Nanoscale Restructuring of Polymer Materials to Produce Single Polymer Composites and Miscible Blends

Abstract
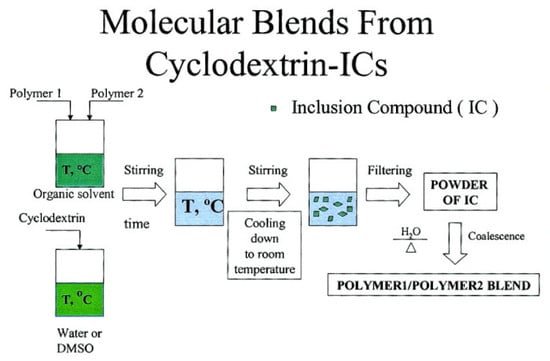
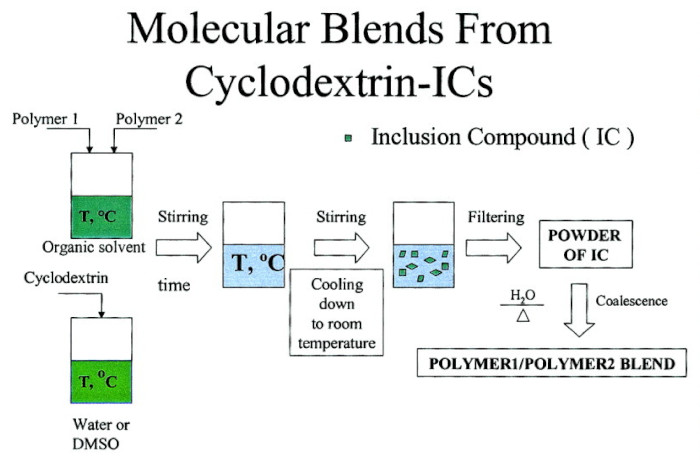
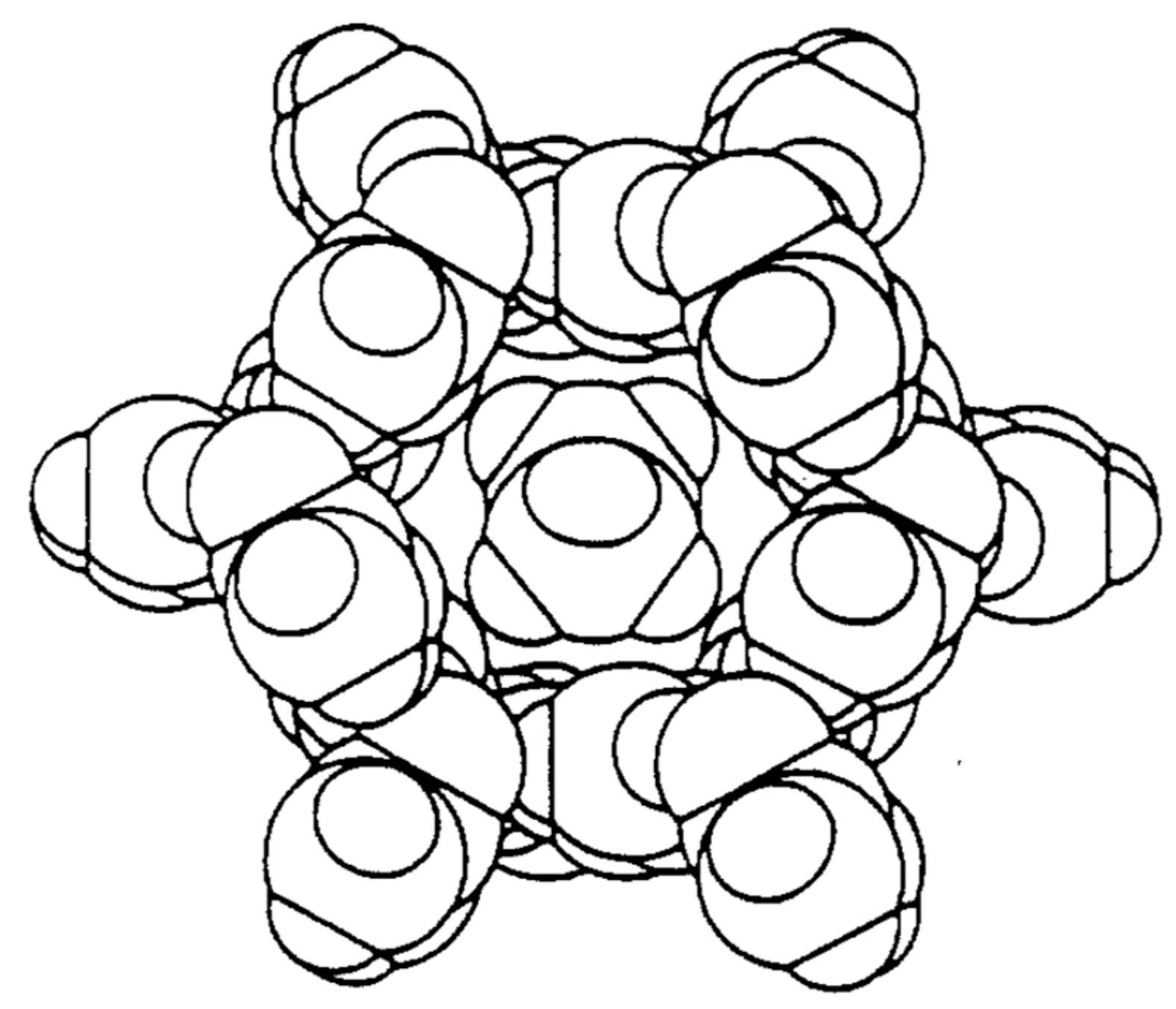
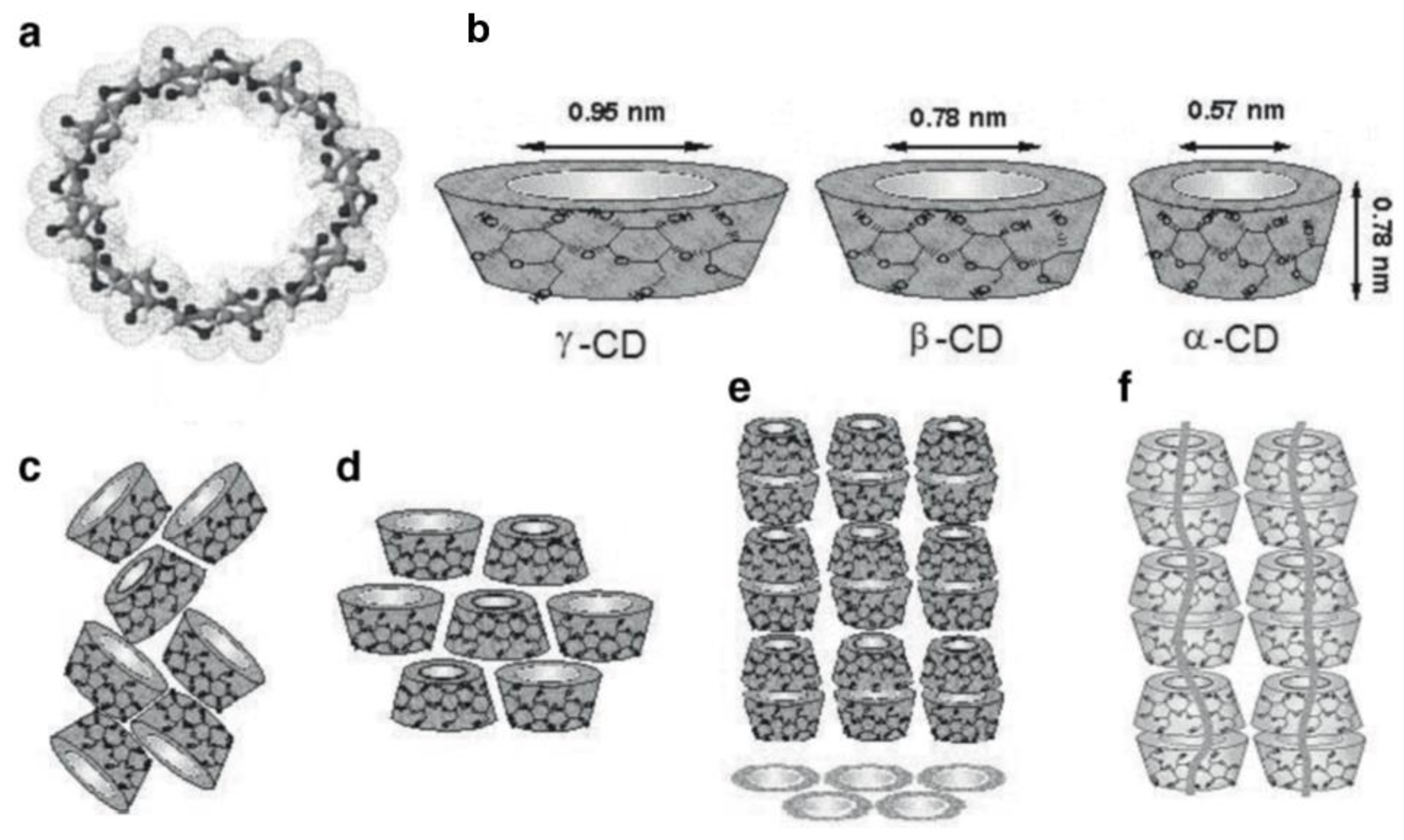
1. Introduction
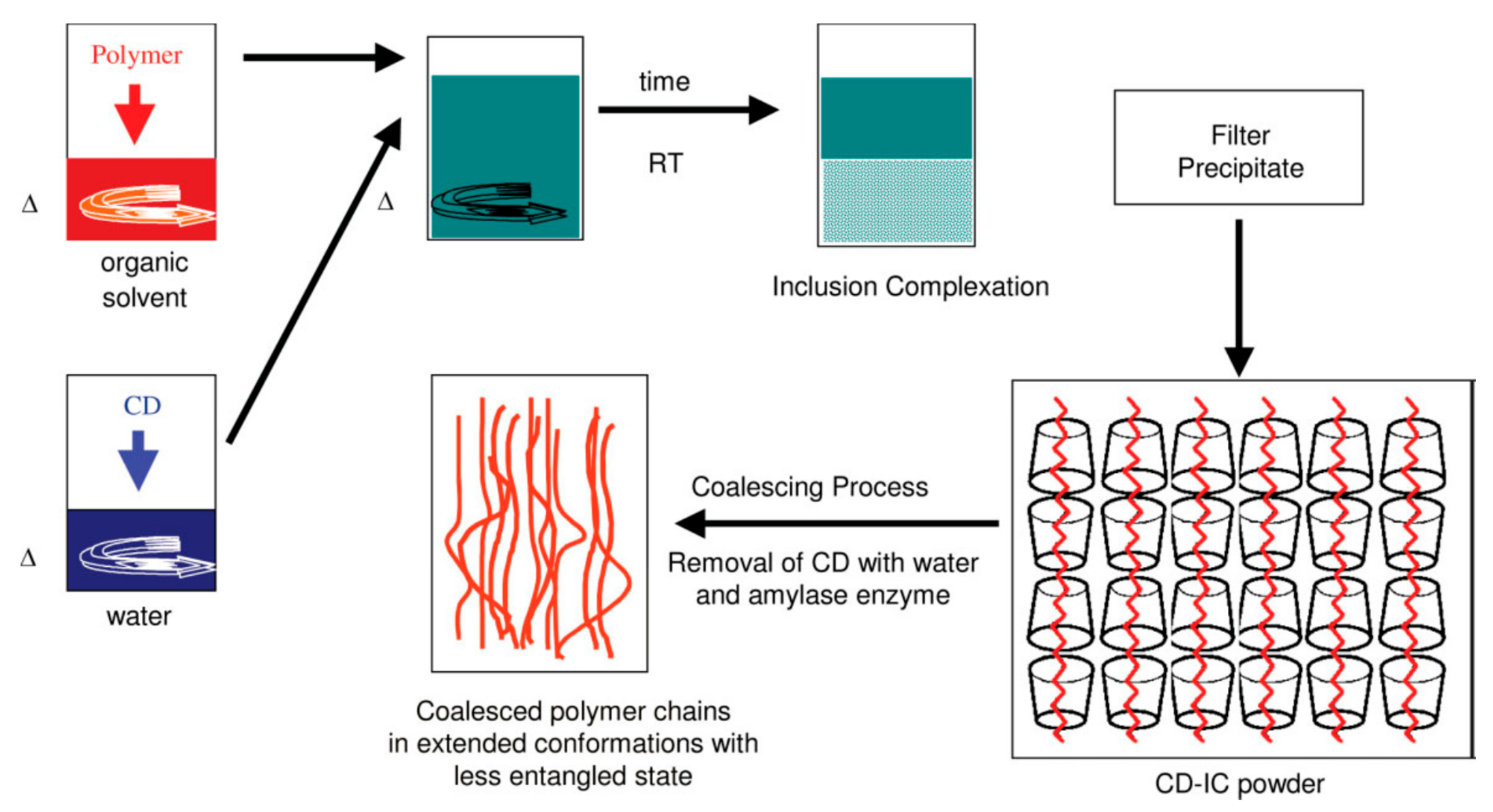
2. Materials and Methods
3. Results and Discussion
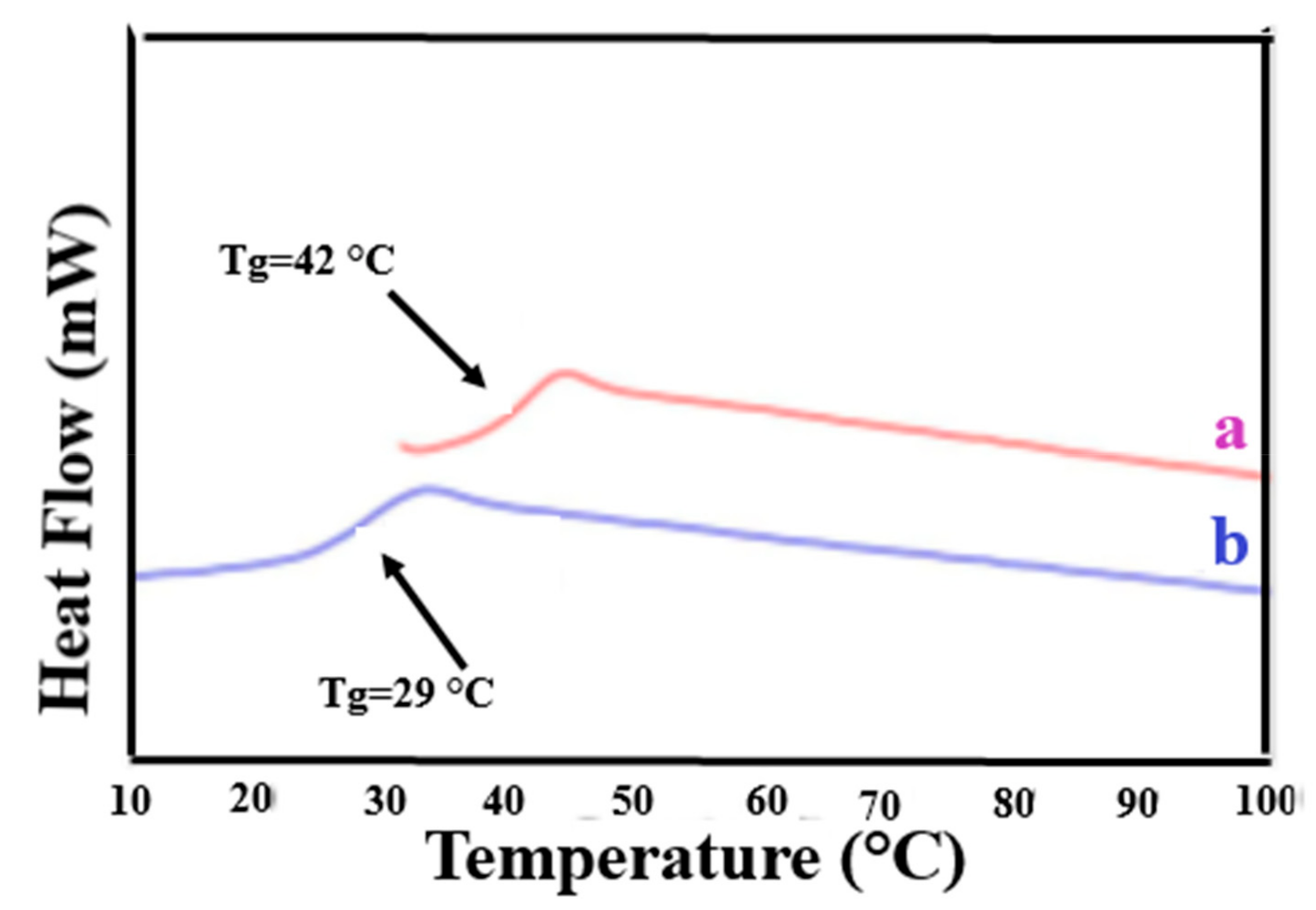
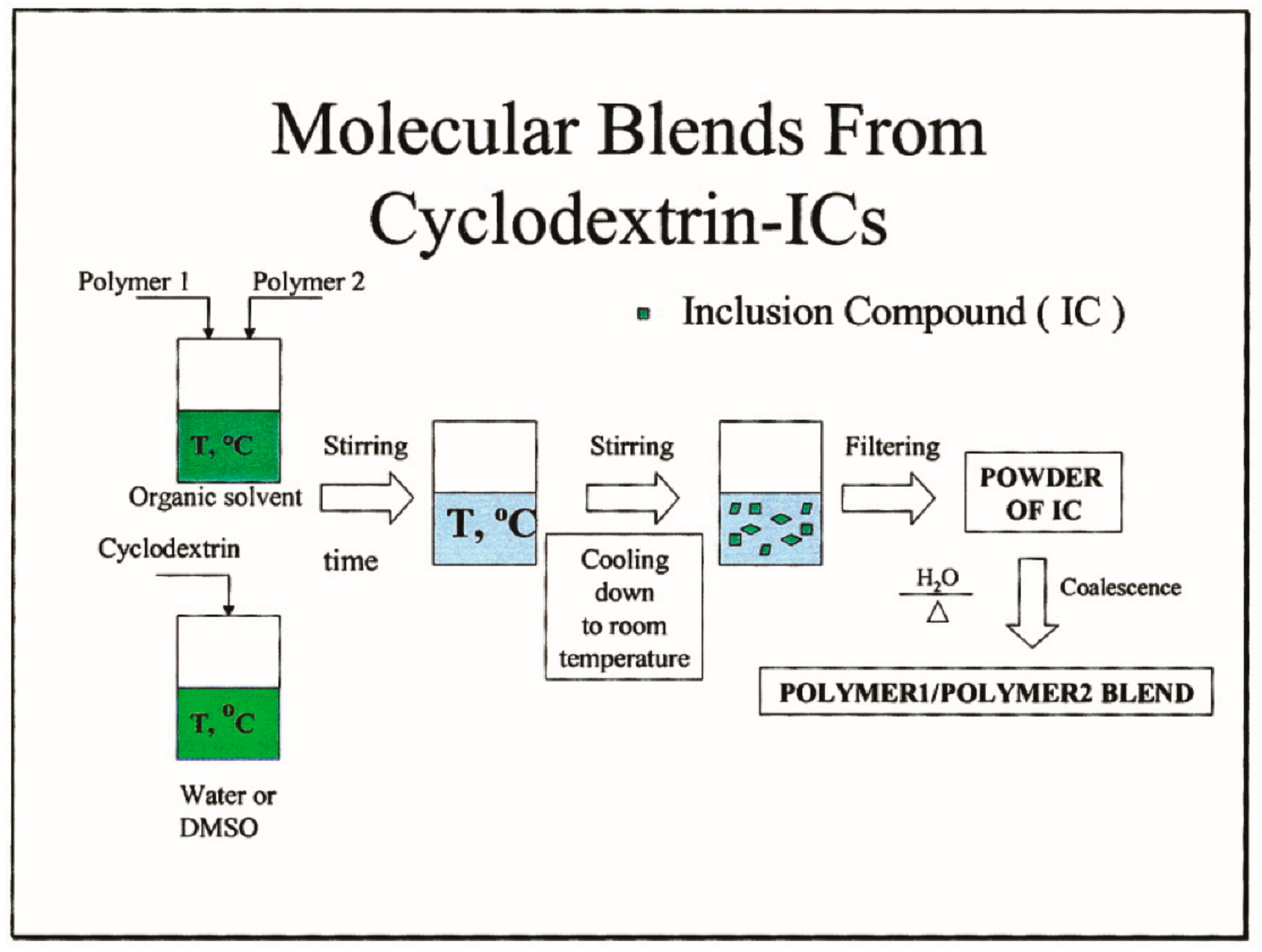
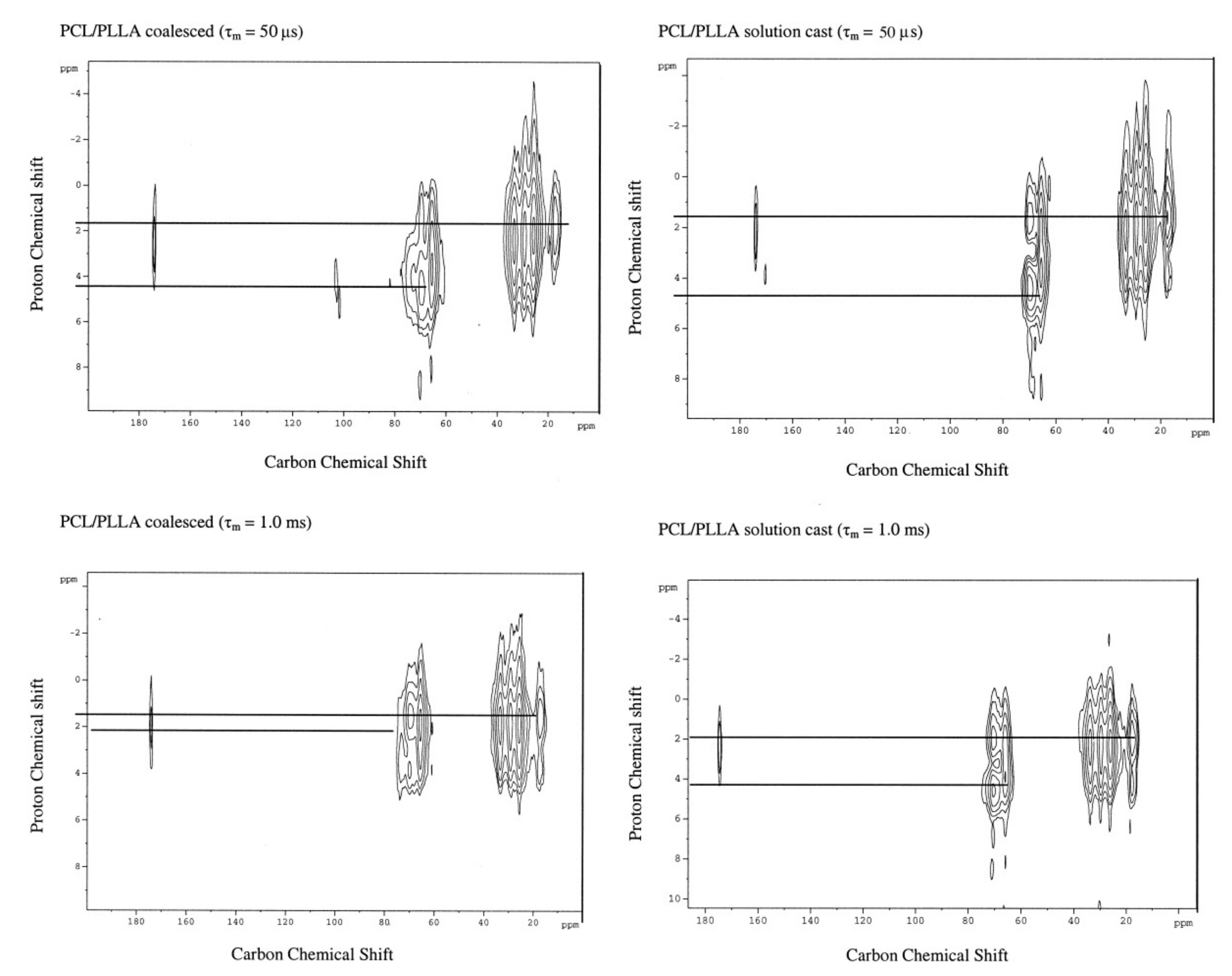
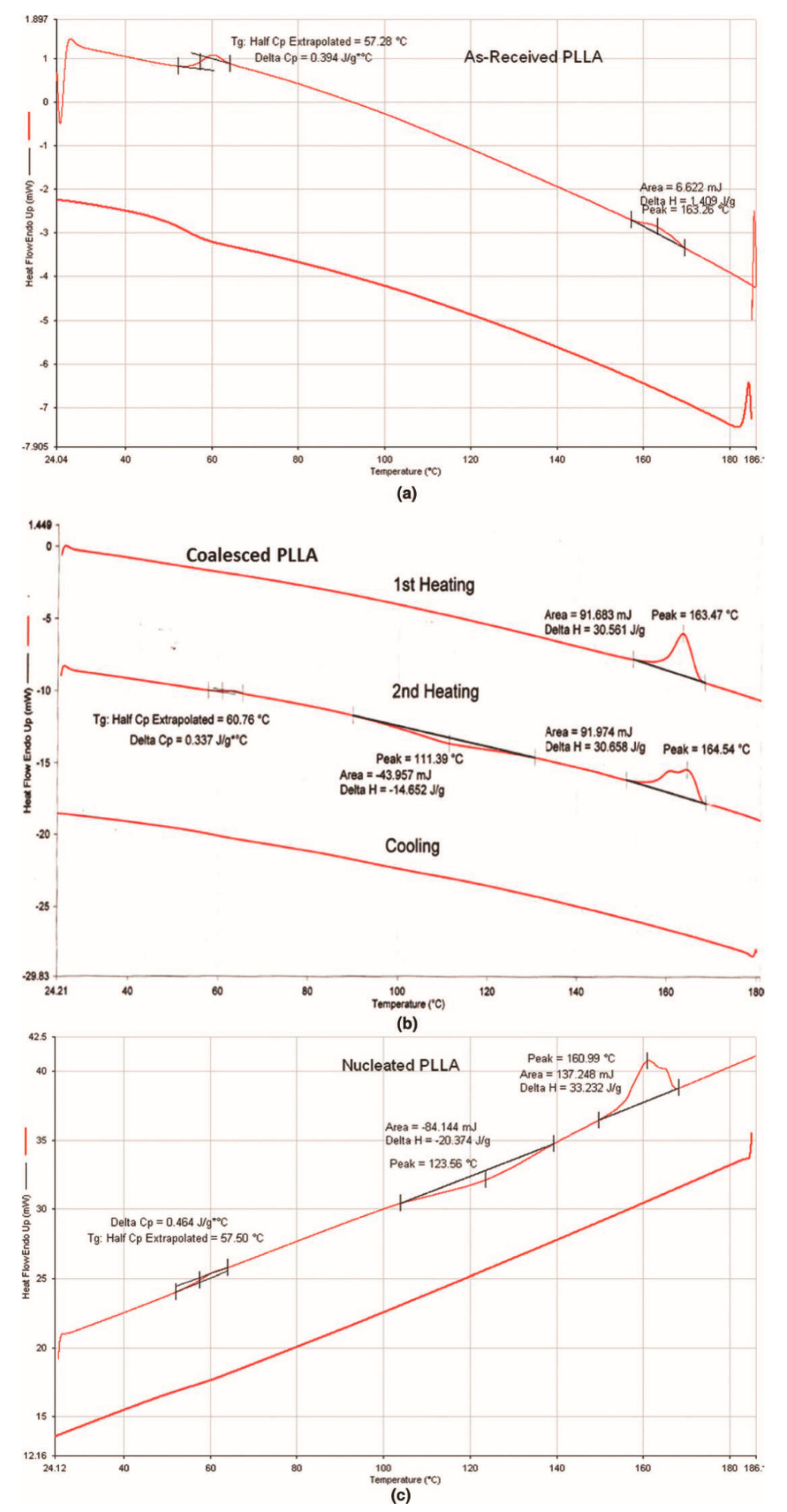
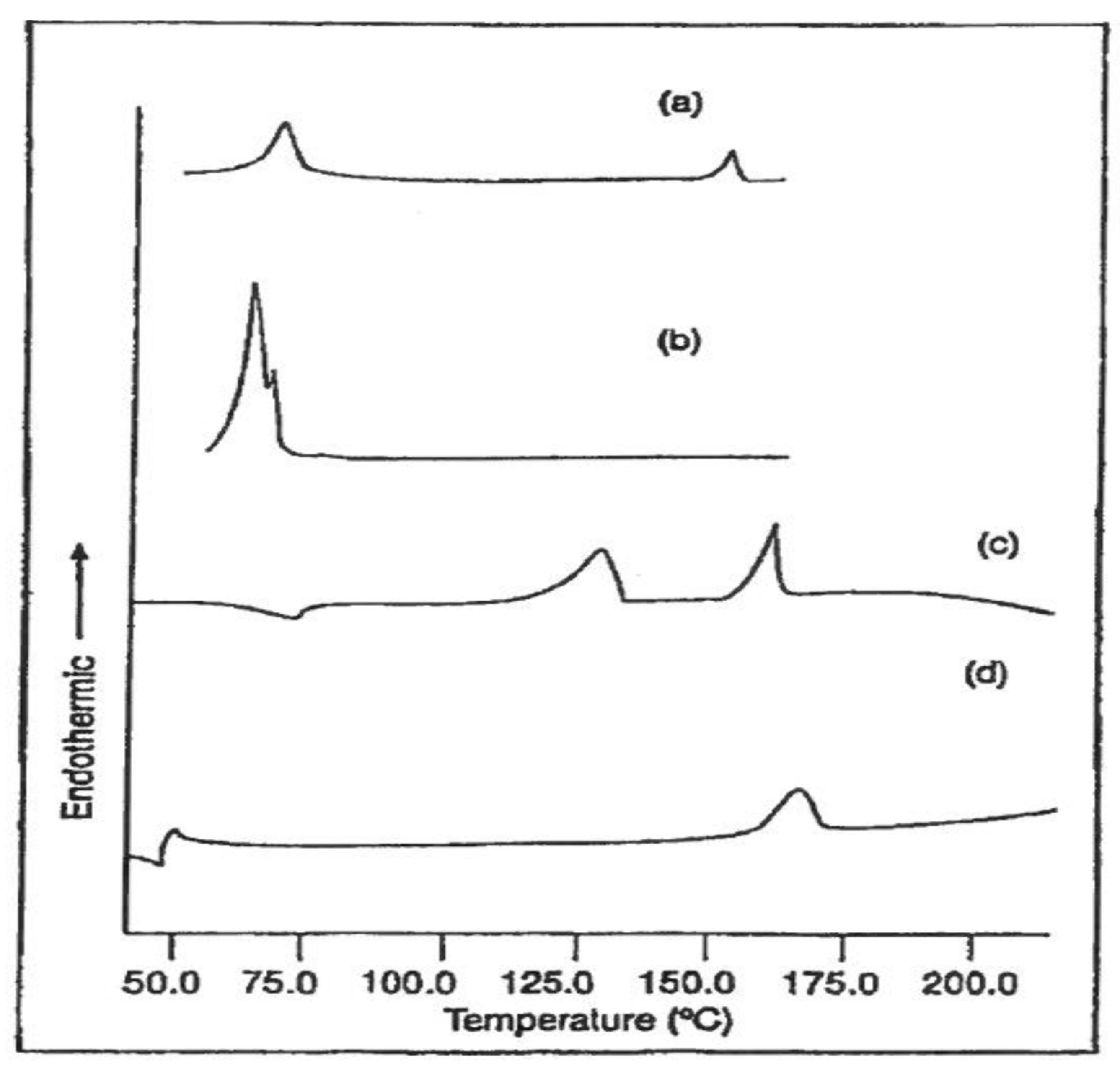
3.1. Compatible Coalesced Polymer Blends
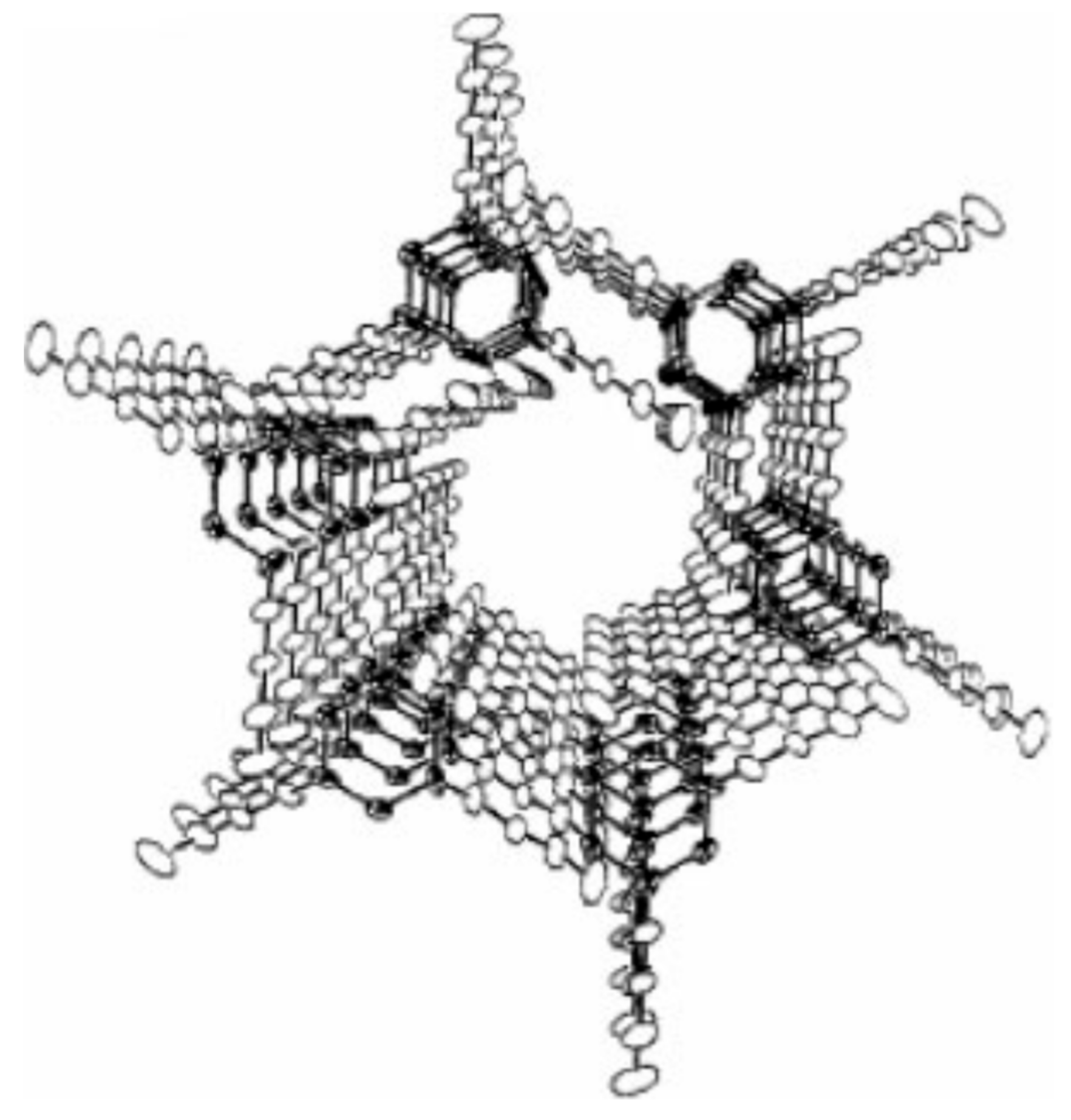
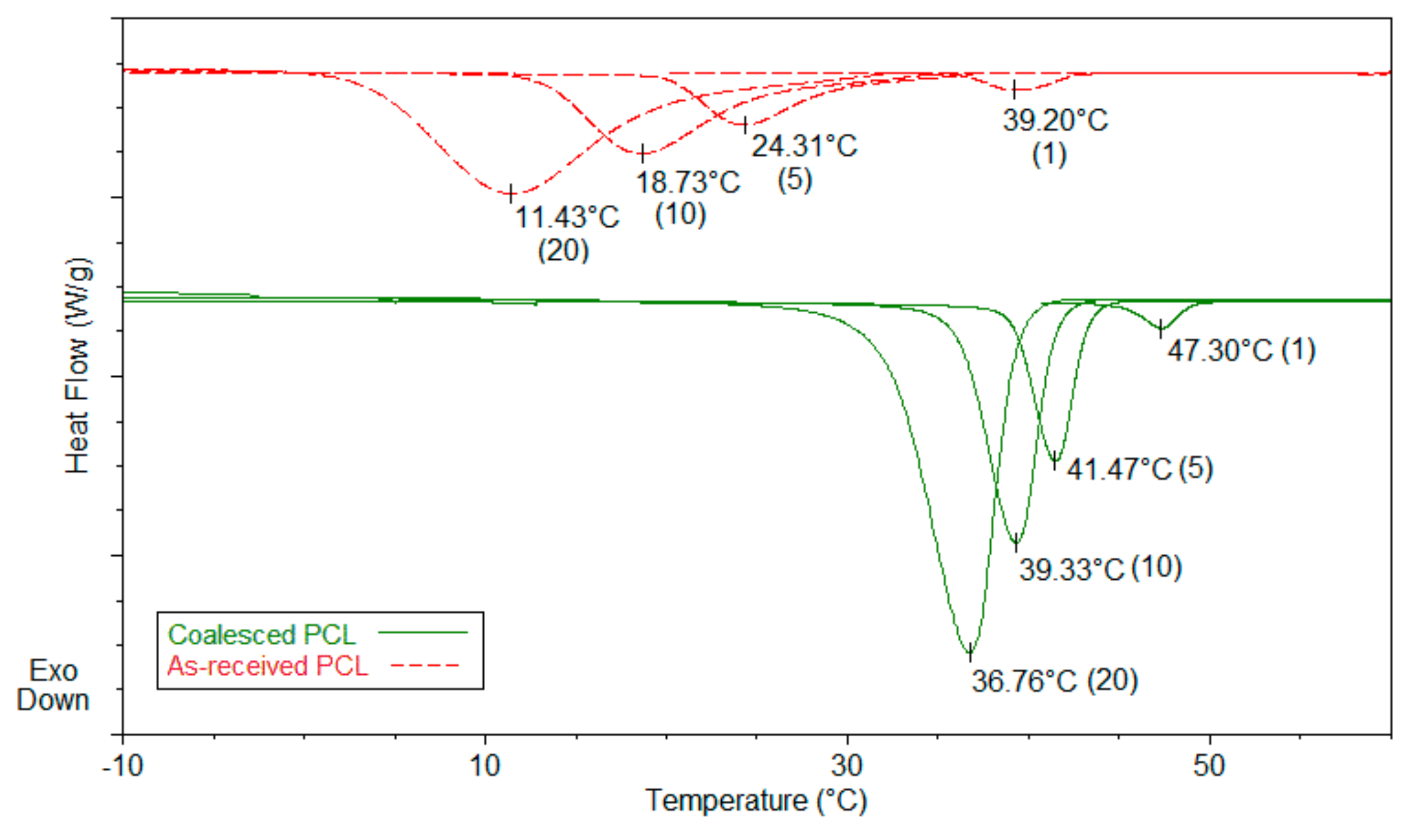
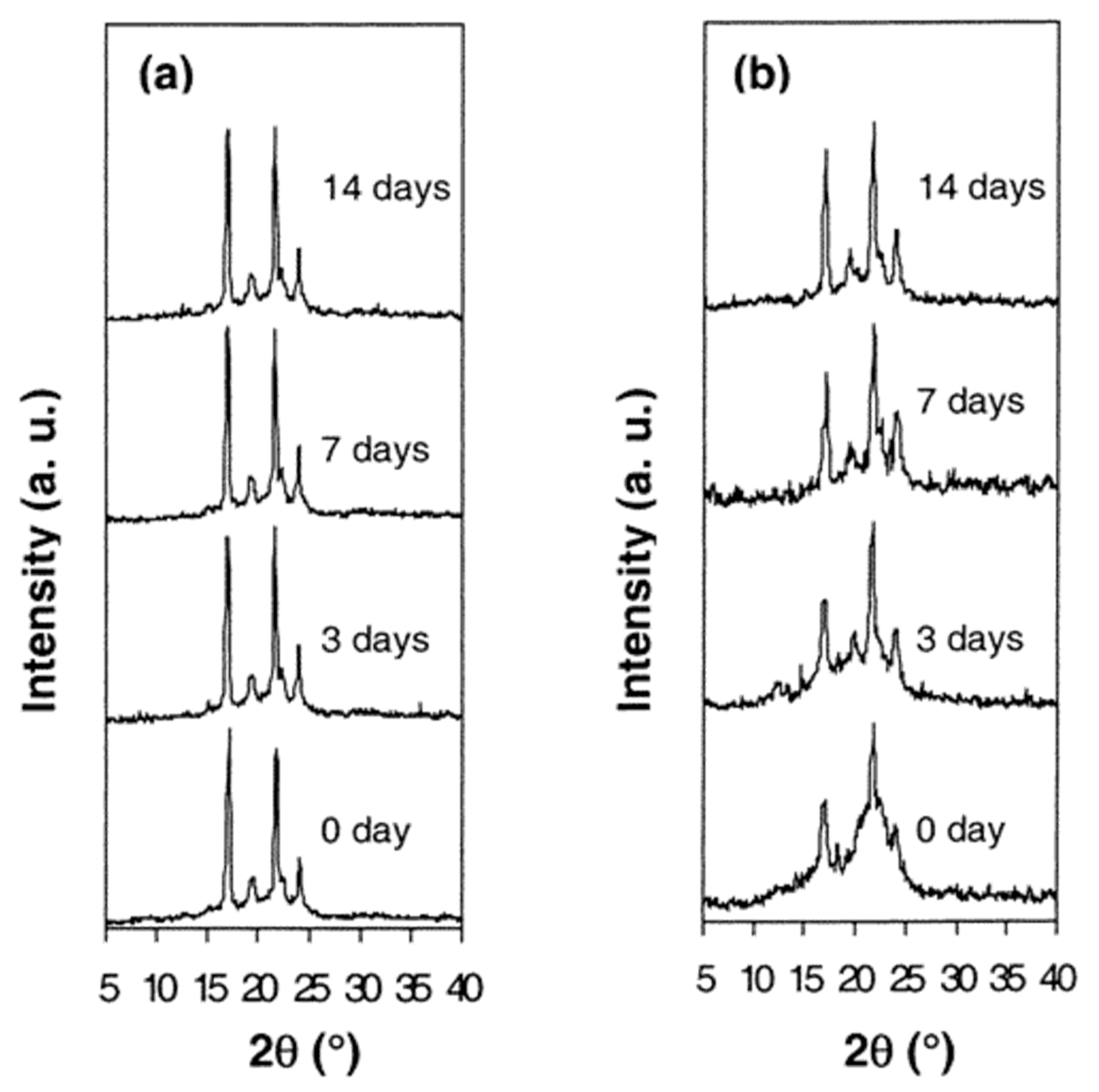
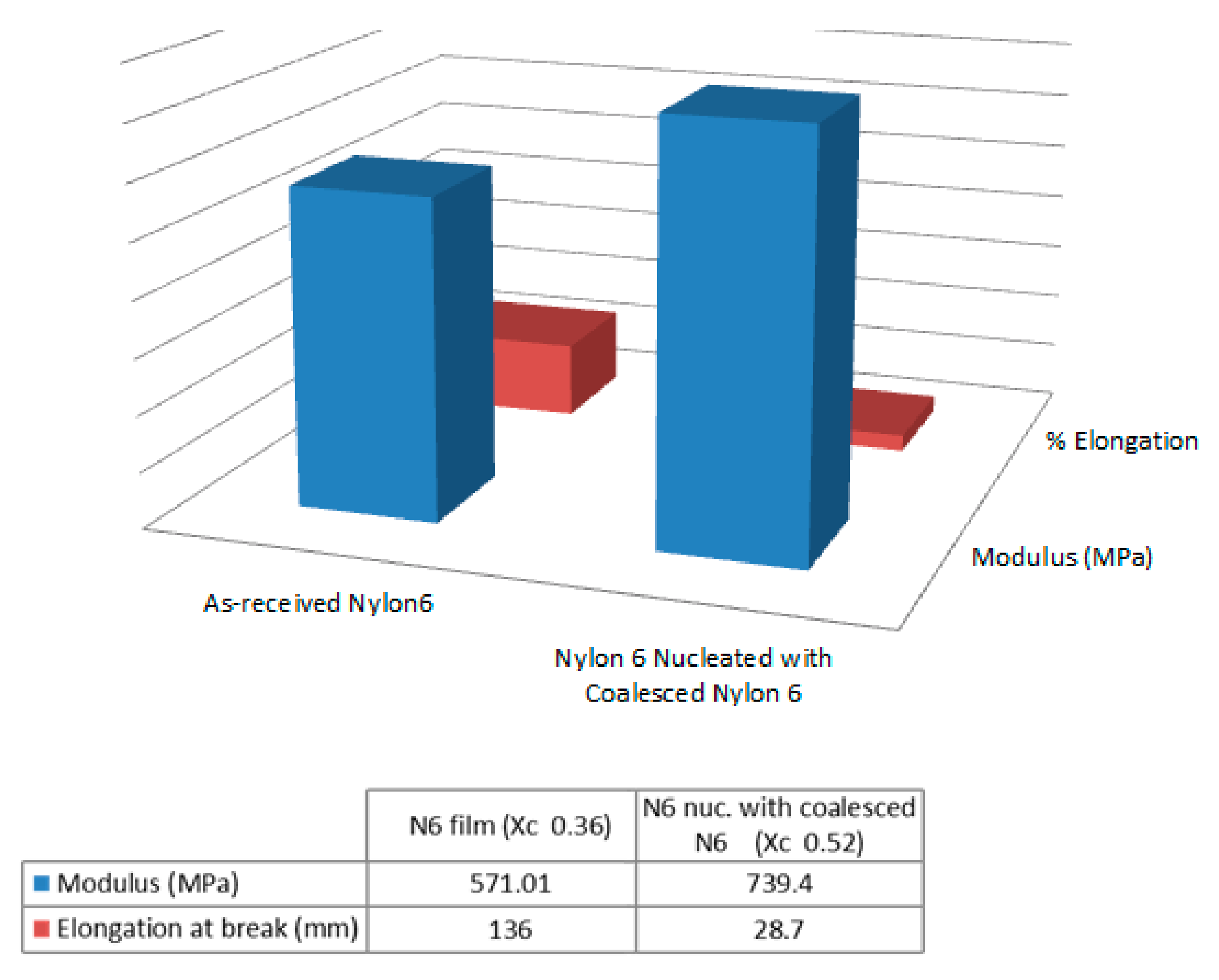
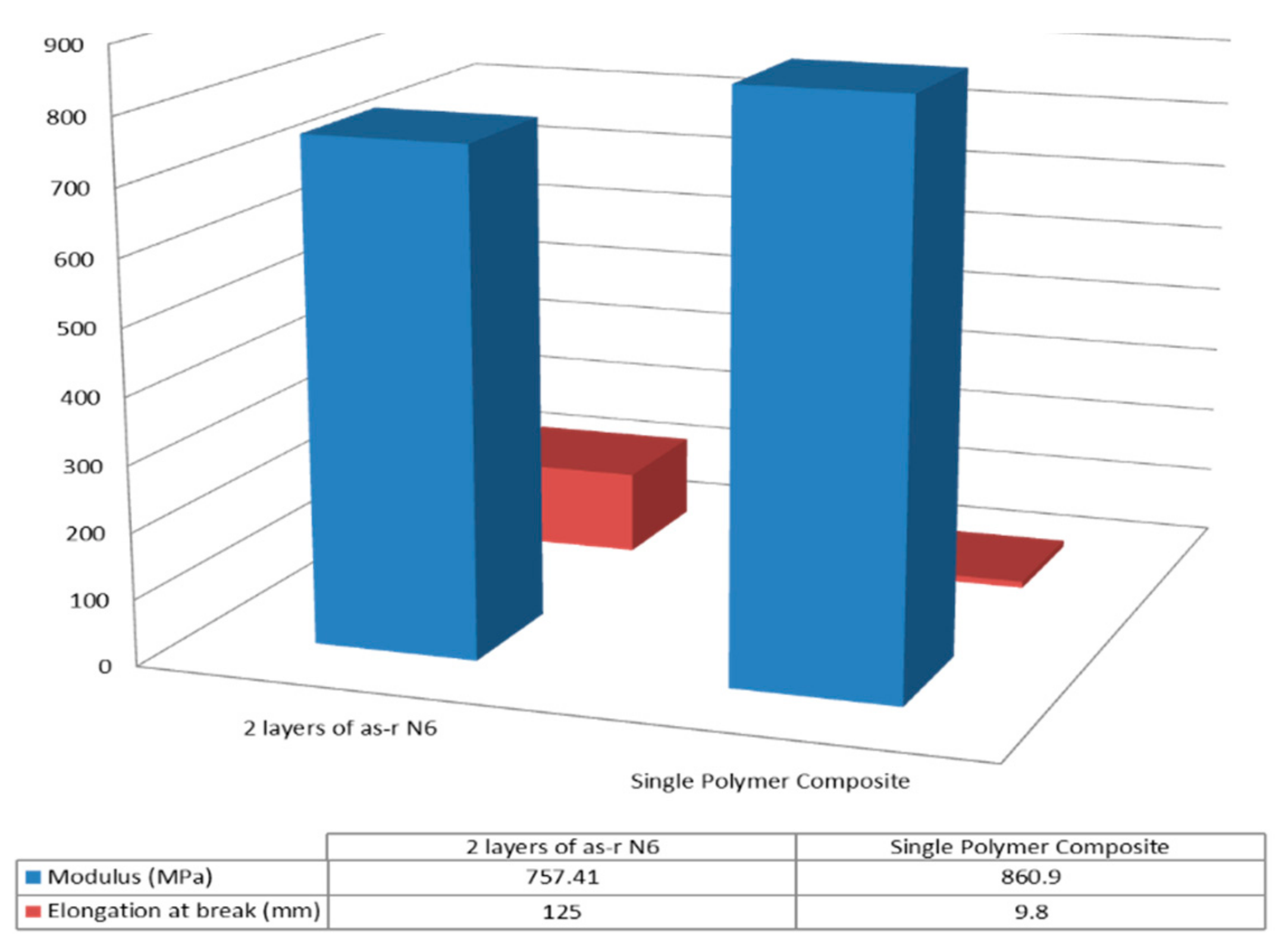
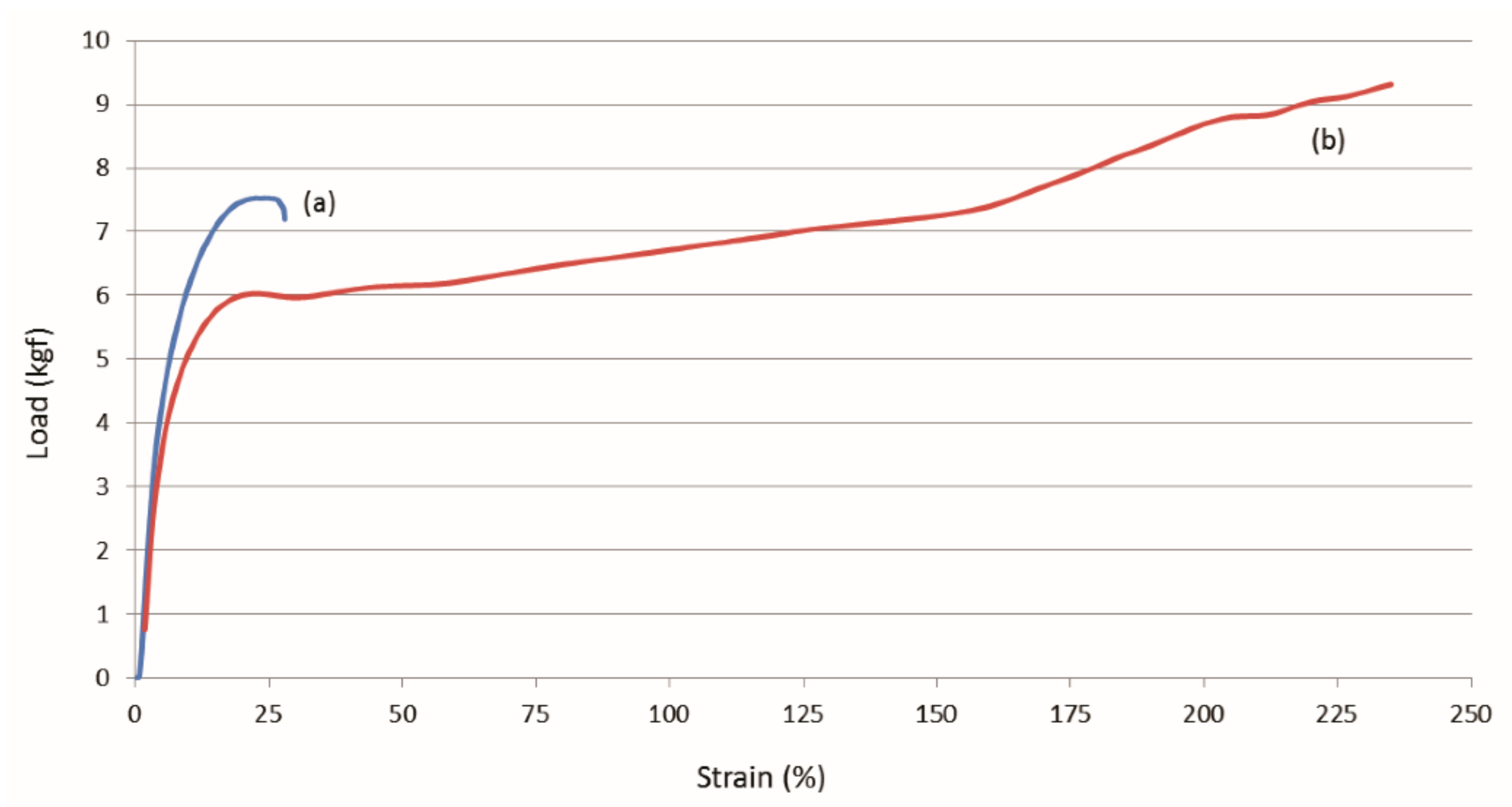
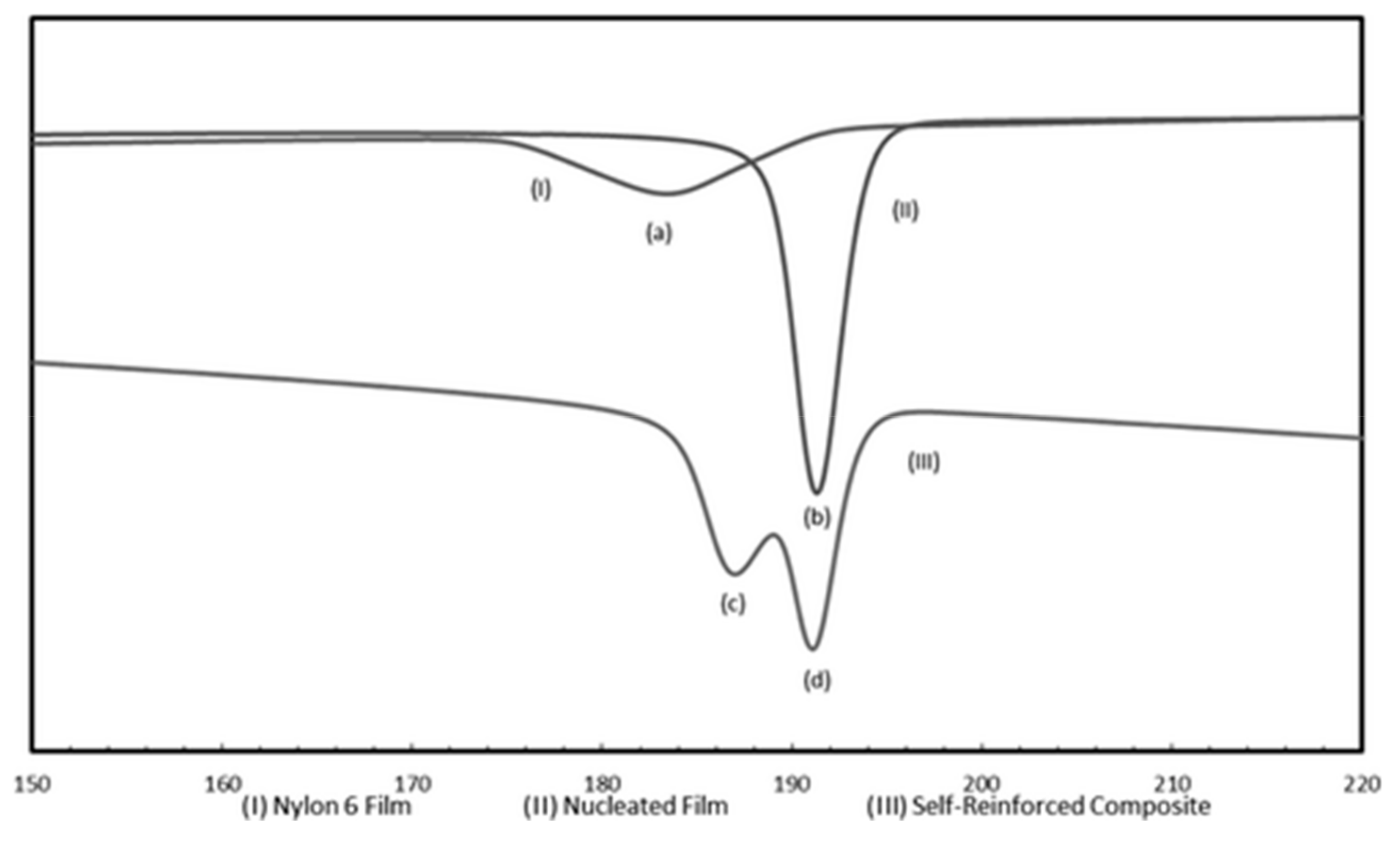
3.2. Single Polymer Composites
4. Summary and Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Harada, A.; Kamachi, M. Complex formation between poly (ethylene glycol) and α-cyclodextrin. Macromolecules 1990, 23, 2821–2823. [Google Scholar] [CrossRef]
- Huang, L.; Tonelli, A.E. Polymer inclusion compounds. J. Macromol. Sci. Part C Polym. Rev. 1998, 38, 781–837. [Google Scholar] [CrossRef]
- Rusa, C.C.; Rusa, M.; Peet, J.; Uyar, T.; Fox, J.; Hunt, M.A.; Wang, X.; Balik, C.M.; Tonelli, A.E. The nanothreading of polymers. J. Incl. Phenom. Macrocyc. Chem. 2006, 55, 185–192. [Google Scholar] [CrossRef]
- Fetterely, L.C. Non-Stoichiometric Compounds; AcademicPress: New York, NY, USA, 1964; 491p. [Google Scholar]
