Structural Analysis of the 42 kDa Parvulin of Trypanosoma brucei

 ,
,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Expression of TbPar42 Constructs
2.2. Nuclear Magnetic Resonance (NMR) Spectroscopy
2.3. NMR Titration Experiments
2.4. EXSY Experiment
2.5. Relaxation Measurements
2.6. Sample Preparation for TbPar42-PPIase Crystallization Experiments
2.7. Data Collection and Structure Determination
2.8. PPIase Activity
3. Results
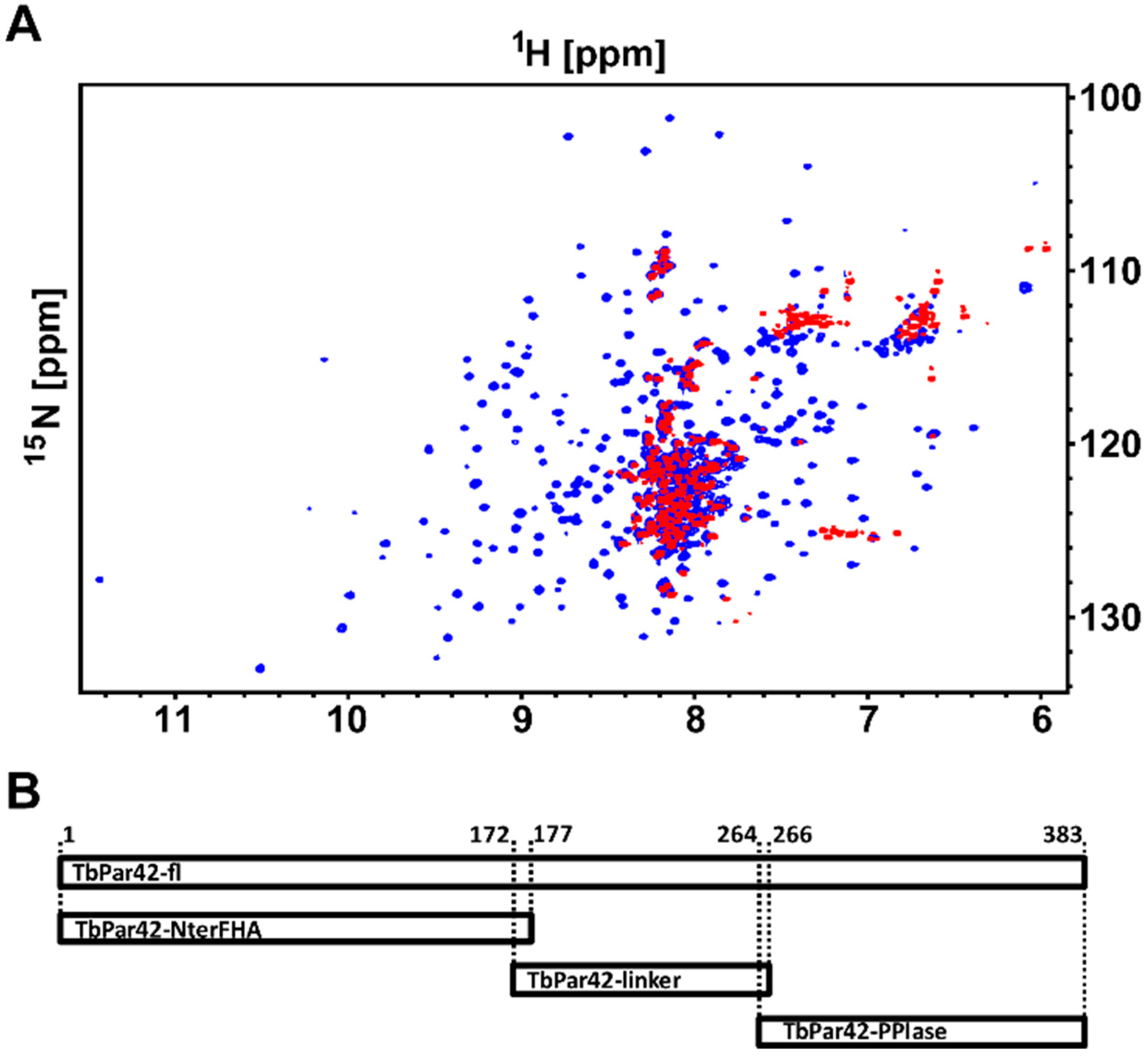
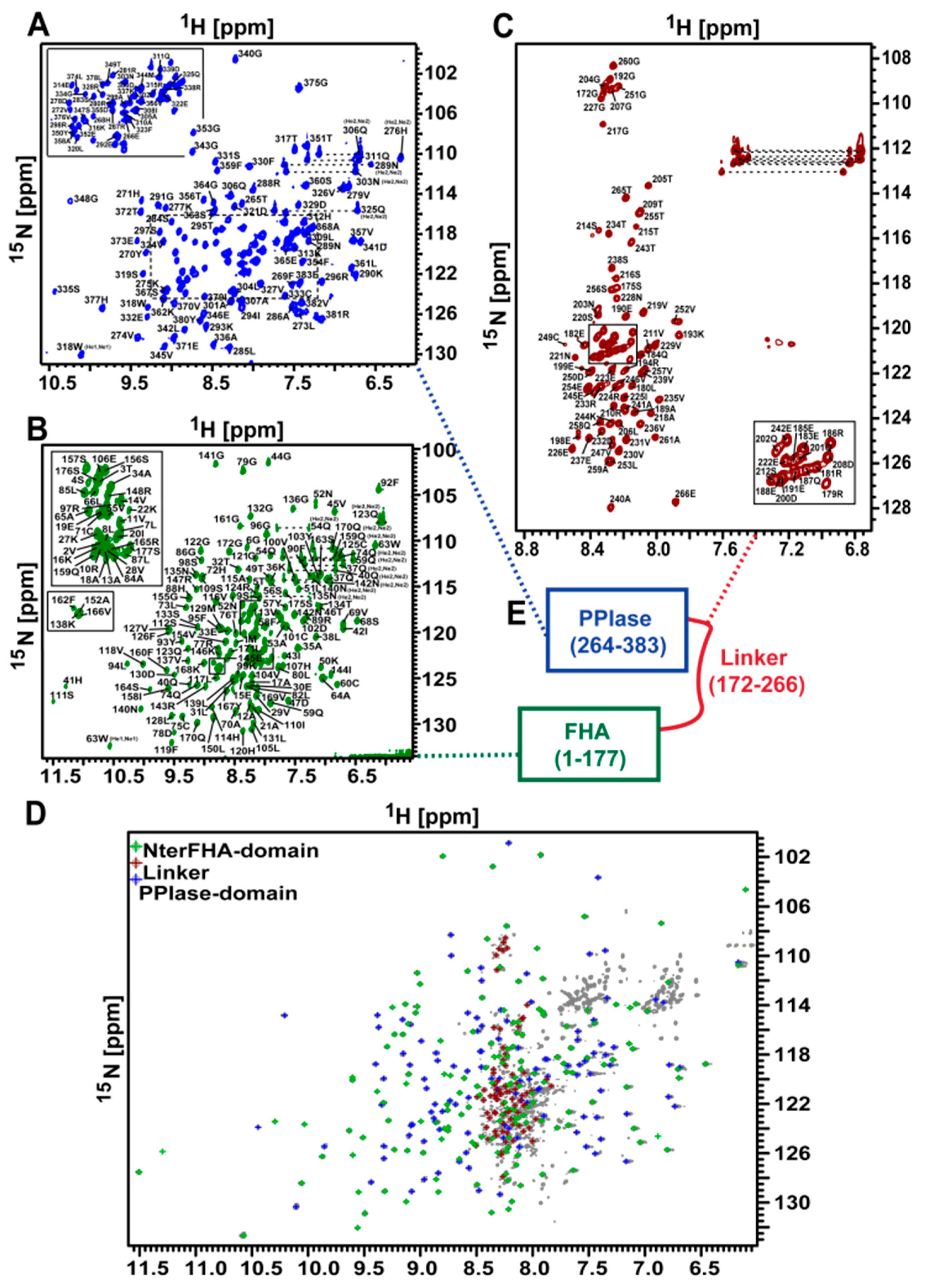
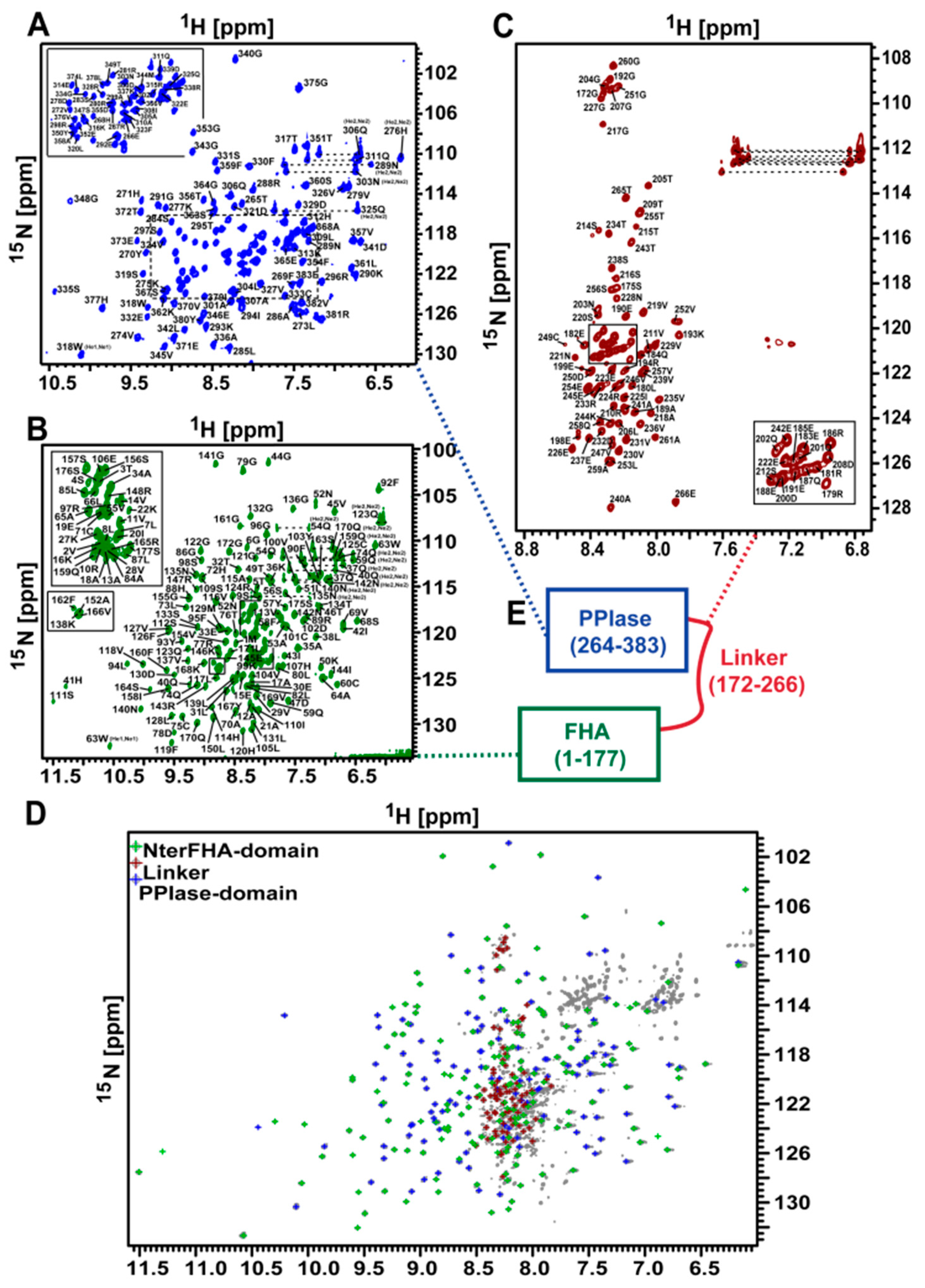
3.1. NMR Measurements of Full-Length TbPar42 (1–383)
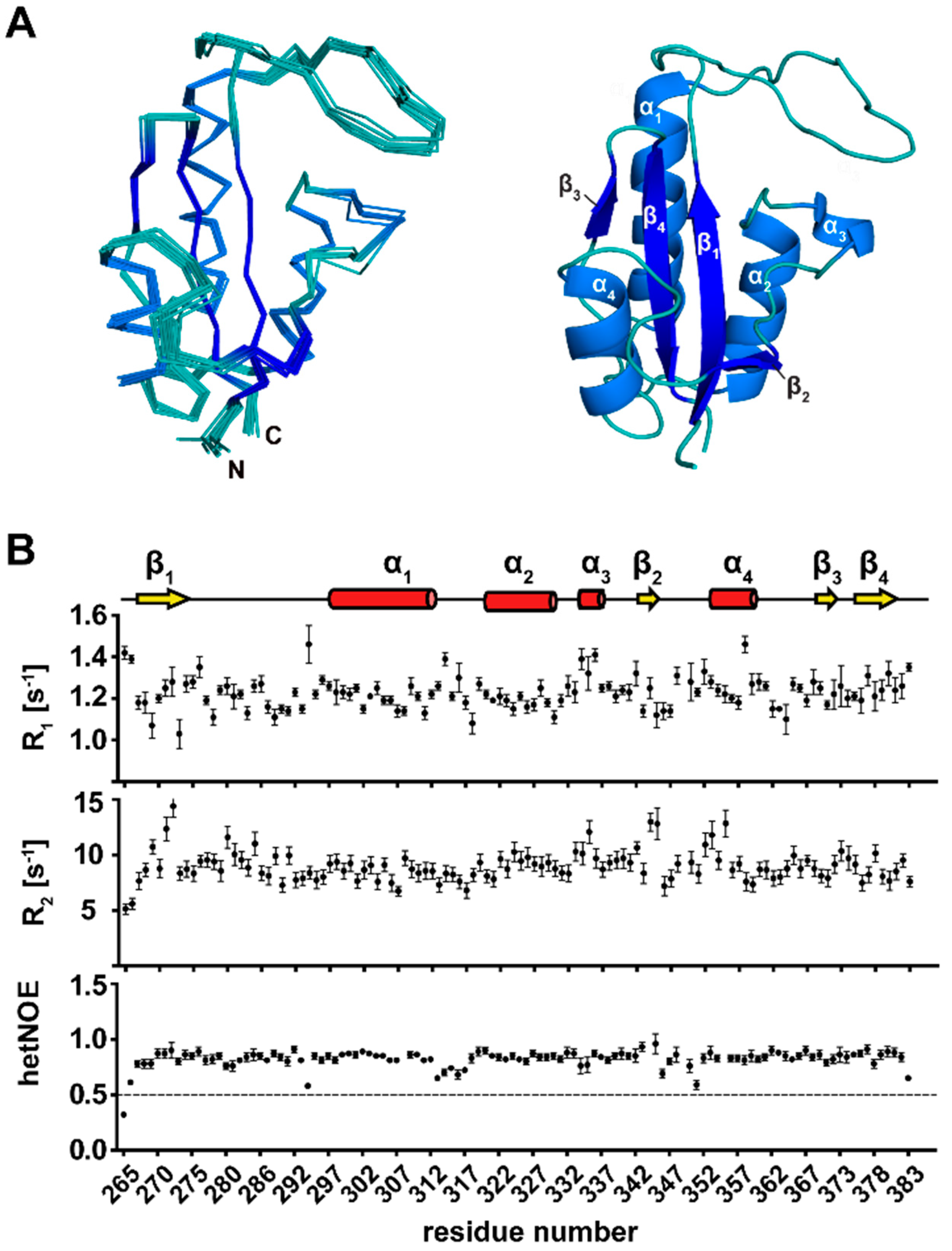
3.2. NMR Structure Calculation of TbPar42-PPIase
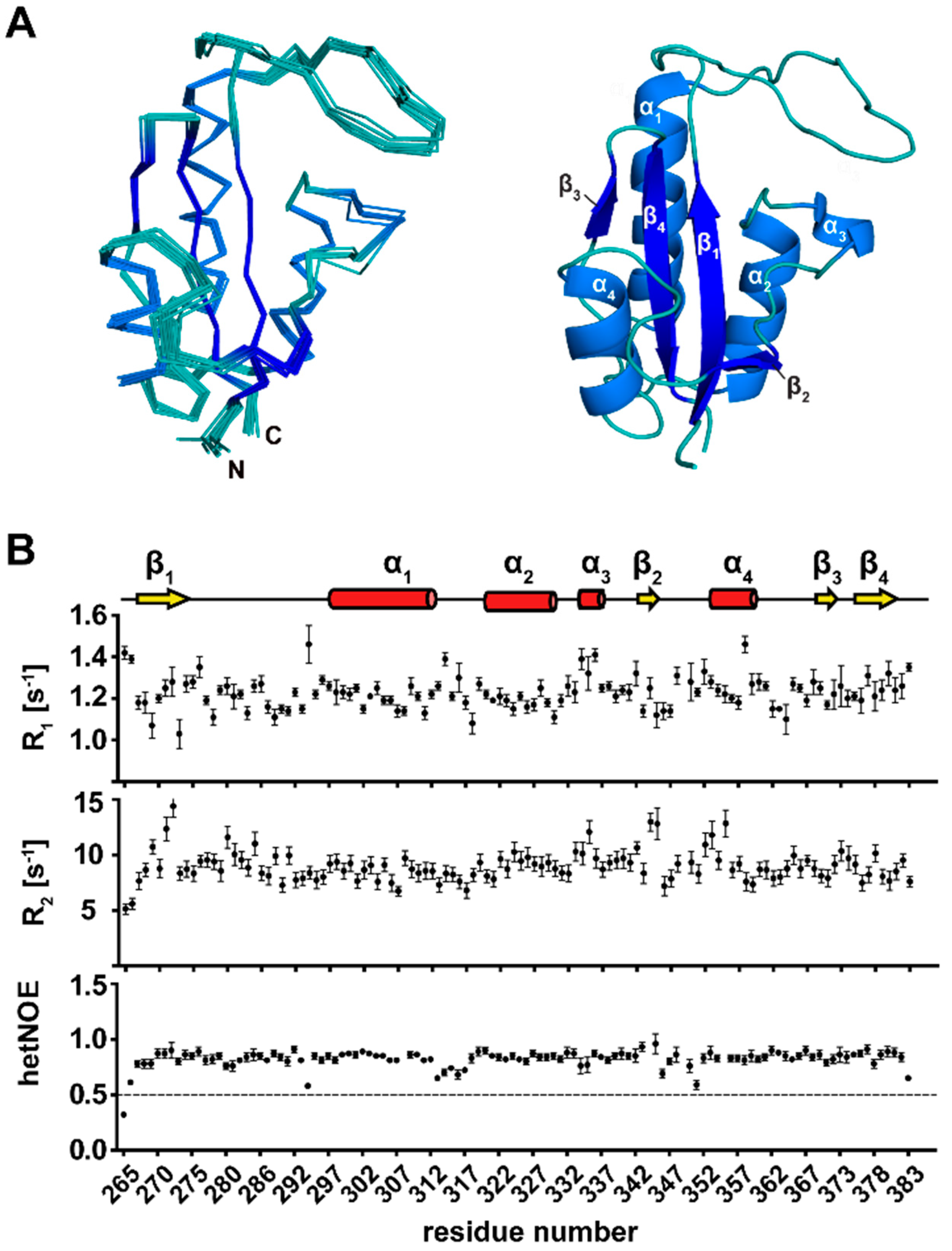
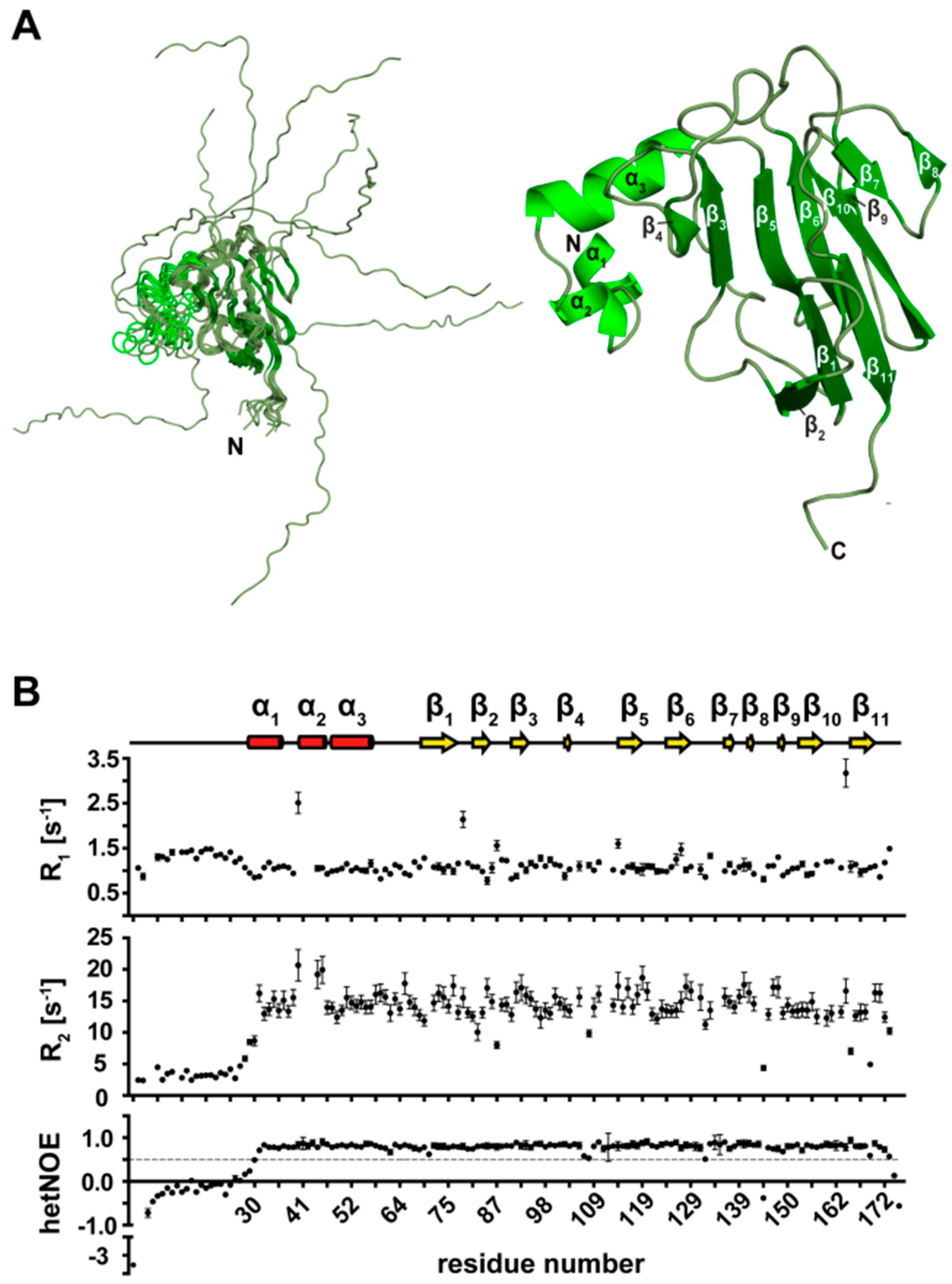
3.3. Secondary Structure Elements of TbPar42-PPIase
3.4. The Tertiary Structure of TbPar42-PPIase Comprises the Typical Parvulin Fold
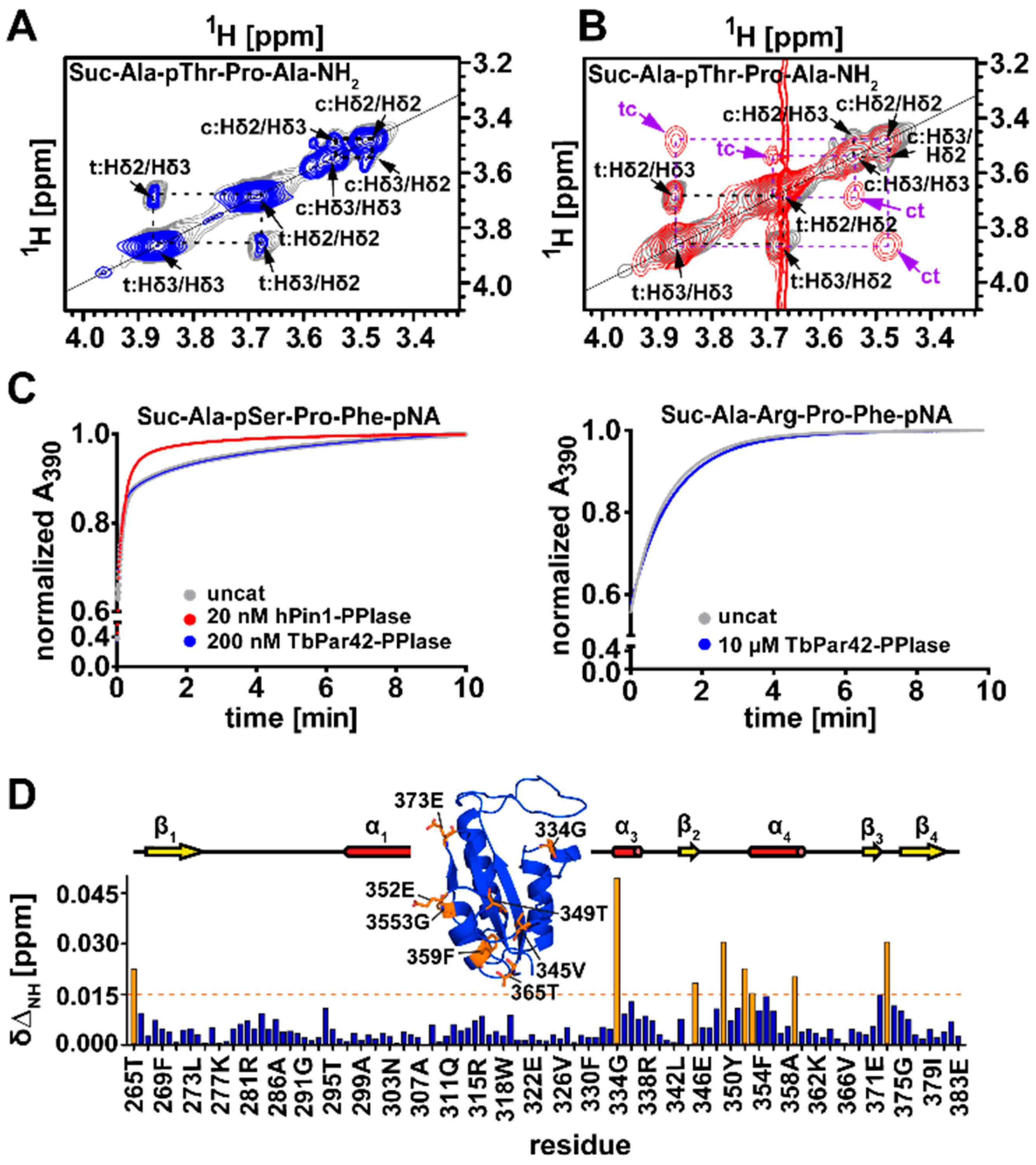
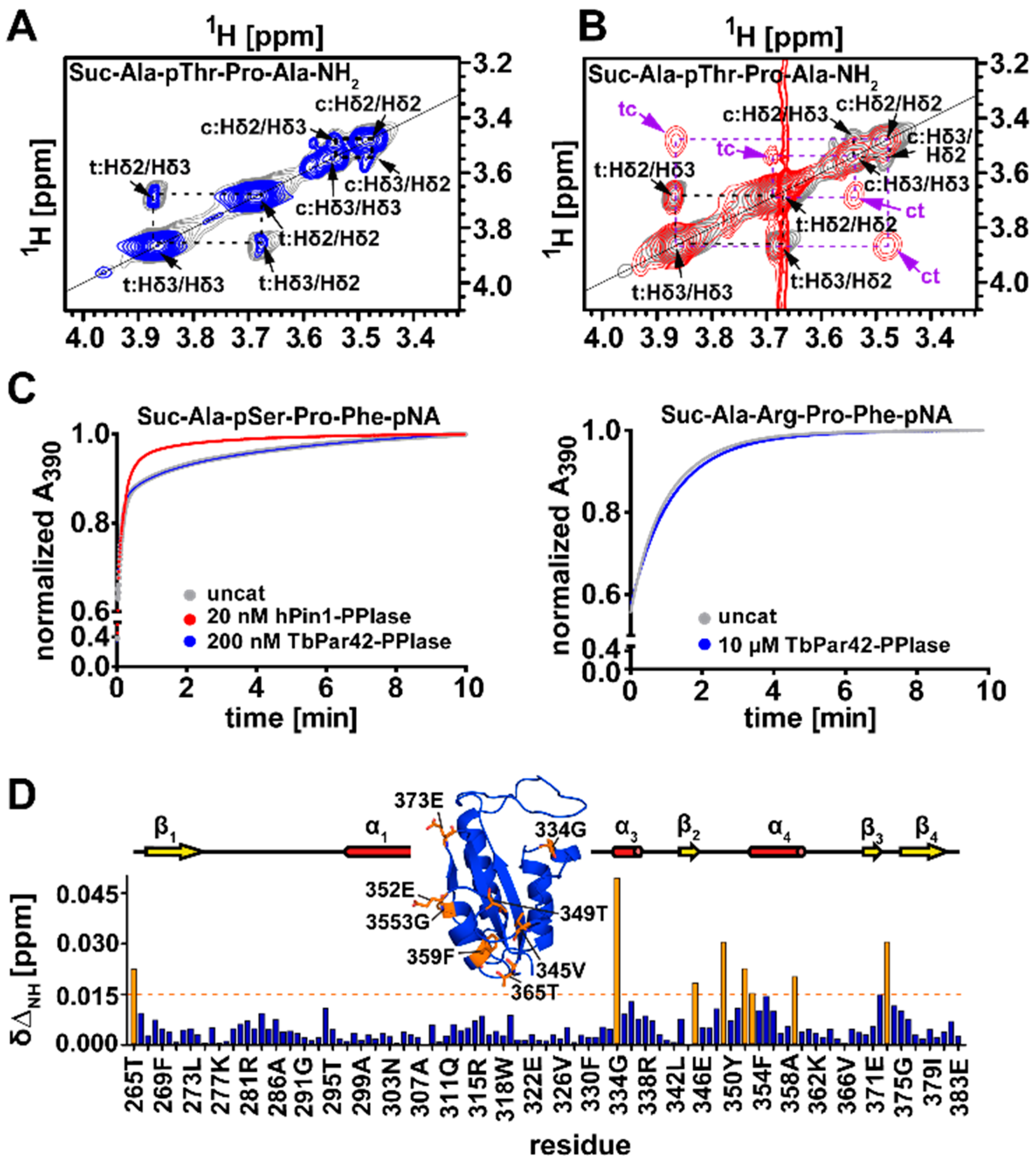
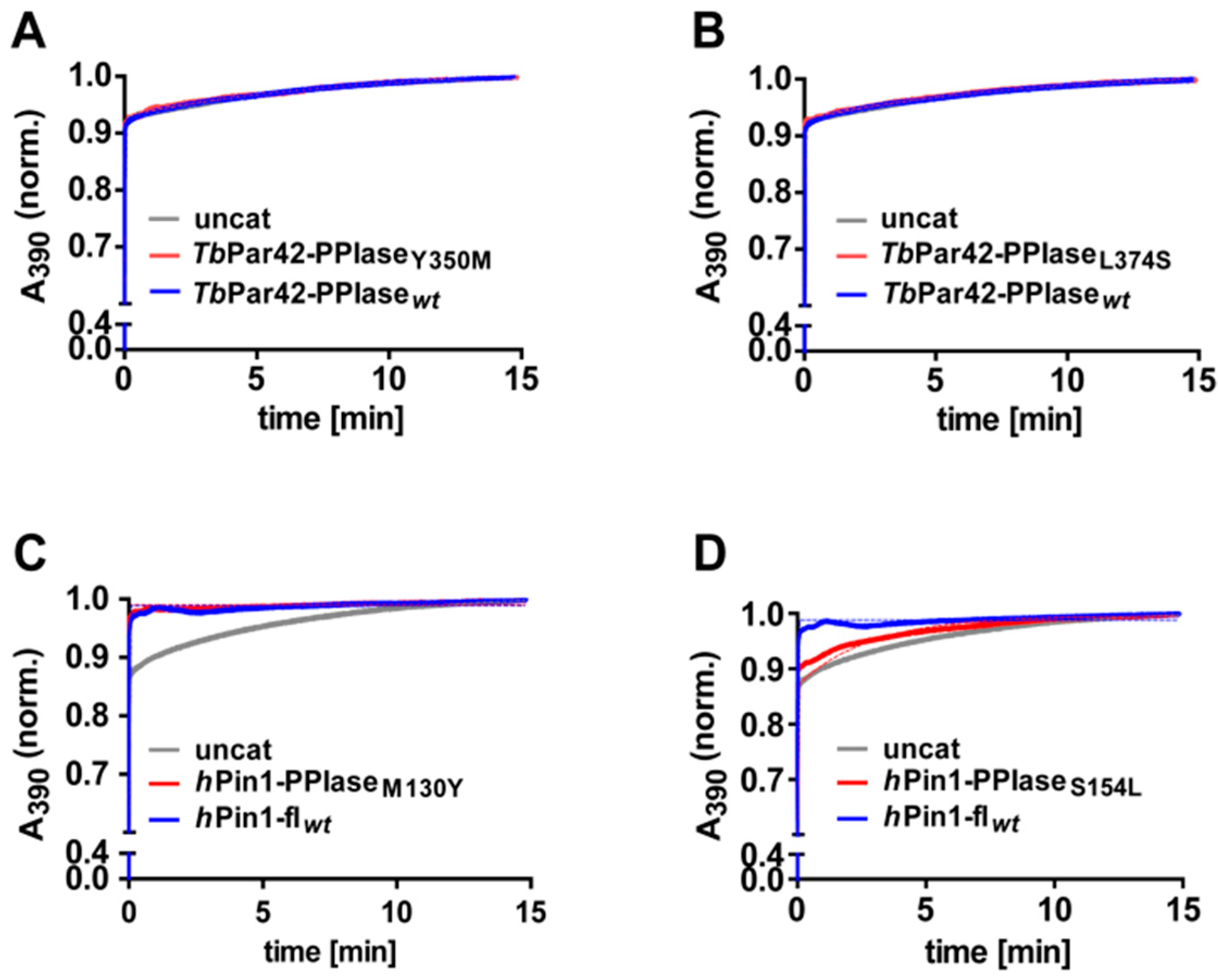
3.5. TbPar42-PPIase Lacks Catalytic PPIase Activity
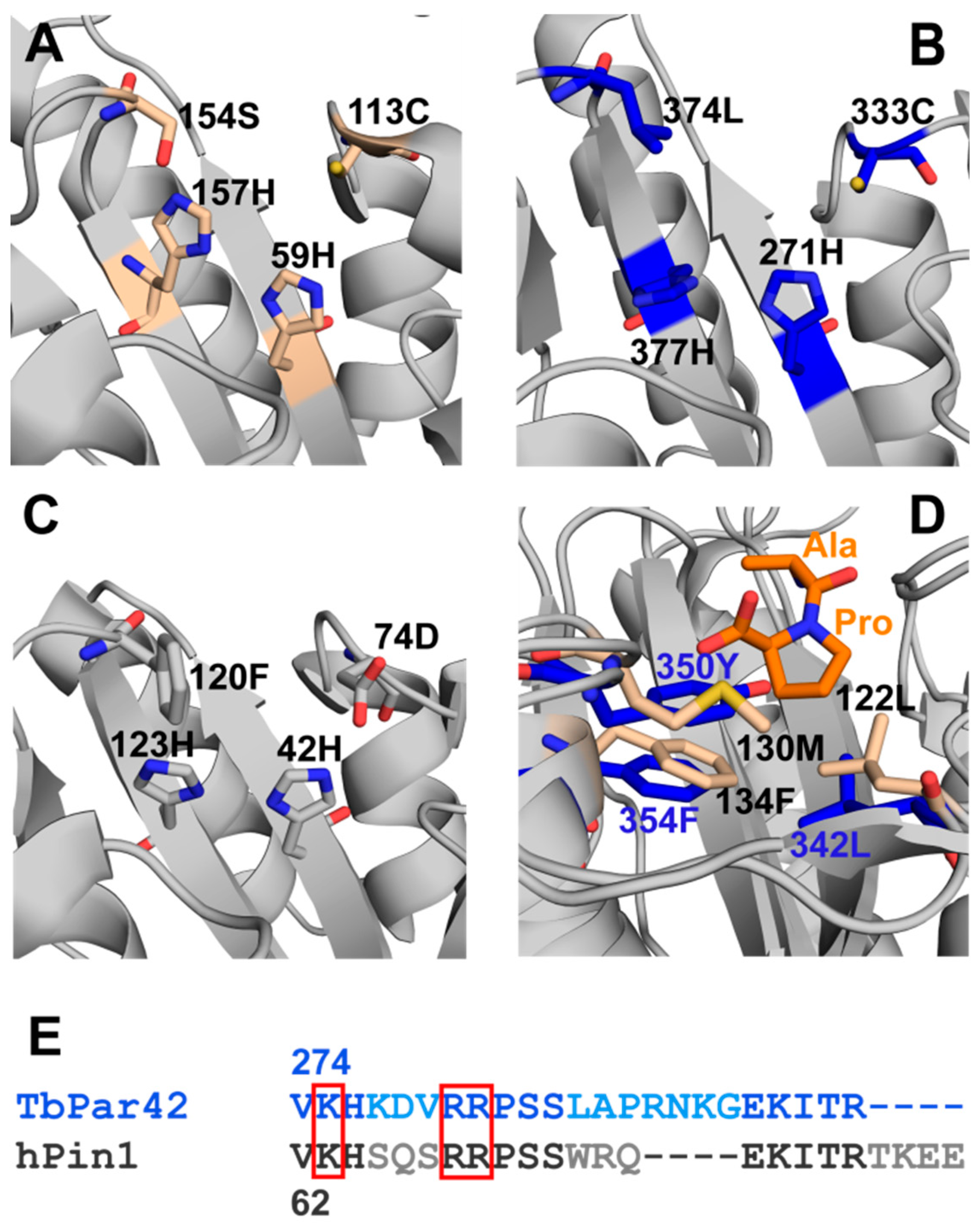
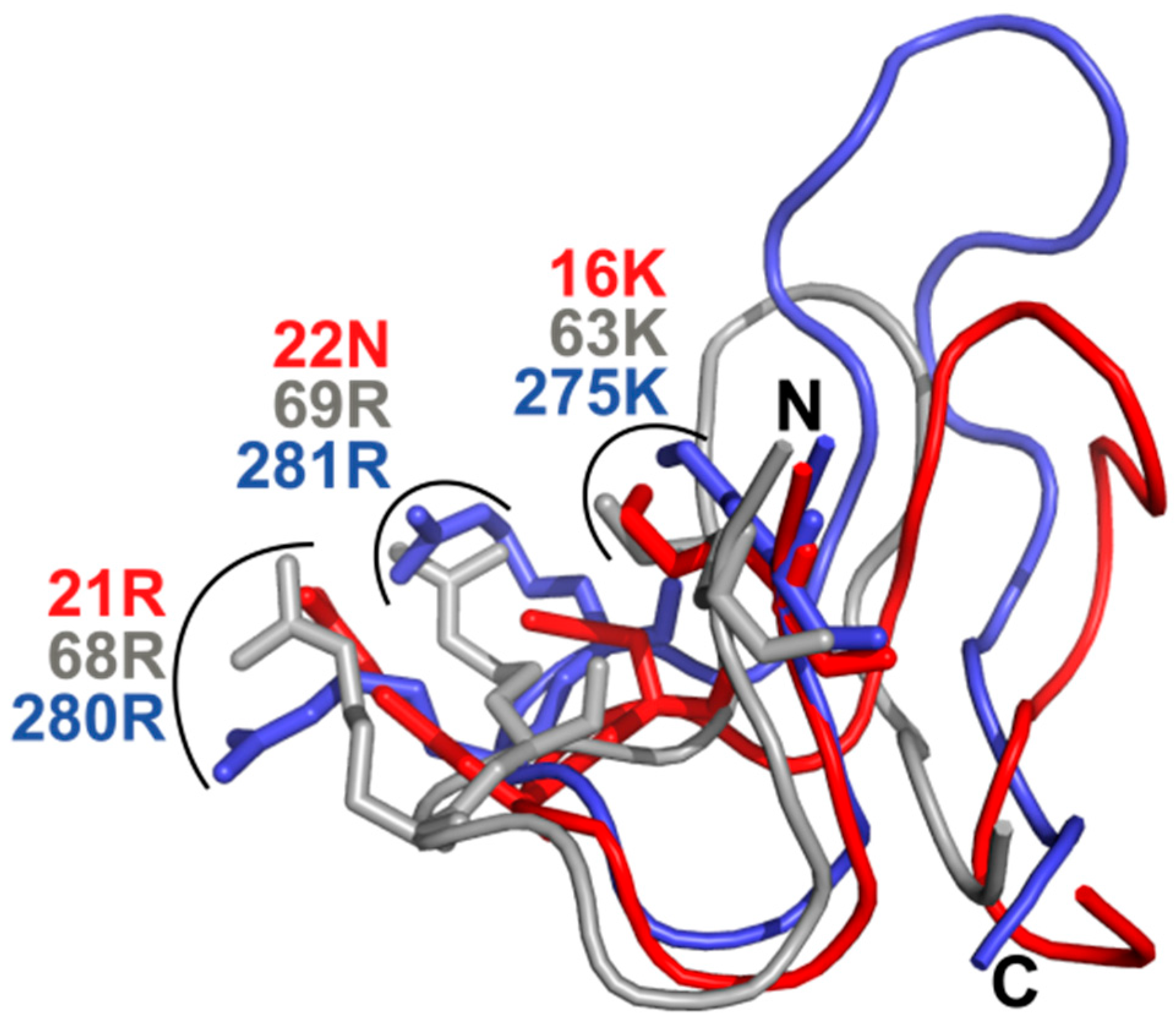
3.6. What Is the Structural Origin of the Lack of Catalytic Activity of TbPar42-PPIase?
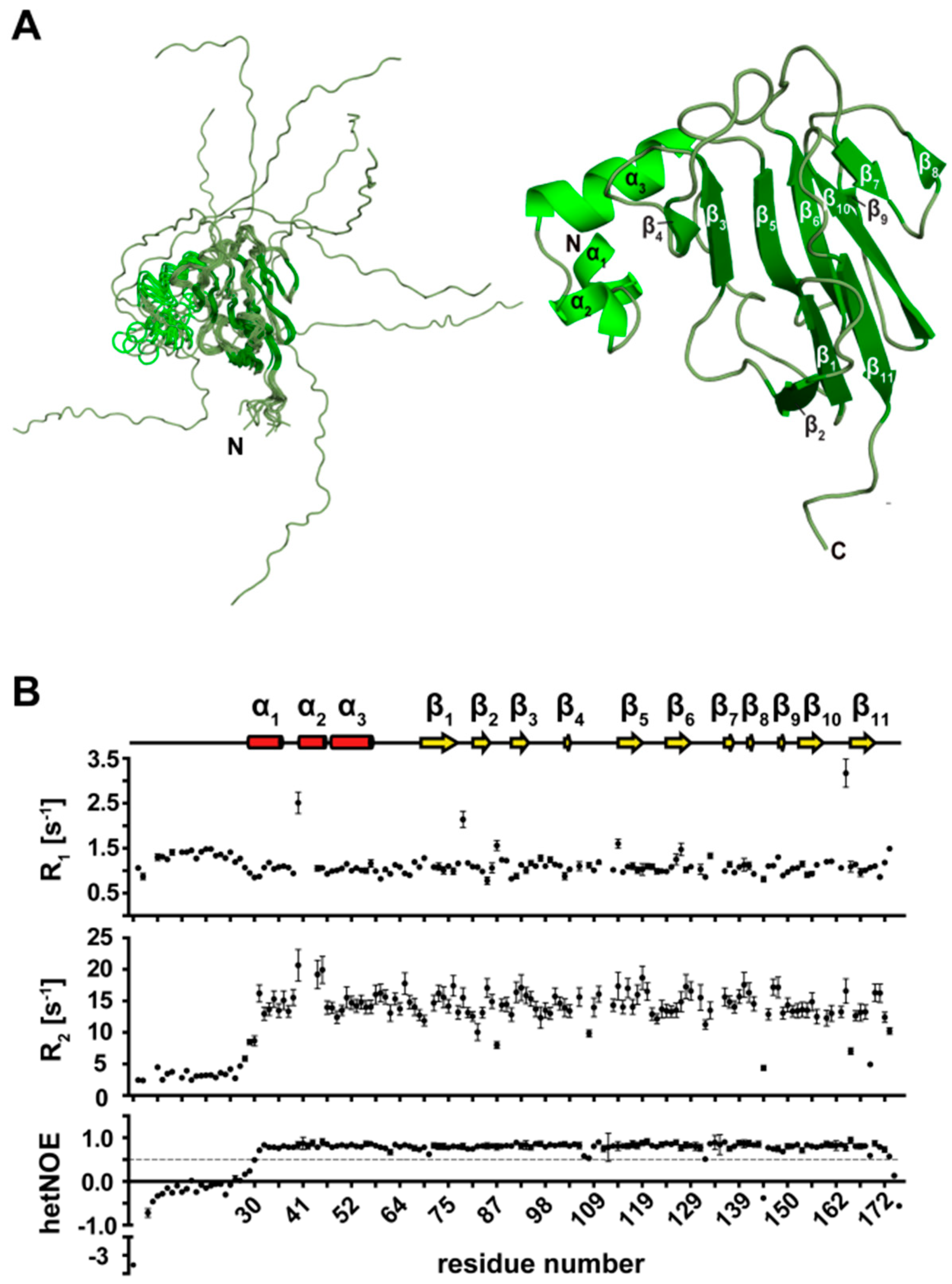
3.7. NMR Structure Calculation of the N-Terminal Extended FHA Domain
3.8. Secondary Structure Elements of the N-Terminal Extended FHA Domain (1–177)
3.9. Tertiary Structure of TbPar42-NterFHA1–177
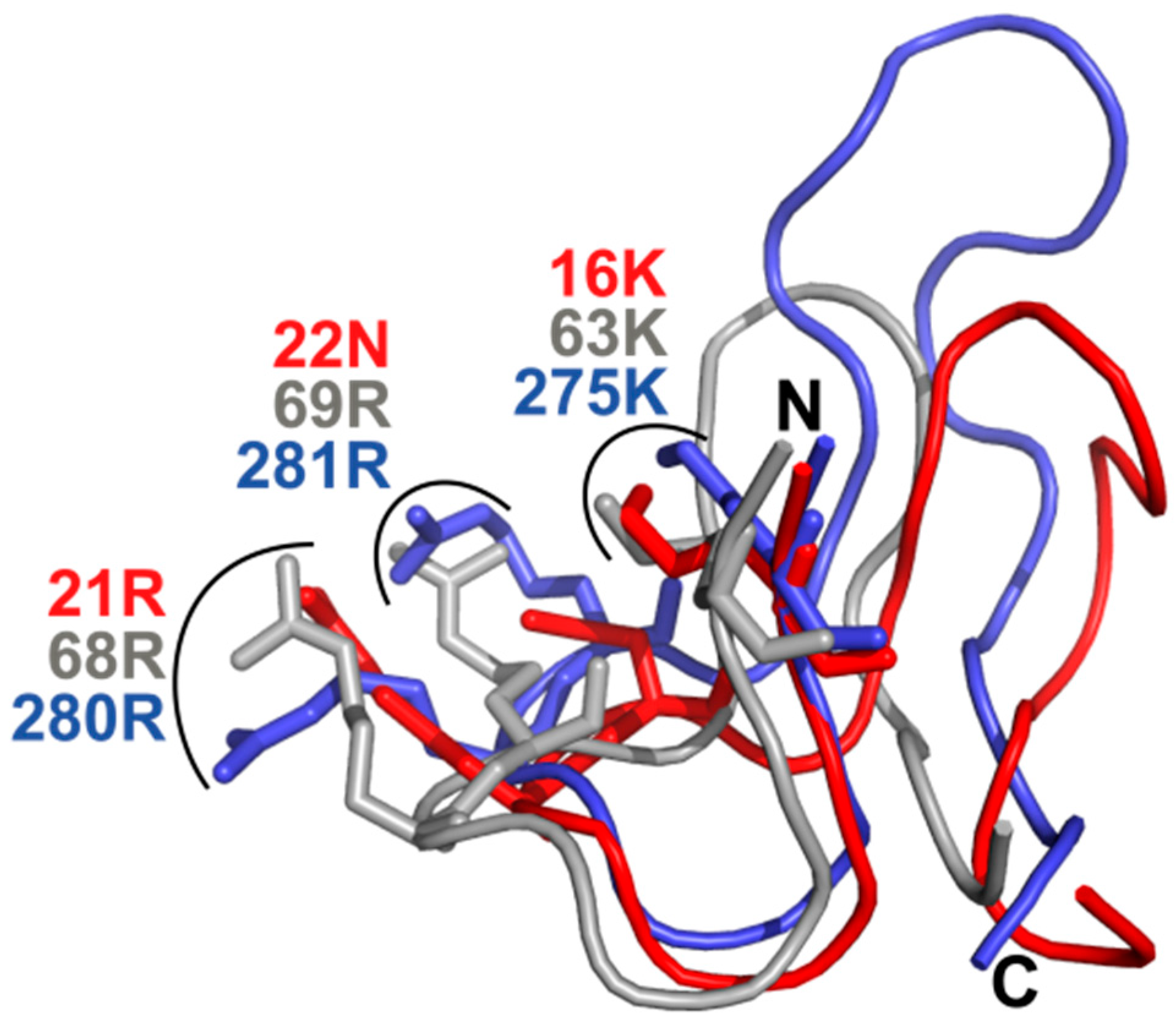
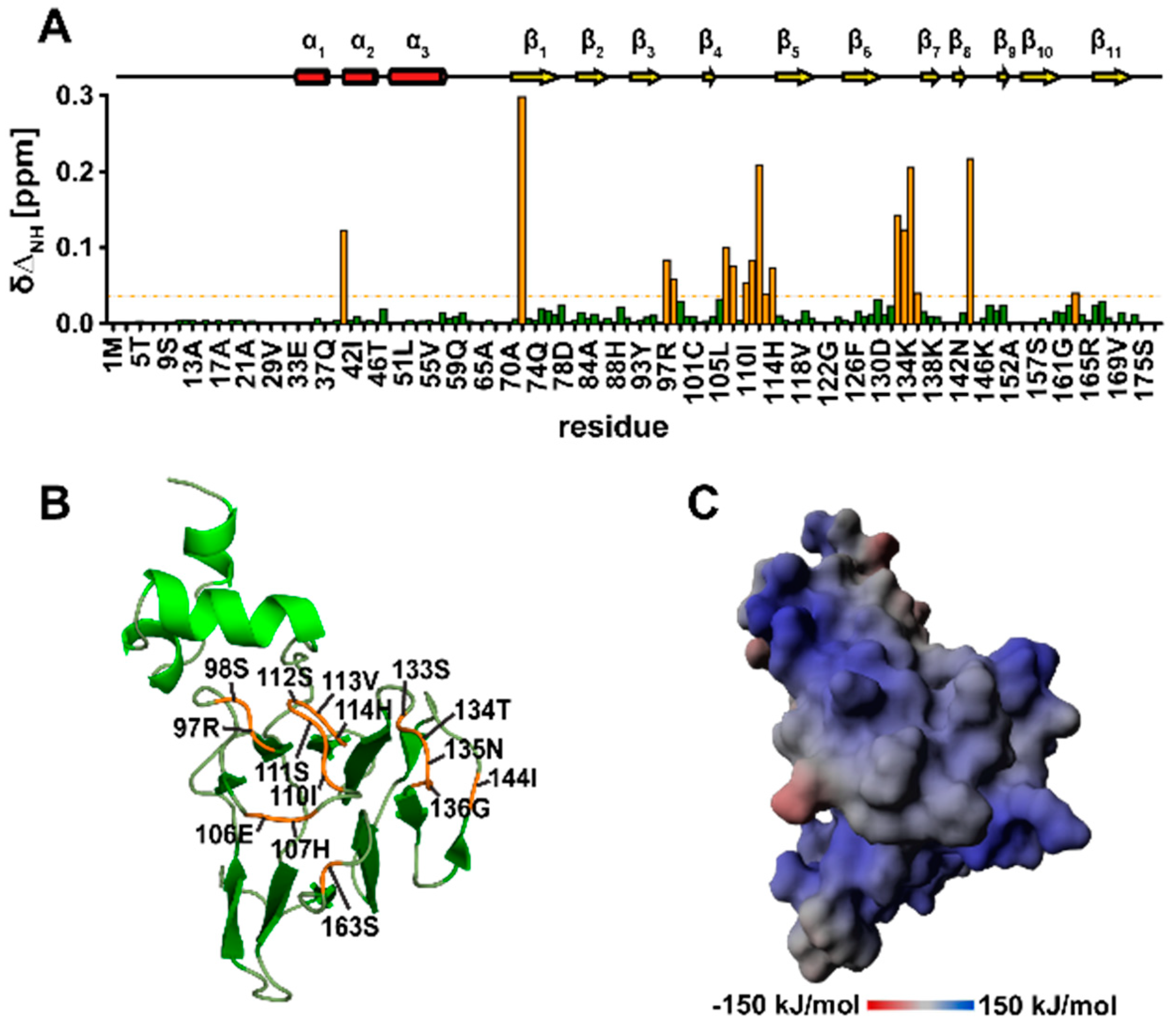
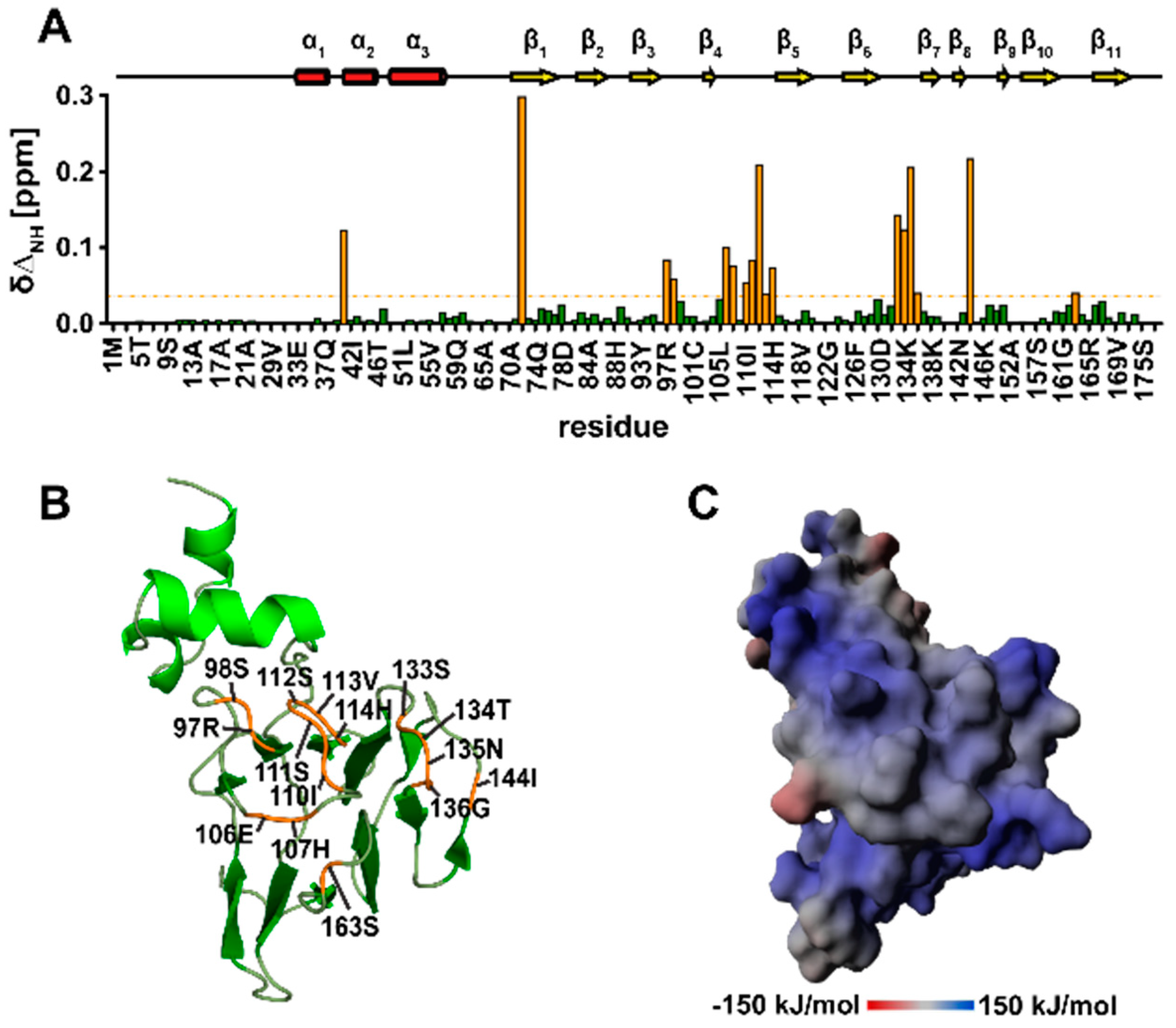
3.10. TbPar42-NterFHA1–177 binds pThr-Pro Moieties within Peptides
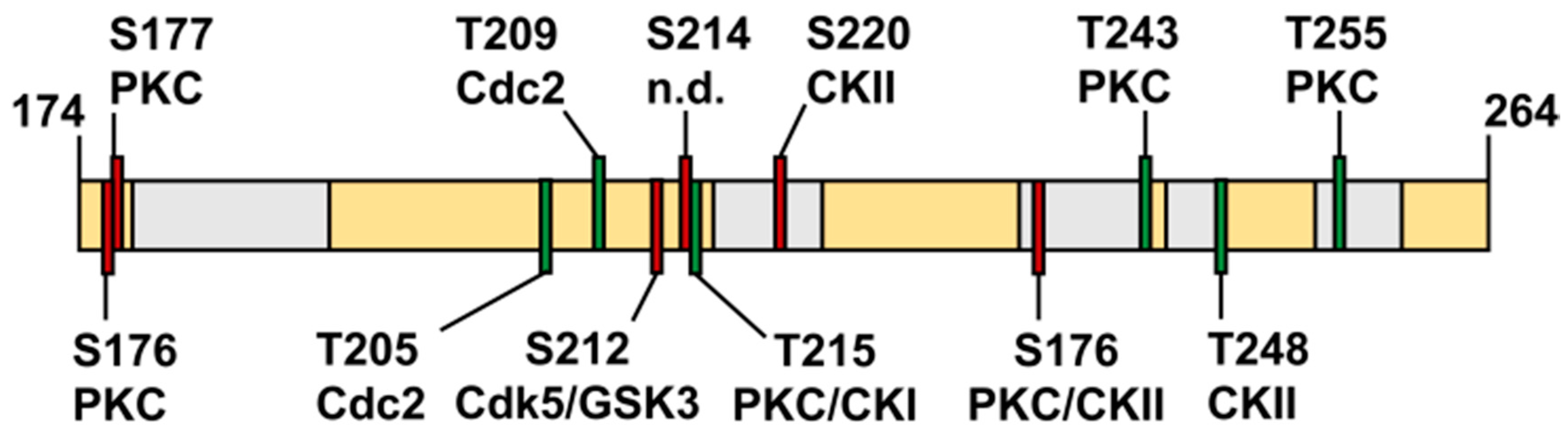
3.11. The Isolated Linker Region between the NterFHA and PPIase Domain Is Flexible and Unfolded
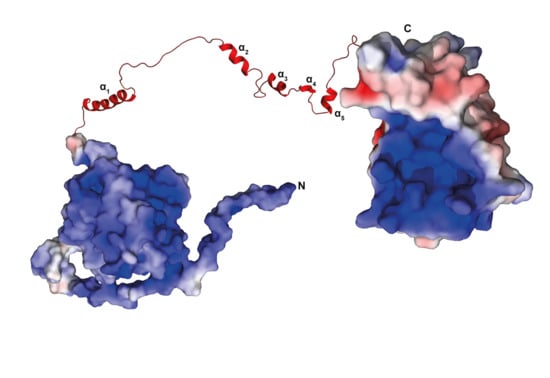
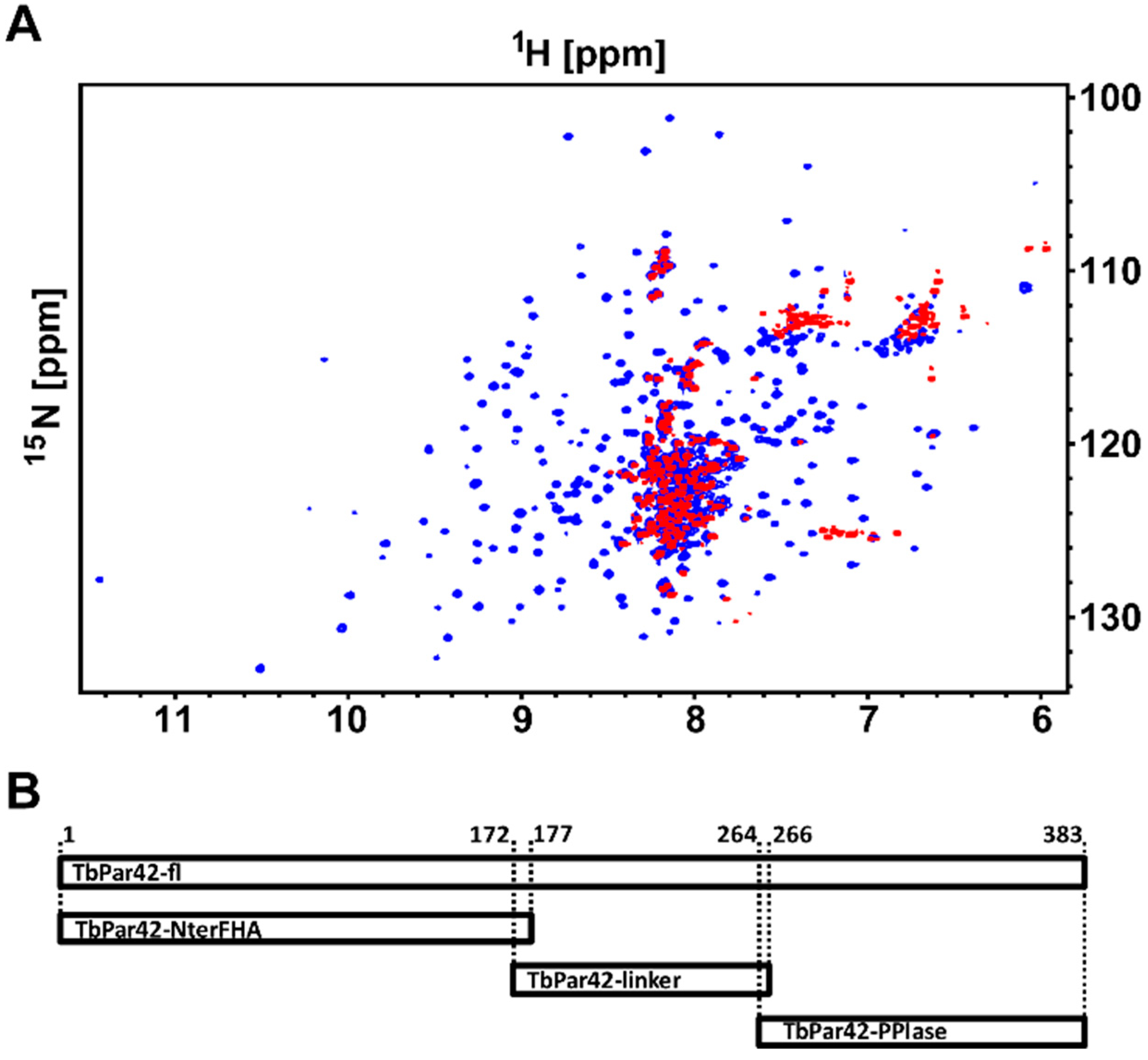
3.12. There Is No Interaction between the Domains of TbPar42
4. Discussion
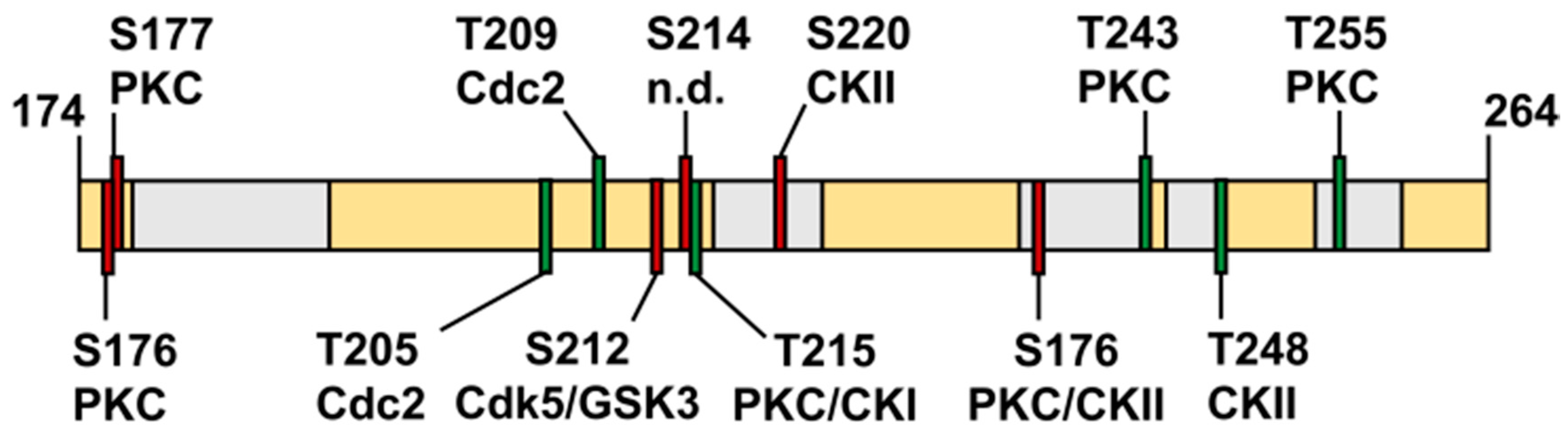
4.1. What is the Cellular Role of the FHA Domain of TbPar42?
4.2. TbPar42 May Interact as a Protein Recruitment Platform
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Goh, J.Y.; Lai, C.-Y.; Tan, L.C.; Yang, D.; He, C.Y.; Liou, Y.-C. Functional characterization of two novel parvulins in Trypanosoma brucei. Febs Lett. 2010, 584, 2901–2908. [Google Scholar] [CrossRef] [PubMed]
