Host-Microbiome Synergistic Control on Sphingolipid Metabolism by Mechanotransduction in Model Arthritis

 , ,
, , 
Abstract
:1. Introduction
2. Materials and Methods
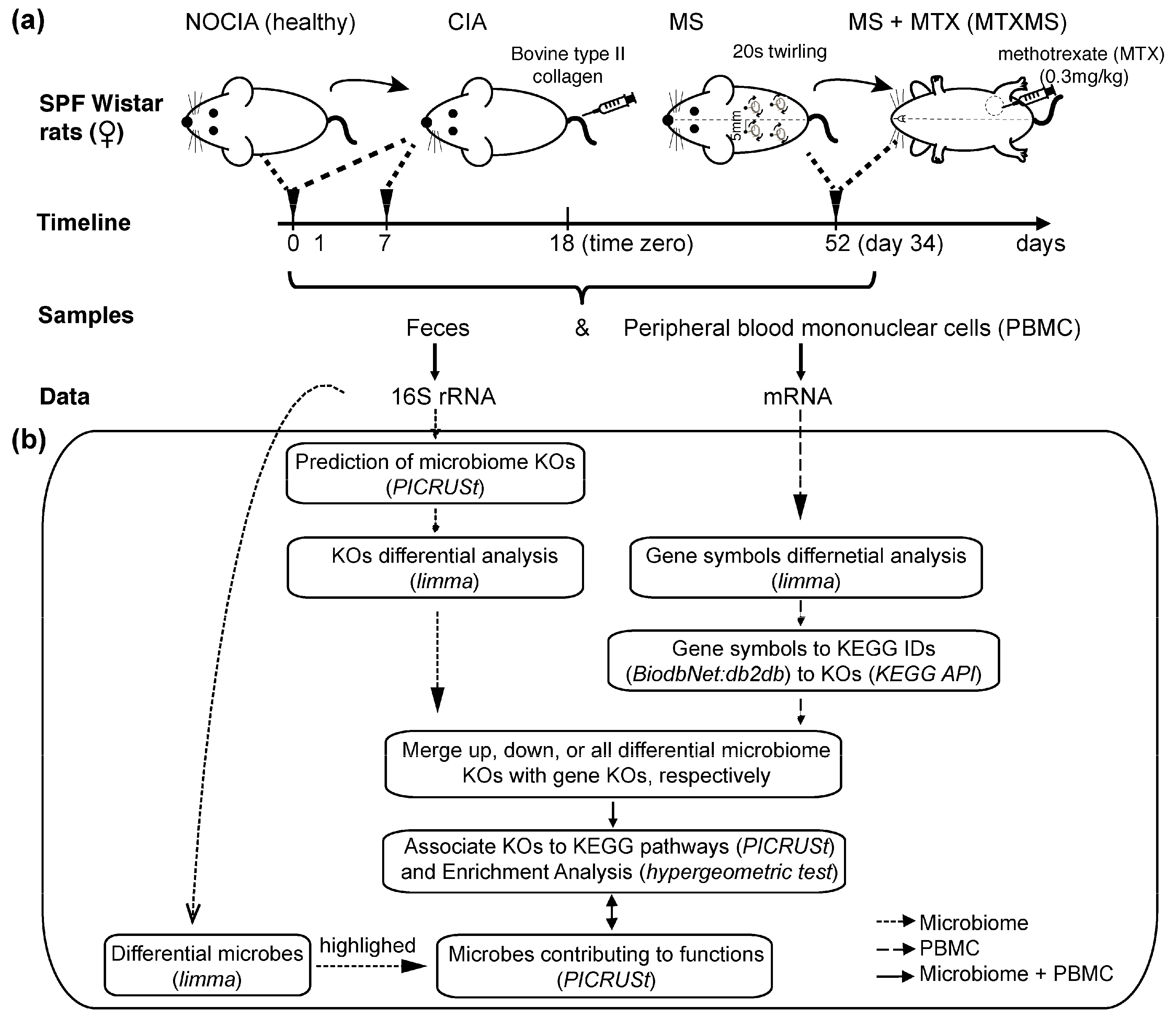
2.1. Animal Models
2.2. Fecal Sample Collection
2.3. Microbial 16S rRNA Sequencing
2.4. General Method
2.4.1. OTUs Expansion in KEGG Orthology
2.4.2. Statistical Analysis
2.4.3. Gene Symbols Conversion into KEGG Orthologies
2.4.4. Functional Analysis
2.5. Data and Materials Availibility
3. Results
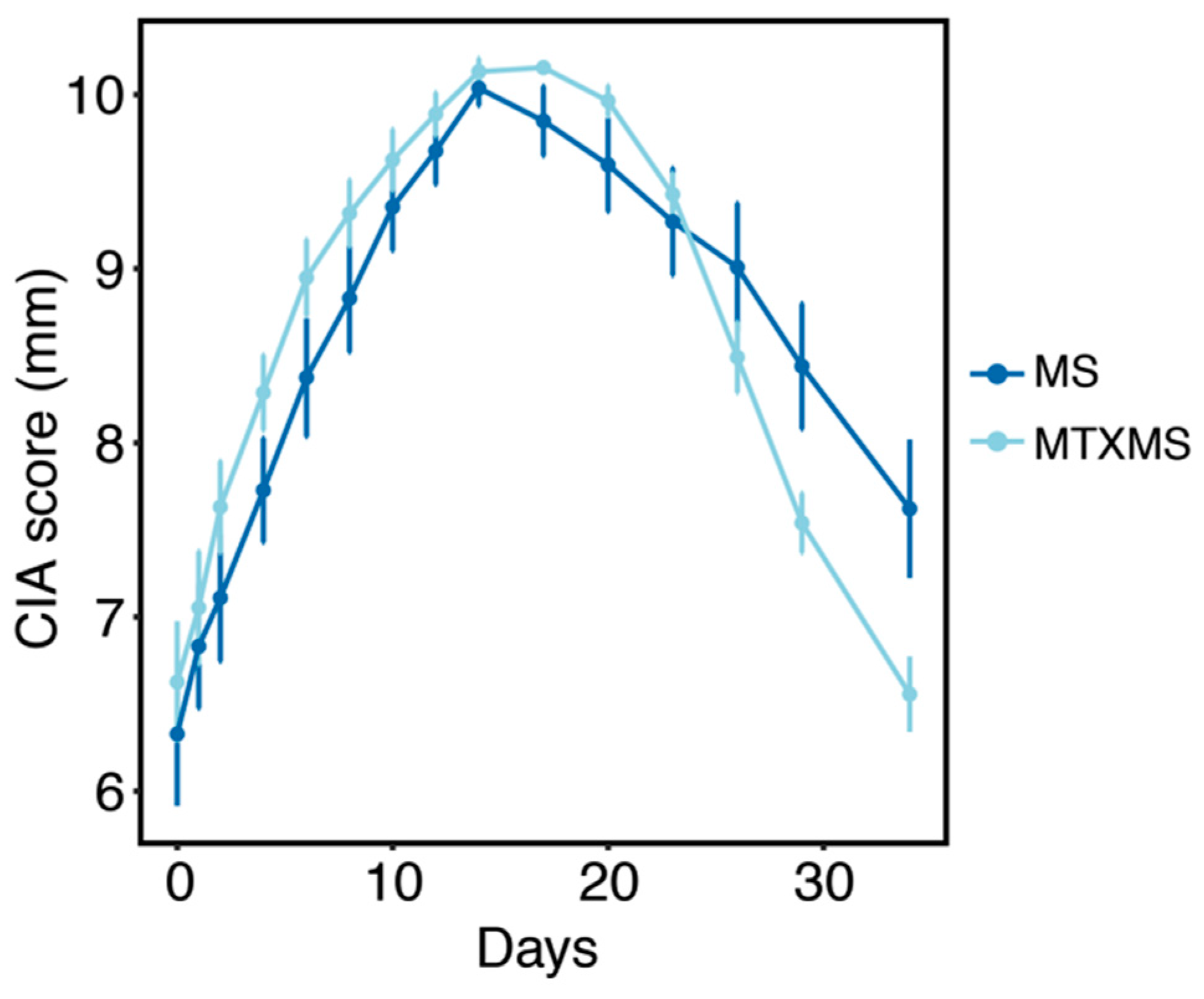
3.1. Collagen Induced Arthritis Phenotype
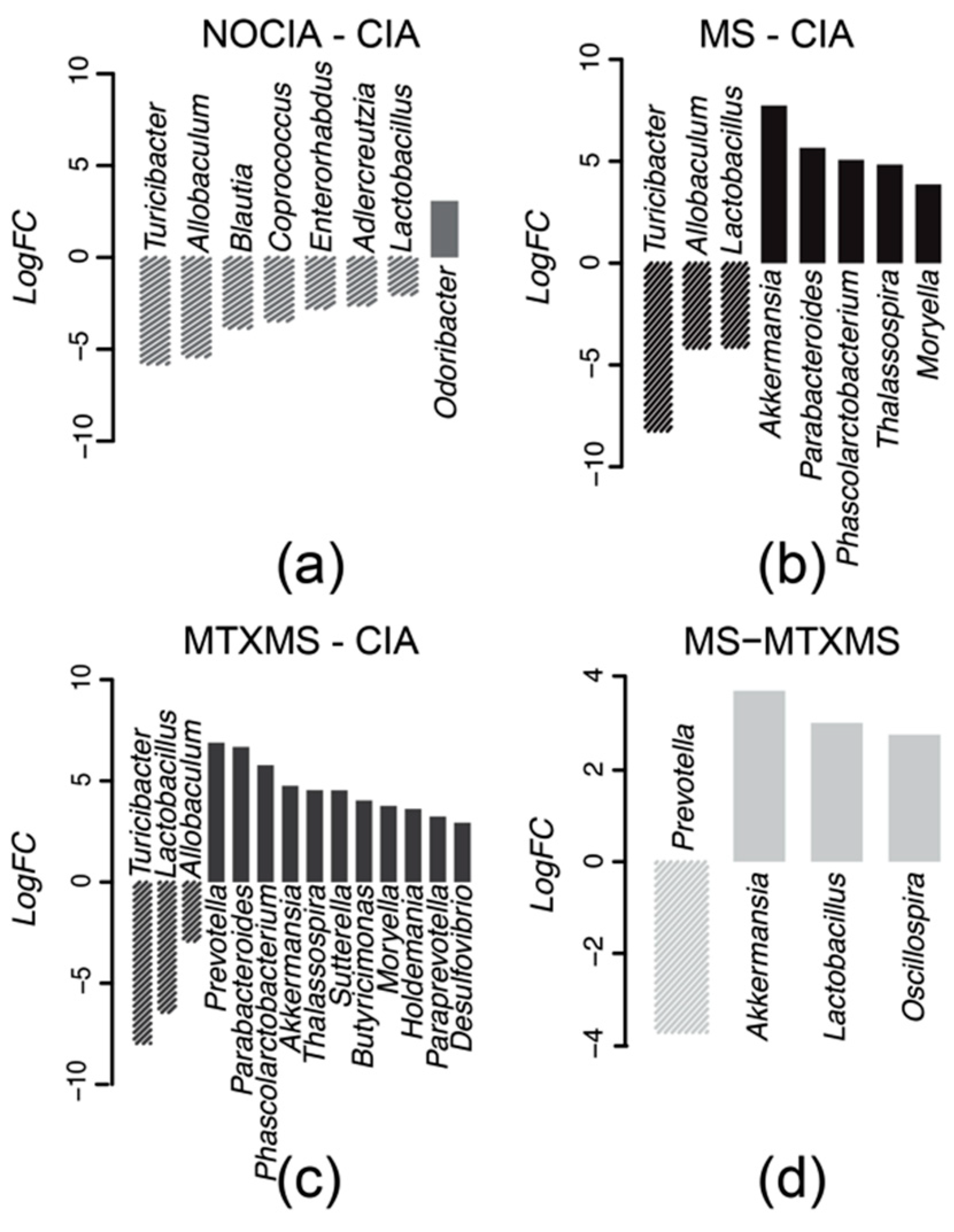
3.2. Microbial Taxa Differential Analysis
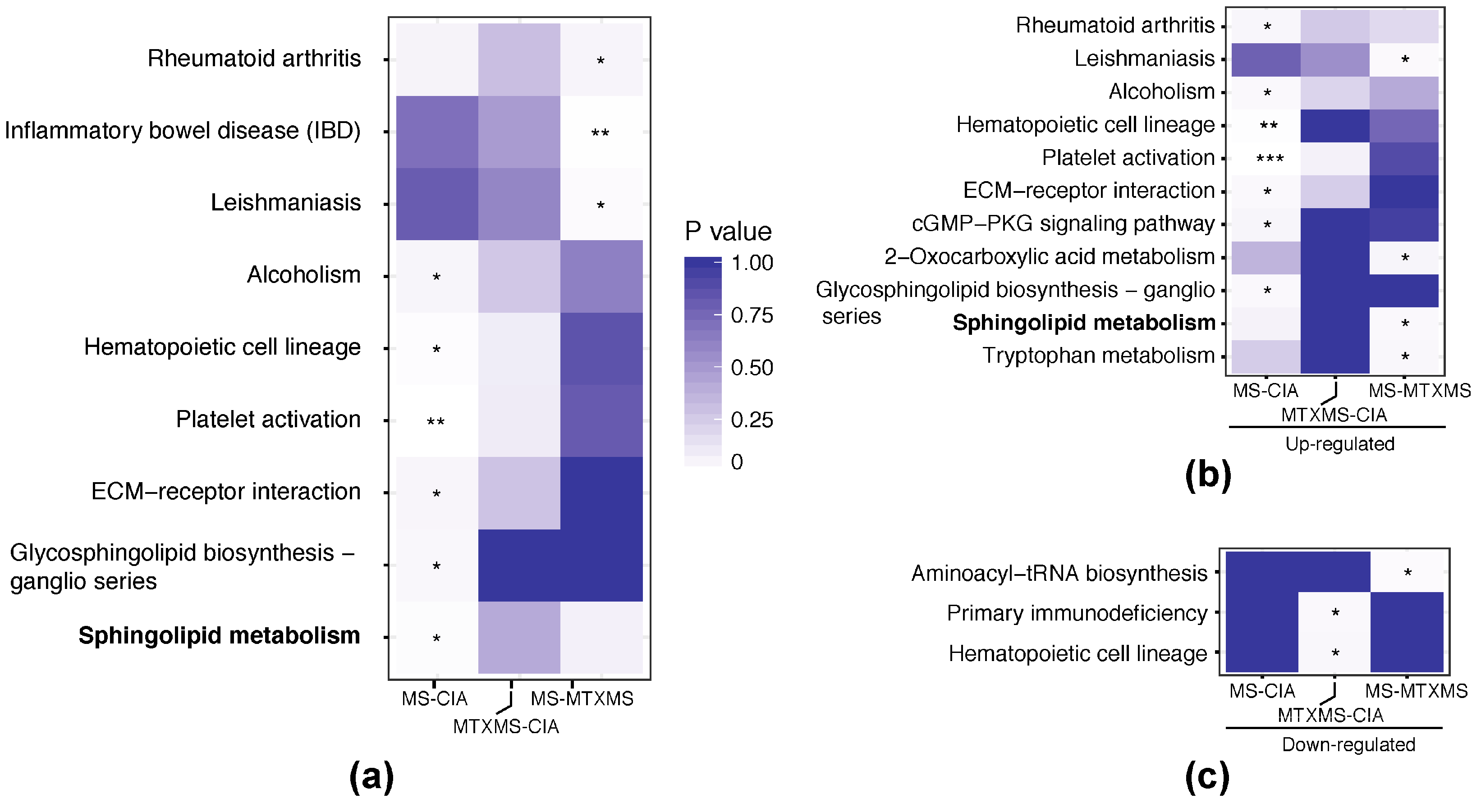
3.3. Single Omic Functional Analysis
3.3.1. Microbiome Side, Gastrointestinal KEGG Orthologies
3.3.2. Host Side, Peripheral Blood Mononuclear Cell KEGG Orthologies
3.4. Host-Microbiome Interaction, KEGG Orthology Joint Analysis
4. Discussion
4.1. Collagen Induced Arthritis Dysbiosis
4.2. The Sphingolipid Metabolism—Etiopathogenesis of Rheumatoid Arthritis
4.3. The Sphingolipid Metabolism—Potential Therapeutic Target of Mechnical Stimulation
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genus | Batch_Current | Batch_Previous | ||||||
|---|---|---|---|---|---|---|---|---|
| MS/CIA | MTXMS/CIA | CIA/NOCIA | MS/MTXMS | MS/CIA | MTX/CIA | CIA/NOCIA | MS/MTX | |
| Lactobacillus | −4.137 * | −6.446 | 2.03 | 3.028 | - | −2.021 | 2.708 | 2.504 |
| Prevotella | - | 6.887 | - | −3.725 | - | 2.108 | - | - |
| Allobaculum | −4.18 | −2.929 | 5.418 | - | 2.757 | 2.429 | - | - |
| Odoribacter | - | - | −3.078 | - | - | - | 2.917 | - |
| Blautia | - | - | 3.862 | - | 4.695 | 3.369 | −3.453 | - |
| Phascolarctobacterium | 5.068 | 5.768 | - | - | - | 3.684 | - | - |
| Adlercreutzia | - | - | 2.6 | - | −2.002 | - | - | - |
References
