Structural Determinants of Isoform Selectivity in PI3K Inhibitors


Abstract
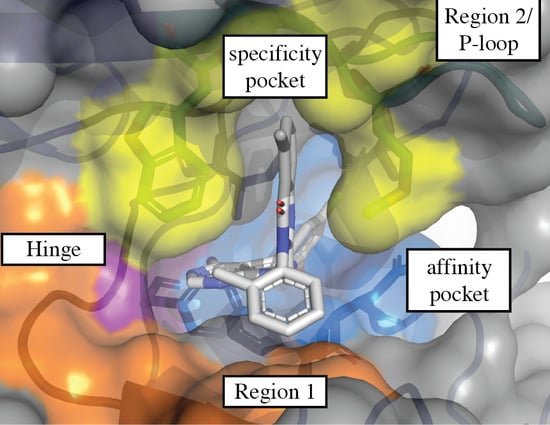
:
1. Introduction
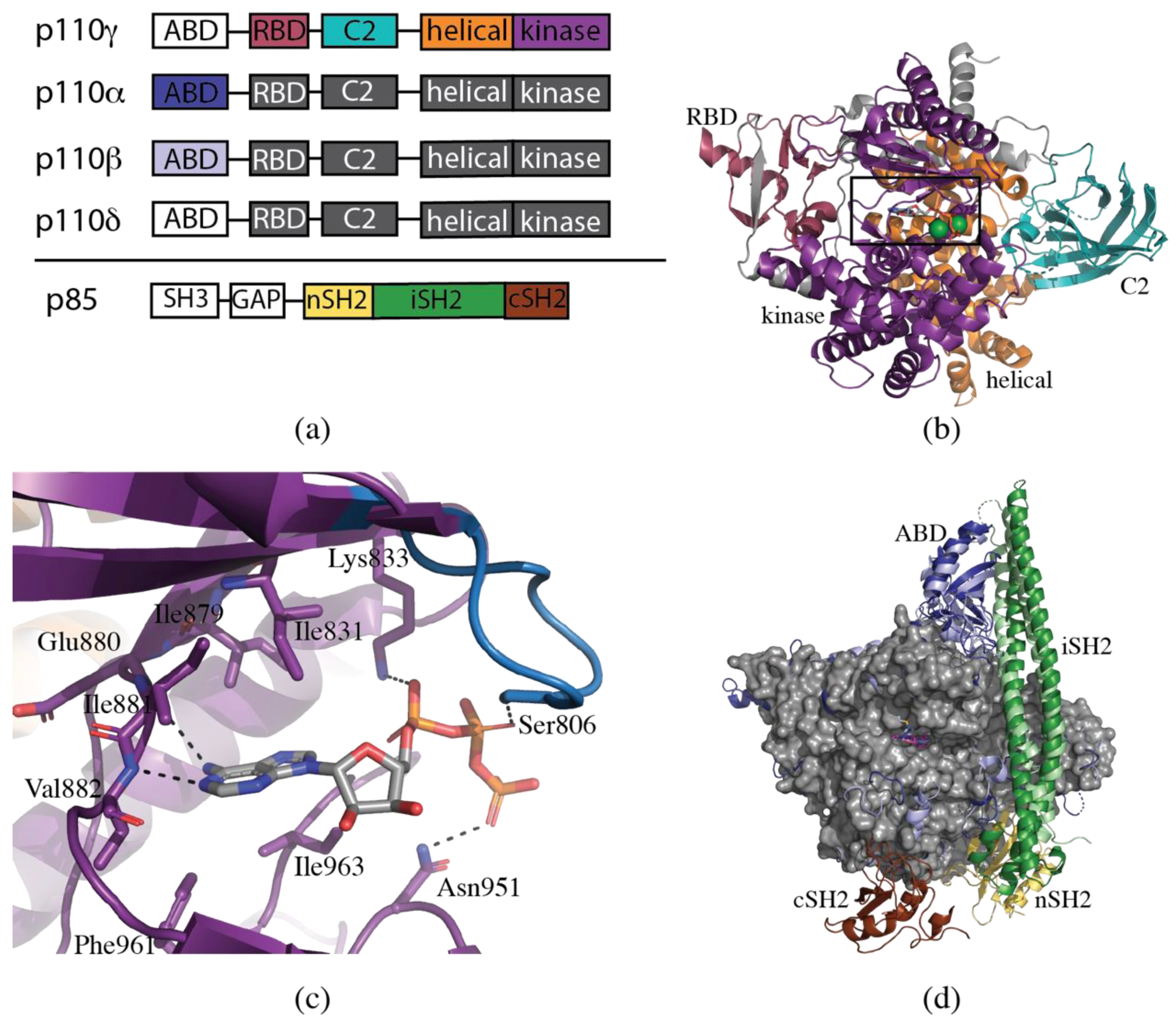
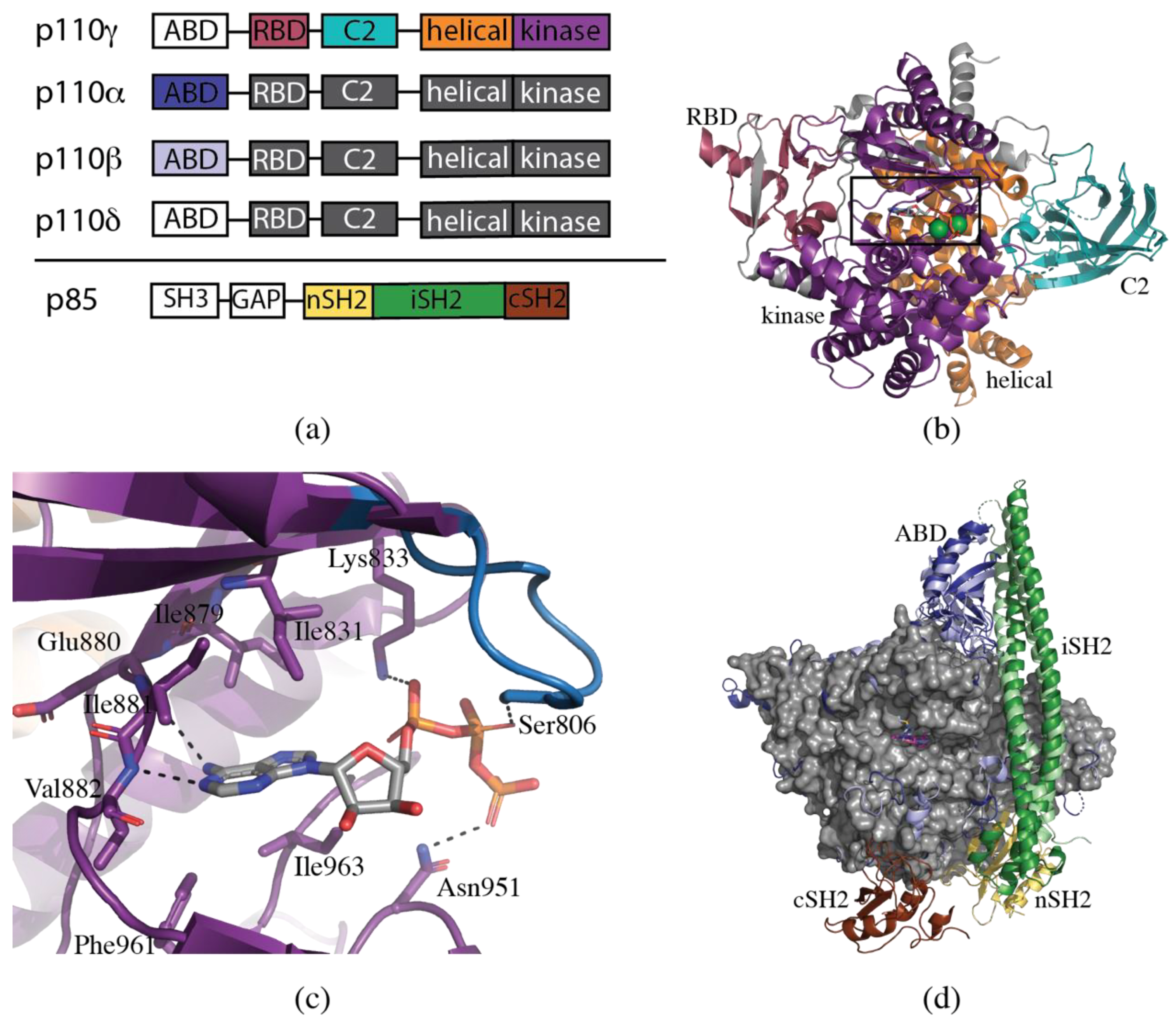
2. Structural Biology of PI3K
3. Structural Determinants of Isoform Selectivity
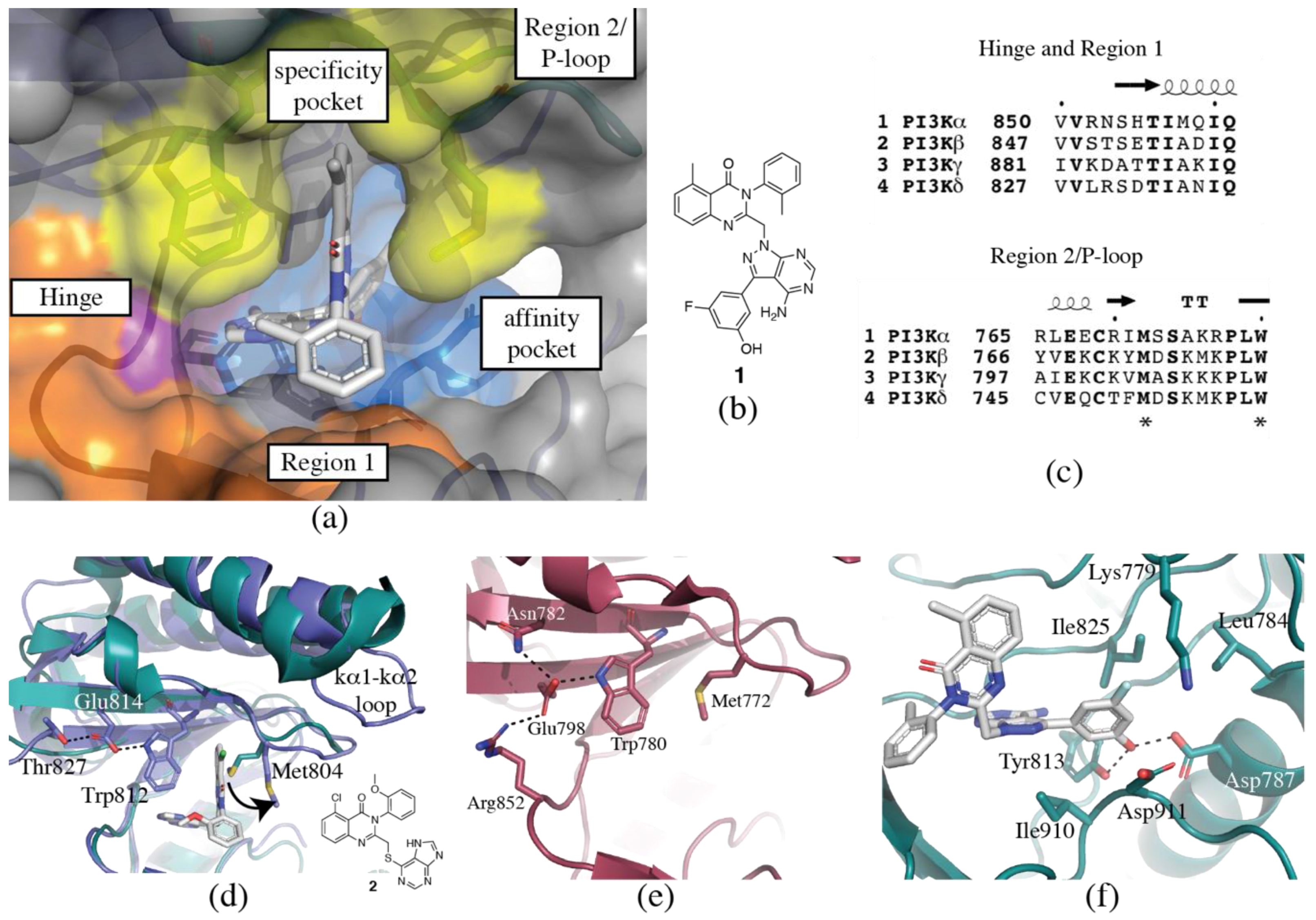
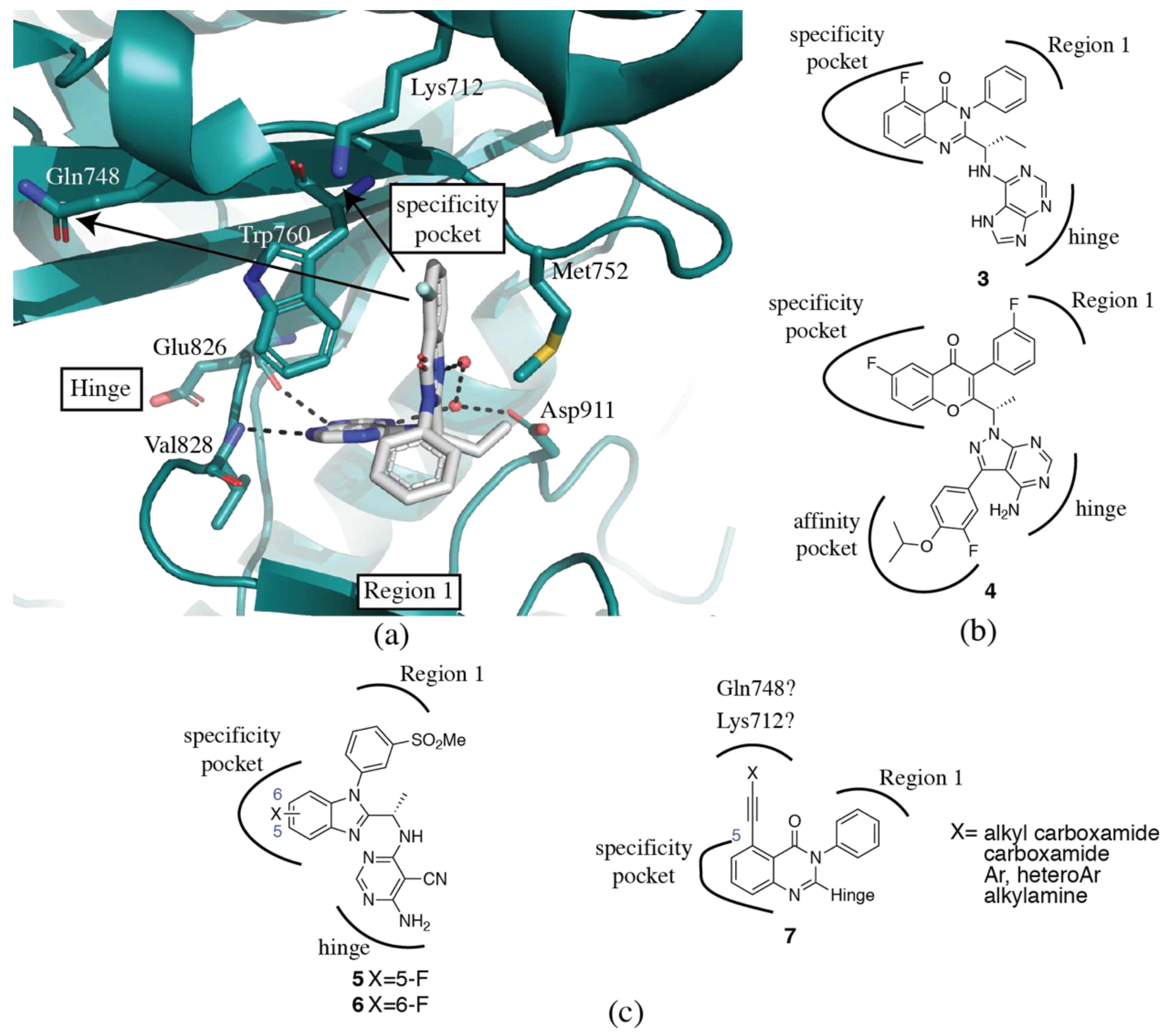
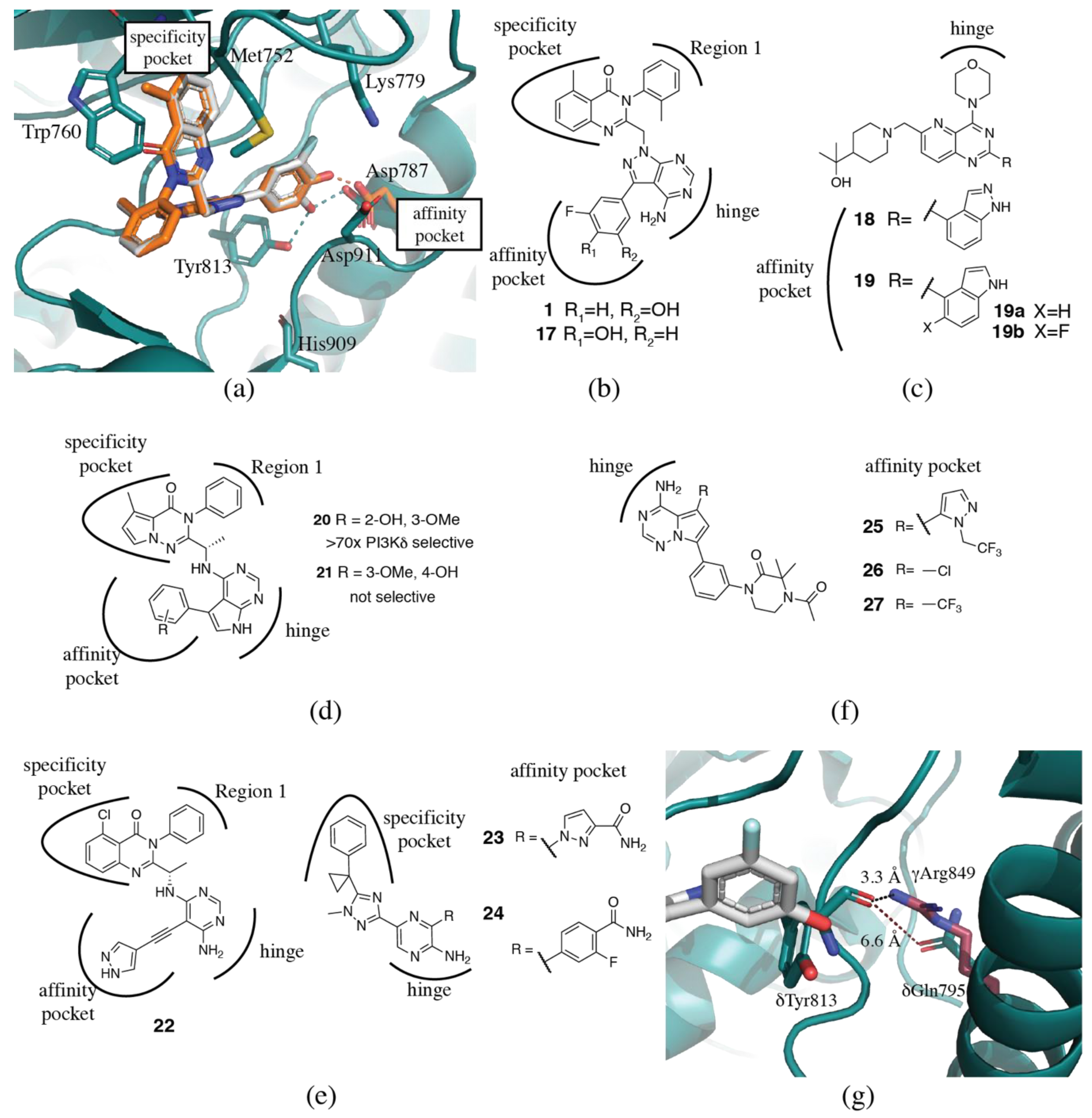
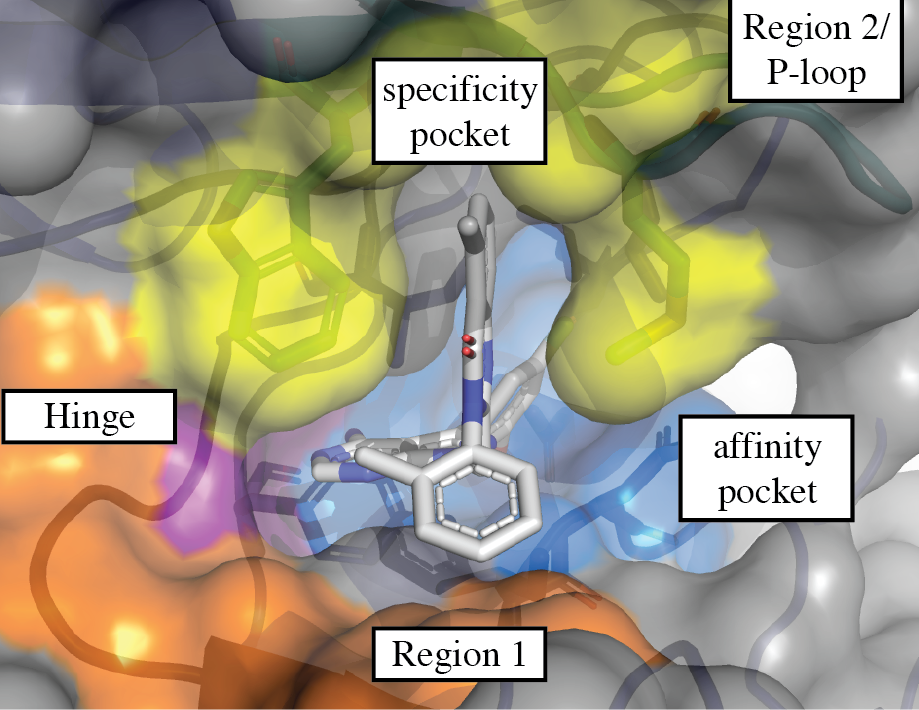
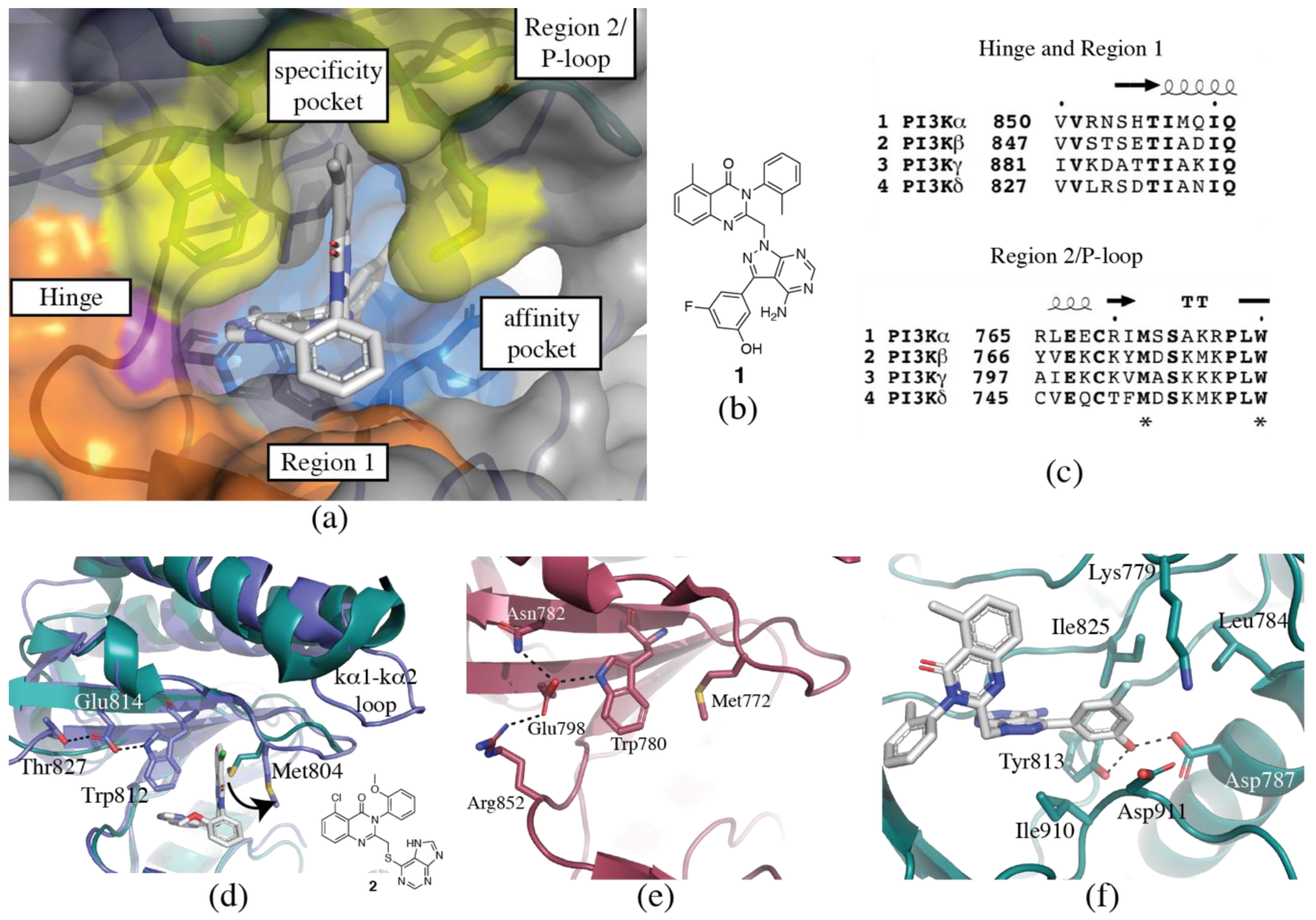
3.1. Regions of the Binding Site
3.1.1. Hinge-Region
3.1.2. Specificity Pocket
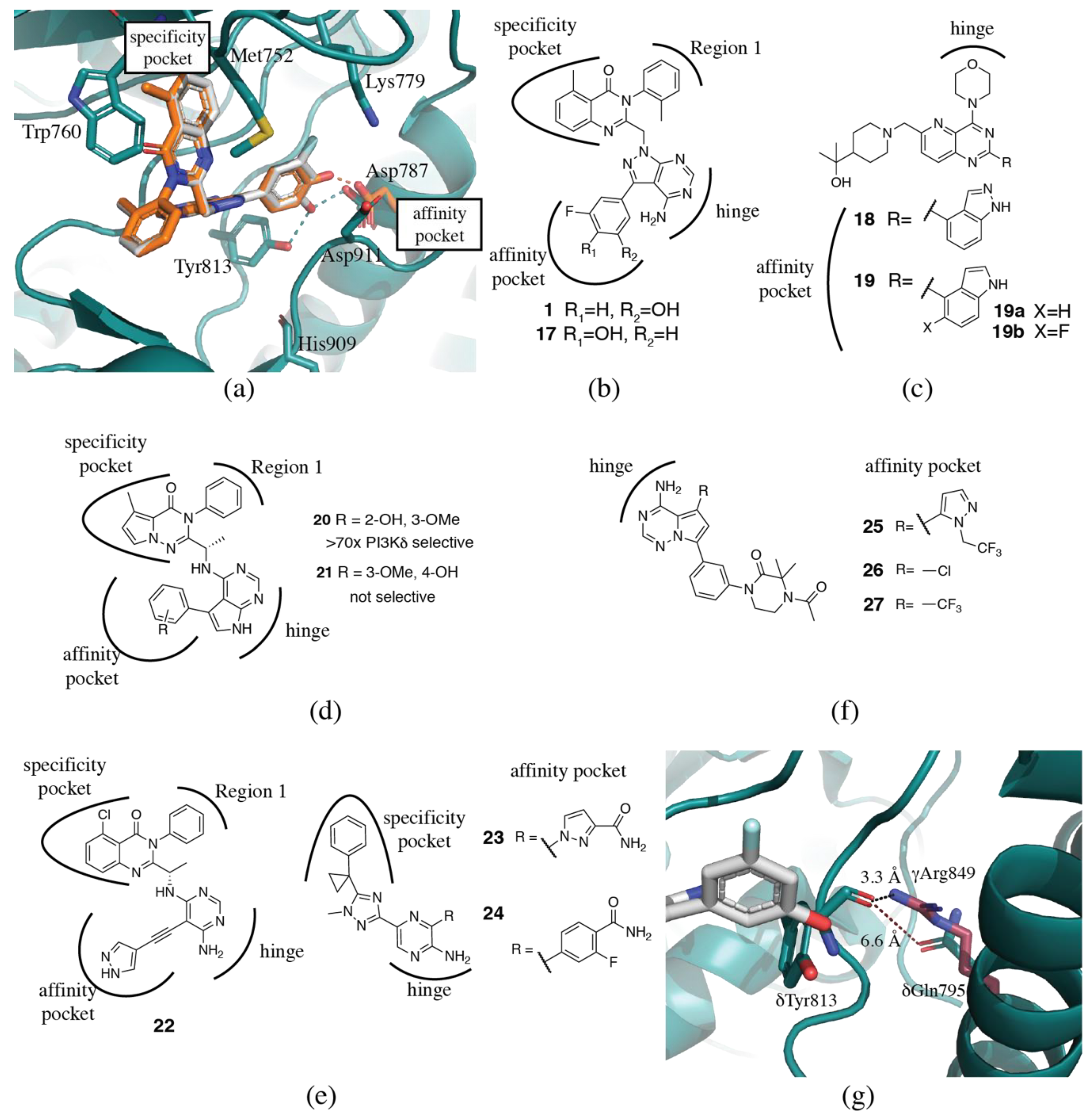
3.1.3. Affinity Pocket
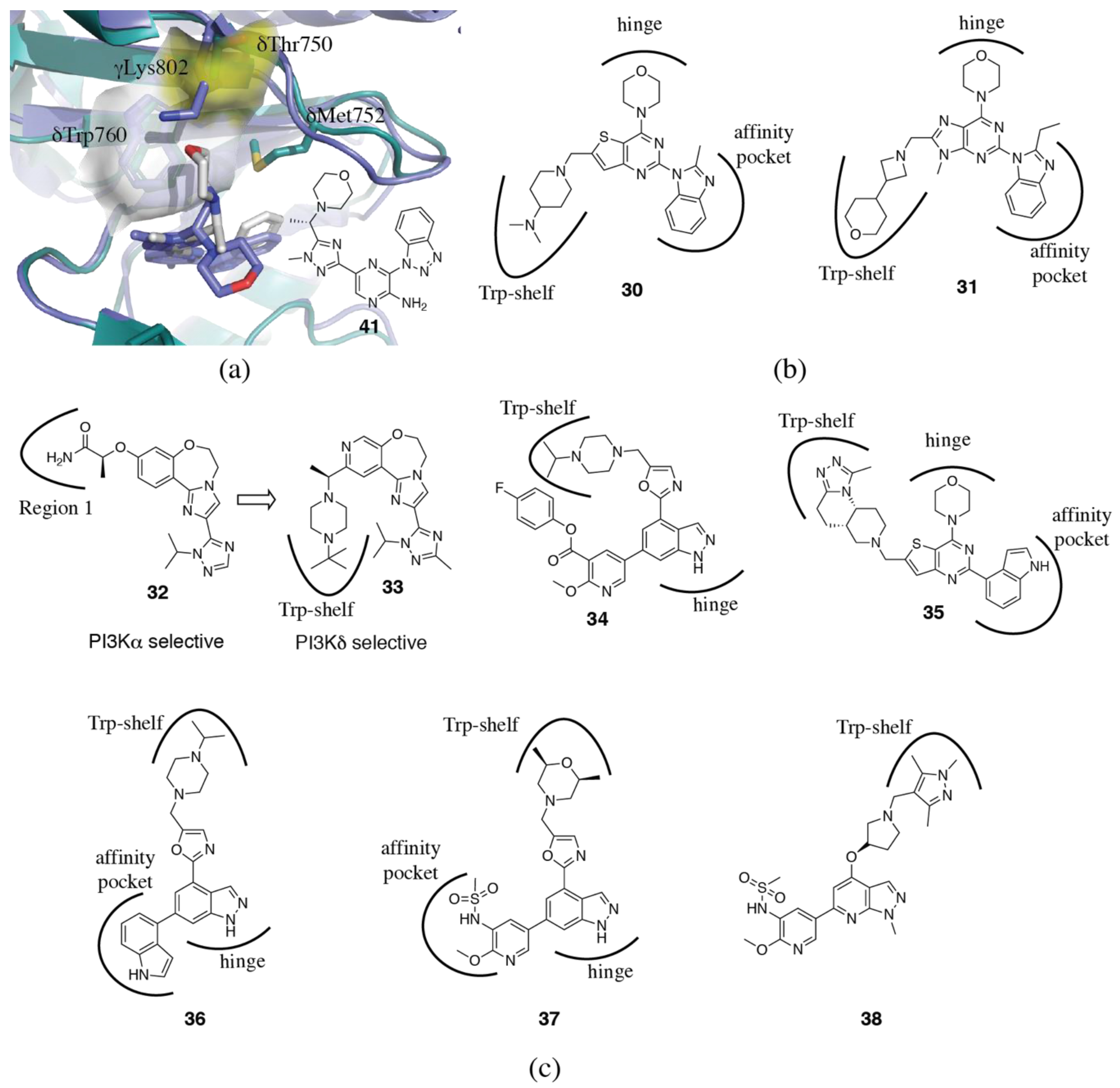
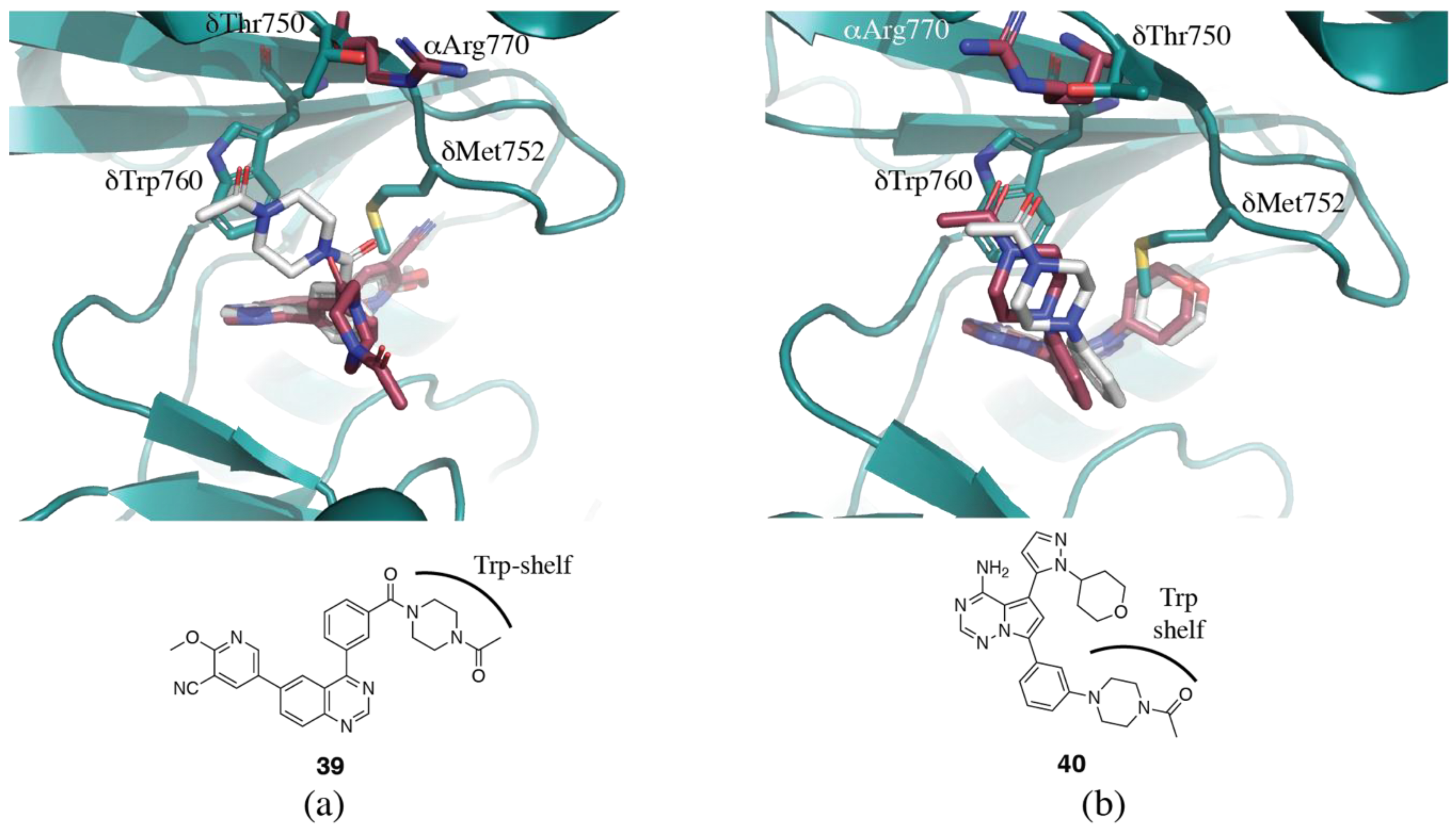
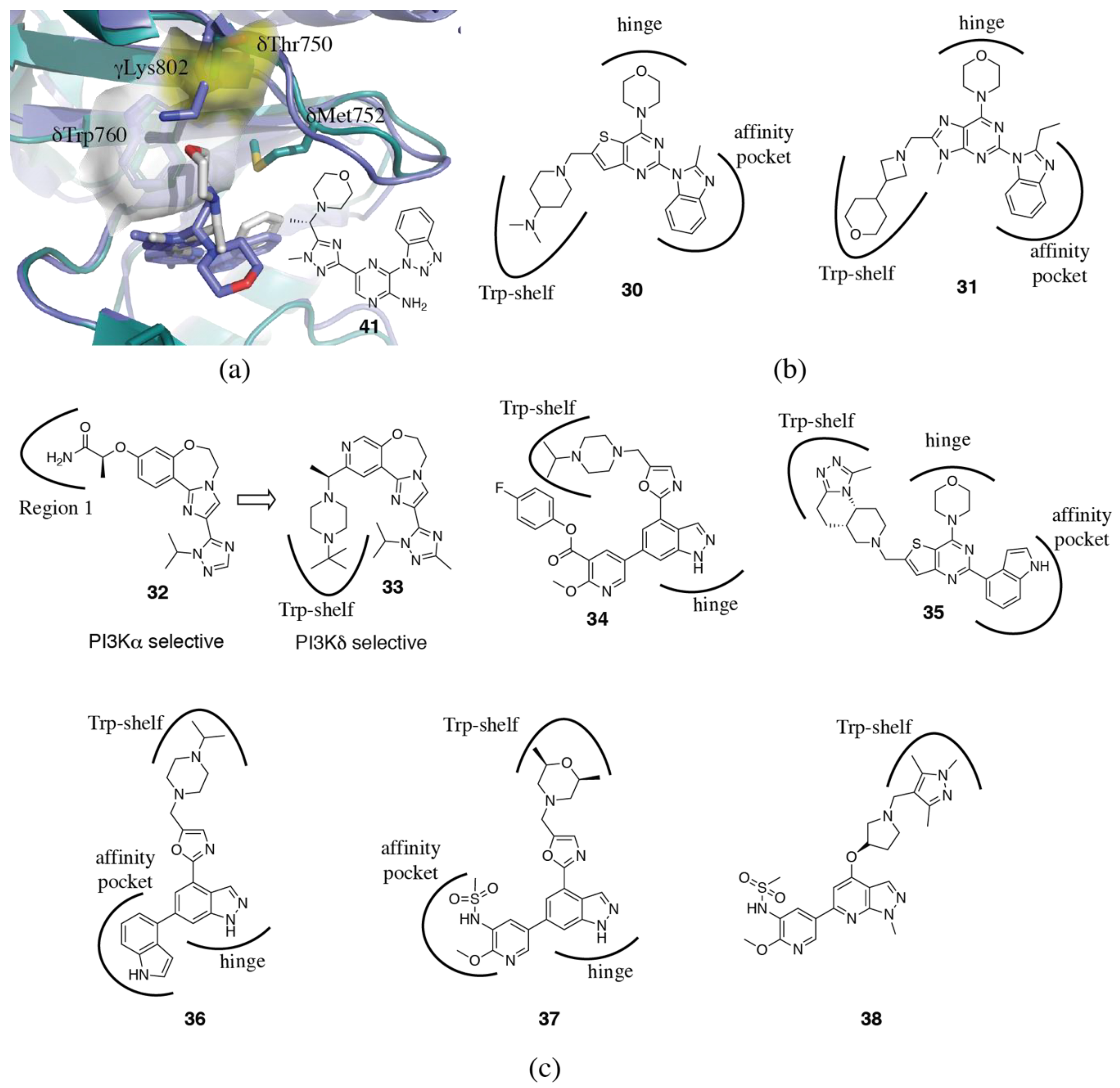
3.1.4. Non-Conserved Regions
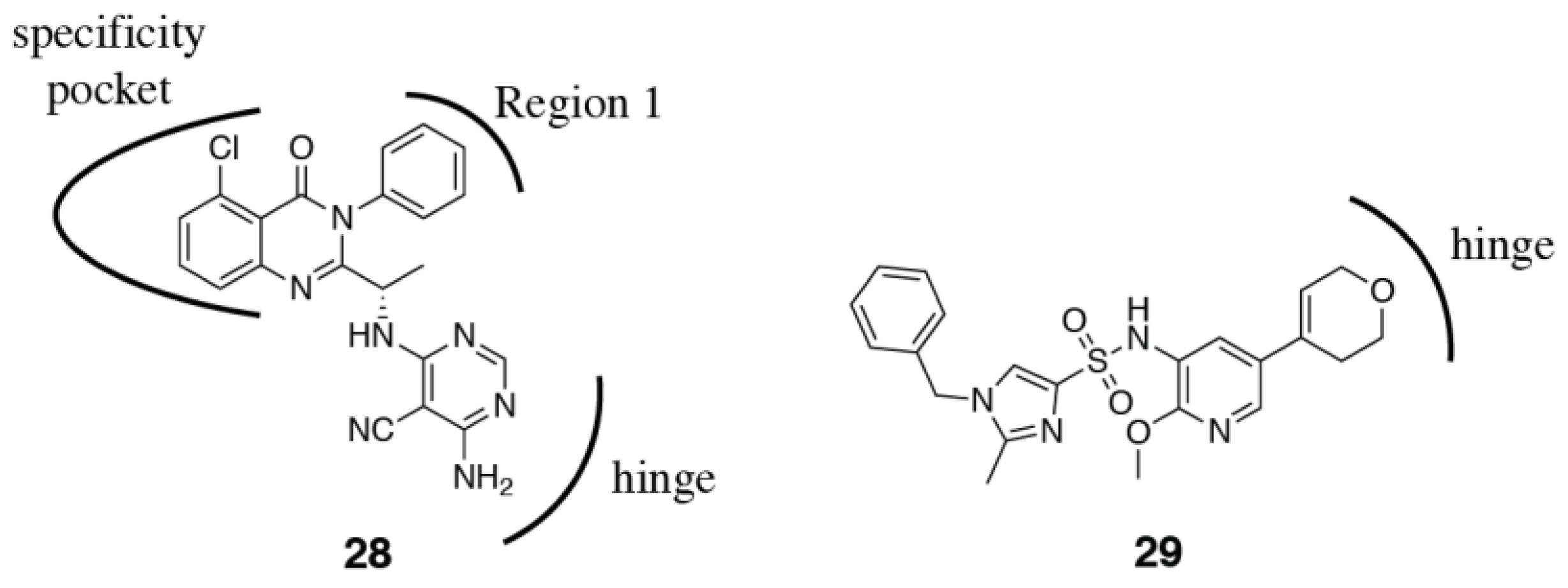
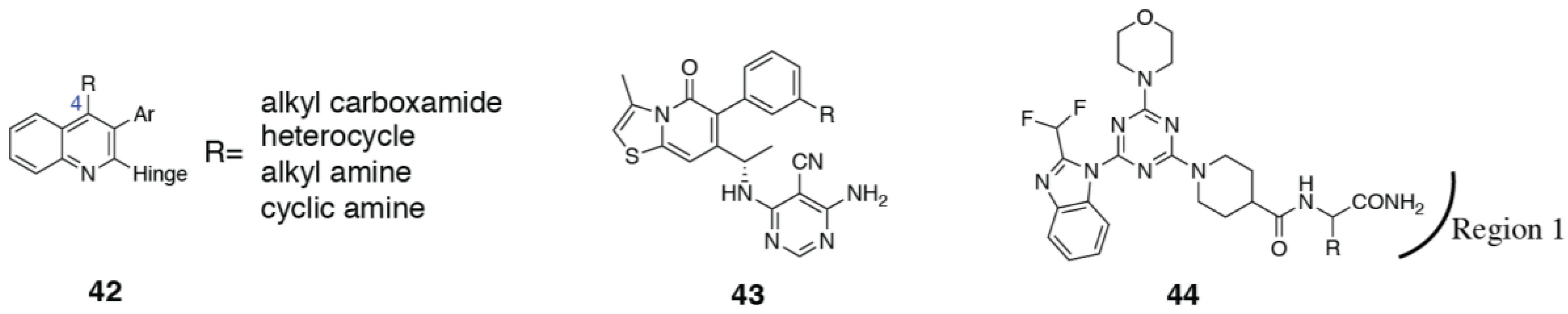
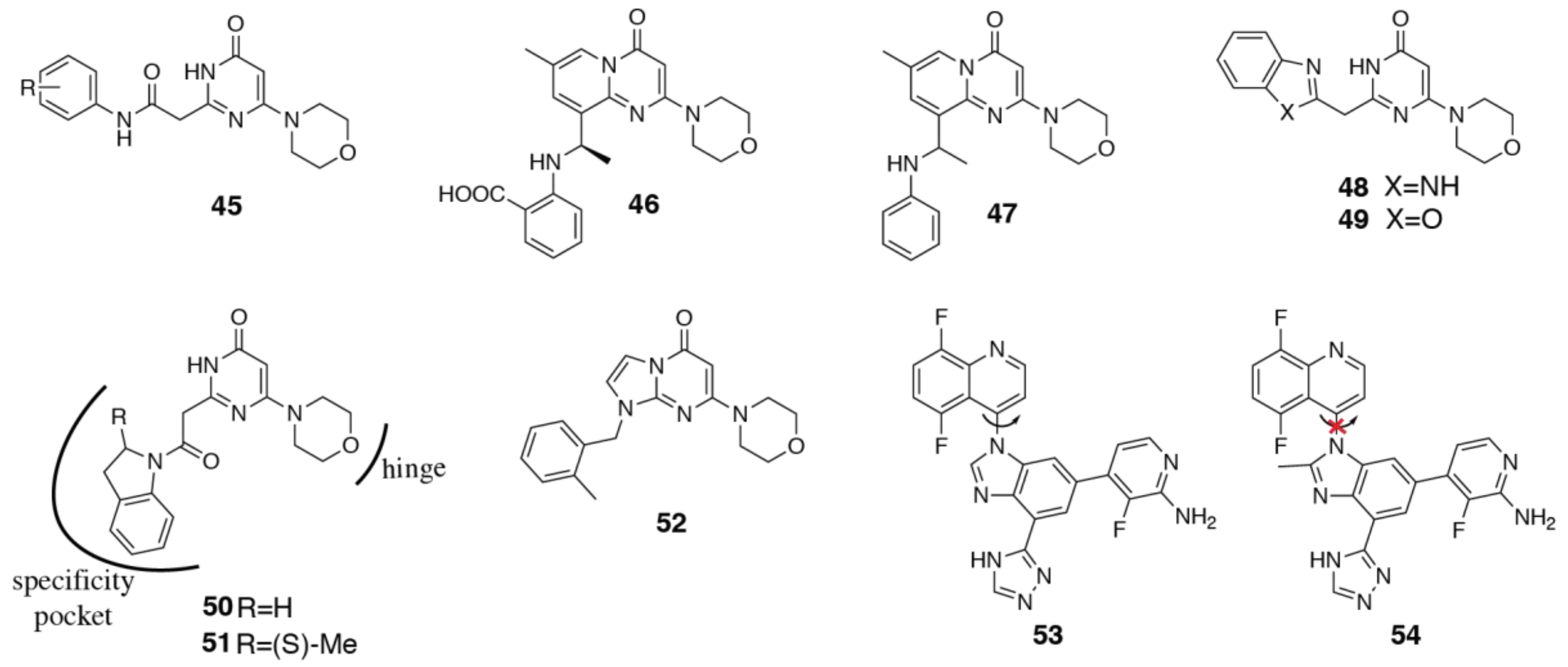
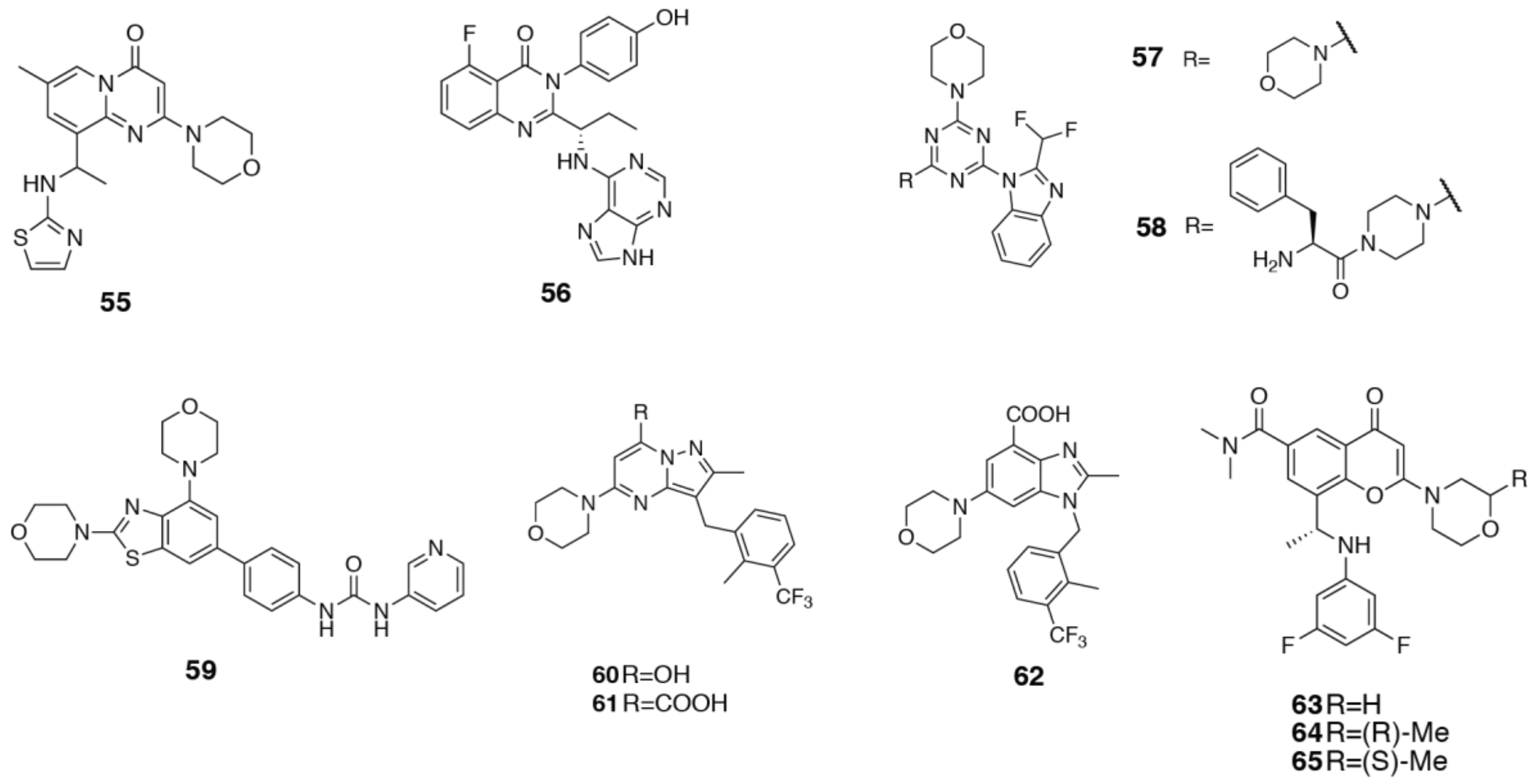
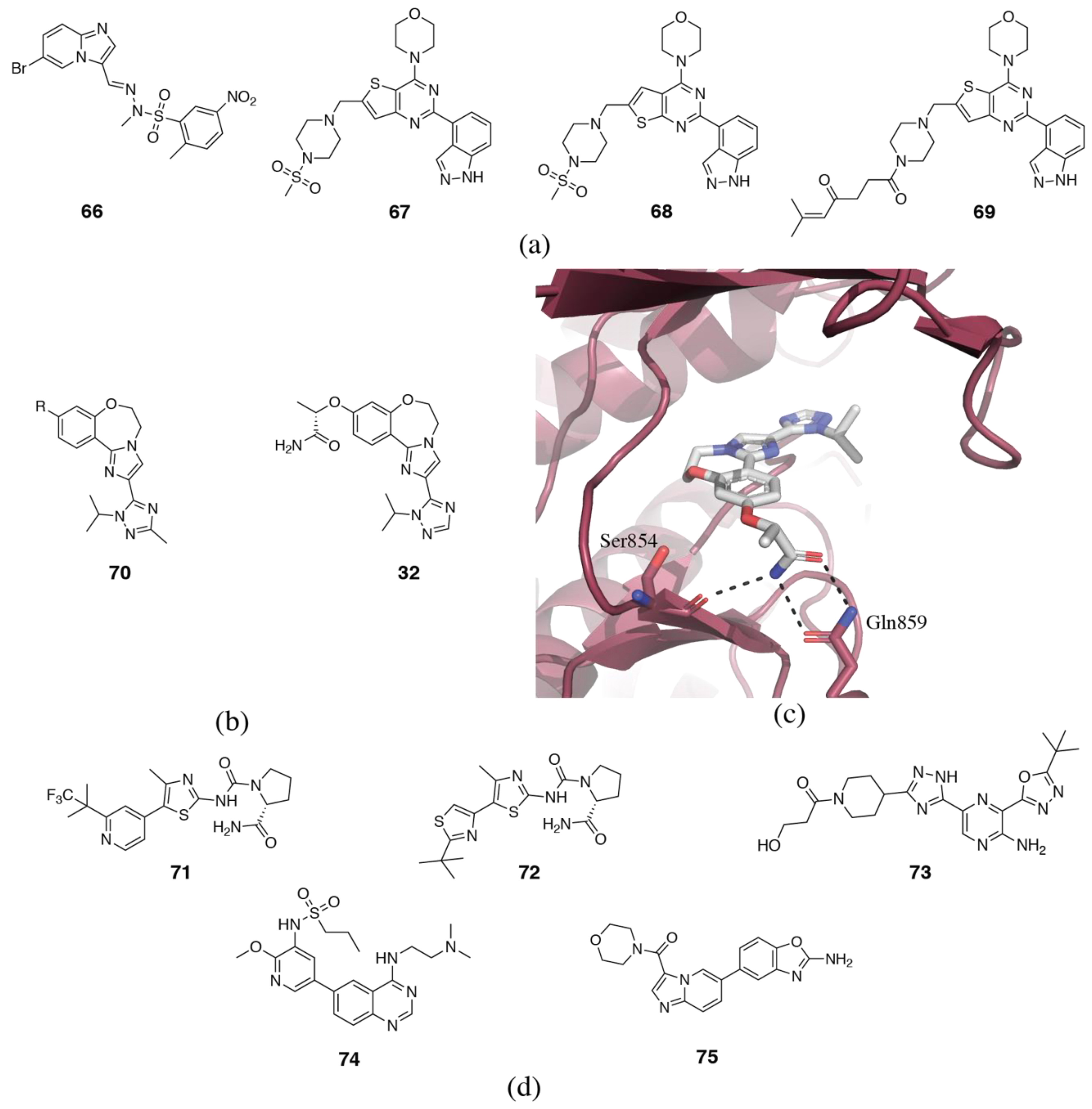
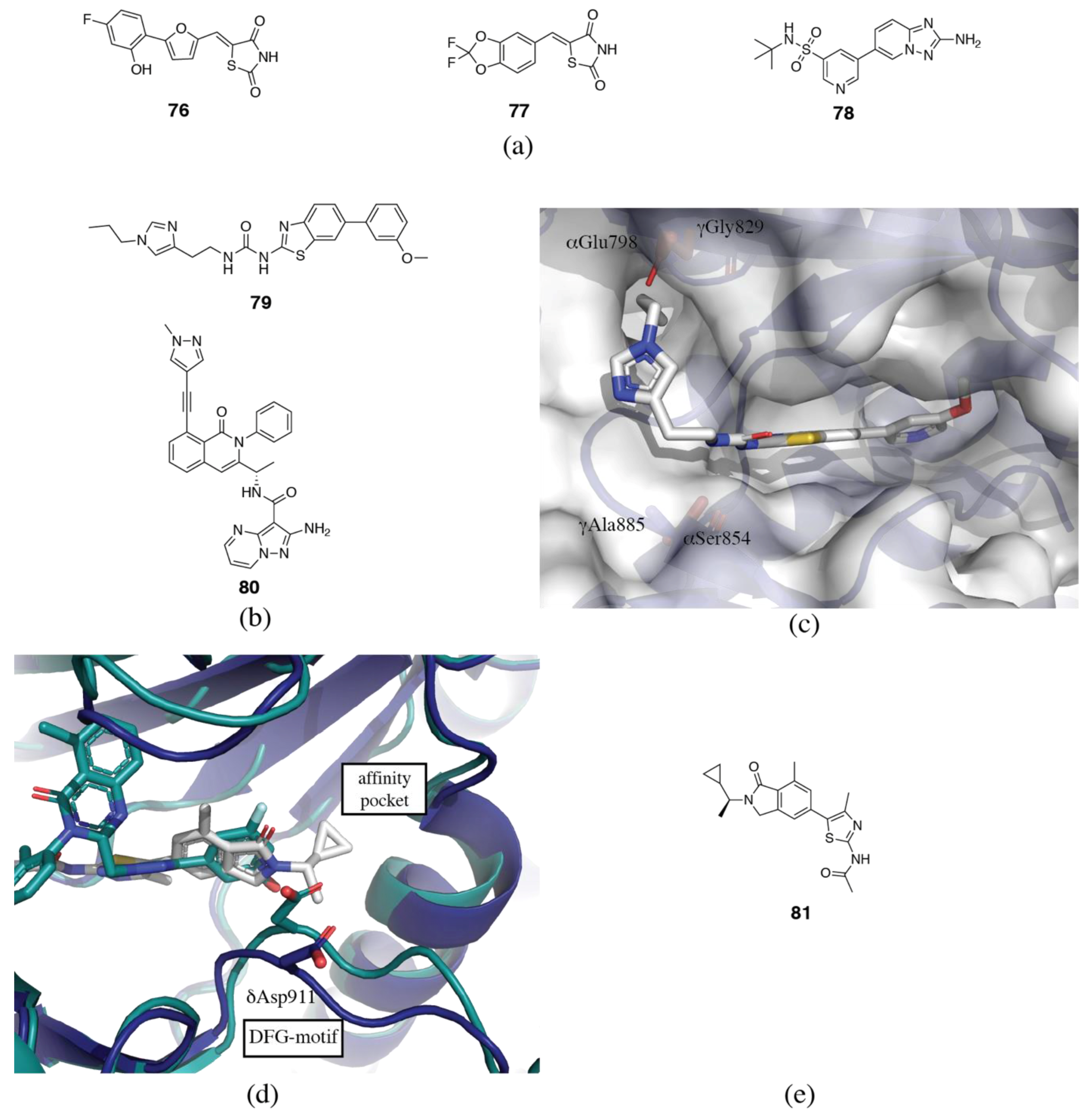
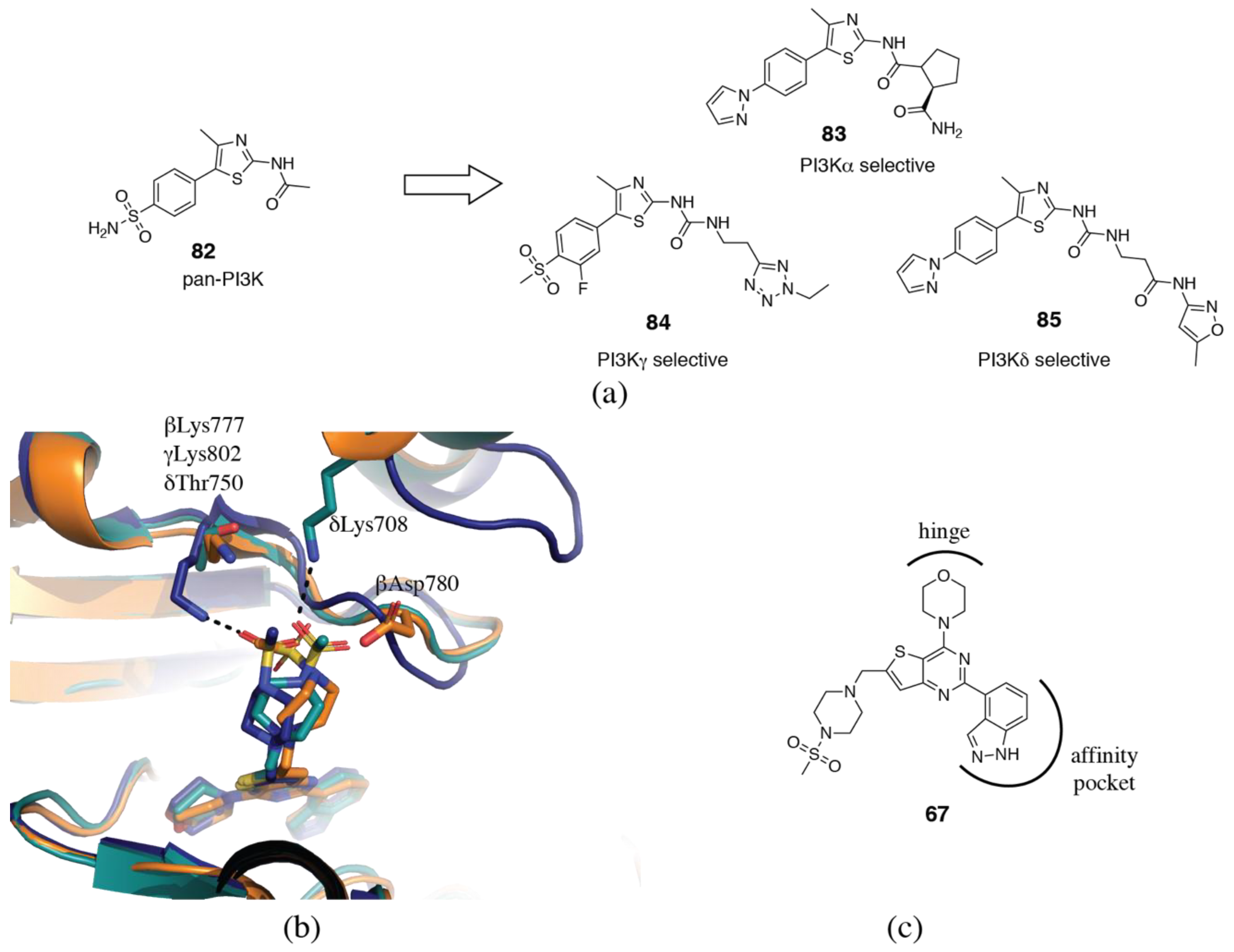
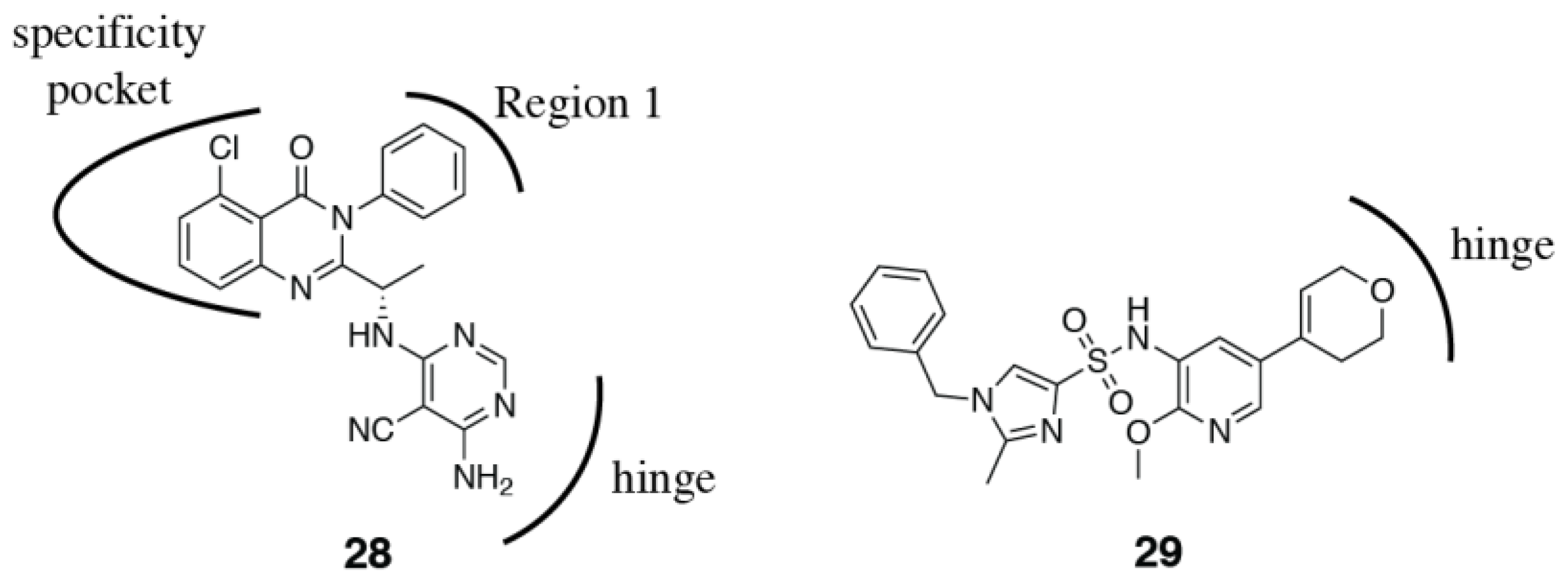
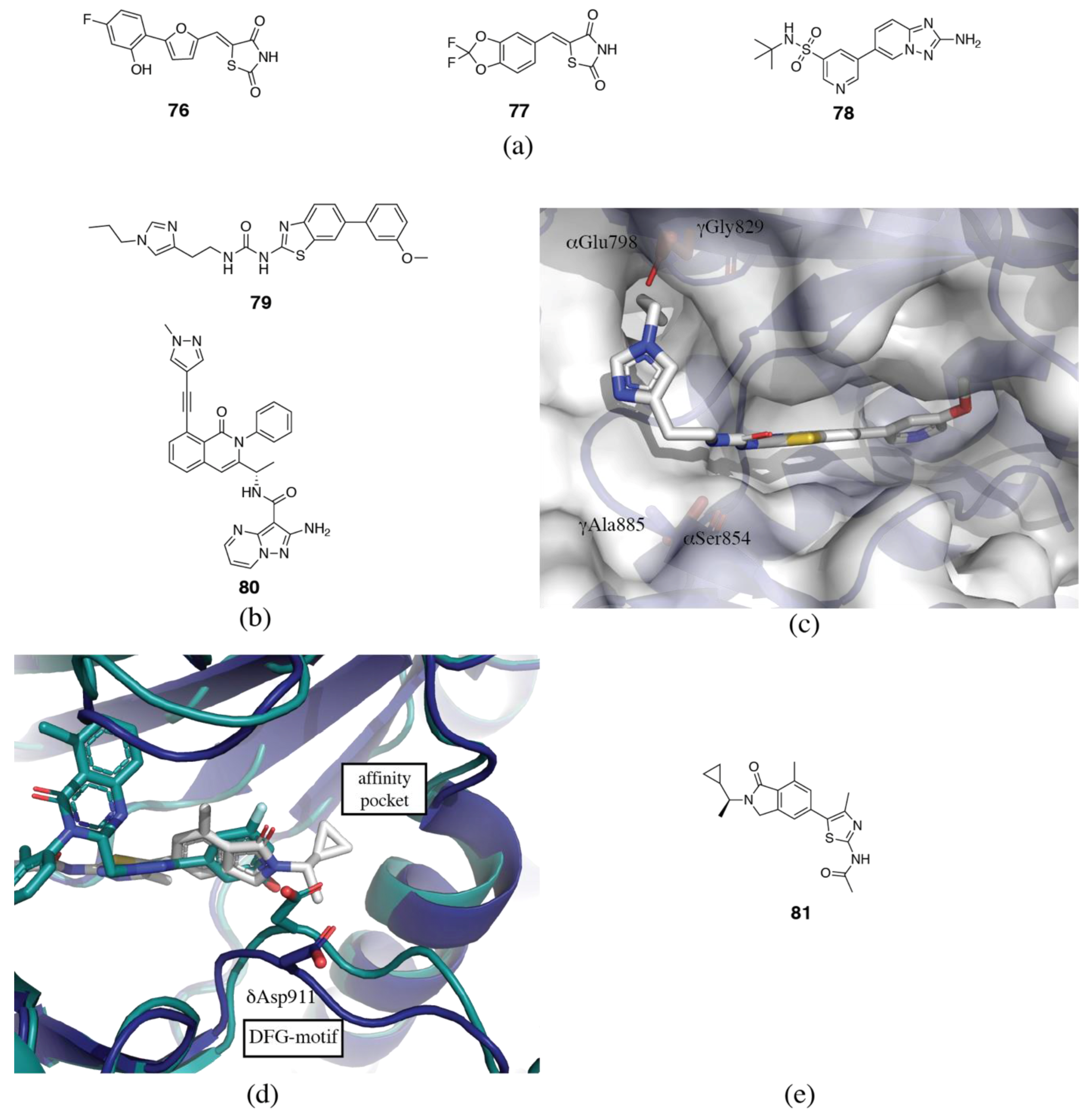
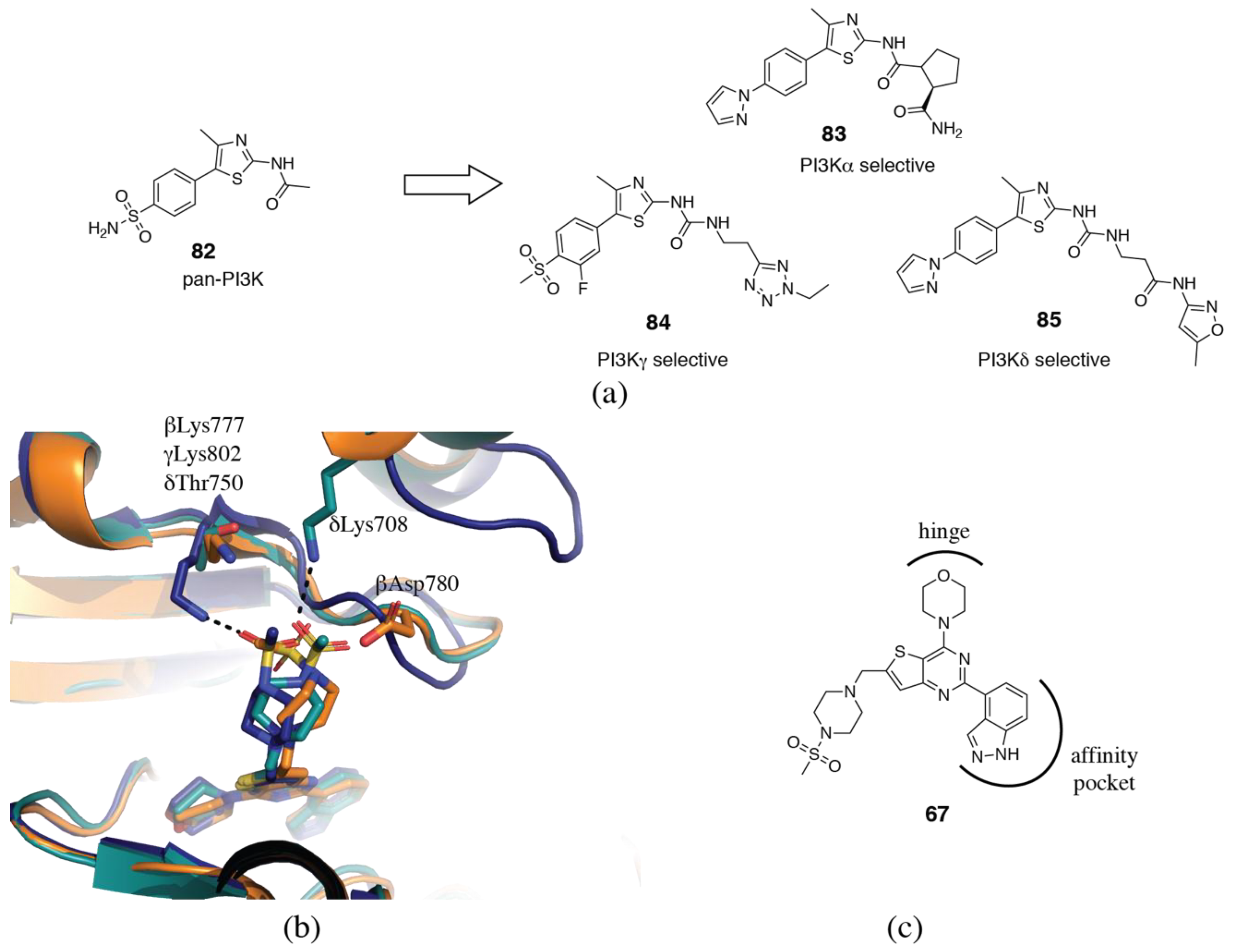
3.2. PI3Kδ Selective Inhibitors
3.3. PI3Kβ Selective Inhibitors
3.4. PI3Kα Selective Inhibitors
3.5. PI3Kγ Selective Inhibitors
3.6. Rational and Irrational Isoform Selectivity
3.7. PI3Kα Oncogenic Mutant Selectivity
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Ferguson, F.M.; Gray, N.S. Kinase inhibitors: The road ahead. Nat. Rev. Drug Discov. 2018, 17, 353–377. [Google Scholar] [CrossRef] [PubMed]
- Maheshwari, S.; Miller, M.S.; O’Meally, R.; Cole, R.N.; Amzel, L.M.; Gabelli, S.B. Kinetic and structural analyses reveal residues in phosphoinositide 3-kinase α that are critical for catalysis and substrate recognition. J. Biol. Chem. 2017, 292, 13541–13550. [Google Scholar] [CrossRef] [PubMed]
- Geering, B.; Cutillas, P.R.; Nock, G.; Gharbi, S.I.; Vanhaesebroeck, B. Class IA phosphoinositide 3-kinases are obligate p85-p110 heterodimers. Proc. Natl. Acad. Sci. USA 2007, 104, 7809–7814. [Google Scholar] [CrossRef] [PubMed]
- Mellor, P.; Furber, L.A.; Nyarko, J.N.K.; Anderson, D.H. Multiple roles for the p85α isoform in the regulation and function of PI3K signalling and receptor trafficking. Biochem. J. 2012, 441, 23–37. [Google Scholar] [CrossRef] [PubMed]
- Suire, S.; Coadwell, J.; Ferguson, G.J.; Davidson, K.; Hawkins, P.; Stephens, L. p84, a New Gβγ-Activated Regulatory Subunit of the Type IB Phosphoinositide 3-Kinase p110γ. Curr. Biol. 2005, 15, 566–570. [Google Scholar] [CrossRef] [PubMed]
- Stephens, L.; Eguinoa, A.; Erdjument-Bromage, H.; Lui, M.; Cooke, F.; Coadwell, J.; Smrcka, A.; Thelen, M.; Cadwallader, K.; Tempst, P.; et al. The Gβγ Sensitivity of a PI3K Is Dependent upon a Tightly Associated Adaptor, p101. Cell 1997, 89, 105–114. [Google Scholar] [CrossRef]
- Shymanets, A.; Vadas, O.; Czupalla, C.; LoPiccolo, J.; Brenowitz, M.; Ghigo, A.; Hirsch, E.; Krause, E.; Wetzker, R.; Williams, R.L.; et al. Different inhibition of Gβγ-stimulated class IB phosphoinositide 3-kinase (PI3K) variants by a monoclonal antibody. Specific function of p101 as a Gβγ-dependent regulator of PI3Kγ enzymatic activity. Biochem. J. 2015, 469, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Thorpe, L.M.; Yuzugullu, H.; Zhao, J.J. PI3K in cancer: Divergent roles of isoforms, modes of activation and therapeutic targeting. Nat. Rev. Cancer 2015, 15, 7–24. [Google Scholar] [CrossRef] [PubMed]
- Knight, Z.A.; Gonzalez, B.; Feldman, M.E.; Zunder, E.R.; Goldenberg, D.D.; Williams, O.; Loewith, R.; Stokoe, D.; Balla, A.; Toth, B.; et al. A Pharmacological Map of the PI3-K Family Defines a Role for p110α in Insulin Signaling. Cell 2006, 125, 733–747. [Google Scholar] [CrossRef] [PubMed]
- Sopasakis, V.R.; Liu, P.; Suzuki, R.; Kondo, T.; Winnay, J.; Tran, T.T.; Asano, T.; Smyth, G.; Sajan, M.P.; Farese, R.V.; et al. Specific roles of the p110alpha isoform of phosphatidylinsositol 3-kinase in hepatic insulin signaling and metabolic regulation. Cell Metab. 2010, 11, 220–230. [Google Scholar] [CrossRef] [PubMed]
- McMullen, J.R.; Shioi, T.; Zhang, L.; Tarnavski, O.; Sherwood, M.C.; Kang, P.M.; Izumo, S. Phosphoinositide 3-kinase(p110α) plays a critical role for the induction of physiological, but not pathological, cardiac hypertrophy. Proc. Natl. Acad. Sci. USA 2003, 100, 12355–12360. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; McMullen, J.R.; Sobkiw, C.L.; Zhang, L.; Dorfman, A.L.; Sherwood, M.C.; Logsdon, M.N.; Horner, J.W.; DePinho, R.A.; Izumo, S.; et al. Class IA Phosphoinositide 3-Kinase Regulates Heart Size and Physiological Cardiac Hypertrophy. Mol. Cell. Biol. 2005, 25, 9491–9502. [Google Scholar] [CrossRef] [PubMed]
- Weeks, K.L.; Bernardo, B.C.; Ooi, J.Y.Y.; Patterson, N.L.; McMullen, J.R. The IGF1-PI3K-Akt Signaling Pathway in Mediating Exercise-Induced Cardiac Hypertrophy and Protection. Adv. Exp. Med. Biol. 2017, 1000, 187–210. [Google Scholar] [PubMed]
- Aoyagi, T.; Matsui, T. Phosphoinositide-3 kinase signaling in cardiac hypertrophy and heart failure. Curr. Pharm. Des. 2011, 17, 1818–1824. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.P.; Schoenwaelder, S.M.; Goncalves, I.; Nesbitt, W.S.; Yap, C.L.; Wright, C.E.; Kenche, V.; Anderson, K.E.; Dopheide, S.M.; Yuan, Y.; et al. PI 3-kinase p110β: A new target for antithrombotic therapy. Nat. Med. 2005, 11, 507–514. [Google Scholar] [CrossRef] [PubMed]
- Nylander, S.; Kull, B.; Björkman, J.A.; Ulvinge, J.C.; Oakes, N.; Emanuelsson, B.M.; Andersson, M.; Skärby, T.; Inghardt, T.; Fjellström, O.; et al. Human target validation of phosphoinositide 3-kinase (PI3K)β: Effects on platelets and insulin sensitivity, using AZD6482 a novel PI3Kβ inhibitor. J. Thromb. Haemost. 2012, 10, 2127–2136. [Google Scholar] [CrossRef] [PubMed]
- Schoenwaelder, S.M.; Ono, A.; Nesbitt, W.S.; Lim, J.; Jarman, K.; Jackson, S.P. Phosphoinositide 3-kinase p110 beta regulates integrin alpha IIb beta 3 avidity and the cellular transmission of contractile forces. J. Biol. Chem. 2010, 285, 2886–2896. [Google Scholar] [CrossRef] [PubMed]
- Kaneda, M.M.; Messer, K.S.; Ralainirina, N.; Li, H.; Leem, C.J.; Gorjestani, S.; Woo, G.; Nguyen, A.V.; Figueiredo, C.C.; Foubert, P.; et al. PI3Kγ is a molecular switch that controls immune suppression. Nature 2016, 539, 437–442. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, P.T.; Stephens, L.R. PI3K signalling in inflammation. BBA Mol. Cell Biol. Lipids 2015, 1851, 882–897. [Google Scholar] [CrossRef] [PubMed]
- Cushing, T.D.; Metz, D.P.; Whittington, D.A.; McGee, L.R. PI3Kδ and PI3Kγ as Targets for Autoimmune and Inflammatory Diseases. J. Med. Chem. 2012, 55, 8559–8581. [Google Scholar] [CrossRef] [PubMed]
- Rowan, W.C.; Smith, J.L.; Affleck, K.; Amour, A. Targeting phosphoinositide 3-kinase δ for allergic asthma. Biochem. Soc. Trans. 2012, 40, 240–245. [Google Scholar] [CrossRef] [PubMed]
- Yoo, E.J.; Ojiaku, C.A.; Sunder, K.; Panettieri, R.A. Phosphoinositide 3-Kinase in Asthma: Novel Roles and Therapeutic Approaches. Am. J. Respir. Cell Mol. Biol. 2017, 56, 700–707. [Google Scholar] [CrossRef] [PubMed]