- Brown, J.F., Jr.; White, D.M. Stereospecific polymerization in thiourea canal complexes. J. Am. Chem. Soc. 1960, 82, 5671–5678. [Google Scholar] [CrossRef]
- White, D.M. Stereospecific polymerization in urea canal complexes. J. Am. Chem. 1960, 82, 5678–5685. [Google Scholar] [CrossRef]
- Farina, M. Polyhydrotriphenylene. Tetrahedron Lett. 1961, 2, 2097–2100. [Google Scholar]
- Sozzani, P.; Comotti, A.; Bracco, S.; Simonutti, R. Cooperation of multiple CH⋯π interactions to stabilize polymers in aromatic nanochannels as indicated by 2D solid state NMR. Chem. Commun. 2004, 7, 768–769. [Google Scholar] [CrossRef] [PubMed]
- Abe, A.; Bracco, S.; Comotti, A.; Corradini, P.; De Jeu, W.H.; De Rosa, C.; Furuya, H.; Hiejima, T.; Kobayashi, Y.; Li, L.; et al. Interphases and Mesophases in Polymer Crystallization II; Springer: Berlin, Germany, 2005; pp. 153–177. [Google Scholar]
- Allcock, H.R.; Levin, M.L. Stereocontrolled polymerization of acrylic monomers within a tris (o-phenylenedioxy) cyclotriphosphazene tunnel clathrate. Macromolecules 1985, 18, 1324–1330. [Google Scholar] [CrossRef]
- Harada, A.; Li, J.; Kamachi, M. Double-stranded inclusion complexes of cyclodextrin threaded on poly (ethylene glycol). Nature 1994, 370, 126–128. [Google Scholar] [CrossRef]
- Shin, I.D.; Huang, L.; Tonelli, A.E. Double-stranded inclusion complexes of cyclodextrin threaded on poly(ethylene glycol). Macromol. Symp. 1999, 138, 21–40. [Google Scholar] [CrossRef]
- Kawaguchi, Y.; Nishiyama, T.; Okada, M.; Kamachi, M.; Harada, A. Complex formation of poly (ε-caprolactone) with cyclodextrins. Macromolecules 2000, 33, 4472–4477. [Google Scholar] [CrossRef]