- Tremaroli, V.; Backhed, F. Functional interactions between the gut microbiota and host metabolism. Nature 2012, 489, 242–249. [Google Scholar] [CrossRef]
- Okada, Y.; Wu, D.; Trynka, G.; Raj, T.; Terao, C.; Ikari, K.; Kochi, Y.; Ohmura, K.; Suzuki, A.; Yoshida, S.; et al. Genetics of rheumatoid arthritis contributes to biology and drug discovery. Nature 2014, 506, 376–381. [Google Scholar] [CrossRef] [PubMed]
- Raychaudhuri, S. Recent advances in the genetics of rheumatoid arthritis. Curr. Opin. Rheumatol. 2010, 22, 109–118. [Google Scholar] [CrossRef]
- Scher, J.U.; Abramson, S.B. The microbiome and rheumatoid arthritis. Nat. Rev. Rheumatol. 2011, 7, 569–578. [Google Scholar] [CrossRef]
- Chen, J.; Wright, K.; Davis, J.M.; Jeraldo, P.; Marietta, E.V.; Murray, J.; Nelson, H.; Matteson, E.L.; Taneja, V. An expansion of rare lineage intestinal microbes characterizes rheumatoid arthritis. Genome Med. 2016, 8, 43. [Google Scholar] [CrossRef]
- Bonneville, M.; Scotet, E.; Peyrat, M.A.; Saulquin, X.; Houssaint, E. Epstein-Barr virus and rheumatoid arthritis. Rev. Rhum 1998, 65, 365–368. [Google Scholar]
- Masuko-Hongo, K.; Kato, T.; Nishioka, K. Virus-associated arthritis. Best Pract. Res. Clin. Rheumatol. 2003, 17, 309–318. [Google Scholar] [CrossRef]
- Edwards, C.J.; Cooper, C. Early environmental factors and rheumatoid arthritis. Clin. Exp. Immunol. 2006, 143, 1–5. [Google Scholar] [CrossRef]
- Tieri, P.; Zhou, X.; Zhu, L.; Nardini, C. Multi-omic landscape of rheumatoid arthritis: Re-evaluation of drug adverse effects. Front. Cell Dev. Biol. 2014, 2, 59. [Google Scholar] [CrossRef] [PubMed]
- Rohlke, F.; Stollman, N. Fecal microbiota transplantation in relapsing Clostridium difficile infection. Ther. Adv. Gastroenterol. 2012, 5, 403–420. [Google Scholar] [CrossRef] [PubMed]
