Getting the Akt Together: Guiding Intracellular Akt Activity by PI3K


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Akt-Intrinsic Regulatory Mechanisms
2.1. Posttranslational Modifications
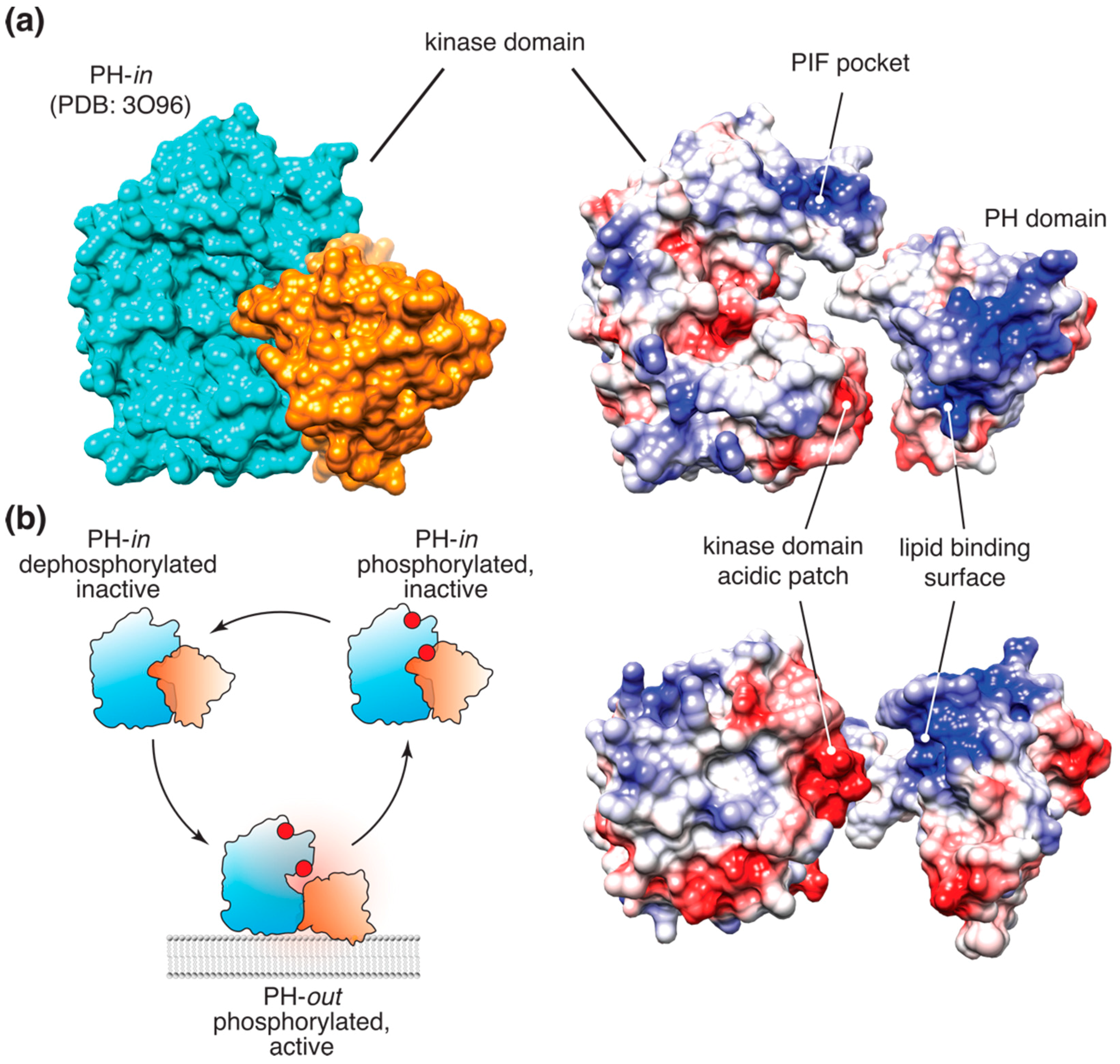
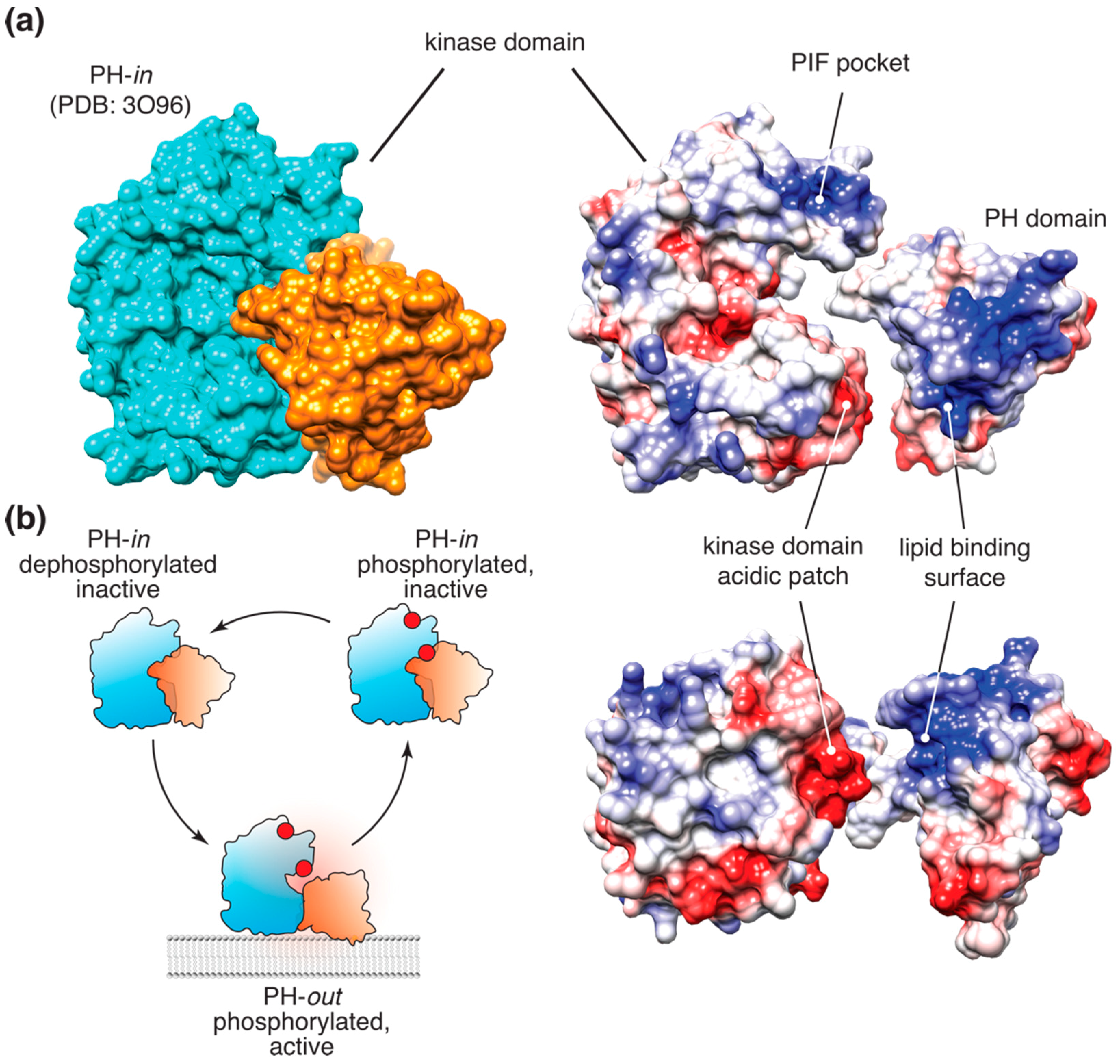
2.2. Akt Allosteric Regulation by Lipid Binding
2.3. Other Possible Mechanisms
3. Cell-Autonomous Akt Control Mechanisms
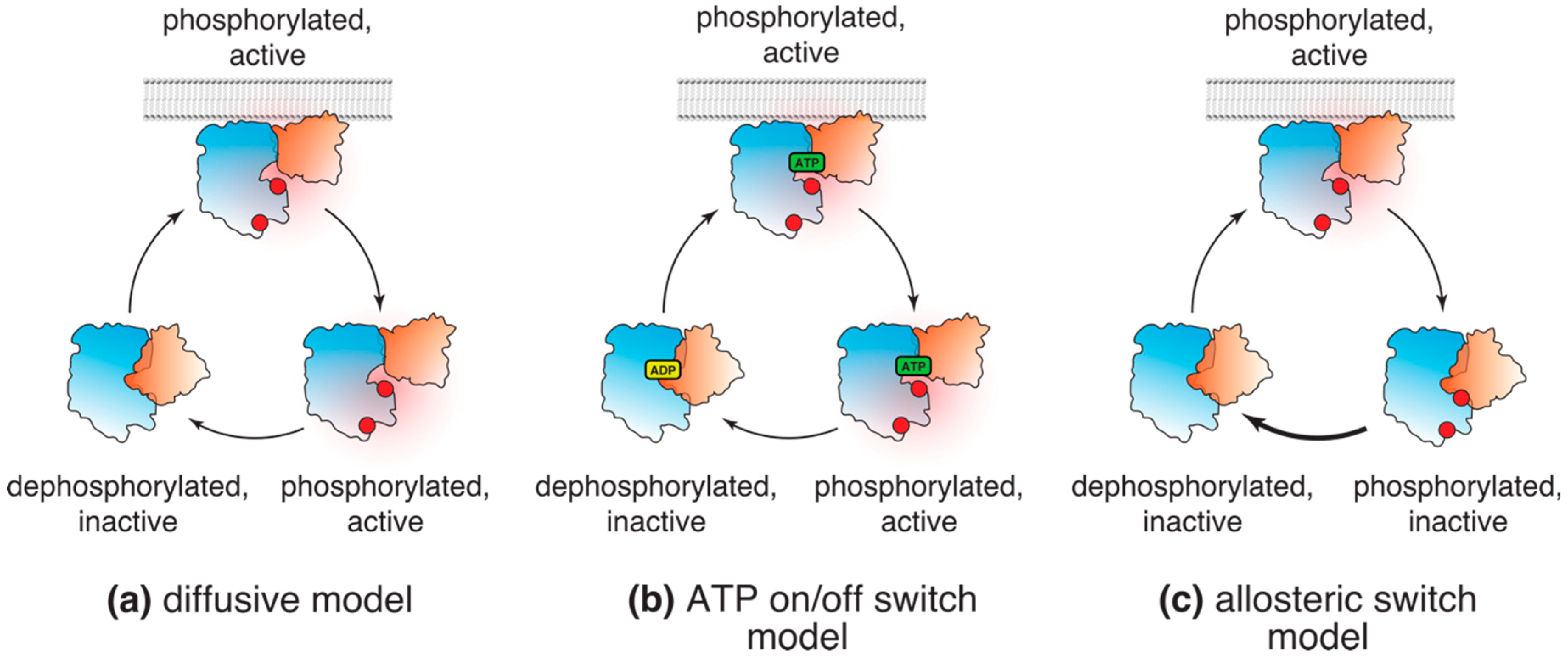
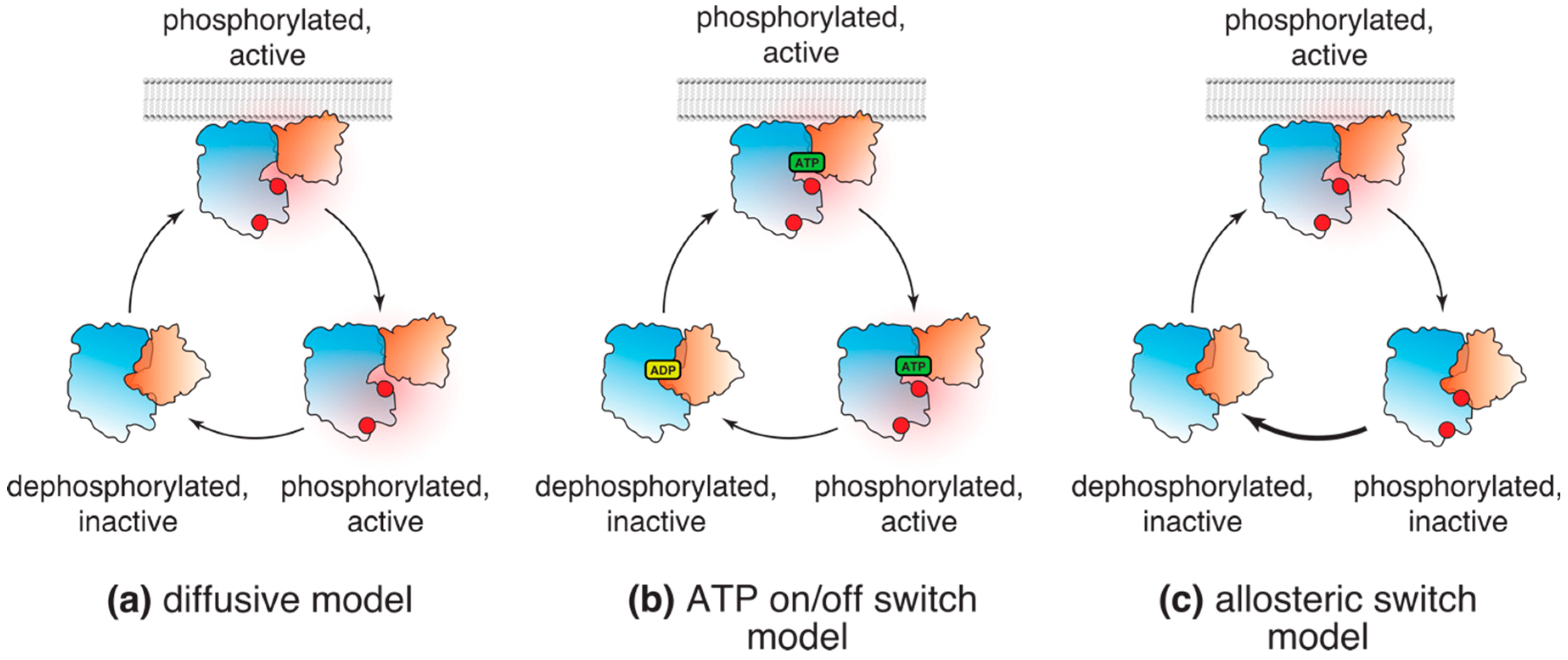
3.1. Classical, or Diffusive, Model
3.2. “ATP On/Off Switch” Model
3.3. Allosteric Switch Model
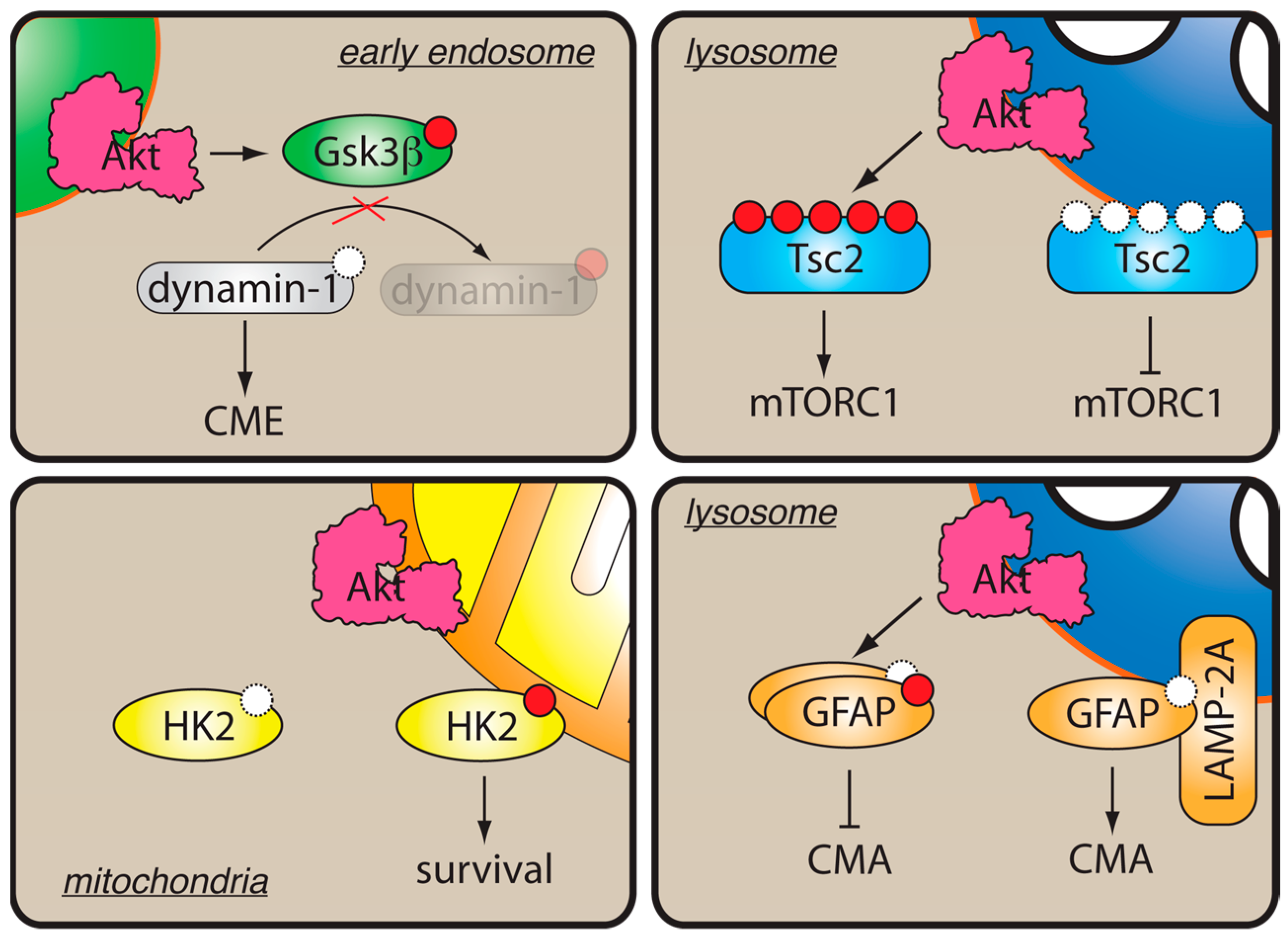
4. Physiological Relevance of PI3K/Akt Coupling
5. Open Questions
Funding
Acknowledgments
Conflicts of Interest
References
- Pearce, L.R.; Komander, D.; Alessi, D.R. The nuts and bolts of AGC protein kinases. Nat. Rev. Mol. Cell Biol. 2010, 11, 9–22. [Google Scholar] [CrossRef]
- Toker, A.; Marmiroli, S. Signaling specificity in the Akt pathway in biology and disease. Adv. Biol. Regul. 2014, 55, 28–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manning, B.D.; Toker, A. AKT/PKB Signaling: Navigating the Network. Cell 2017, 169, 381–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manning, B.D.; Cantley, L.C. AKT/PKB signaling: Navigating downstream. Cell 2007, 129, 1261–1274. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, S.J.; Yang, G.; Yang, P.; Fazakerley, D.J.; Stöckli, J.; Yang, J.Y.; James, D.E. Dynamic adipocyte phosphoproteome reveals that Akt directly regulates mTORC2. Cell Metab. 2013, 17, 1009–1020. [Google Scholar] [CrossRef] [PubMed]