- Harris, K.D.; Jonsen, P. 2H NMR investigation of the dynamic behaviour of n-hexadecane in its urea inclusion compound. Chem. Phys. Lett. 1989, 154, 593–598. [Google Scholar] [CrossRef]
- Tonelli, A.E. Molecular processing of polymers with cyclodextrins. Adv. Polym. Sci. 2009, 222, 115–173. [Google Scholar]
- Hunt, M.A.; Rusa, C.C.; Tonelli, A.E.; Balik, C.M. Structure and stability of columnar cyclomaltooctaose (α-cyclodextrin) hydrate. Carbohydr. Res. 2004, 339, 2805–2810. [Google Scholar] [CrossRef] [PubMed]
- Hunt, M.A.; Rusa, C.C.; Tonelli, A.E.; Balik, C.M. Structure and stability of columnar cyclomaltooctaose (γ-cyclodextrin) hydrate. Carbohydr. Res. 2005, 340, 1631–1637. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Mirau, P.A.; Tonelli, A.E. Chain conformations and dynamics of crystalline polymers as observed in their inclusion compounds by solid-state NMR. Prog. Polym. Sci. 2002, 27, 357–401. [Google Scholar] [CrossRef]
- Rusa, C.C.; Wei, M.; Bullions, T.A.; Shuai, X.; Uyar, T.; Tonelli, A.E. Nanostructuring polymers with cyclodextrins. Polym. Adv. Technol. 2005, 16, 269–275. [Google Scholar] [CrossRef]
- Tonelli, A.E. Nanostructuring and functionalizing polymers with cyclodextrin. Polymer 2008, 9, 1725–1736. [Google Scholar] [CrossRef]
- Tonelli, A.E. Restructuring polymers via nanoconfinement and subsequent release. Beilstein J. Org. Chem. 2012, 8, 1318–1332. [Google Scholar] [CrossRef]
- Tonelli, A.E. Non-stoichiometric polymer-cyclodextrin inclusion compounds: Con-straints placed on un-included chain portions tethered at both ends and their relation to polymer brushes. Polymers 2019, 6, 2166. [Google Scholar] [CrossRef]