- Kellermayer, R.; Nagy-Szakal, D.; Harris, R.A.; Luna, R.A.; Pitashny, M.; Schady, D.; Mir, S.A.V.; Lopez, M.E.; Gilger, M.A.; Belmont, J.; et al. Serial Fecal Microbiota Transplantation Alters Mucosal Gene Expression in Pediatric Ulcerative Colitis. Am. J. Gastroenterol. 2015, 110, 604–606. [Google Scholar] [CrossRef] [PubMed]
- Cezar, C.A.; Roche, E.T.; Vandenburgh, H.H.; Duda, G.N.; Walsh, C.J.; Mooney, D.J. Biologic-free mechanically induced muscle regeneration. Proc. Natl. Acad. Sci. USA 2016, 113, 1534–1539. [Google Scholar] [CrossRef] [PubMed]
- Trentham, D.E.; Townes, A.S.; Kang, A.H. Autoimmunity to type II collagen an experimental model of arthritis. J. Exp. Med. 1977, 146, 857–868. [Google Scholar] [CrossRef] [PubMed]
- Koopman, F.A.; Chavan, S.S.; Miljko, S.; Grazio, S.; Sokolovic, S.; Schuurman, P.R.; Mehta, A.D.; Levine, Y.A.; Faltys, M.; Zitnik, R.; et al. Vagus nerve stimulation inhibits cytokine production and attenuates disease severity in rheumatoid arthritis. Proc. Natl. Acad. Sci. USA 2016, 113, 8284–8289. [Google Scholar] [CrossRef] [PubMed]
- Langevin, H.M.; Bouffard, N.A.; Badger, G.J.; Churchill, D.L.; Howe, A.K. Subcutaneous tissue fibroblast cytoskeletal remodeling induced by acupuncture: Evidence for a mechanotransduction-based mechanism. J. Cell. Physiol. 2006, 207, 767–774. [Google Scholar] [CrossRef] [PubMed]
- Nardini, C.; Devescovi, V.; Liu, Y.; Zhou, X.; Lu, Y.; Dent, J.E. Systemic Wound Healing Associated with local sub-Cutaneous Mechanical Stimulation. Sci. Rep. 2016, 6, 39043. [Google Scholar] [CrossRef] [PubMed]
- Rogers, P.A.; Schoen, A.M.; Limehouse, J. Acupuncture for immune-mediated disorders. Literature review and clinical applications. Probl. Vet. Med. 1992, 4, 162–193. [Google Scholar]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glockner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef]
- DeSantis, T.Z.; Hugenholtz, P.; Larsen, N.; Rojas, M.; Brodie, E.L.; Keller, K.; Huber, T.; Dalevi, D.; Hu, P.; Andersen, G.L. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 2006, 72, 5069–5072. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Pena, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef]