- Lucas, C.L.; Kuehn, H.S.; Zhao, F.; Niemela, J.E.; Deenick, E.K.; Palendira, U.; Avery, D.T.; Moens, L.; Cannons, J.L.; Biancalana, M.; et al. Dominant-activating germline mutations in the gene encoding the PI(3)K catalytic subunit p110δ result in T cell senescence and human immunodeficiency. Nat. Immunol. 2014, 15, 88–97. [Google Scholar] [CrossRef] [PubMed]
- Angulo, I.; Vadas, O.; Garçon, F.; Banham-Hall, E.; Plagnol, V.; Leahy, T.R.; Baxendale, H.; Coulter, T.; Curtis, J.; Wu, C.; et al. Phosphoinositide 3-kinase δ gene mutation predisposes to respiratory infection and airway damage. Science 2013, 342, 866–871. [Google Scholar] [CrossRef] [PubMed]
- Kracker, S.; Curtis, J.; Ibrahim, M.A.A.; Sediva, A.; Salisbury, J.; Campr, V.; Debré, M.; Edgar, J.D.M.; Imai, K.; Picard, C.; et al. Occurrence of B-cell lymphomas in patients with activated phosphoinositide 3-kinase δ syndrome. J. Allergy Clin. Immunol. 2014, 134, 233–236. [Google Scholar] [CrossRef] [PubMed]
- Coulter, T.I.; Chandra, A.; Bacon, C.M.; Babar, J.; Curtis, J.; Screaton, N.; Goodlad, J.R.; Farmer, G.; Steele, C.L.; Leahy, T.R.; et al. Clinical spectrum and features of activated phosphoinositide 3-kinase δ syndrome: A large patient cohort study. J. Allergy Clin. Immunol. 2017, 139, 597–606. [Google Scholar] [CrossRef] [PubMed]
- Elkaim, E.; Neven, B.; Bruneau, J.; Mitsui-Sekinaka, K.; Stanislas, A.; Heurtier, L.; Lucas, C.L.; Matthews, H.; Deau, M.-C.; Sharapova, S.; et al. Clinical and immunologic phenotype associated with activated phosphoinositide 3-kinase δ syndrome 2: A cohort study. J. Allergy Clin. Immunol. 2016, 138, 210–218. [Google Scholar] [CrossRef] [PubMed]
- Samuels, Y.; Wang, Z.; Bardelli, A.; Silliman, N.; Ptak, J.; Szabo, S.; Yan, H.; Gazdar, A.; Powell, S.M.; Riggins, G.J.; et al. High frequency of mutations of the PIK3CA gene in human cancers. Science 2004, 304, 554. [Google Scholar] [CrossRef] [PubMed]
- Samuels, Y.; Waldman, T. Oncogenic Mutations of PIK3CA in Human Cancers. Curr. Top. Microbiol. Immunol. 2011, 347, 21–41. [Google Scholar]
- Kang, S.; Seo, S.S.; Chang, H.J.; Yoo, C.W.; Park, S.Y.; Dong, S.M. Mutual exclusiveness between PIK3CA and KRAS mutations in endometrial carcinoma. Int. J. Gynecol. Cancer 2008, 18, 1339–1343. [Google Scholar] [CrossRef] [PubMed]
- Hayes, M.P.; Wang, H.; Espinal-Witter, R.; Douglas, W.; Solomon, G.J.; Baker, S.J.; Ellenson, L.H. PIK3CA and PTEN Mutations in Uterine Endometrioid Carcinoma and Complex Atypical Hyperplasia. Clin. Cancer Res. 2006, 12, 5932–5935. [Google Scholar] [CrossRef] [PubMed]
- Oda, K.; Stokoe, D.; Taketani, Y.; McCormick, F. High Frequency of Coexistent Mutations of PIK3CA and PTEN Genes in Endometrial Carcinoma. Cancer Res. 2005, 65, 10669–10673. [Google Scholar] [CrossRef] [PubMed]
- American Association for Cancer Research. Alpelisib Extends PFS in PIK3CA-Mutant Breast Cancer. Cancer Discov. 2019, 9, 6–7. [Google Scholar]
- Kang, S.; Denley, A.; Vanhaesebroeck, B.; Vogt, P.K. Oncogenic transformation induced by the p110beta, -gamma, and -delta isoforms of class I phosphoinositide 3-kinase. Proc. Natl. Acad. Sci. USA 2006, 103, 1289–1294. [Google Scholar] [CrossRef] [PubMed]
- Denley, A.; Kang, S.; Karst, U.; Vogt, P.K. Oncogenic signaling of class I PI3K isoforms. Oncogene 2008, 27, 2561–2574. [Google Scholar] [CrossRef] [PubMed]
- Berenjeno, I.M.; Guillermet-Guibert, J.; Pearce, W.; Gray, A.; Fleming, S.; Vanhaesebroeck, B. Both p110α and p110β isoforms of PI3K can modulate the impact of loss-of-function of the PTEN tumour suppressor. Biochem. J. 2012, 442, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Wee, S.; Wiederschain, D.; Maira, S.-M.; Loo, A.; Miller, C.; Stegmeier, F.; Yao, Y.-M.; Lengauer, C. PTEN-deficient cancers depend on PIK3CB. Proc. Natl. Acad. Sci. USA 2008, 105, 13057–13062. [Google Scholar] [CrossRef] [PubMed]
- Dbouk, H.A.; Pang, H.; Fiser, A.; Backer, J.M. A biochemical mechanism for the oncogenic potential of the p110β catalytic subunit of phosphoinositide 3-kinase. Proc. Natl. Acad. Sci. USA 2010, 107, 19897–19902. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, S.; Wongvipat, J.; Trigwell, C.B.; Hancox, U.; Carver, B.S.; Rodrik-Outmezguine, V.; Will, M.; Yellen, P.; de Stanchina, E.; Baselga, J.; et al. Feedback suppression of PI3Kα signaling in PTEN-mutated tumors is relieved by selective inhibition of PI3Kβ. Cancer Cell 2015, 27, 109–122. [Google Scholar] [CrossRef] [PubMed]
- Lynch, J.T.; Polanska, U.M.; Hancox, U.; Delpuech, O.; Maynard, J.; Trigwell, C.; Eberlein, C.; Lenaghan, C.; Polanski, R.; Avivar-Valderas, A.; et al. Combined Inhibition of PI3Kβ and mTOR Inhibits Growth of PTEN-null Tumors. Mol. Cancer Ther. 2018, 17, 2309–2319. [Google Scholar] [CrossRef] [PubMed]
- Mateo, J.; Ganji, G.; Lemech, C.; Burris, H.A.; Han, S.-W.; Swales, K.; Decordova, S.; DeYoung, M.P.; Smith, D.A.; Kalyana-Sundaram, S.; et al. A First-Time-in-Human Study of GSK2636771, a Phosphoinositide 3 Kinase Beta-Selective Inhibitor, in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2017, 19, 5981–5992. [Google Scholar] [CrossRef] [PubMed]
- Bédard, P.L.; Davies, M.A.; Kopetz, S.; Juric, D.; Shapiro, G.I.; Luke, J.J.; Spreafico, A.; Wu, B.; Castell, C.; Gomez, C.; et al. First-in-human trial of the PI3Kβ-selective inhibitor SAR260301 in patients with advanced solid tumors. Cancer 2018, 124, 315–324. [Google Scholar] [CrossRef] [PubMed]
- Lampson, B.L.; Brown, J.R. PI3Kδ-selective and PI3Kα/δ-combinatorial inhibitors in clinical development for B-cell non-Hodgkin lymphoma. Expert Opin. Investig. Drugs 2017, 26, 1267–1279. [Google Scholar] [CrossRef] [PubMed]
- Chiu, H.; Mallya, S.; Nguyen, P.; Mai, A.; Jackson, L.V.; Winkler, D.G.; DiNitto, J.P.; Brophy, E.E.; McGovern, K.; Kutok, J.L.; et al. The Selective Phosphoinoside-3-Kinase p110δ Inhibitor IPI-3063 Potently Suppresses B Cell Survival, Proliferation, and Differentiation. Front. Immunol. 2017, 8, 747. [Google Scholar] [CrossRef] [PubMed]
- Fruman, D.A.; Rommel, C. PI3Kδ Inhibitors in Cancer: Rationale and Serendipity Merge in the Clinic. Cancer Discov. 2011, 1, 562–572. [Google Scholar] [CrossRef] [PubMed]
- Miller, B.W.; Przepiorka, D.; Claro, R.A.; Lee, K.; Nie, L.; Simpson, N.; Gudi, R.; Saber, H.; Shord, S.; Bullock, J.; et al. FDA Approval: Idelalisib Monotherapy for the Treatment of Patients with Follicular Lymphoma and Small Lymphocytic Lymphoma. Clin. Cancer Res. 2015, 21, 1525–1529. [Google Scholar] [CrossRef] [PubMed]
- Furman, R.R.; Sharman, J.P.; Coutre, S.E.; Cheson, B.D.; Pagel, J.M.; Hillmen, P.; Barrientos, J.C.; Zelenetz, A.D.; Kipps, T.J.; Flinn, I.; et al. Idelalisib and rituximab in relapsed chronic lymphocytic leukemia. N. Engl. J. Med. 2014, 370, 997–1007. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Modi, P.; Newcomb, T.; Quéva, C.; Gandhi, V. Idelalisib: First-in-Class PI3K Delta Inhibitor for the Treatment of Chronic Lymphocytic Leukemia, Small Lymphocytic Leukemia, and Follicular Lymphoma. Clin. Cancer Res. 2015, 21, 1537–1542. [Google Scholar] [CrossRef] [PubMed]
- Perry, M.W.D.; Abdulai, R.; Mogemark, M.; Petersen, J.; Thomas, M.J.; Valastro, B.; Westin Eriksson, A. Evolution of PI3Kγ and δ Inhibitors for Inflammatory and Autoimmune Diseases. J. Med. Chem. 2018. [Google Scholar] [CrossRef] [PubMed]
- Cheah, C.Y.; Fowler, N.H. Idelalisib in the management of lymphoma. Blood 2016, 128, 331–336. [Google Scholar] [CrossRef] [PubMed]