- Hers, I.; Vincent, E.E.; Tavaré, J.M. Akt signalling in health and disease. Cell. Signal. 2011, 23, 1515–1527. [Google Scholar] [CrossRef] [PubMed]
- Risso, G.; Blaustein, M.; Pozzi, B.; Mammi, P.; Srebrow, A. Akt/PKB: One kinase, many modifications. Biochem. J. 2015, 468, 203–214. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Cron, P.; Good, V.M.; Thompson, V.; Hemmings, B.A.; Barford, D. Crystal structure of an activated Akt/protein kinase B ternary complex with GSK3-peptide and AMP-PNP. Nat. Struct. Biol. 2002, 9, 940–944. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Cron, P.; Thompson, V.; Good, V.M.; Hess, D.; Hemmings, B.A.; Barford, D. Molecular mechanism for the regulation of protein kinase B/Akt by hydrophobic motif phosphorylation. Mol. Cell 2002, 9, 1227–1240. [Google Scholar] [CrossRef]
- Sarbassov, D.D.; Guertin, D.A.; Ali, S.M.; Sabatini, D.M. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 2005, 307, 1098–1101. [Google Scholar] [CrossRef]
- Guertin, D.A.; Stevens, D.M.; Thoreen, C.C.; Burds, A.A.; Kalaany, N.Y.; Moffat, J.; Brown, M.; Fitzgerald, K.J.; Sabatini, D.M. Ablation in Mice of the mTORC Components raptor, rictor, or mLST8 Reveals that mTORC2 Is Required for