- Rossello-Mora, R.; Amann, R. The species concept for prokaryotes. FEMS Microbiol. Rev. 2001, 25, 39–67. [Google Scholar] [CrossRef] [PubMed]
- Langille, M.G.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Vega Thurber, R.L.; Knight, R.; et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Smyth, G.K. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat. Appl. Genet. Mol. Biol. 2004, 3, 3. [Google Scholar] [CrossRef] [PubMed]
- Law, C.W.; Chen, Y.; Shi, W.; Smyth, G.K. Voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 2014, 15, R29. [Google Scholar] [CrossRef] [PubMed]
- Jung, K.; Friede, T.; Beissbarth, T. Reporting FDR analogous confidence intervals for the log fold change of differentially expressed genes. BMC Bioinform. 2011, 12, 288. [Google Scholar] [CrossRef] [PubMed]
- Mudunuri, U.; Che, A.; Yi, M.; Stephens, R.M. bioDBnet: The biological database network. Bioinformatics 2009, 25, 555–556. [Google Scholar] [CrossRef]
- Hosack, D.A.; Dennis, G., Jr.; Sherman, B.T.; Lane, H.C.; Lempicki, R.A. Identifying biological themes within lists of genes with EASE. Genome Biol. 2003, 4, R70. [Google Scholar] [CrossRef]
- Huang da, W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 37, 1–13. [Google Scholar] [CrossRef]
- Scher, J.U.; Sczesnak, A.; Longman, R.S.; Segata, N.; Ubeda, C.; Bielski, C.; Rostron, T.; Cerundolo, V.; Pamer, E.G.; Abramson, S.B.; et al. Expansion of intestinal Prevotella copri correlates with enhanced susceptibility to arthritis. eLife 2013, 2, e01202. [Google Scholar] [CrossRef]
- Friman, C.; Eronen, I.; Videman, T. Plasma Glycosaminoglycans in Experimental Osteoarthritis Caused by Immobilization. J. Rheumatol. 1982, 9, 292–294. [Google Scholar] [PubMed]