- Blair, H.A. Duvelisib: First Global Approval. Drugs 2018, 78, 1847–1853. [Google Scholar] [CrossRef] [PubMed]
- Flinn, I.W.; Hillmen, P.; Montillo, M.; Nagy, Z.; Illés, Á.; Etienne, G.; Delgado, J.; Kuss, B.J.; Tam, C.S.; Gasztonyi, Z.; et al. The phase 3 DUO trial: Duvelisib versus ofatumumab in relapsed and refractory CLL/SLL. Blood 2018, 23, 2446–2455. [Google Scholar] [CrossRef] [PubMed]
- Markham, A. Copanlisib: First Global Approval. Drugs 2017, 77, 2057–2062. [Google Scholar] [CrossRef] [PubMed]
- Krause, G.; Hassenrück, F.; Hallek, M. Copanlisib for treatment of B-cell malignancies: The development of a PI3K inhibitor with considerable differences to idelalisib. Drug Des. Dev. Ther. 2018, 12, 2577–2590. [Google Scholar] [CrossRef] [PubMed]
- Mensah, F.A.; Blaize, J.-P.; Bryan, L.J. Spotlight on copanlisib and its potential in the treatment of relapsed/refractory follicular lymphoma: Evidence to date. Oncotargets Ther. 2018, 11, 4817–4827. [Google Scholar] [CrossRef] [PubMed]
- Scott, W.J.; Hentemann, M.F.; Rowley, R.B.; Bull, C.O.; Jenkins, S.; Bullion, A.M.; Johnson, J.; Redman, A.; Robbins, A.H.; Esler, W.; et al. Discovery and SAR of Novel 2,3-Dihydroimidazo[1,2-c]quinazoline PI3K Inhibitors: Identification of Copanlisib (BAY 80-6946). ChemMedChem 2016, 11, 1517–1530. [Google Scholar] [CrossRef] [PubMed]
- Evans, C.A.; Liu, T.; Lescarbeau, A.; Nair, S.J.; Grenier, L.; Pradeilles, J.A.; Glenadel, Q.; Tibbitts, T.; Rowley, A.M.; DiNitto, J.P.; et al. Discovery of a Selective Phosphoinositide-3-Kinase (PI3K)-γ Inhibitor (IPI-549) as an Immuno-Oncology Clinical Candidate. ACS Med. Chem. Lett. 2016, 7, 862–867. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, T.; Irie-Sasaki, J.; Jones, R.G.; Oliveira-dos-Santos, A.J.; Stanford, W.L.; Bolon, B.; Wakeham, A.; Itie, A.; Bouchard, D.; Kozieradzki, I.; et al. Function of PI3Kgamma in thymocyte development, T cell activation, and neutrophil migration. Science 2000, 287, 1040–1046. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, E.; Katanaev, V.L.; Garlanda, C.; Azzolino, O.; Pirola, L.; Silengo, L.; Sozzani, S.; Mantovani, A.; Altruda, F.; Wymann, M.P. Central role for G protein-coupled phosphoinositide 3-kinase gamma in inflammation. Science 2000, 287, 1049–1053. [Google Scholar] [CrossRef] [PubMed]
- Schmid, M.C.; Avraamides, C.J.; Dippold, H.C.; Franco, I.; Foubert, P.; Ellies, L.G.; Acevedo, L.M.; Manglicmot, J.R.E.; Song, X.; Wrasidlo, W.; et al. Receptor tyrosine kinases and TLR/IL1Rs unexpectedly activate myeloid cell PI3kγ, a single convergent point promoting tumor inflammation and progression. Cancer Cell 2011, 19, 715–727. [Google Scholar] [CrossRef] [PubMed]
- Joshi, S.; Singh, A.R.; Zulcic, M.; Durden, D.L. A macrophage-dominant PI3K isoform controls hypoxia-induced HIF1α and HIF2α stability and tumor growth, angiogenesis, and metastasis. Mol. Cancer Res. 2014, 12, 1520–1531. [Google Scholar] [CrossRef] [PubMed]
- Gunderson, A.J.; Kaneda, M.M.; Tsujikawa, T.; Nguyen, A.V.; Affara, N.I.; Ruffell, B.; Gorjestani, S.; Liudahl, S.M.; Truitt, M.; Olson, P.; et al. Bruton Tyrosine Kinase-Dependent Immune Cell Cross-talk Drives Pancreas Cancer. Cancer Discov. 2016, 6, 270–285. [Google Scholar] [CrossRef] [PubMed]
- Rivera, L.B.; Meyronet, D.; Hervieu, V.; Frederick, M.J.; Bergsland, E.; Bergers, G. Intratumoral myeloid cells regulate responsiveness and resistance to antiangiogenic therapy. Cell Rep. 2015, 11, 577–591. [Google Scholar] [CrossRef] [PubMed]
- Garces, A.E.; Stocks, M.J. Class 1 PI3K Clinical Candidates and Recent Inhibitor Design Strategies: A Medicinal Chemistry Perspective. J. Med. Chem. 2018. [Google Scholar] [CrossRef] [PubMed]
- Furet, P.; Guagnano, V.; Fairhurst, R.A.; Imbach-Weese, P.; Bruce, I.; Knapp, M.; Fritsch, C.; Blasco, F.; Blanz, J.; Aichholz, R.; et al. Discovery of NVP-BYL719 a potent and selective phosphatidylinositol-3 kinase alpha inhibitor selected for clinical evaluation. Bioorg. Med. Chem. Lett. 2013, 23, 3741–3748. [Google Scholar] [CrossRef] [PubMed]
- So, L.; Yea, S.S.; Oak, J.S.; Lu, M.; Manmadhan, A.; Ke, Q.H.; Janes, M.R.; Kessler, L.V.; Kucharski, J.M.; Li, L.-S.; et al. Selective Inhibition of Phosphoinositide 3-Kinase p110α Preserves Lymphocyte Function. J. Biol. Chem. 2013, 288, 5718–5731. [Google Scholar] [CrossRef] [PubMed]
- Heffron, T.P.; Heald, R.A.; Ndubaku, C.; Wei, B.; Augistin, M.; Do, S.; Edgar, K.; Eigenbrot, C.; Friedman, L.; Gancia, E.; et al. The Rational Design of Selective Benzoxazepin Inhibitors of the α-Isoform of Phosphoinositide 3-Kinase Culminating in the Identification of (S)-2-((2-(1-Isopropyl-1H-1,2,4-triazol-5-yl)-5,6-dihydrobenzo[f]imidazo[1,2-d][1,4]oxazepin-9-yl)oxy)propanamide (GDC-0326). J. Med. Chem. 2016, 59, 985–1002. [Google Scholar] [PubMed]
- Liu, X.; Xu, Y.; Wang, Y.; Chen, Y.; Wang, B.; Wang, Y.; Chen, Y.; Tan, C.; Hu, L.; Ma, Q.; et al. Decrease in phosphorylated ERK indicates the therapeutic efficacy of a clinical PI3Kα-selective inhibitor CYH33 in breast cancer. Cancer Lett. 2018, 433, 273–282. [Google Scholar] [CrossRef] [PubMed]
- Barlaam, B.; Cosulich, S.; Degorce, S.; Fitzek, M.; Green, S.; Hancox, U.; Lambert-van der Brempt, C.; Lohmann, J.-J.; Maudet, M.; Morgentin, R.; et al. Discovery of (R)-8-(1-(3,5-Difluorophenylamino)ethyl)-N,N-dimethyl-2-morpholino-4-oxo-4H-chromene-6-carboxamide (AZD8186): A Potent and Selective Inhibitor of PI3Kβ and PI3Kδ for the Treatment of PTEN-Deficient Cancers. J. Med. Chem. 2015, 58, 943–962. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Duan, S.; Dong, L.; Wang, Y.; Hu, Q.; Liu, C.; Forrest, M.L.; Holzbeierlein, J.M.; Han, S.; Li, B. Characterization of a novel p110β-specific inhibitor BL140 that overcomes MDV3100-resistance in castration-resistant prostate cancer cells. Prostate 2017, 77, 1187–1198. [Google Scholar] [CrossRef] [PubMed]
- Certal, V.; Carry, J.-C.; Halley, F.; Virone-Oddos, A.; Thompson, F.; Filoche-Rommé, B.; El-Ahmad, Y.; Karlsson, A.; Charrier, V.; Delorme, C.; et al. Discovery and Optimization of Pyrimidone Indoline Amide PI3Kβ Inhibitors for the Treatment of Phosphatase and Tensin Homologue (PTEN)-Deficient Cancers. J. Med. Chem. 2014, 57, 903–920. [Google Scholar] [CrossRef] [PubMed]
- Cushing, T.D.; Hao, X.; Shin, Y.; Andrews, K.; Brown, M.; Cardozo, M.; Chen, Y.; Duquette, J.; Fisher, B.; Gonzalez-Lopez de Turiso, F.; et al. Discovery and in Vivo Evaluation of (S)-N-(1-(7-Fluoro-2-(pyridin-2-yl)quinolin-3-yl)ethyl)-9H-purin-6-amine (AMG319) and Related PI3Kδ Inhibitors for Inflammation and Autoimmune Disease. J. Med. Chem. 2015, 58, 480–511. [Google Scholar] [CrossRef] [PubMed]
- Down, K.; Amour, A.; Baldwin, I.R.; Cooper, A.W.J.; Deakin, A.M.; Felton, L.M.; Guntrip, S.B.; Hardy, C.; Harrison, Z.A.; Jones, K.L.; et al. Optimization of Novel Indazoles as Highly Potent and Selective Inhibitors of Phosphoinositide 3-Kinase δ for the Treatment of Respiratory Disease. J. Med. Chem. 2015, 58, 7381–7399. [Google Scholar] [CrossRef] [PubMed]
- Somoza, J.R.; Koditek, D.; Villaseñor, A.G.; Novikov, N.; Wong, M.H.; Liclican, A.; Xing, W.; Lagpacan, L.; Wang, R.; Schultz, B.E.; et al. Structural, Biochemical, and Biophysical Characterization of Idelalisib Binding to Phosphoinositide 3-Kinase δ. J. Biol. Chem. 2015, 290, 8439–8446. [Google Scholar] [CrossRef] [PubMed]