- Joijode, A.S.; Antony, G.J.; Tonelli, A.E. Glass-transition temperatures of nano-structured amorphous bulk polymers and their blends. J. Polym. Sci. Part B Polym. Phys. 2013, 51, 1041–1050. [Google Scholar] [CrossRef]
- Uyar, T.; Rusa, C.C.; Hunt, M.A.; Aslan, E.; Hacaloglu, J.; Tonelli, A.E. Reorganization and improvement of bulk polymers by processing with their cyclodextrin inclusion compounds. Polymer 2005, 46, 4762–4775. [Google Scholar] [CrossRef]
- Williamson, B.R.; Krishnaswamy, R.; Tonelli, A.E. Physical properties of poly (ɛ-caprolactone) coalesced from its α-cyclodextrin inclusion compound. Polymer 2011, 52, 4517–4527. [Google Scholar] [CrossRef]
- Tonelli, A.E. Organizational stabilities of bulk neat and well-mixed, blended polymer samples coalesced from their crystalline inclusion compounds formed with cyclodextrins. J. Polym. Sci. Part. B Polym. Phys. 2009, 47, 1543–1553. [Google Scholar] [CrossRef]
- Gurarslan, A.; Joijode, A.S.; Tonelli, A.E. Polymers coalesced from their cyclodextrin inclusion complexes: What can they tell us about the morphology of melt-crystallized polymers? J. Polym. Sci. Part B Polym. Phys. 2012, 50, 813–823. [Google Scholar] [CrossRef]
- Rusa, C.C.; Wei, M.; Shuai, X.; Bullions, T.A.; Wang, X.; Rusa, M.; Uyar, T.; Tonelli, A.E. Molecular mixing of incompatible polymers through formation of and coalescence from their common crystalline cyclodextrin inclusion compounds. J. Polym. Sci. Part B Polym. Phys. 2004, 42, 4207–4224. [Google Scholar] [CrossRef]