- Reddy, G.K.; Dhar, S.C. Metabolism of Glycosaminoglycans in Tissues of Adjuvant Arthritic Rat. Mol. Cell. Biochem. 1991, 106, 117–124. [Google Scholar]
- Pirkmajer, S.; Kulkarni, S.S.; Tom, R.Z.; Ross, F.A.; Hawley, S.A.; Hardie, D.G.; Zierath, J.R.; Chibalin, A.V. Methotrexate promotes glucose uptake and lipid oxidation in skeletal muscle via AMPK activation. Diabetes 2015, 64, 360–369. [Google Scholar] [CrossRef] [PubMed]
- Gergely, P.; Nuesslein-Hildesheim, B.; Guerini, D.; Brinkmann, V.; Traebert, M.; Bruns, C.; Pan, S.; Gray, N.S.; Hinterding, K.; Cooke, N.G.; et al. The selective sphingosine 1-phosphate receptor modulator BAF312 redirects lymphocyte distribution and has species-specific effects on heart rate. Br. J. Pharmacol. 2012, 167, 1035–1047. [Google Scholar] [CrossRef] [PubMed]
- Tolle, M.; Levkau, B.; Kleuser, B.; van der Giet, M. Sphingosine-1-phosphate and FTY720 as anti-atherosclerotic lipid compounds. Eur. J. Clin. Investig. 2007, 37, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Bornigen, D.; Morgan, X.C.; Franzosa, E.A.; Ren, B.; Xavier, R.J.; Garrett, W.S.; Huttenhower, C. Functional profiling of the gut microbiome in disease-associated inflammation. Genome Med. 2013, 5, 65. [Google Scholar] [CrossRef]
- Gomez, A.; Luckey, D.; Yeoman, C.J.; Marietta, E.V.; Berg Miller, M.E.; Murray, J.A.; White, B.A.; Taneja, V. Loss of sex and age driven differences in the gut microbiome characterize arthritis-susceptible 0401 mice but not arthritis-resistant 0402 mice. PLoS ONE 2012, 7, e36095. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zou, Q.; Zeng, B.; Fang, Y.; Wei, H. Analysis of fecal Lactobacillus community structure in patients with early rheumatoid arthritis. Curr. Microbiol. 2013, 67, 170–176. [Google Scholar] [CrossRef]
- Kano, H.; Kaneko, T.; Kaminogawa, S. Oral intake of Lactobacillus delbrueckii subsp. bulgaricus OLL1073R-1 prevents collagen-induced arthritis in mice. J. Food Prot. 2002, 65, 153–160. [Google Scholar] [CrossRef]