Signaling to Akt-FOXO and PKCα, but Not S6K1. Dev. Cell 2006, 11, 859–871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, N.; Salguero, A.L.; Liu, A.Z.; Chen, Z.; Dempsey, D.R.; Ficarro, S.B.; Alexander, W.M.; Marto, J.A.; Li, Y.; Amzel, L.M.; et al. Akt Kinase Activation Mechanisms Revealed Using Protein Semisynthesis. Cell 2018, 174, 897–907.e14. [Google Scholar] [CrossRef] [PubMed]
- Biondi, R.M.; Kieloch, A.; Currie, R.A.; Deak, M.; Alessi, D.R. The PIF-binding pocket in PDK1 is essential for activation of S6K and SGK, but not PKB. EMBO J. 2001, 20, 4380–4390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ebner, M.; Lučić, I.; Leonard, T.A.; Yudushkin, I. PI(3,4,5)P3 Engagement Restricts Akt Activity to Cellular Membranes. Mol. Cell 2017, 65, 416–431.e6. [Google Scholar] [CrossRef] [PubMed]
- Arencibia, J.M.; Pastor-Flores, D.; Bauer, A.F.; Schulze, J.O.; Biondi, R.M. AGC protein kinases: From structural mechanism of regulation to allosteric drug development for the treatment of human diseases. Biochim. Biophys. Acta 2013, 1834, 1302–1321. [Google Scholar] [CrossRef] [PubMed]
- Balzano, D.; Fawal, M.-A.; Velázquez, J.V.; Santiveri, C.M.; Yang, J.; Pastor, J.; Campos-Olivas, R.; Djouder, N.; Lietha, D. Alternative Activation Mechanisms of Protein Kinase B Trigger Distinct Downstream Signaling Responses. J. Biol. Chem. 2015, 290, 24975–24985. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Begley, M.; Michowski, W.; Inuzuka, H.; Ginzberg, M.; Gao, D.; Tsou, P.; Gan, W.; Papa, A.; Kim, B.M.; et al. Cell-cycle-regulated activation of Akt kinase by phosphorylation at its carboxyl terminus. Nature 2014, 508, 541–545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahajan, K.; Mahajan, N.P. PI3K-independent AKT activation in cancers: A treasure trove for novel therapeutics. J. Cell. Physiol. 2012, 227, 3178–3184. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.; Chakraborty, A.A.; Liu, P.; Gan, W.; Zheng, X.; Inuzuka, H.; Wang, B.; Zhang, J.; Zhang, L.; Yuan, M.; et al. pVHL suppresses kinase activity of Akt in a proline-hydroxylation-dependent manner. Science 2016, 353, 929–932. [Google Scholar] [CrossRef] [Green Version]
- Yoon, S.-O.; Shin, S.; Karreth, F.A.; Buel, G.R.; Jedrychowski, M.P.; Plas, D.R.; Dedhar, S.; Gygi, S.P.; Roux, P.P.; Dephoure, N.; et al. Focal Adhesion- and IGF1R-Dependent Survival and Migratory Pathways Mediate Tumor Resistance to mTORC1/2 Inhibition. Mol. Cell 2017, 67, 512–527.e4. [Google Scholar] [CrossRef]
- Stokoe, D.; Stephens, L.R.; Copeland, T.; Gaffney, P.R.; Reese, C.B.; Painter, G.F.; Holmes, A.B.; McCormick, F.; Hawkins, P.T. Dual role of phosphatidylinositol-3,4,5-trisphosphate in the activation of protein kinase B. Science 1997, 277, 567–570. [Google Scholar] [CrossRef]