- Erben, E.D.; Valguarnera, E.; Nardelli, S.; Chung, J.; Daum, S.; Potenza, M.; Schenkman, S.; Téllez-Iñón, M.T. Identification of an atypical peptidyl-prolyl cis/trans isomerase from trypanosomatids. Biochim. Biophys. Acta 2010, 1803, 1028–1037. [Google Scholar] [CrossRef] [PubMed]
- Vallon, O. Chlamydomonas Immunophilins and Parvulins: Survey and Critical Assessment of Gene Models. Eukaryot. Cell 2005, 4, 230–241. [Google Scholar] [CrossRef] [PubMed]
- Andreotti, A.H. Native state proline isomerization: An intrinsic molecular switch. Biochemistry 2003, 42, 9515–9524. [Google Scholar] [CrossRef] [PubMed]
- Hammet, A.; Pike, B.L.; McNees, C.J.; Conlan, L.A.; Tenis, N.; Heierhorst, J. FHA domains as phospho-threonine binding modules in cell signaling. IUBMB Life 2003, 55, 23–27. [Google Scholar] [CrossRef] [PubMed]
- Coquelle, N.; Glover, J.N.M. FHA domain pThr binding specificity: it’s all about me. Structure 2010, 18, 1549–1550. [Google Scholar] [CrossRef] [PubMed]
- Mahajan, A.; Yuan, C.; Lee, H.; Chen, E.S.-W.; Wu, P.-Y.; Tsai, M.-D. Structure and function of the phosphothreonine-specific FHA domain. Sci. Signal. 2008, 1, re12. [Google Scholar] [CrossRef] [PubMed]
- Scholz, C.; Rahfeld, J.; Fischer, G.; Schmid, F.X. Catalysis of protein folding by parvulin. J. Mol. Biol. 1997, 273, 752–762. [Google Scholar] [CrossRef] [PubMed]
- Matena, A.; Rehic, E.; Hönig, D.; Kamba, B.; Bayer, P. Structure and function of the human parvulins Pin1 and Par14/17. Biol. Chem. 2018, 399, 101–125. [Google Scholar] [CrossRef] [PubMed]
- Holm, L.; Laakso, L.M. Dali server update. Nucleic Acids Res. 2016, 44, W351–W355. [Google Scholar] [CrossRef] [PubMed]
- Grum, D.; van den Boom, J.; Neumann, D.; Matena, A.; Link, N.M.; Mueller, J.W. A heterodimer of human 3′-phospho-adenosine-5′-phosphosulphate (PAPS) synthases is a new sulphate activating complex. Biochem. Biophys. Res. Commun. 2010, 395, 420–425. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.; Bigam, C.; Yao, J.; Abildgaard, F.; Dyson, H.J.; Oldfield, E.; Markley, J.; Sykes, B. 1H, 13C and 15N chemical shift referencing in biomolecular NMR. J. Biomol. NMR 1995, 6, 135–140. [Google Scholar] [CrossRef] [PubMed]
- Markley, J.L.; Bax, A.; Arata, Y.; Hilbers, C.W.; Kaptein, R.; Sykes, B.D.; Wright, P.E.; Wüthrich, K. Recommendations for the presentation of NMR structures of proteins and nucleic acids. IUPAC-IUBMB-IUPAB Inter-Union Task Group on the Standardization of Data Bases of Protein and Nucleic Acid Structures Determined by NMR Spectroscopy. J. Biomol. NMR 1998, 12, 1–23. [Google Scholar] [CrossRef] [PubMed]