- Forbes, J.D.; Van Domselaar, G.; Bernstein, C.N. The Gut Microbiota in Immune-Mediated Inflammatory Diseases. Front. Microbiol. 2016, 7, 1081. [Google Scholar] [CrossRef]
- Kolter, T.; Sandhoff, K. Sphingolipid metabolism diseases. Biochim. Biophys. Acta 2006, 1758, 2057–2079. [Google Scholar] [CrossRef]
- Maceyka, M.; Spiegel, S. Sphingolipid metabolites in inflammatory disease. Nature 2014, 510, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Dorsam, G.; Graeler, M.H.; Seroogy, C.; Kong, Y.; Voice, J.K.; Goetzl, E.J. Transduction of multiple effects of sphingosine 1-phosphate (S1P) on T cell functions by the S1P1 G protein-coupled receptor. J. Immunol. 2003, 171, 3500–3507. [Google Scholar] [CrossRef] [PubMed]
- Ishii, M.; Egen, J.G.; Klauschen, F.; Meier-Schellersheim, M.; Saeki, Y.; Vacher, J.; Proia, R.L.; Germain, R.N. Sphingosine-1-phosphate mobilizes osteoclast precursors and regulates bone homeostasis. Nature 2009, 458, 524–528. [Google Scholar] [CrossRef] [PubMed]
- Paquin-Proulx, D.; Gibbs, A.; Bachle, S.M.; Checa, A.; Introini, A.; Leeansyah, E.; Wheelock, C.E.; Nixon, D.F.; Broliden, K.; Tjernlund, A.; et al. Innate Invariant NKT Cell Recognition of HIV-1-Infected Dendritic Cells Is an Early Detection Mechanism Targeted by Viral Immune Evasion. J. Immunol. 2016, 197, 1843–1851. [Google Scholar] [CrossRef] [PubMed]
- Kato, M.; Muto, Y.; Tanaka-Bandoh, K.; Watanabe, K.; Ueno, K. Sphingolipid composition in Bacteroides species. Anaerobe 1995, 1, 135–139. [Google Scholar] [CrossRef] [PubMed]
- An, D.; Oh, S.F.; Olszak, T.; Neves, J.F.; Avci, F.Y.; Erturk-Hasdemir, D.; Lu, X.; Zeissig, S.; Blumberg, R.S.; Kasper, D.L. Sphingolipids from a symbiotic microbe regulate homeostasis of host intestinal natural killer T cells. Cell 2014, 156, 123–133. [Google Scholar] [CrossRef]