- Rusa, C.C.; Tonelli, A.E. Polymer/polymer inclusion compounds as a novel approach to obtaining a PLLA/PCL intimately compatible blend. Macromolecules 2000, 33, 5321–5324. [Google Scholar] [CrossRef]
- Wei, M.; Tonelli, A.E. Complex formation of poly (ε-caprolactone) with cyclodextrins. Macromolecules 2001, 34, 4061–4065. [Google Scholar] [CrossRef]
- Shuai, X.; Porbeni, F.E.; Wei, M.; Bullions, T.; Tonelli, A.E. Formation of inclusion complexes of poly (3-hydroxybutyrate)s with cyclodextrins. 1. Immobilization of atactic poly (R,S-3-hydroxybutyrate) and miscibility enhancement between poly (R,S-3-hydroxybutyrate) and poly (ε-caprolactone). Macromolecules 2002, 35, 3126–3132. [Google Scholar] [CrossRef]
- Bullions, T.A.; Edeki, E.M.; Porbeni, F.E.; Wei, M.; Shuai, X.; Rusa, C.C.; Tonelli, A.E. Intimate blend of poly (ethylene terephthalate) and poly (ethylene 2,6-naphthalate) via formation with and coalescence from their common inclusion compound with γ-cyclodextrin. J. Polym. Sci. Part B Polym. Phys. 2003, 41, 139–148. [Google Scholar] [CrossRef]
- Rusa, C.C.; Uyar, T.; Rusa, M.; Hunt, M.A.; Wang, X.; Tonelli, A.E. An intimate polycarbonate/poly (methyl methacrylate)/poly (vinyl acetate) ternary blend via coalescence from their common inclusion compound with γ-cyclodextrin. J. Polym. Sci. Part B Polym. Phys. 2004, 42, 4182–4194. [Google Scholar] [CrossRef]
- Wei, M.; Shin, I.D.; Urban, B.; Tonelli, A.E. Partial miscibility in a nylon-6/nylon-66 blend coalesced from their common α-cyclodextrin inclusion complex. J. Polym. Sci. Part B Polym. Phys. 2004, 42, 1369–1378. [Google Scholar] [CrossRef]