- López-Méndez, B.; Güntert, P. Automated protein structure determination from NMR spectra. J. Am. Chem. Soc. 2006, 128, 13112–13122. [Google Scholar] [CrossRef] [PubMed]
- Cornilescu, G.; Delaglio, F.; Bax, A. Protein backbone angle restraints from searching a database for chemical shift and sequence homology. J. Biomol. NMR 1999, 13, 289–302. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, T.; Güntert, P.; Wüthrich, K. Protein NMR structure determination with automated NOE assignment using the new software CANDID and the torsion angle dynamics algorithm DYANA. J. Mol. Biol. 2002, 319, 209–227. [Google Scholar] [CrossRef]
- Güntert, P. Automated NMR structure calculation with CYANA. Methods Mol. Biol. 2004, 278, 353–378. [Google Scholar] [CrossRef] [PubMed]
- Wagner, G.; Neuhaus, D.; Wörgötter, E.; Vašák, M.; Kägi, J.H.R.; Wüthrich, K. Nuclear magnetic resonance identification of “half-turn” and 310-helix secondary structure in rabbit liver metallothionein-2. J. Mol. Biol. 1986, 187, 131–135. [Google Scholar] [CrossRef]
- Wishart, D.S.; Sykes, B.D.; Richards, F.M. Relationship between nuclear magnetic resonance chemical shift and protein secondary structure. J. Mol. Biol. 1991, 222, 311–333. [Google Scholar] [CrossRef]
- Wishart, D.S.; Sykes, B.D.; Richards, F.M. The chemical shift index: A fast and simple method for the assignment of protein secondary structure through NMR spectroscopy. Biochemistry 1992, 31, 1647–1651. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Sykes, B.D. The 13C chemical-shift index: A simple method for the identification of protein secondary structure using 13C chemical-shift data. J. Biomol. NMR 1994, 4, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Ayed, A.; Mulder, F.A.; Yi, G.S.; Lu, Y.; Kay, L.E.; Arrowsmith, C.H. Latent and active p53 are identical in conformation. Nat. Struct. Biol. 2001, 8, 756–760. [Google Scholar] [CrossRef] [PubMed]
- Delano, W.L. The PyMOL Molecular Graphics System. 2002. Available online: http://www.pymol.org (accessed on February 2019).
- Kay, L.E.; Nicholson, L.K.; Delaglio, F.; Bax, A.; Torchia, D.A. Pulse sequences for removal of the effects of cross correlation between dipolar and chemical-shift anisotropy relaxation mechanisms on the measurement of heteronuclear T1 and T2 values in proteins. J. Magn. Reson. 1992, 97, 359–375. [Google Scholar] [CrossRef]