- Selvanantham, T.; Lin, Q.; Guo, C.X.; Surendra, A.; Fieve, S.; Escalante, N.K.; Guttman, D.S.; Streuther, C.J.; Robertson, S.J.; Philpott, D.J.; et al. NKT Cell-Deficient Mice Harbor an Altered Microbiota That Fuels Intestinal Inflammation during Chemically Induced Colitis. J. Immunol. 2016, 197, 4464–4472. [Google Scholar] [CrossRef] [PubMed]
- Parietti, V.; Chifflot, H.; Sibilia, J.; Muller, S.; Monneaux, F. Rituximab treatment overcomes reduction of regulatory iNKT cells in patients with rheumatoid arthritis. Clin. Immunol. 2010, 134, 331–339. [Google Scholar] [CrossRef]
- Mangat, P.; Wegner, N.; Venables, P.J.; Potempa, J. Bacterial and human peptidylarginine deiminases: Targets for inhibiting the autoimmune response in rheumatoid arthritis? Arthritis Res. Ther. 2010, 12, 209. [Google Scholar] [CrossRef] [PubMed]
- Boulter, E.; Estrach, S.; Tissot, F.S.; Hennrich, M.L.; Tosello, L.; Cailleteau, L.; de la Ballina, L.R.; Pisano, S.; Gavin, A.C.; Feral, C.C. Cell metabolism regulates integrin mechanosensing via an SLC3A2-dependent sphingolipid biosynthesis pathway. Nat. Commun. 2018, 9, 4862. [Google Scholar] [CrossRef] [PubMed]
- Cannon, J.P.; Lee, T.A.; Bolanos, J.T.; Danziger, L.H. Pathogenic relevance of Lactobacillus: A retrospective review of over 200 cases. Eur. J. Clin. Microbiol. Infect. Dis. 2005, 24, 31–40. [Google Scholar] [CrossRef]
- Wattam, A.R.; Abraham, D.; Dalay, O.; Disz, T.L.; Driscoll, T.; Gabbard, J.L.; Gillespie, J.J.; Gough, R.; Hix, D.; Kenyon, R.; et al. PATRIC, the bacterial bioinformatics database and analysis resource. Nucleic Acids Res. 2014, 42, D581–D591. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Nardini, C. A method for automated pathogenic content estimation with application to rheumatoid arthritis. BMC Syst. Biol. 2016, 10, 107. [Google Scholar] [CrossRef] [PubMed]


| KEGG Pathway | CIA vs. NOCIA | MS vs. CIA | MTXMS vs. CIA | |||
|---|---|---|---|---|---|---|