- Hoegenauer, K.; Soldermann, N.; Zécri, F.; Strang, R.S.; Graveleau, N.; Wolf, R.M.; Cooke, N.G.; Smith, A.B.; Hollingworth, G.J.; Blanz, J.; et al. Discovery of CDZ173 (Leniolisib), Representing a Structurally Novel Class of PI3K Delta-Selective Inhibitors. ACS Med. Chem. Lett. 2017, 8, 975–980. [Google Scholar] [CrossRef] [PubMed]
- Winkler, D.G.; Faia, K.L.; DiNitto, J.P.; Ali, J.A.; White, K.F.; Brophy, E.E.; Pink, M.M.; Proctor, J.L.; Lussier, J.; Martin, C.M.; et al. PI3K-δ and PI3K-γ Inhibition by IPI-145 Abrogates Immune Responses and Suppresses Activity in Autoimmune and Inflammatory Disease Models. Chem. Biol. 2013, 20, 1364–1374. [Google Scholar] [CrossRef] [PubMed]
- Walker, E.H.; Perisic, O.; Ried, C.; Stephens, L.; Williams, R.L. Structural insights into phosphoinositide 3-kinase catalysis and signalling. Nature 1999, 402, 313–320. [Google Scholar] [CrossRef] [PubMed]
- Nassar, N.; Horn, G.; Herrmann, C.A.; Scherer, A.; McCormick, F.; Wittinghofer, A. The 2.2 Å crystal structure of the Ras-binding domain of the serine/threonine kinase c-Raf1 in complex with RaplA and a GTP analogue. Nature 1995, 375, 554–560. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Hofer, F.; Martin, G.S.; Kim, S.-H. Structural basis for the interaction of Ras with RaIGDS. Nat. Struct. Mol. Biol. 1998, 5, 422–426. [Google Scholar] [CrossRef]
- Essen, L.-O.; Perisic, O.; Lynch, D.E.; Katan, M.; Williams, R.L. A Ternary Metal Binding Site in the C2 Domain of Phosphoinositide-Specific Phospholipase C-δ1. Biochemistry 1997, 36, 2753–2762. [Google Scholar] [CrossRef] [PubMed]
- Walker, E.H.; Pacold, M.E.; Perisic, O.; Stephens, L.; Hawkins, P.T.; Wymann, M.P.; Williams, R.L. Structural determinants of phosphoinositide 3-kinase inhibition by wortmannin, LY294002, quercetin, myricetin, and staurosporine. Mol. Cell 2000, 6, 909–919. [Google Scholar] [CrossRef]
- Huang, C.-H.; Mandelker, D.; Schmidt-Kittler, O.; Samuels, Y.; Velculescu, V.E.; Kinzler, K.W.; Vogelstein, B.; Gabelli, S.B.; Amzel, L.M. The Structure of a Human p110α/p85α Complex Elucidates the Effects of Oncogenic PI3Kα Mutations. Science 2007, 318, 1744–1748. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Wjasow, C.; Backer, J.M. Regulation of the P85/P110α Phosphatidylinositol 3′-Kinase: Distinct roles for the N-terminal and C-terminal SH2 domains. J. Biol. Chem. 1998, 273, 30199–30203. [Google Scholar] [CrossRef] [PubMed]
- Mandelker, D.; Gabelli, S.B.; Schmidt-Kittler, O.; Zhu, J.; Cheong, I.; Huang, C.H.; Kinzler, K.W.; Vogelstein, B.; Amzel, L.M. A frequent kinase domain mutation that changes the interaction between PI3Kα and the membrane. Proc. Natl. Acad. Sci. USA 2009, 106, 16996–17001. [Google Scholar] [CrossRef] [PubMed]
- Berndt, A.; Miller, S.; Williams, O.; Le, D.D.; Houseman, B.T.; Pacold, J.I.; Gorrec, F.; Hon, W.-C.; Ren, P.; Liu, Y.; et al. The p110δ structure: Mechanisms for selectivity and potency of new PI(3)K inhibitors. Nat. Chem. Biol. 2010, 6, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Vadas, O.; Perisic, O.; Anderson, K.E.; Clark, J.; Hawkins, P.T.; Stephens, L.R.; Williams, R.L. Structure of Lipid Kinase p110β/p85β Elucidates an Unusual SH2-Domain-Mediated Inhibitory Mechanism. Mol. Cell 2011, 41, 567–578. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Henrick, K.; Nakamura, H. Announcing the worldwide Protein Data Bank. Nat. Struct. Biol. 2003, 10, 980. [Google Scholar] [CrossRef] [PubMed]
- Hon, W.-C.; Berndt, A.; Williams, R.L. Regulation of lipid binding underlies the activation mechanism of class IA PI3-kinases. Oncogene 2012, 31, 3655–3666. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Knapp, M.; Crawford, K.; Warne, R.; Elling, R.; Yan, K.; Doyle, M.; Pardee, G.; Zhang, L.; Ma, S.; et al. Rationally Designed PI3Kα Mutants to Mimic ATR and Their Use to Understand Binding Specificity of ATR Inhibitors. J. Mol. Biol. 2017, 429, 1684–1704. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Zhang, X.; Chen, Y.; Lu, S.; Peng, Y.; Wang, X.; Guo, C.; Zhou, A.; Zhang, J.; Luo, Y.; et al. Crystal Structures of PI3Kα Complexed with PI103 and Its Derivatives: New Directions for Inhibitors Design. ACS Med. Chem. Lett. 2014, 5, 138–142. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Medeiros, P.F.; Raha, K.; Elkins, P.; Lind, K.E.; Lehr, R.; Adams, N.D.; Burgess, J.L.; Schmidt, S.J.; Knight, S.D.; et al. Discovery of a Potent Class of PI3Kα Inhibitors with Unique Binding Mode via Encoded Library Technology (ELT). ACS Med. Chem. Lett. 2015, 6, 531–536. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Deng, Y.-L.; Bergqvist, S.; Falk, M.D.; Liu, W.; Timofeevski, S.; Brooun, A. Engineering of an isolated p110α subunit of PI3Kα permits crystallization and provides a platform for structure-based drug design. Protein Sci. 2014, 23, 1332–1340. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Shi, Q.; Marcoux, D.; Batt, D.G.; Cornelius, L.; Qin, L.-Y.; Ruan, Z.; Neels, J.; Beaudoin-Bertrand, M.; Srivastava, A.S.; et al. Identification of a Potent, Selective, and Efficacious Phosphatidylinositol 3-Kinase δ (PI3Kδ) Inhibitor for the Treatment of Immunological Disorders. J. Med. Chem. 2017, 60, 5193–5208. [Google Scholar] [CrossRef] [PubMed]
- Qin, L.-Y.; Ruan, Z.; Cherney, R.J.; Dhar, T.G.M.; Neels, J.; Weigelt, C.A.; Sack, J.S.; Srivastava, A.S.; Cornelius, L.A.M.; Tino, J.A.; et al. Discovery of 7-(3-(piperazin-1-yl)phenyl)pyrrolo[2,1-f][1,2,4]triazin-4-amine derivatives as highly potent and selective PI3Kδ inhibitors. Bioorg. Med. Chem. Lett. 2017, 27, 855–861. [Google Scholar] [CrossRef] [PubMed]
- Castanedo, G.M.; Blaquiere, N.; Beresini, M.; Bravo, B.; Brightbill, H.; Chen, J.; Cui, H.-F.; Eigenbrot, C.; Everett, C.; Feng, J.; et al. Structure-Based Design of Tricyclic NF-κB Inducing Kinase (NIK) Inhibitors That Have High Selectivity over Phosphoinositide-3-kinase (PI3K). J. Med. Chem. 2017, 60, 627–640. [Google Scholar] [CrossRef] [PubMed]
- Frazzetto, M.; Suphioglu, C.; Zhu, J.; Schmidt-Kittler, O.; Jennings, I.G.; Cranmer, S.L.; Jackson, S.P.; Kinzler, K.W.; Vogelstein, B.; Thompson, P.E. Dissecting Isoform Selectivity of PI3 Kinase Inhibitors. The Role of Non-conserved Residues in the Catalytic Pocket. Biochem. J. 2008, 414, 383–390. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.; Amran, S.I.; Thompson, P.E.; Jennings, I.G. Isoform-Selective Inhibition of Phosphoinositide 3-Kinase: Identification of a New Region of Nonconserved Amino Acids Critical for p110α Inhibition. Mol. Pharmacol. 2011, 80, 657–664. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.; Miller, M.S.; Jennings, I.G.; Thompson, P.E. Mechanisms of PI3Kβ-Selective Inhibition Revealed by Reciprocal Mutagenesis. ACS Chem. Biol. 2013, 8, 679–683. [Google Scholar] [CrossRef] [PubMed]
- Patel, L.; Chandrasekhar, J.; Evarts, J.; Forseth, K.; Haran, A.C.; Ip, C.; Kashishian, A.; Kim, M.; Koditek, D.; Koppenol, S.; et al. Discovery of Orally Efficacious Phosphoinositide 3-Kinase δ Inhibitors with Improved Metabolic Stability. J. Med. Chem. 2016, 59, 9228–9242. [Google Scholar] [CrossRef] [PubMed]