- Kabsch, W. Integration, scaling, space-group assignment and post-refinement. Acta Crystallogr. Sect. D 2010, 66, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Kabsch, W. XDS. Acta Crystallogr. Sect. D 2010, 66, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Vagin, A.; Teplyakov, A. MOLREP: An Automated Program for Molecular Replacement. J. Appl. Cryst. 1997, 30, 1022–1025. [Google Scholar] [CrossRef]
- Murshudov, G.N.; Vagin, A.A.; Dodson, E.J. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. Sect. D 1997, 53, 240–255. [Google Scholar] [CrossRef] [PubMed]
- Adams, P.D.; Afonine, P.V.; Bunkóczi, G.; Chen, V.B.; Davis, I.W.; Echols, N.; Headd, J.J.; Hung, L.-W.; Kapral, G.J.; Grosse-Kunstleve, R.W.; et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. Sect. D 2010, 66, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Emsley, P.; Cowtan, K. Coot: Model-building tools for molecular graphics. Acta Crystallogr. Sect. D 2004, 60, 2126–2132. [Google Scholar] [CrossRef] [PubMed]
- Fischer, G.; Bang, H.; Mech, C. Nachweis einer Enzymkatalyse für die cis-trans-Isomerisierung der Peptidbindung in prolinhaltigen Peptiden. Biomed. Biochim. Acta 1984, 43, 1101–1111. [Google Scholar] [PubMed]
- Kofron, J.L.; Kuzmic, P.; Kishore, V.; Colón-Bonilla, E.; Rich, D.H. Determination of kinetic constants for peptidyl prolyl cis-trans isomerases by an improved spectrophotometric assay. Biochemistry 1991, 30, 6127–6134. [Google Scholar] [CrossRef] [PubMed]
- Fanghänel, J.; Fischer, G. Insights into the catalytic mechanism of peptidyl prolyl cis/trans isomerases. Front. Biosci. 2004, 9, 3453–3478. [Google Scholar] [CrossRef] [PubMed]
- Ranganathan, R.; Lu, K.P.; Hunter, T.; Noel, J.P. Structural and Functional Analysis of the Mitotic Rotamase Pin1 Suggests Substrate Recognition Is Phosphorylation Dependent. Cell 1997, 89, 875–886. [Google Scholar] [CrossRef]
- Mueller, J.W.; Link, N.M.; Matena, A.; Hoppstock, L.; Rüppel, A.; Bayer, P.; Blankenfeldt, W. Crystallographic proof for an extended hydrogen-bonding network in small prolyl isomerases. J. Am. Chem. Soc. 2011, 133, 20096–20099. [Google Scholar] [CrossRef] [PubMed]