- Alessi, D.R.; Deak, M.; Casamayor, A.; Caudwell, F.B.; Morrice, N.; Norman, D.G.; Gaffney, P.; Reese, C.B.; MacDougall, C.N.; Harbison, D.; et al. 3-Phosphoinositide-dependent protein kinase-1 (PDK1): Structural and functional homology with the Drosophila DSTPK61 kinase. Curr. Biol. 1997, 7, 776–789. [Google Scholar] [CrossRef]
- Ebner, M.; Sinkovics, B.; Szczygieł, M.; Ribeiro, D.W.; Yudushkin, I. Localization of mTORC2 activity inside cells. J. Cell Biol. 2017, 216, 343–353. [Google Scholar] [CrossRef] [Green Version]
- Bellacosa, A.; Chan, T.O.; Ahmed, N.N.; Datta, K.; Malstrom, S.; Stokoe, D.; McCormick, F.; Feng, J.; Tsichlis, P. Akt activation by growth factors is a multiple-step process: The role of the PH domain. Oncogene 1998, 17, 313–325. [Google Scholar] [CrossRef] [PubMed]
- Andjelković, M.; Alessi, D.R.; Meier, R.; Fernandez, A.; Lamb, N.J.; Frech, M.; Cron, P.; Cohen, P.; Lucocq, J.M.; Hemmings, B.A. Role of translocation in the activation and function of protein kinase B. J. Biol. Chem. 1997, 272, 31515–31524. [Google Scholar] [CrossRef] [PubMed]
- Sundaresan, N.R.; Pillai, V.B.; Wolfgeher, D.; Samant, S.; Vasudevan, P.; Parekh, V.; Raghuraman, H.; Cunningham, J.M.; Gupta, M.; Gupta, M.P. The Deacetylase SIRT1 Promotes Membrane Localization and Activation of Akt and PDK1 During Tumorigenesis and Cardiac Hypertrophy. Sci. Signal. 2011, 4, ra46. [Google Scholar] [CrossRef]