- Perreault, S.; Chandrasekhar, J.; Cui, Z.-H.; Evarts, J.; Hao, J.; Kaplan, J.A.; Kashishian, A.; Keegan, K.S.; Kenney, T.; Koditek, D.; et al. Discovery of a Phosphoinositide 3-Kinase (PI3K) β/δ Inhibitor for the Treatment of Phosphatase and Tensin Homolog (PTEN) Deficient Tumors: Building PI3Kβ Potency in a PI3Kδ-Selective Template by Targeting Nonconserved Asp856. J. Med. Chem. 2017, 60, 1555–1567. [Google Scholar] [CrossRef] [PubMed]
- Patel, L.; Chandrasekhar, J.; Evarts, J.; Haran, A.C.; Ip, C.; Kaplan, J.A.; Kim, M.; Koditek, D.; Lad, L.; Lepist, E.-I.; et al. 2,4,6-Triaminopyrimidine as a Novel Hinge Binder in a Series of PI3Kδ Selective Inhibitors. J. Med. Chem. 2016, 59, 3532–3548. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Lopez de Turiso, F.; Shin, Y.; Brown, M.; Cardozo, M.; Chen, Y.; Fong, D.; Hao, X.; He, X.; Henne, K.; Hu, Y.-L.; et al. Discovery and in Vivo Evaluation of Dual PI3Kβ/δ Inhibitors. J. Med. Chem. 2012, 55, 7667–7685. [Google Scholar] [CrossRef] [PubMed]
- Shin, Y.; Suchomel, J.; Cardozo, M.; Duquette, J.; He, X.; Henne, K.; Hu, Y.-L.; Kelly, R.C.; McCarter, J.; McGee, L.R.; et al. Discovery, Optimization, and in Vivo Evaluation of Benzimidazole Derivatives AM-8508 and AM-9635 as Potent and Selective PI3Kδ Inhibitors. J. Med. Chem. 2016, 59, 431–447. [Google Scholar] [CrossRef] [PubMed]
- Wei, M.; Zhang, X.; Wang, X.; Song, Z.; Ding, J.; Meng, L.-H.; Zhang, A. SAR study of 5-alkynyl substituted quinazolin-4(3H)-ones as phosphoinositide 3-kinase delta (PI3Kδ) inhibitors. Eur. J. Med. Chem. 2017, 125, 1156–1171. [Google Scholar] [CrossRef] [PubMed]
- Williams, O.; Houseman, B.T.; Kunkel, E.J.; Aizenstein, B.; Hoffman, R.; Knight, Z.A.; Shokat, K.M. Discovery of Dual Inhibitors of the Immune Cell PI3Ks p110δ and p110γ: A Prototype for New Anti-inflammatory Drugs. Chem. Biol. 2010, 17, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Sutherlin, D.P.; Baker, S.; Bisconte, A.; Blaney, P.M.; Brown, A.; Chan, B.K.; Chantry, D.; Castanedo, G.; DePledge, P.; Goldsmith, P.; et al. Potent and selective inhibitors of PI3Kδ: Obtaining isoform selectivity from the affinity pocket and tryptophan shelf. Bioorg. Med. Chem. Lett. 2012, 22, 4296–4302. [Google Scholar] [CrossRef] [PubMed]
- Erra, M.; Taltavull, J.; Bernal, F.J.; Caturla, J.F.; Carrascal, M.; Pagès, L.; Mir, M.; Espinosa, S.; Gràcia, J.; Domínguez, M.; et al. Discovery of a Novel Inhaled PI3Kδ Inhibitor for the Treatment of Respiratory Diseases. J. Med. Chem. 2018, 61, 9551–9567. [Google Scholar] [CrossRef] [PubMed]
- Terstiege, I.; Perry, M.; Petersen, J.; Tyrchan, C.; Svensson, T.; Lindmark, H.; Öster, L. Discovery of triazole aminopyrazines as a highly potent and selective series of PI3Kδ inhibitors. Bioorg. Med. Chem. Lett. 2017, 27, 679–687. [Google Scholar] [CrossRef] [PubMed]
- Barton, N.; Convery, M.; Cooper, A.W.J.; Down, K.; Hamblin, J.N.; Inglis, G.; Peace, S.; Rowedder, J.; Rowland, P.; Taylor, J.A.; et al. Discovery of Potent, Efficient, and Selective Inhibitors of Phosphoinositide 3-Kinase δ through a Deconstruction and Regrowth Approach. J. Med. Chem. 2018, 61, 11061–11073. [Google Scholar] [CrossRef] [PubMed]
- Safina, B.S.; Sweeney, Z.K.; Li, J.; Chan, B.K.; Bisconte, A.; Carrera, D.; Castanedo, G.; Flagella, M.; Heald, R.; Lewis, C.; et al. Identification of GNE-293, a potent and selective PI3Kδ inhibitor: Navigating in vitro genotoxicity while improving potency and selectivity. Bioorg. Med. Chem. Lett. 2013, 23, 4953–4959. [Google Scholar] [CrossRef] [PubMed]
- Murray, J.M.; Sweeney, Z.K.; Chan, B.K.; Balazs, M.; Bradley, E.; Castanedo, G.; Chabot, C.; Chantry, D.; Flagella, M.; Goldstein, D.M.; et al. Potent and Highly Selective Benzimidazole Inhibitors of PI3-Kinase Delta. J. Med. Chem. 2012, 55, 7686–7695. [Google Scholar] [CrossRef] [PubMed]
- Safina, B.S.; Elliott, R.L.; Forrest, A.K.; Heald, R.A.; Murray, J.M.; Nonomiya, J.; Pang, J.; Salphati, L.; Seward, E.M.; Staben, S.T.; et al. Design of Selective Benzoxazepin PI3Kδ Inhibitors Through Control of Dihedral Angles. Acs Med. Chem. Lett. 2017, 8, 936–940. [Google Scholar] [CrossRef] [PubMed]
- Dalton, S.E.; Dittus, L.; Thomas, D.A.; Convery, M.A.; Nunes, J.; Bush, J.T.; Evans, J.P.; Werner, T.; Bantscheff, M.; Murphy, J.A.; et al. Selectively Targeting the Kinome-Conserved Lysine of PI3Kδ as a General Approach to Covalent Kinase Inhibition. J. Am. Chem. Soc. 2018, 140, 932–939. [Google Scholar] [CrossRef] [PubMed]
- Schwehm, C.; Kellam, B.; Garces, A.E.; Hill, S.J.; Kindon, N.D.; Bradshaw, T.D.; Li, J.; Macdonald, S.J.F.; Rowedder, J.E.; Stoddart, L.A.; et al. Design and Elaboration of a Tractable Tricyclic Scaffold To Synthesize Druglike Inhibitors of Dipeptidyl Peptidase-4 (DPP-4), Antagonists of the C–C Chemokine Receptor Type 5 (CCR5), and Highly Potent and Selective Phosphoinositol-3 Kinase δ (PI3Kδ) Inhibitors. J. Med. Chem. 2017, 60, 1534–1554. [Google Scholar] [PubMed]
- Hamajima, T.; Takahashi, F.; Kato, K.; Mukoyoshi, K.; Yoshihara, K.; Yamaki, S.; Sugano, Y.; Moritomo, A.; Yamagami, K.; Yokoo, K.; et al. Discovery and biological evaluation of novel pyrazolopyridine derivatives as potent and orally available PI3Kδ inhibitors. Bioorg. Med. Chem. 2018, 26, 2410–2419. [Google Scholar] [CrossRef] [PubMed]
- Hoegenauer, K.; Soldermann, N.; Stauffer, F.; Furet, P.; Graveleau, N.; Smith, A.B.; Hebach, C.; Hollingworth, G.J.; Lewis, I.; Gutmann, S.; et al. Discovery and Pharmacological Characterization of Novel Quinazoline-Based PI3K Delta-Selective Inhibitors. ACS Med. Chem. Lett. 2016, 7, 762–767. [Google Scholar] [CrossRef] [PubMed]
- Bhide, R.S.; Neels, J.; Qin, L.-Y.; Ruan, Z.; Stachura, S.; Weigelt, C.; Sack, J.S.; Stefanski, K.; Gu, X.; Xie, J.H.; et al. Discovery and SAR of pyrrolo[2,1-f][1,2,4]triazin-4-amines as potent and selective PI3Kδ inhibitors. Bioorg. Med. Chem. Lett. 2016, 26, 4256–4260. [Google Scholar] [CrossRef] [PubMed]
- Marcoux, D.; Qin, L.-Y.; Ruan, Z.; Shi, Q.; Ruan, Q.; Weigelt, C.; Qiu, H.; Schieven, G.; Hynes, J.; Bhide, R.; et al. Identification of highly potent and selective PI3Kδ inhibitors. Bioorg. Med. Chem. Lett. 2017, 27, 2849–2853. [Google Scholar] [CrossRef] [PubMed]
- Bui, M.; Hao, X.; Shin, Y.; Cardozo, M.; He, X.; Henne, K.; Suchomel, J.; McCarter, J.; McGee, L.R.; San Miguel, T.; et al. Synthesis and SAR study of potent and selective PI3Kδ inhibitors. Bioorg. Med. Chem. Lett. 2015, 25, 1104–1109. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Lopez de Turiso, F.; Hao, X.; Shin, Y.; Bui, M.; Campuzano, I.D.G.; Cardozo, M.; Dunn, M.C.; Duquette, J.; Fisher, B.; Foti, R.S.; et al. Discovery and in Vivo Evaluation of the Potent and Selective PI3Kδ Inhibitors 2-((1S)-1-((6-Amino-5-cyano-4-pyrimidinyl)amino)ethyl)-6-fluoro-N-methyl-3-(2-pyridinyl)-4-quinolinecarboxamide (AM-0687) and 2-((1S)-1-((6-Amino-5-cyano-4-pyrimidinyl)amino)ethyl)-5-fluoro-N-methyl-3-(2-pyridinyl)-4-quinolinecarboxamide (AM-1430). J. Med. Chem. 2016, 59, 7252–7267. [Google Scholar] [PubMed]
- Perry, M.W.D.; Björhall, K.; Bonn, B.; Carlsson, J.; Chen, Y.; Eriksson, A.; Fredlund, L.; Hao, H.; Holden, N.S.; Karabelas, K.; et al. Design and Synthesis of Soluble and Cell-Permeable PI3Kδ Inhibitors for Long-Acting Inhaled Administration. J. Med. Chem. 2017, 60, 5057–5071. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.S.; Mountford, S.J.; Pinson, J.-A.; Zheng, Z.; Künzli, M.; Patel, V.; Hogg, S.J.; Shortt, J.; Jennings, I.G.; Thompson, P.E. Development of single and mixed isoform selectivity PI3Kδ inhibitors by targeting Asn836 of PI3Kδ. Bioorg. Med. Chem. Lett. 2016, 26, 4790–4794. [Google Scholar] [CrossRef] [PubMed]