- Bayer, E.; Goettsch, S.; Mueller, J.W.; Griewel, B.; Guiberman, E.; Mayr, L.M.; Bayer, P. Structural analysis of the mitotic regulator hPin1 in solution: Insights into domain architecture and substrate binding. J. Biol. Chem. 2003, 278, 26183–26193. [Google Scholar] [CrossRef] [PubMed]
- Heikkinen, O.; Seppala, R.; Tossavainen, H.; Heikkinen, S.; Koskela, H.; Permi, P.; Kilpeläinen, I. Solution structure of the parvulin-type PPIase domain of Staphylococcus aureus PrsA—Implications for the catalytic mechanism of parvulins. BMC Struct. Biol. 2009, 9, 17. [Google Scholar] [CrossRef] [PubMed]
- Daum, S.; Erdmann, F.; Fischer, G.; Féaux de Lacroix, B.; Hessamian-Alinejad, A.; Houben, S.; Frank, W.; Braun, M. Aryl indanyl ketones: Efficient inhibitors of the human peptidyl prolyl cis/trans isomerase Pin1. Angew. Chem. 2006, 45, 7454–7458. [Google Scholar] [CrossRef] [PubMed]
- Behrsin, C.D.; Bailey, M.L.; Bateman, K.S.; Hamilton, K.S.; Wahl, L.M.; Brandl, C.J.; Shilton, B.H.; Litchfield, D.W. Functionally important residues in the peptidyl-prolyl isomerase Pin1 revealed by unigenic evolution. J. Mol. Biol. 2007, 365, 1143–1162. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Wu, X.; Peng, Y.; Goh, J.Y.; Liou, Y.-C.; Lin, D.; Zhao, Y. Solution structural analysis of the single-domain parvulin TbPin1. PLoS ONE 2012, 7, e43017. [Google Scholar] [CrossRef] [PubMed]
- Weininger, U.; Jakob, R.P.; Kovermann, M.; Balbach, J.; Schmid, F.X. The prolyl isomerase domain of PpiD from Escherichia coli shows a parvulin fold but is devoid of catalytic activity. Protein Sci. 2010, 19, 6–18. [Google Scholar] [CrossRef] [PubMed]
- Behrens, S.; Maier, R.; de Cock, H.; Schmid, F.X.; Gross, C.A. The SurA periplasmic PPIase lacking its parvulin domains functions in vivo and has chaperone activity. EMBO J. 2001, 20, 285–294. [Google Scholar] [CrossRef] [PubMed]
- Durocher, D.; Taylor, I.A.; Sarbassova, D.; Haire, L.F.; Westcott, S.L.; Jackson, S.P.; Smerdon, S.J.; Yaffe, M.B. The Molecular Basis of FHA Domain:Phosphopeptide Binding Specificity and Implications for Phospho-Dependent Signaling Mechanisms. Mol. Cell 2000, 6, 1169–1182. [Google Scholar] [CrossRef]
- Xu, Q.; Deller, M.C.; Nielsen, T.K.; Grant, J.C.; Lesley, S.A.; Elsliger, M.-A.; Deacon, A.M.; Wilson, I.A. Structural insights into the recognition of phosphopeptide by the FHA domain of kanadaptin. PLoS ONE 2014, 9, e107309. [Google Scholar] [CrossRef] [PubMed]