- Yang, W.-L.; Jin, G.; Li, C.-F.; Jeong, Y.S.; Moten, A.; Xu, D.; Feng, Z.; Chen, W.; Cai, Z.; Darnay, B.; et al. Cycles of Ubiquitination and Deubiquitination Critically Regulate Growth Factor-Mediated Activation of Akt Signaling. Sci. Signal. 2013, 6, ra3. [Google Scholar] [CrossRef] [PubMed]
- Malstrom, S.; Tili, E.; Kappes, D.; Ceci, J.D.; Tsichlis, P.N. Tumor induction by an Lck-MyrAkt transgene is delayed by mechanisms controlling the size of the thymus. Proc. Natl. Acad. Sci. USA 2001, 98, 14967–14972. [Google Scholar] [CrossRef] [Green Version]
- Carpten, J.D.; Faber, A.L.; Horn, C.; Donoho, G.P.; Briggs, S.L.; Robbins, C.M.; Hostetter, G.; Boguslawski, S.; Moses, T.Y.; Savage, S.; et al. A transforming mutation in the pleckstrin homology domain of AKT1 in cancer. Nature 2007, 448, 439–444. [Google Scholar] [CrossRef]
- Calleja, V.; Alcor, D.; Laguerre, M.; Park, J.; Vojnovic, B.; Hemmings, B.A.; Downward, J.; Parker, P.J.; Larijani, B. Intramolecular and intermolecular interactions of protein kinase B define its activation in vivo. PLoS Biol. 2007, 5, e95. [Google Scholar] [CrossRef]
- Calleja, V.; Laguerre, M.; Larijani, B. 3-D structure and dynamics of protein kinase B-new mechanism for the allosteric regulation of an AGC kinase. J. Chem. Biol. 2009, 2, 11–25. [Google Scholar] [CrossRef] [PubMed]