- Certal, V.; Halley, F.; Virone-Oddos, A.; Thompson, F.; Filoche-Rommé, B.; El-Ahmad, Y.; Carry, J.-C.; Delorme, C.; Karlsson, A.; Abecassis, P.-Y.; et al. Preparation and optimization of new 4-(morpholin-4-yl)-(6-oxo-1,6-dihydropyrimidin-2-yl)amide derivatives as PI3Kβ inhibitors. Bioorg. Med. Chem. Lett. 2012, 22, 6381–6384. [Google Scholar] [CrossRef] [PubMed]
- Ni, J.; Liu, Q.; Xie, S.; Carlson, C.; Von, T.; Vogel, K.; Riddle, S.; Benes, C.; Eck, M.; Roberts, T.; et al. Functional Characterization of an Isoform-Selective Inhibitor of PI3K-p110β as a Potential Anticancer Agent. Cancer Discov. 2012, 2, 425–433. [Google Scholar] [CrossRef] [PubMed]
- Certal, V.; Halley, F.; Virone-Oddos, A.; Delorme, C.; Karlsson, A.; Rak, A.; Thompson, F.; Filoche-Rommé, B.; El-Ahmad, Y.; Carry, J.-C.; et al. Discovery and Optimization of New Benzimidazole- and Benzoxazole-Pyrimidone Selective PI3Kβ Inhibitors for the Treatment of Phosphatase and TENsin homologue (PTEN)-Deficient Cancers. J. Med. Chem. 2012, 55, 4788–4805. [Google Scholar] [CrossRef] [PubMed]
- Giordanetto, F.; Wållberg, A.; Cassel, J.; Ghosal, S.; Kossenjans, M.; Yuan, Z.-Q.; Wang, X.; Liang, L. Discovery of 4-morpholino-pyrimidin-6-one and 4-morpholino-pyrimidin-2-one-containing Phosphoinositide 3-kinase (PI3K) p110β isoform inhibitors through structure-based fragment optimisation. Bioorg. Med. Chem. Lett. 2012, 22, 6665–6670. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Erhard, K.; Hardwicke, M.A.; Luengo, J.I.; Mack, J.F.; McSurdy-Freed, J.; Plant, R.; Raha, K.; Rominger, C.M.; Sanchez, R.M.; et al. Synthesis and structure–activity relationships of imidazo[1,2-a]pyrimidin-5(1H)-ones as a novel series of beta isoform selective phosphatidylinositol 3-kinase inhibitors. Bioorg. Med. Chem. Lett. 2012, 22, 2230–2234. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, R.M.; Erhard, K.; Hardwicke, M.A.; Lin, H.; McSurdy-Freed, J.; Plant, R.; Raha, K.; Rominger, C.M.; Schaber, M.D.; Spengler, M.D.; et al. Synthesis and structure–activity relationships of 1,2,4-triazolo[1,5-a]pyrimidin-7(3H)-ones as novel series of potent β isoform selective phosphatidylinositol 3-kinase inhibitors. Bioorg. Med. Chem. Lett. 2012, 22, 3198–3202. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekhar, J.; Dick, R.; Van Veldhuizen, J.; Koditek, D.; Lepist, E.-I.; McGrath, M.E.; Patel, L.; Phillips, G.; Sedillo, K.; Somoza, J.R.; et al. Atropisomerism by Design: Discovery of a Selective and Stable Phosphoinositide 3-Kinase (PI3K) β Inhibitor. J. Med. Chem. 2018, 61, 6858–6868. [Google Scholar] [CrossRef] [PubMed]
- Robinson, D.; Bertrand, T.; Carry, J.-C.; Halley, F.; Karlsson, A.; Mathieu, M.; Minoux, H.; Perrin, M.-A.; Robert, B.; Schio, L.; et al. Differential Water Thermodynamics Determine PI3K-Beta/Delta Selectivity for Solvent-Exposed Ligand Modifications. J. Chem. Inf. Model. 2016, 56, 886–894. [Google Scholar] [CrossRef] [PubMed]
- Pinson, J.-A.; Zheng, Z.; Miller, M.S.; Chalmers, D.K.; Jennings, I.G.; Thompson, P.E. L-Aminoacyl-triazine Derivatives Are Isoform-Selective PI3Kβ Inhibitors That Target Nonconserved Asp862 of PI3Kβ. ACS Med. Chem. Lett. 2013, 4, 206–210. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.; Pinson, J.A.; Mountford, S.J.; Orive, S.; Schoenwaelder, S.M.; Shackleford, D.; Powell, A.; Nelson, E.M.; Hamilton, J.R.; Jackson, S.; et al. Discovery and antiplatelet activity of a selective PI3Kβ inhibitor (MIPS-9922). Eur. J. Med. Chem. 2016, 122, 339–351. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.; Cao, R.; Liu, X.; Luo, X.; Zhong, W. Design, Synthesis and Biological Evaluation of Novel Benzothiazole Derivatives as Selective PI3Kβ Inhibitors. Molecules 2016, 21, 876. [Google Scholar] [CrossRef] [PubMed]
- Giordanetto, F.; Wållberg, A.; Ghosal, S.; Iliefski, T.; Cassel, J.; Yuan, Z.-Q.; von Wachenfeldt, H.; Andersen, S.M.; Inghardt, T.; Tunek, A.; et al. Discovery of phosphoinositide 3-kinases (PI3K) p110β isoform inhibitor 4-[2-hydroxyethyl(1-naphthylmethyl)amino]-6-[(2S)-2-methylmorpholin-4-yl]-1H-pyrimidin-2-one, an effective antithrombotic agent without associated bleeding and insulin resistance. Bioorg. Med. Chem. Lett. 2012, 22, 6671–6676. [Google Scholar] [CrossRef] [PubMed]
- Giordanetto, F.; Barlaam, B.; Berglund, S.; Edman, K.; Karlsson, O.; Lindberg, J.; Nylander, S.; Inghardt, T. Discovery of 9-(1-phenoxyethyl)-2-morpholino-4-oxo-pyrido[1,2-a]pyrimidine-7-carboxamides as oral PI3Kβ inhibitors, useful as antiplatelet agents. Bioorg. Med. Chem. Lett. 2014, 24, 3936–3943. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Schulz, M.J.; Xie, R.; Zeng, J.; Luengo, J.I.; Squire, M.D.; Tedesco, R.; Qu, J.; Erhard, K.; Mack, J.F.; et al. Rational Design, Synthesis, and SAR of a Novel Thiazolopyrimidinone Series of Selective PI3K-beta Inhibitors. ACS Med. Chem. Lett. 2012, 3, 524–529. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Moore, M.L.; Erhard, K.; Hardwicke, M.A.; Lin, H.; Luengo, J.I.; McSurdy-Freed, J.; Plant, R.; Qu, J.; Raha, K.; et al. [3a,4]-Dihydropyrazolo[1,5a]pyrimidines: Novel, Potent, and Selective Phosphatidylinositol-3-kinase β Inhibitors. ACS Med. Chem. Lett. 2013, 4, 230–234. [Google Scholar] [CrossRef] [PubMed]
- Greshock, J. Abstract IA17: Exploiting the synthetic lethal properties of selective PI3K-β inhibition in PTEN deficient cells with GSK2636771. Mol. Cancer Ther. 2013, 12, IA17. [Google Scholar] [CrossRef]
- Hayakawa, M.; Kawaguchi, K.; Kaizawa, H.; Koizumi, T.; Ohishi, T.; Yamano, M.; Okada, M.; Ohta, M.; Tsukamoto, S.; Raynaud, F.I.; et al. Synthesis and biological evaluation of sulfonylhydrazone-substituted imidazo [1, 2-a] pyridines as novel PI3 kinase p110α inhibitors. Bioorg. Med. Chem. 2007, 15, 5837–5844. [Google Scholar] [CrossRef] [PubMed]
- Schmidt-Kittler, O.; Zhu, J.; Yang, J.; Liu, G.; Hendricks, W.; Lengauer, C.; Gabelli, S.B.; Kinzler, K.W.; Vogelstein, B.; Huso, D.L.; et al. PI3Kα inhibitors that inhibit metastasis. Oncotarget 2010, 1, 339. [Google Scholar] [PubMed]
- Kendall, J.D.; O’Connor, P.D.; Marshall, A.J.; Frédérick, R.; Marshall, E.S.; Lill, C.L.; Lee, W.-J.; Kolekar, S.; Chao, M.; Malik, A.; et al. Discovery of pyrazolo[1,5-a]pyridines as p110α-selective PI3 kinase inhibitors. Bioorg. Med. Chem. 2012, 20, 69–85. [Google Scholar] [CrossRef] [PubMed]
- Kendall, J.D.; Giddens, A.C.; Tsang, K.Y.; Frédérick, R.; Marshall, E.S.; Singh, R.; Lill, C.L.; Lee, W.-J.; Kolekar, S.; Chao, M.; et al. Novel pyrazolo[1,5-a]pyridines as p110α-selective PI3 kinase inhibitors: Exploring the benzenesulfonohydrazide SAR. Bioorg. Med. Chem. 2012, 20, 58–68. [Google Scholar] [CrossRef] [PubMed]
- Kendall, J.D.; Rewcastle, G.W.; Frédérick, R.; Mawson, C.; Denny, W.A.; Marshall, E.S.; Baguley, B.C.; Chaussade, C.; Jackson, S.P.; Shepherd, P.R. Synthesis, biological evaluation and molecular modelling of sulfonohydrazides as selective PI3K p110alpha inhibitors. Bioorg. Med. Chem. 2007, 15, 7677–7687. [Google Scholar] [CrossRef] [PubMed]
- Kendall, J.D.; Giddens, A.C.; Tsang, K.Y.; Marshall, E.S.; Lill, C.L.; Lee, W.-J.; Kolekar, S.; Chao, M.; Malik, A.; Yu, S.; et al. Novel pyrazolo[1,5-a]pyridines with improved aqueous solubility as p110α-selective PI3 kinase inhibitors. Bioorg. Med. Chem. Lett. 2017, 27, 187–190. [Google Scholar] [CrossRef] [PubMed]