- Pennell, S.; Westcott, S.; Ortiz-Lombardía, M.; Patel, D.; Li, J.; Nott, T.J.; Mohammed, D.; Buxton, R.S.; Yaffe, M.B.; Verma, C.; et al. Structural and functional analysis of phosphothreonine-dependent FHA domain interactions. Structure 2010, 18, 1587–1595. [Google Scholar] [CrossRef] [PubMed]
- Lange, O.F.; Rossi, P.; Sgourakis, N.G.; Song, Y.; Lee, H.-W.; Aramini, J.M.; Ertekin, A.; Xiao, R.; Acton, T.B.; Montelione, G.T.; et al. Determination of solution structures of proteins up to 40 kDa using CS-Rosetta with sparse NMR data from deuterated samples. Proc. Natl. Acad. Sci. USA 2012, 109, 10873–10878. [Google Scholar] [CrossRef] [PubMed]
- Morris, E.R.; Chevalier, D.; Walker, J.C. DAWDLE, a forkhead-associated domain gene, regulates multiple aspects of plant development. Plant Physiol. 2006, 141, 932–941. [Google Scholar] [CrossRef] [PubMed]
- Yu, B.; Bi, L.; Zheng, B.; Ji, L.; Chevalier, D.; Agarwal, M.; Ramachandran, V.; Li, W.; Lagrange, T.; Walker, J.C.; et al. The FHA domain proteins DAWDLE in Arabidopsis and SNIP1 in humans act in small RNA biogenesis. Proc. Natl. Acad. Sci. USA 2008, 105, 10073–10078. [Google Scholar] [CrossRef] [PubMed]
- Fujiyama-Nakamura, S.; Yoshikawa, H.; Homma, K.; Hayano, T.; Tsujimura-Takahashi, T.; Izumikawa, K.; Ishikawa, H.; Miyazawa, N.; Yanagida, M.; Miura, Y.; et al. Parvulin (Par14), a peptidyl-prolyl cis-trans isomerase, is a novel rRNA processing factor that evolved in the metazoan lineage. Mol. Cell. Proteom. 2009, 8, 1552–1565. [Google Scholar] [CrossRef] [PubMed]
- Kim, R.H.; Flanders, K.C.; Birkey Reffey, S.; Anderson, L.A.; Duckett, C.S.; Perkins, N.D.; Roberts, A.B. SNIP1 inhibits NF-kappa B signaling by competing for its binding to the C/H1 domain of CBP/p300 transcriptional co-activators. J. Biol. Chem. 2001, 276, 46297–46304. [Google Scholar] [CrossRef] [PubMed]
- Kim, R.H.; Wang, D.; Tsang, M.; Martin, J.; Huff, C.; de Caestecker, M.P.; Parks, W.T.; Meng, X.; Lechleider, R.J.; Wang, T.; et al. A novel Smad nuclear interacting protein, SNIP1, suppresses p300-dependent TGF-β signal transduction. Genes Dev. 2000, 14, 1605–1616. [Google Scholar] [PubMed]
- Zhou, X.Z.; Lu, K.P. The isomerase PIN1 controls numerous cancer-driving pathways and is a unique drug target. Nat. Rev. Cancer 2016, 16, 463–478. [Google Scholar] [CrossRef] [PubMed]