- Uyar, T.; Rusa, C.C.; Wang, X.; Rusa, M.; Hacaloglu, J.; Tonelli, A.E. Intimate blending of binary polymer systems from their common cyclodextrin inclusion compounds. J. Polym. Sci. Part B Polym. Phys. 2005, 43, 2578–2593. [Google Scholar] [CrossRef]
- Jia, X.; Wang, X.; Tonelli, A.E.; White, J.L. Two-dimensional spin diffusion NMR reveals differential mixing in biodegradable polymer blends. Macromolecules 2005, 38, 2775–2780. [Google Scholar] [CrossRef]
- White, J.L.; Mirau, P.A. Heteronuclear correlation in solid polymers: identification of hydrogen bond donors and acceptors in miscible polymer blends. Macromolecules 1994, 27, 1648–1650. [Google Scholar] [CrossRef]
- Burum, D.P.; Bielecki, A. An improved experiment for heteronuclear-correlation 2D-NMR in solids. J. Magn. Reson. 1991, 94, 645. [Google Scholar] [CrossRef]
- Caravatti, P.; Braunschweiler, L.; Ernst, R.R. Heteronuclear correlation spectroscopy in rotating solids. Chem. Phys. Lett. 1983, 100, 305–310. [Google Scholar] [CrossRef]
- Jia, X.; Wolak, J.; Wang, X.; White, J.L. Independent calibration of 1H spin-diffusion coefficients in amorphous polymers by intramolecular polarization transfer. Macromolecules 2003, 36, 712–718. [Google Scholar] [CrossRef]
- Shuai, X.; Porbeni, F.E.; Wei, M.; Shin, I.D.; Tonelli, A.E. Formation of and coalescence from the inclusion complex of a biodegradable block copolymer and r-Cyclodextrin. 2: A novel way to regulate the biodegradation behavior of biodegradable block copolymers. Biomacromolecules 2002, 3, 201–207. [Google Scholar] [CrossRef]