- Hayakawa, M.; Kaizawa, H.; Kawaguchi, K.; Ishikawa, N.; Koizumi, T.; Ohishi, T.; Yamano, M.; Okada, M.; Ohta, M.; Tsukamoto, S.; et al. Synthesis and biological evaluation of imidazo[1,2-a]pyridine derivatives as novel PI3 kinase p110α inhibitors. Bioorg. Med. Chem. 2007, 15, 403–412. [Google Scholar] [CrossRef] [PubMed]
- Frédérick, R.; Denny, W.A. Phosphoinositide-3-kinases (PI3Ks): Combined Comparative Modeling and 3D-QSAR To Rationalize the Inhibition of p110α. J. Chem. Inf. Model. 2008, 48, 629–638. [Google Scholar] [CrossRef] [PubMed]
- Han, M.; Zhang, J.Z.H. Class I Phospho-inositide-3-kinases (PI3Ks) Isoform-Specific Inhibition Study by the Combination of Docking and Molecular Dynamics Simulation. J. Chem. Inf. Model. 2010, 50, 136–145. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, Y.; Zhang, F. Pharmacophore modeling and 3D-QSAR analysis of phosphoinositide 3-kinase p110α inhibitors. J. Mol. Model. 2010, 16, 1449–1460. [Google Scholar] [CrossRef] [PubMed]
- Sabbah, D.A.; Vennerstrom, J.L.; Zhong, H. Docking Studies on Isoform-Specific Inhibition of Phosphoinositide-3-Kinases. J. Chem. Inf. Model. 2010, 50, 1887–1898. [Google Scholar] [CrossRef] [PubMed]
- Tesch, R.; Becker, C.; Müller, M.P.; Beck, M.E.; Quambusch, L.; Getlik, M.; Lategahn, J.; Uhlenbrock, N.; Costa, F.N.; Polêto, M.D.; et al. An Unusual Intramolecular Halogen Bond Guides Conformational Selection. Angew. Chem. Int. Ed. 2018, 57, 9970–9975. [Google Scholar] [CrossRef] [PubMed]
- Heffron, T.P.; Wei, B.; Olivero, A.; Staben, S.T.; Tsui, V.; Do, S.; Dotson, J.; Folkes, A.J.; Goldsmith, P.; Goldsmith, R.; et al. Rational Design of Phosphoinositide 3-Kinase α Inhibitors That Exhibit Selectivity over the Phosphoinositide 3-Kinase β Isoform. J. Med. Chem. 2011, 54, 7815–7833. [Google Scholar] [CrossRef] [PubMed]
- Nacht, M.; Qiao, L.; Sheets, M.P.; St. Martin, T.; Labenski, M.; Mazdiyasni, H.; Karp, R.; Zhu, Z.; Chaturvedi, P.; Bhavsar, D.; et al. Discovery of a Potent and Isoform-Selective Targeted Covalent Inhibitor of the Lipid Kinase PI3Kα. J. Med. Chem. 2013, 56, 712–721. [Google Scholar] [CrossRef] [PubMed]
- Staben, S.T.; Ndubaku, C.; Blaquiere, N.; Belvin, M.; Bull, R.J.; Dudley, D.; Edgar, K.; Gray, D.; Heald, R.; Heffron, T.P.; et al. Discovery of thiazolobenzoxepin PI3-kinase inhibitors that spare the PI3-kinase β isoform. Bioorg. Med. Chem. Lett. 2013, 23, 2606–2613. [Google Scholar] [CrossRef] [PubMed]
- Ndubaku, C.; Heffron, T.P.; Staben, S.T.; Baumgardner, M.; Blaquiere, N.; Bradley, E.; Bull, R.; Do, S.; Dotson, J.; Dudley, D.; et al. Discovery of 2-{3-[2-(1-isopropyl-3-methyl-1H-1,2-4-triazol-5-yl)-5,6-dihydrobenzo[f]imidazo[1,2-d][1,4]oxazepin-9-yl]-1H-pyrazol-1-yl}-2-methylpropanamide (GDC-0032): A β-sparing phosphoinositide 3-kinase (PI3K) inhibitor with high unbound exposure and robust in vivo anti-tumor activity. J. Med. Chem. 2013, 56, 4597–4610. [Google Scholar] [PubMed]
- Bruce, I.; Akhlaq, M.; Bloomfield, G.C.; Budd, E.; Cox, B.; Cuenoud, B.; Finan, P.; Gedeck, P.; Hatto, J.; Hayler, J.F.; et al. Development of isoform selective PI3-kinase inhibitors as pharmacological tools for elucidating the PI3K pathway. Bioorg. Med. Chem. Lett. 2012, 22, 5445–5450. [Google Scholar] [CrossRef] [PubMed]
- Gerspacher, M.; Fairhurst, R.A.; Mah, R.; Roehn-Carnemolla, E.; Furet, P.; Fritsch, C.; Guthy, D.A. Discovery of a novel tricyclic 4H-thiazolo[5′,4′:4,5]pyrano[2,3-c]pyridine-2-amino scaffold and its application in a PI3Kα inhibitor with high PI3K isoform selectivity and potent cellular activity. Bioorg. Med. Chem. Lett. 2015, 25, 3582–3584. [Google Scholar] [CrossRef] [PubMed]
- Jamieson, S.; Flanagan, J.U.; Kolekar, S.; Buchanan, C.; Kendall, J.D.; Lee, W.; Rewcastle, G.W.; Denny, W.A.; Singh, R.; Dickson, J.; et al. A drug targeting only p110α can block phosphoinositide 3-kinase signalling and tumour growth in certain cell types. Biochem. J. 2011, 438, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.; Amran, S.I.; Zhu, J.; Schmidt-Kittler, O.; Kinzler, K.W.; Vogelstein, B.; Shepherd, P.R.; Thompson, P.E.; Jennings, I.G. Definition of the binding mode of a new class of phosphoinositide 3-kinase α-selective inhibitors using in vitro mutagenesis of non-conserved amino acids and kinetic analysis. Biochem. J. 2012, 444, 529–535. [Google Scholar] [CrossRef] [PubMed]
- Barlaam, B.; Cosulich, S.; Fitzek, M.; Germain, H.; Green, S.; Hanson, L.L.; Harris, C.S.; Hancox, U.; Hudson, K.; Lambert-van der Brempt, C.; et al. Discovery of a novel aminopyrazine series as selective PI3Kα inhibitors. Bioorg. Med. Chem. Lett. 2017, 27, 3030–3035. [Google Scholar] [CrossRef] [PubMed]
- Barlaam, B.; Cosulich, S.; Fitzek, M.; Green, S.; Harris, C.S.; Hudson, K.; Lambert-van der Brempt, C.; Ouvry, G.; Page, K.; Ruston, L.; et al. Design of selective PI3Kα inhibitors starting from a promiscuous pan kinase scaffold. Bioorg. Med. Chem. Lett. 2015, 25, 2679–2685. [Google Scholar] [CrossRef] [PubMed]
- Barlaam, B.; Cosulich, S.; Delouvrié, B.; Ellston, R.; Fitzek, M.; Germain, H.; Green, S.; Hancox, U.; Harris, C.S.; Hudson, K.; et al. Discovery of 1-(4-(5-(5-amino-6-(5-tert-butyl-1,3,4-oxadiazol-2-yl)pyrazin-2-yl)-1-ethyl-1,2,4-triazol-3-yl)piperidin-1-yl)-3-hydroxypropan-1-one (AZD8835): A potent and selective inhibitor of PI3Kα and PI3Kδ for the treatment of cancers. Bioorg. Med. Chem. Lett. 2015, 25, 5155–5162. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.-H.; Ding, H.-W.; Liu, D.-D.; Song, H.-R.; Xu, Y.-N.; Wang, J. Novel 4-aminoquinazoline derivatives induce growth inhibition, cell cycle arrest and apoptosis via PI3Kα inhibition. Bioorg. Med. Chem. 2018, 26, 1675–1685. [Google Scholar] [CrossRef] [PubMed]
- Camps, M.; Rückle, T.; Ji, H.; Ardissone, V.; Rintelen, F.; Shaw, J.; Ferrandi, C.; Chabert, C.; Gillieron, C.; Françon, B.; et al. Blockade of PI3Kγ suppresses joint inflammation and damage in mouse models of rheumatoid arthritis. Nat. Med. 2005, 11, 936–943. [Google Scholar] [CrossRef] [PubMed]
- Pomel, V.; Klicic, J.; Covini, D.; Church, D.D.; Shaw, J.P.; Roulin, K.; Burgat-Charvillon, F.; Valognes, D.; Camps, M.; Chabert, C.; et al. Furan-2-ylmethylene Thiazolidinediones as Novel, Potent, and Selective Inhibitors of Phosphoinositide 3-Kinase γ. J. Med. Chem. 2006, 49, 3857–3871. [Google Scholar] [CrossRef] [PubMed]
- Collier, P.N.; Martinez-Botella, G.; Cornebise, M.; Cottrell, K.M.; Doran, J.D.; Griffith, J.P.; Mahajan, S.; Maltais, F.; Moody, C.S.; Huck, E.P.; et al. Structural Basis for Isoform Selectivity in a Class of Benzothiazole Inhibitors of Phosphoinositide 3-Kinase γ. J. Med. Chem. 2015, 58, 517–521. [Google Scholar] [CrossRef] [PubMed]
- Bell, K.; Sunose, M.; Ellard, K.; Cansfield, A.; Taylor, J.; Miller, W.; Ramsden, N.; Bergamini, G.; Neubauer, G. SAR studies around a series of triazolopyridines as potent and selective PI3Kγ inhibitors. Bioorg. Med. Chem. Lett. 2012, 22, 5257–5263. [Google Scholar] [CrossRef] [PubMed]
- Bergamini, G.; Bell, K.; Shimamura, S.; Werner, T.; Cansfield, A.; Müller, K.; Perrin, J.; Rau, C.; Ellard, K.; Hopf, C.; et al. A selective inhibitor reveals PI3Kγ dependence of TH17 cell differentiation. Nat. Chem. Biol. 2012, 8, 576–582. [Google Scholar] [CrossRef] [PubMed]