- Nakatsu, Y.; Matsunaga, Y.; Yamamotoya, T.; Ueda, K.; Inoue, Y.; Mori, K.; Sakoda, H.; Fujishiro, M.; Ono, H.; Kushiyama, A.; et al. Physiological and Pathogenic Roles of Prolyl Isomerase Pin1 in Metabolic Regulations via Multiple Signal Transduction Pathway Modulations. Int. J. Mol. Sci. 2016, 17. [Google Scholar] [CrossRef] [PubMed]
- Ryo, A.; Suizu, F.; Yoshida, Y.; Perrem, K.; Liou, Y.-C.; Wulf, G.; Rottapel, R.; Yamaoka, S.; Lu, K.P. Regulation of NF-kappaB signaling by Pin1-dependent prolyl isomerization and ubiquitin-mediated proteolysis of p65/RelA. Mol. Cell 2003, 12, 1413–1426. [Google Scholar] [CrossRef]
- Wijnands, S.P.W.; Engelen, W.; Lafleur, R.P.M.; Meijer, E.W.; Merkx, M. Controlling protein activity by dynamic recruitment on a supramolecular polymer platform. Nat. Commun. 2018, 9, 65. [Google Scholar] [CrossRef] [PubMed]
- Thiele, A.; Krentzlin, K.; Erdmann, F.; Rauh, D.; Hause, G.; Zerweck, J.; Kilka, S.; Pösel, S.; Fischer, G.; Schutkowski, M.; et al. Parvulin 17 promotes microtubule assembly by its peptidyl-prolyl cis/trans isomerase activity. J. Mol. Biol. 2011, 411, 896–909. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Nakatsu, Y.; Shinjo, T.; Guo, Y.; Sakoda, H.; Yamamotoya, T.; Otani, Y.; Okubo, H.; Kushiyama, A.; Fujishiro, M.; et al. Par14 protein associates with insulin receptor substrate 1 (IRS-1), thereby enhancing insulin-induced IRS-1 phosphorylation and metabolic actions. J. Biol. Chem. 2013, 288, 20692–20701. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.-L.; Lin, H.-P.; Zhou, W.-J.; He, C.-X.; Zhang, Z.-Y.; Cheng, Z.-L.; Song, J.-B.; Liu, P.; Chen, X.-Y.; Xia, Y.-K.; et al. SNIP1 Recruits TET2 to Regulate c-MYC Target Genes and Cellular DNA Damage Response. Cell Rep. 2018, 25, 1485–1500. [Google Scholar] [CrossRef] [PubMed]
- Blom, N.; Gammeltoft, S.; Brunak, S. Sequence and structure-based prediction of eukaryotic protein phosphorylation sites. J. Mol. Biol. 1999, 294, 1351–1362. [Google Scholar] [CrossRef] [PubMed]
- Dunker, A.K.; Obradovic, Z.; Romero, P.; Garner, E.C.; Brown, C.J. Intrinsic protein disorder in complete genomes. Genome Inform. 2000, 11, 161–171. [Google Scholar]
- Romero, P.; Obradovic, Z.; Li, X.; Garner, E.C.; Brown, C.J.; Dunker, A.K. Sequence complexity of disordered protein. Proteins 2001, 42, 38–48. [Google Scholar] [CrossRef]
- Dyson, H.J.; Wright, P.E. Intrinsically unstructured proteins and their functions. Nat. Rev. Mol. Cell Biol. 2005, 6, 197–208. [Google Scholar] [CrossRef] [PubMed]