| Up | Down | Up | Down | Up | Down | |
| Galactose metabolism | 0.7426 | 0.0437 | 0.0194 | 0.4272 | 0.3884 | 0.0357 |
| Sphingolipid metabolism | 1 | 0.0098 | 0.0209 | 1 | 0.6777 | 1 |
| Butirosin and neomycin biosynthesis | 1 | 0.0067 | 0.0097 | 1 | 0.1246 | 1 |
| Enrichment by p values (p ≤ 0.05 in bold) | ||||||
| KEGG Pathway | Gene | Microbe | Comparison |
|---|---|---|---|
| Sphingolipid metabolism | Acer2, Asah1, B4galt6, Sptlc1 | Akkermansia, Parabacteroides, Lactobacillus, Turicibacter, Allobaculum, Moryella | MS-CIA_ALL |
| Acer2, Asah1, B4galt6 | Akkermansia, Parabacteroides, Lactobacillus, Turicibacter, Allobaculum, Moryella | MS-CIA_UP | |
| B4galt6, Ppap2c, Sptlc1 | Akkermansia, Prevotella, Lactobacillus, Oscillospira | MS-MTXMS_ALL; MS-MTXMS_UP | |
| Acer2 | Blautia, Lactobacillus, Turicibacter, Coprococcus, Odoribacter | NOCIA-CIA_ALL; NOCIA-CIA_UP |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, X.; Devescovi, V.; Liu, Y.; Dent, J.E.; Nardini, C. Host-Microbiome Synergistic Control on Sphingolipid Metabolism by Mechanotransduction in Model Arthritis. Biomolecules 2019, 9, 144. https://doi.org/10.3390/biom9040144
Zhou X, Devescovi V, Liu Y, Dent JE, Nardini C. Host-Microbiome Synergistic Control on Sphingolipid Metabolism by Mechanotransduction in Model Arthritis. Biomolecules. 2019; 9(4):144. https://doi.org/10.3390/biom9040144
Chicago/Turabian StyleZhou, Xiaoyuan, Valentina Devescovi, Yuanhua Liu, Jennifer E. Dent, and Christine Nardini. 2019. "Host-Microbiome Synergistic Control on Sphingolipid Metabolism by Mechanotransduction in Model Arthritis" Biomolecules 9, no. 4: 144. https://doi.org/10.3390/biom9040144
APA StyleZhou, X., Devescovi, V., Liu, Y., Dent, J. E., & Nardini, C. (2019). Host-Microbiome Synergistic Control on Sphingolipid Metabolism by Mechanotransduction in Model Arthritis. Biomolecules, 9(4), 144. https://doi.org/10.3390/biom9040144