- Gurarslan, A.; Caydamli, Y.; Shen, J.; Tse, S.; Yetukuri, M.; Tonelli, A.E. Coalesced poly (ε-caprolactone) fibers are stronger. Biomacromolecules 2015, 16, 890–893. [Google Scholar] [CrossRef]
- Gurarslan, A.; Shen, J.; Tonelli, A.E. Single-component poly (ε-caprolactone) composites. Polymer 2013, 54, 5747–5753. [Google Scholar] [CrossRef]
- Mohan, A.; Gurarslan, A.; Joyner, X.; Child, R.; Tonelli, A.E. Melt-crystallized nylon-6 nucleated by the constrained chains of its non-stoichiometric cyclodextrin inclusion compounds and the nylon-6 coalesced from them. Polymer 2011, 52, 1055–1062. [Google Scholar] [CrossRef]
- Gurarslan, A.; Tonelli, A.E. Single component polymer composites. Macromolecules 2011, 44, 3856–3861. [Google Scholar] [CrossRef]
- Huang, L.; Vasanthan, N.; Tonelli, A.E. Polymer-polymer composites fabricated by the in situ release and coalescence of polymer chains from their inclusion compounds with urea into a carrier polymer phase. J. Appl. Polym. Sci. 1997, 64, 281–287. [Google Scholar] [CrossRef]
- Huang, L.; Gerber, M.; Taylor, H.; Lu, J.; Tapaszi, E.; Wutkowski, M.; Hill, M.; Lewis, C.; Harvey, A.; Herndon, A.; et al. Creation of novel polymer materials by processing with inclusion compounds. In Macromolecular Symposia; John Wiley & Sons: Hoboken, NJ, USA, 2001; pp. 129–144. [Google Scholar]


















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Modulus (MPa) | Elongation at Break (mm) |
|---|---|---|
| asr-PCL 80 | 344.7 ± 34 | 247.5 ± 38 (495%) |
| nuc-PCL 80 (2 wt% c-PCL80, 98 wt% asr-PCL 80) | 369.7 ± 40 | 220.6 ± 65 (441%) |
| asr-PCL70-90 | 158.6 ± 8 | 445.5 ± 17 (891%) |
| neat c-PCL70-90 | 210.5 ± 11 | 420.6 ± 36 (841%) |