- Tompa, P. Intrinsically disordered proteins: A 10-year recap. Trends Biochem. Sci. 2012, 37, 509–516. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. Disorder in the lifetime of a protein. Intrinsically Disord. Proteins 2013, 1, e26782. [Google Scholar] [CrossRef] [PubMed]
- Trusch, F.; Matena, A.; Vuk, M.; Koerver, L.; Knævelsrud, H.; Freemont, P.S.; Meyer, H.; Bayer, P. The N-terminal Region of the Ubiquitin Regulatory X (UBX) Domain-containing Protein 1 (UBXD1) Modulates Interdomain Communication within the Valosin-containing Protein p97. J. Biol. Chem. 2015, 290, 29414–29427. [Google Scholar] [CrossRef] [PubMed]
- Komiya, Y.; Habas, R. Wnt signal transduction pathways. Organogenesis 2008, 4, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Marinou, K.; Christodoulides, C.; Antoniades, C.; Koutsilieris, M. Wnt signaling in cardiovascular physiology. Trends Endocrinol. Metab. 2012, 23, 628–636. [Google Scholar] [CrossRef] [PubMed]
- Ryo, A.; Liou, Y.-C.; Lu, K.P.; Wulf, G. Prolyl isomerase Pin1: A catalyst for oncogenesis and a potential therapeutic target in cancer. J. Cell Sci. 2003, 116, 773–783. [Google Scholar] [CrossRef] [PubMed]
- Burgardt, N.I.; Schmidt, A.; Manns, A.; Schutkowski, A.; Jahreis, G.; Lin, Y.-J.; Schulze, B.; Masch, A.; Lücke, C.; Weiwad, M. Parvulin 17-catalyzed Tubulin Polymerization Is Regulated by Calmodulin in a Calcium-dependent Manner. J. Biol. Chem. 2015, 290, 16708–16722. [Google Scholar] [CrossRef] [PubMed]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rehic, E.; Hoenig, D.; Kamba, B.E.; Goehring, A.; Hofmann, E.; Gasper, R.; Matena, A.; Bayer, P. Structural Analysis of the 42 kDa Parvulin of Trypanosoma brucei. Biomolecules 2019, 9, 93. https://doi.org/10.3390/biom9030093
Rehic E, Hoenig D, Kamba BE, Goehring A, Hofmann E, Gasper R, Matena A, Bayer P. Structural Analysis of the 42 kDa Parvulin of Trypanosoma brucei. Biomolecules. 2019; 9(3):93. https://doi.org/10.3390/biom9030093
Chicago/Turabian StyleRehic, Edisa, Dana Hoenig, Bianca E. Kamba, Anna Goehring, Eckhard Hofmann, Raphael Gasper, Anja Matena, and Peter Bayer. 2019. "Structural Analysis of the 42 kDa Parvulin of Trypanosoma brucei" Biomolecules 9, no. 3: 93. https://doi.org/10.3390/biom9030093
APA StyleRehic, E., Hoenig, D., Kamba, B. E., Goehring, A., Hofmann, E., Gasper, R., Matena, A., & Bayer, P. (2019). Structural Analysis of the 42 kDa Parvulin of Trypanosoma brucei. Biomolecules, 9(3), 93. https://doi.org/10.3390/biom9030093