- Sunose, M.; Bell, K.; Ellard, K.; Bergamini, G.; Neubauer, G.; Werner, T.; Ramsden, N. Discovery of 5-(2-amino-[1,2,4]triazolo[1,5-a]pyridin-7-yl)-N-(tert-butyl)pyridine-3-sulfonamide (CZC24758), as a potent, orally bioavailable and selective inhibitor of PI3K for the treatment of inflammatory disease. Bioorg. Med. Chem. Lett. 2012, 22, 4613–4618. [Google Scholar] [CrossRef] [PubMed]
- Ellard, K.; Sunose, M.; Bell, K.; Ramsden, N.; Bergamini, G.; Neubauer, G. Discovery of novel PI3Kγ/δ inhibitors as potential agents for inflammation. Bioorg. Med. Chem. Lett. 2012, 22, 4546–4549. [Google Scholar] [CrossRef] [PubMed]
- Collier, P.N.; Panchagnula, A.; O’Dowd, H.; Le Tiran, A.; Aronov, A.M. Synthesis of a 6-Aza-Isoindolinone-Based Inhibitor of Phosphoinositide 3-Kinase γ via Ruthenium-Catalyzed [2 + 2 + 2] Cyclotrimerization. ACS Med. Chem. Lett. 2019, 10, 117–120. [Google Scholar] [CrossRef] [PubMed]
- Collier, P.N.; Messersmith, D.; Le Tiran, A.; Bandarage, U.K.; Boucher, C.; Come, J.; Cottrell, K.M.; Damagnez, V.; Doran, J.D.; Griffith, J.P.; et al. Discovery of Highly Isoform Selective Thiazolopiperidine Inhibitors of Phosphoinositide 3-Kinase γ. J. Med. Chem. 2015, 58, 5684–5688. [Google Scholar] [CrossRef] [PubMed]
- Come, J.H.; Collier, P.N.; Henderson, J.A.; Pierce, A.C.; Davies, R.J.; Le Tiran, A.; O’Dowd, H.; Bandarage, U.K.; Cao, J.; Deininger, D.; et al. Design and Synthesis of a Novel Series of Orally Bioavailable, CNS-Penetrant, Isoform Selective Phosphoinositide 3-Kinase γ (PI3Kγ) Inhibitors with Potential for the Treatment of Multiple Sclerosis (MS). J. Med. Chem. 2018, 61, 5245–5256. [Google Scholar] [CrossRef] [PubMed]
- Pemberton, N.; Mogemark, M.; Arlbrandt, S.; Bold, P.; Cox, R.J.; Gardelli, C.; Holden, N.S.; Karabelas, K.; Karlsson, J.; Lever, S.; et al. Discovery of Highly Isoform Selective Orally Bioavailable Phosphoinositide 3-Kinase (PI3K)-γ Inhibitors. J. Med. Chem. 2018, 61, 5435–5441. [Google Scholar] [CrossRef] [PubMed]
- Folkes, A.J.; Ahmadi, K.; Alderton, W.K.; Alix, S.; Baker, S.J.; Box, G.; Chuckowree, I.S.; Clarke, P.A.; Depledge, P.; Eccles, S.A.; et al. The Identification of 2-(1H-Indazol-4-yl)-6-(4-methanesulfonyl-piperazin-1-ylmethyl)-4-morpholin-4-yl-thieno[3,2-d]pyrimidine (GDC-0941) as a Potent, Selective, Orally Bioavailable Inhibitor of Class I PI3 Kinase for the Treatment of Cancer. J. Med. Chem. 2008, 51, 5522–5532. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Sobkiw, C.L.; Hirshman, M.F.; Logsdon, M.N.; Li, T.Q.; Goodyear, L.J.; Cantley, L.C. Loss of class IA PI3K signaling in muscle leads to impaired muscle growth, insulin response, and hyperlipidemia. Cell Metab. 2006, 3, 355–366. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, C.M.; Kondo, T.; Sajan, M.; Luo, J.; Bronson, R.; Asano, T.; Farese, R.; Cantley, L.C.; Kahn, C.R. Divergent regulation of hepatic glucose and lipid metabolism by phosphoinositide 3-kinase via Akt and PKCλ/ζ. Cell Metab. 2006, 3, 343–353. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Gong, G.Q.; Zhou, Y.; Lee, W.-J.; Buchanan, C.M.; Denny, W.A.; Rewcastle, G.W.; Kendall, J.D.; Dickson, J.M.J.; Flanagan, J.U.; et al. High-throughput screening campaigns against a PI3Kα isoform bearing the H1047R mutation identified potential inhibitors with novel scaffolds. Acta Pharmacol. Sin. 2018, 39, 1816–1822. [Google Scholar] [CrossRef] [PubMed]
- Sabbah, D.A.; Simms, N.A.; Wang, W.; Dong, Y.; Ezell, E.L.; Brattain, M.G.; Vennerstrom, J.L.; Zhong, H.A. N-Phenyl-4-hydroxy-2-quinolone-3-carboxamides as selective inhibitors of mutant H1047R phosphoinositide-3-kinase (PI3Kα). Bioorg. Med. Chem. 2012, 20, 7175–7183. [Google Scholar] [CrossRef] [PubMed]
- Gkeka, P.; Papafotika, A.; Christoforidis, S.; Cournia, Z. Exploring a Non-ATP Pocket for Potential Allosteric Modulation of PI3Kα. J. Phys. Chem. B 2015, 119, 1002–1016. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.S.; Maheshwari, S.; McRobb, F.M.; Kinzler, K.W.; Amzel, L.M.; Vogelstein, B.; Gabelli, S.B. Identification of allosteric binding sites for PI3Kα oncogenic mutant specific inhibitor design. Bioorg. Med. Chem. 2017, 25, 1481–1486. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
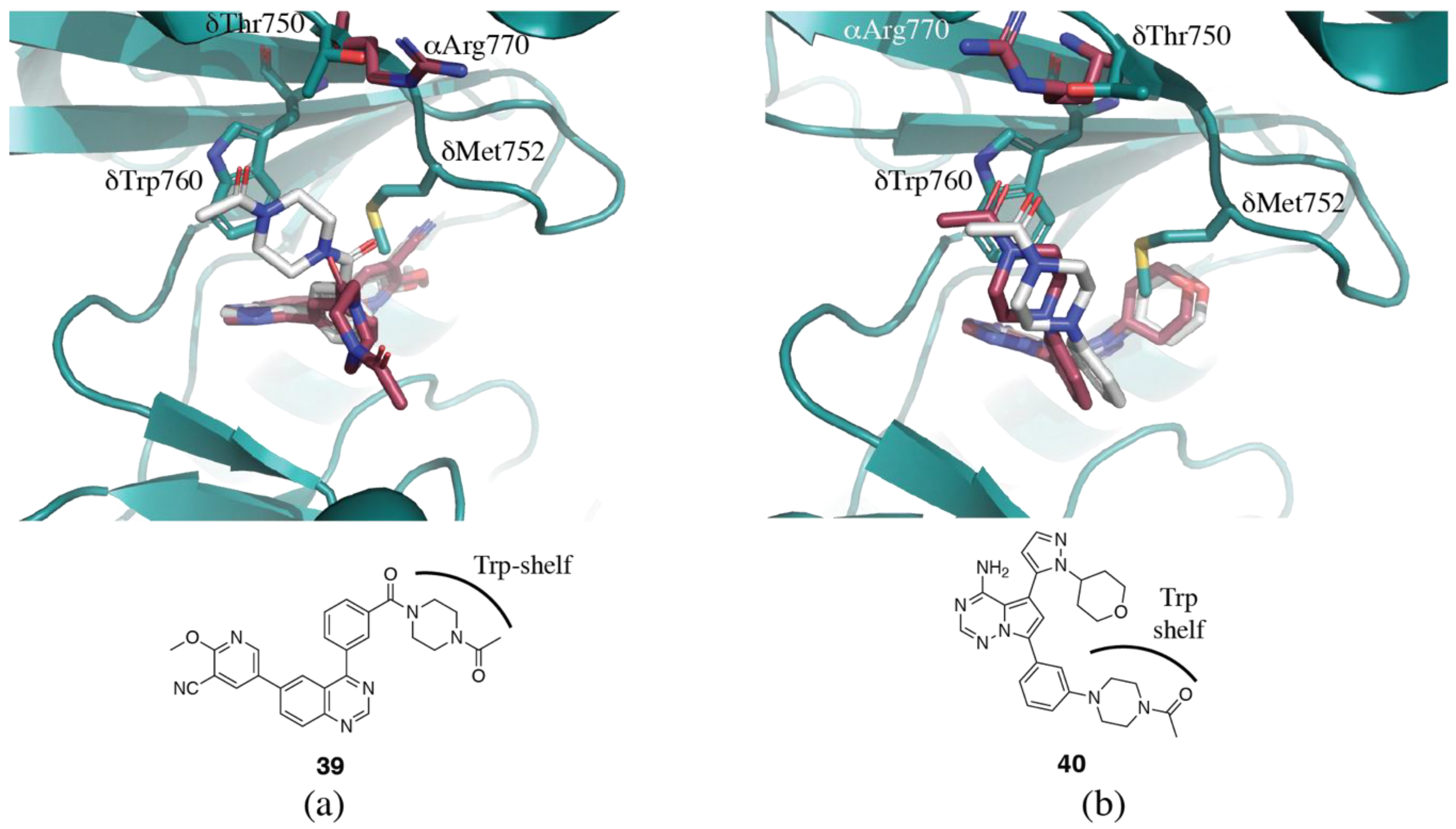{kind=link}
{kind=link}
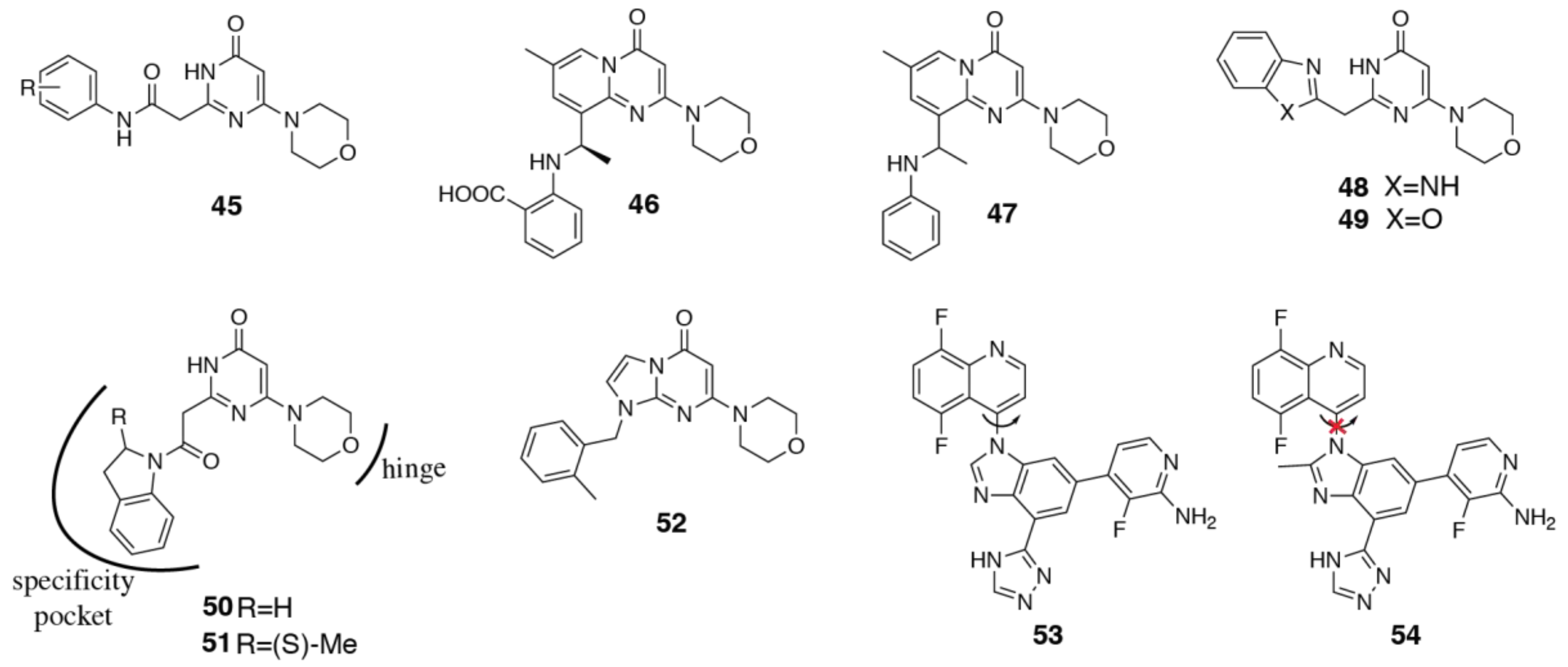{kind=link}
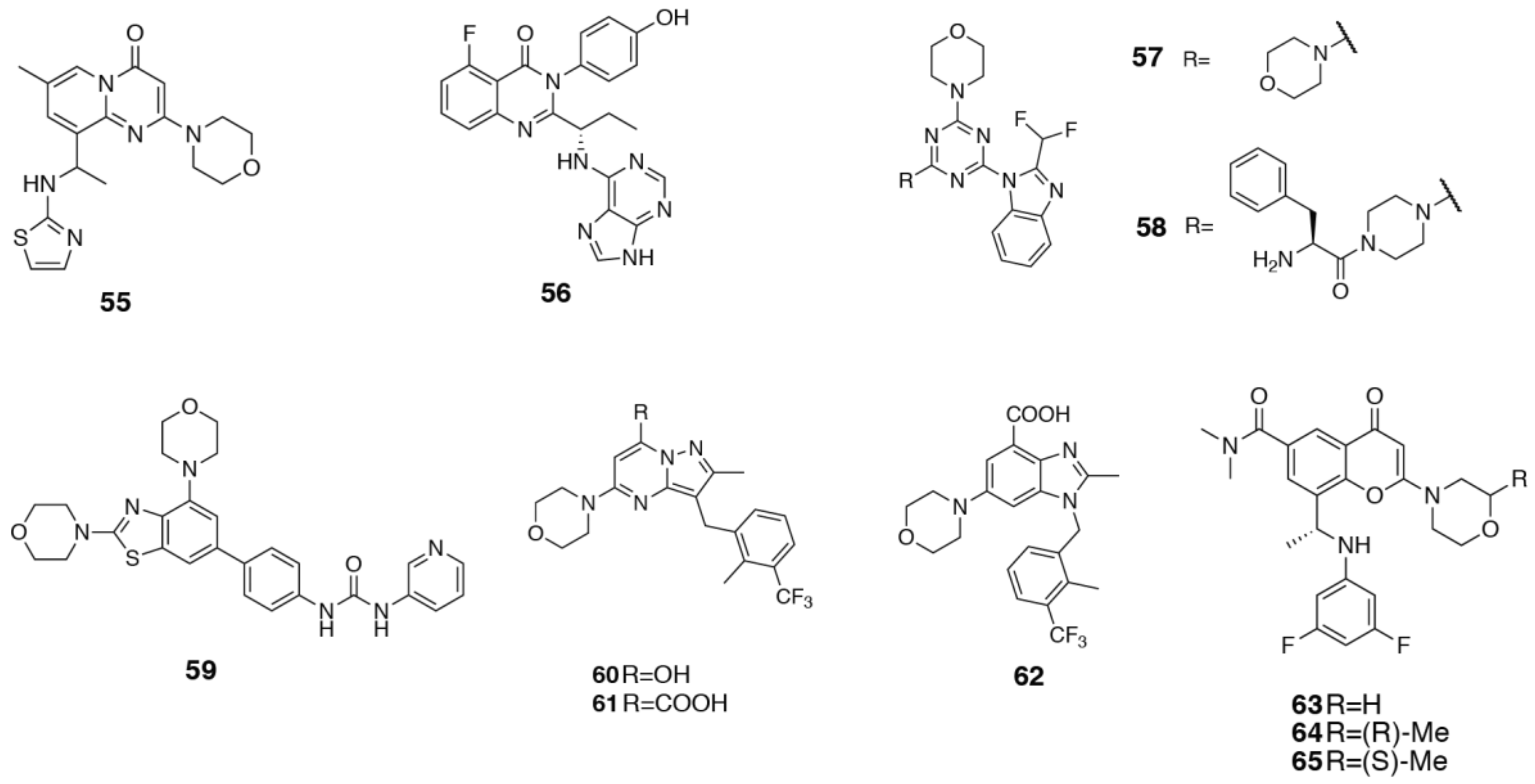{kind=link}
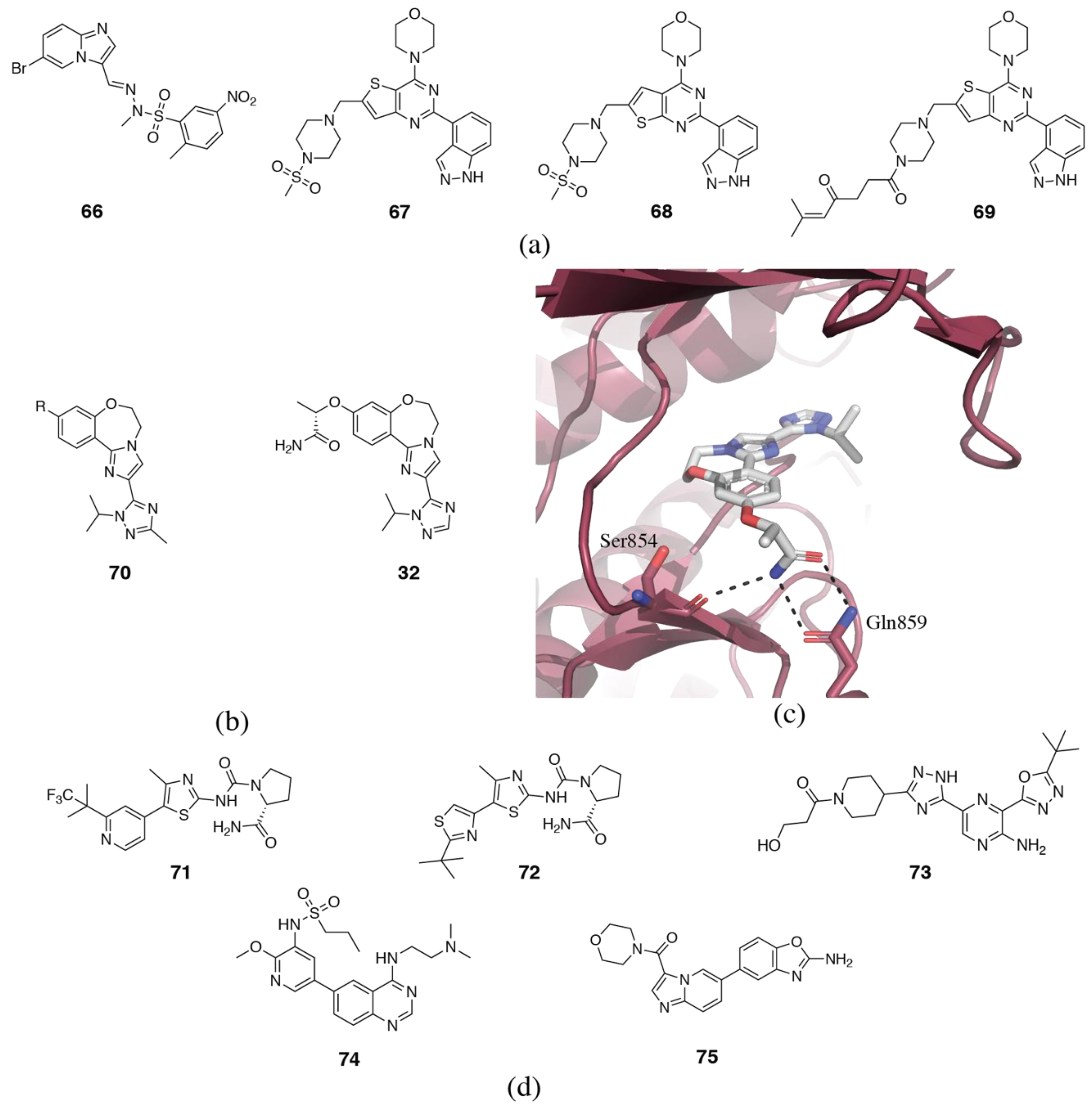{kind=link}
{kind=link}
{kind=link}
| Inhibitor | Other Names | Target | PDB ID (isoform) | IC50 (nM) | |||
|---|---|---|---|---|---|---|---|
| PI3Kα | PI3Kβ | PI3Kγ | PI3Kδ | ||||
| Alpelisib [65] | NVP-BYL719 | α | 4JPS (α) | 5 | 1200 | 250 | 290 |
| Serabelisib [66] | MLN1117; INK1117; TAK-117 | α | - | 15 | 4500 | 1900 | 13,900 |
| GDC-0326 [67] | α | 5DXT (α) | 0.2 1 | 26.6 | 4 | 10.2 | |
| CYH33 [68] | α | - | nd | nd | nd | nd | |
| AZD8186 [69] | β | - | 35 | 4 | 675 | 12 | |
| GSK2636771 [70] | β | - | 35,400 | 20 | nd | 40 | |
| SAR260301 [71] | β | 4BFR (β) | 1539 | 23 | 10,000 | 468 | |
| IPI-549 [57] | γ | - | 3200 | 3500 | 16 | >8400 | |
| AMG319 [72] | δ | 4WWN (γ) | 33,000 | 2700 | 850 | 18 | |
| GSK2292767 [73] | δ | 5AE9 (δ) | 501 | 630 | 501 | 0.079 | |
| Idelalisib [74] | CAL-101, GS-1101 | δ | 4XE0 (δ) | 8600 | 4000 | 2100 | 19 |
| Leniolisib [75] | CDZ173 | δ | 5O83 (δ) | 244 | 424 | 2230 | 11 |
| Nemiralisib [73] | GSK2269557 | δ | 5AE8 (δ) | 5011 | 1584 | 6309 | 0.12 |
| Umbralisib [49] | TGR-1202 | δ | - | >1400 | >756 | >120 | 14 |
| Parsaclisib [49] | INCB050465 | δ | - | nd | nd | nd | 1 |
| Duvelisib [76] | IPI-145 | γ/δ | - | 1602 | 85 | 27 | 2.5 |
| RV1729 [49] | γ/δ | - | 192 | - | 25 | 12 | |
| RV6153 [49] | γ/δ | - | 14,000 | 5000 | 28 | 2.5 | |
| Tenalisib [49] | RP6530 | γ/δ | - | 10,000 | 4000 | 33 | 25 |
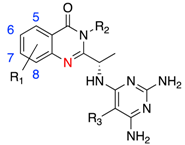
| Compound | R1 | R2 | R3 | IC50 (nM) | Fold Selectivity | ||
|---|---|---|---|---|---|---|---|
| PI3Kδ | α/δ | β/δ | γ/δ | ||||
| 81 | 5-Cl |  | CN | 0.6 | 1716 | 166 | 120 |
| 91 | 8-Cl | 6 | 517 | 65 | 183 | ||
| 101 | 5,8-Cl | 0.7 | 1857 | 108 | 470 | ||
| 111 | 5-F,8-Cl | 0.4 | 1925 | 115 | 127 | ||
| 122 | 6-F | 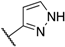 | Cl | 9.8 | 8 | 4 | 238 |
| 132 | 6-F,8-Cl | 5.3 | 109 | 1.5 | >357 | ||
| 143 | H | 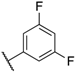 | CN | 15 | 530 | 290 | 74 |
| 153 | 6-F | 4.8 | 600 | 520 | 160 | ||
| 163 | 5-Cl, 6-F | 2.9 | 780 | 470 | 74 | ||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miller, M.S.; Thompson, P.E.; Gabelli, S.B. Structural Determinants of Isoform Selectivity in PI3K Inhibitors. Biomolecules 2019, 9, 82. https://doi.org/10.3390/biom9030082
Miller MS, Thompson PE, Gabelli SB. Structural Determinants of Isoform Selectivity in PI3K Inhibitors. Biomolecules. 2019; 9(3):82. https://doi.org/10.3390/biom9030082
Chicago/Turabian StyleMiller, Michelle S., Philip E. Thompson, and Sandra B. Gabelli. 2019. "Structural Determinants of Isoform Selectivity in PI3K Inhibitors" Biomolecules 9, no. 3: 82. https://doi.org/10.3390/biom9030082
APA StyleMiller, M. S., Thompson, P. E., & Gabelli, S. B. (2019). Structural Determinants of Isoform Selectivity in PI3K Inhibitors. Biomolecules, 9(3), 82. https://doi.org/10.3390/biom9030082