| Sample | Modulus (MPa) | Elongation at Break (mm) |
|---|---|---|
| asr/asr-PCL 80-film | 317.5 ± 24 | 270.3 ± 30 (540%) |
| asr/nuc-PCL 80-film | 340.7 ± 28 | 253.6 ± 15 (507%) |
| asr/asr-PCL 70–90 | 217 ± 24 | 415 ± 52 (830%) |
| asr/c-PCL 70–90 from urea | 273 ± 31 | 404 ± 35 (808%) |
| Sample Name | Average Thickness (mm) | Moisture Vapor Permeability (g/m2/24 h) |
|---|---|---|
| PCL film | 0.022 | 375 |
| Dipped PCL film | 0.010 | 440 |
| PCL-Urea film | 0.139 | 413 |
| Dipped PCL-urea film | 0.313 | 747 |
| PCL-IC film | 0.054 | 418 |
| Dipped PCL-IG film | 0.076 | 583 |
| PLLA film | 0.024 | 173 |
| Dipped PLLA film | 0.041 | 187 |
| PLLA-urea film | 0.180 | 207 |
| Dipped PLLA-urea film | 0.155 | 540 |
| PLLA-IC film | 0.045 | 183 |
| Dipped PLLA-IC film | 0.052 | 236 |
© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tonelli, A.E. Nanoscale Restructuring of Polymer Materials to Produce Single Polymer Composites and Miscible Blends. Biomolecules 2019, 9, 240. https://doi.org/10.3390/biom9060240
Tonelli AE. Nanoscale Restructuring of Polymer Materials to Produce Single Polymer Composites and Miscible Blends. Biomolecules. 2019; 9(6):240. https://doi.org/10.3390/biom9060240
Chicago/Turabian StyleTonelli, Alan E. 2019. "Nanoscale Restructuring of Polymer Materials to Produce Single Polymer Composites and Miscible Blends" Biomolecules 9, no. 6: 240. https://doi.org/10.3390/biom9060240
APA StyleTonelli, A. E. (2019). Nanoscale Restructuring of Polymer Materials to Produce Single Polymer Composites and Miscible Blends. Biomolecules, 9(6), 240. https://doi.org/10.3390/biom9060240