Scholar] [CrossRef]
- Hassan, S.T.; Žemlička, M. Plant-Derived Urease Inhibitors as Alternative Chemotherapeutic Agents. Arch Pharm. 2016, 349, 507–522. [Google Scholar] [CrossRef] [PubMed]
- Modolo, L.V.; de Souza, A.X.; Horta, L.P.; Araujo, D.P.; de Fátima, Â. An overview on the potential of natural products as ureases inhibitors: A review. J Adv Res. 2015, 6, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Rego, Y.F.; Queiroz, M.P.; Brito, T.O.; Carvalho, P.G.; de Queiroz, V.T.; de Fátima, Â.; Macedo, F., Jr. A review on the development of urease inhibitors as antimicrobial agents against pathogenic bacteria. J. Adv. Res. 2018, 13, 69–100. [Google Scholar] [CrossRef] [PubMed]
- Hameed, A.; Al-Rashida, M.; Uroos, M.; Qazi, S.U.; Naz, S.; Ishtiaq, M.; Khan, K.M. A patent update on therapeutic applications of urease inhibitors (2012-2018). Expert Opin. Ther. Pat. 2019, 29, 181–189. [Google Scholar] [CrossRef]
- Hassan, S.T.; Berchová-Bímová, K.; Petráš, J. Plumbagin, a Plant-Derived Compound, Exhibits Antifungal Combinatory Effect with Amphotericin B against Candida albicans Clinical Isolates and Anti-hepatitis C Virus Activity. Phytother. Res. 2016, 30, 1487–1492. [Google Scholar] [CrossRef] [PubMed]
- Calpena, E.; Deshpande, A.A.; Yap, S.; Kumar, A.; Manning, N.J.; Bachhawat, A.K.; Espinós, C. New insights into the genetics of 5-oxoprolinase deficiency and further evidence that it is a benign biochemical condition. Eur J Pediatr. 2015, 174, 407–411. [Google Scholar] [CrossRef]
- Mayatepek, E.; Meissner, T.; Gröbe, H. Acute metabolic crisis with extreme deficiency of glutathione in combination with decreased levels of leukotriene C4 in a patient with glutathione synthetase deficiency. J. Inherit. Metab. Dis. 2004, 27, 297–299. [Google Scholar] [CrossRef]
- Li, X.; Ding, Y.; Liu, Y.; Ma, Y.; Song, J.; Wang, Q.; Yang, Y. Five Chinese patients with 5-oxoprolinuria due to glutathione synthetase and 5-oxoprolinase deficiencies. Brain Dev. 2015, 37, 952–959. [Google Scholar] [CrossRef]
- Kirchmair, J.; Göller, A.H.; Lang, D.; Kunze, J.; Testa, B.; Wilson, I.D.; Glen, R.C.; Schneider, G. Predicting drug metabolism: experiment and/or computation? Nat. Rev. Drug Discov. 2015, 14, 387–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Natesh, R.; Schwager, S.L.; Evans, H.R.; Sturrock, E.D.; Acharya, K.R. Structural details on the binding of antihypertensive drugs captopril and enalaprilat to human testicular angiotensin i-converting enzyme. Biochemistry 2004, 43, 8718–8724. [Google Scholar] [CrossRef] [PubMed]
- Balasubramanian, A.; Ponnuraj, K. Crystal structure of the first plant urease from jack bean: 83 years of journey from its first crystal to molecular structure. J. Mol. Biol. 2010, 400, 274–283. [Google Scholar] [CrossRef]
- Farrugia, M.A.; Macomber, L.; Hausinger, R.P. Biosynthesis of the urease metallocenter. J. Biol. Chem. 2013, 288, 3178–3185. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Inhibitor | IC50 (µM) |
|---|---|
| pGlu | 5.23 ± 1.33 |
| Sildenafil citrate | 7.14 ± 1.52 |
| Inhibitors | % Inhibition |
|---|---|
| pGlu | 98.2 ± 1.12 |
| Captopril | 99.6 ± 1.64 |
| ACE-catalyzed reaction (no inhibition) | Nd |
| IC50 (µg/mL) | ||
|---|---|---|
| Compound | HeLa-R2 | MRC-5 |
| pGlu | 96.42 ± 0.92 | 97.21 ± 0.61 |
| Cisplatin | 6.32 ± 0.62 | 9.14 ± 0.42 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Šudomová, M.; Hassan, S.T.S.; Khan, H.; Rasekhian, M.; Nabavi, S.M. A Multi-Biochemical and In Silico Study on Anti-Enzymatic Actions of Pyroglutamic Acid against PDE-5, ACE, and Urease Using Various Analytical Techniques: Unexplored Pharmacological Properties and Cytotoxicity Evaluation. Biomolecules 2019, 9, 392. https://doi.org/10.3390/biom9090392
Šudomová M, Hassan STS, Khan H, Rasekhian M, Nabavi SM. A Multi-Biochemical and In Silico Study on Anti-Enzymatic Actions of Pyroglutamic Acid against PDE-5, ACE, and Urease Using Various Analytical Techniques: Unexplored Pharmacological Properties and Cytotoxicity Evaluation. Biomolecules. 2019; 9(9):392. https://doi.org/10.3390/biom9090392
Chicago/Turabian StyleŠudomová, Miroslava, Sherif T. S. Hassan, Haroon Khan, Mahsa Rasekhian, and Seyed Mohammad Nabavi. 2019. "A Multi-Biochemical and In Silico Study on Anti-Enzymatic Actions of Pyroglutamic Acid against PDE-5, ACE, and Urease Using Various Analytical Techniques: Unexplored Pharmacological Properties and Cytotoxicity Evaluation" Biomolecules 9, no. 9: 392. https://doi.org/10.3390/biom9090392
APA StyleŠudomová, M., Hassan, S. T. S., Khan, H., Rasekhian, M., & Nabavi, S. M. (2019). A Multi-Biochemical and In Silico Study on Anti-Enzymatic Actions of Pyroglutamic Acid against PDE-5, ACE, and Urease Using Various Analytical Techniques: Unexplored Pharmacological Properties and Cytotoxicity Evaluation. Biomolecules, 9(9), 392. https://doi.org/10.3390/biom9090392