- Calleja, V.; Laguerre, M.; Parker, P.J.; Larijani, B. Role of a novel PH-kinase domain interface in PKB/Akt regulation: Structural mechanism for allosteric inhibition. PLoS Biol. 2009, 7, e17. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.-I.; Voegtli, W.C.; Sturgis, H.L.; Dizon, F.P.; Vigers, G.P.A.; Brandhuber, B.J. Crystal structure of human AKT1 with an allosteric inhibitor reveals a new mode of kinase inhibition. PLoS ONE 2010, 5, e12913. [Google Scholar] [CrossRef] [PubMed]
- Parikh, C.; Janakiraman, V.; Wu, W.-I.; Foo, C.K.; Kljavin, N.M.; Chaudhuri, S.; Stawiski, E.; Lee, B.; Lin, J.; Li, H.; et al. Disruption of PH-kinase domain interactions leads to oncogenic activation of AKT in human cancers. Proc. Natl. Acad. Sci. USA 2012, 109, 19368–19373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landgraf, K.E.; Pilling, C.; Falke, J.J. Molecular mechanism of an oncogenic mutation that alters membrane targeting: Glu17Lys modifies the PIP lipid specificity of the AKT1 PH domain. Biochemistry 2008, 47, 12260–12269. [Google Scholar] [CrossRef] [PubMed]
- Sato, S.; Fujita, N.; Tsuruo, T. Modulation of Akt kinase activity by binding to Hsp90. Proc. Natl. Acad. Sci. USA 2000, 97, 10832–10837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theodoraki, M.A.; Kunjappu, M.; Sternberg, D.W.; Caplan, A.J. Akt shows variable sensitivity to an Hsp90 inhibitor depending on cell context. Exp. Cell Res. 2007, 313, 3851–3858. [Google Scholar] [CrossRef] [Green Version]
- Taipale, M.; Krykbaeva, I.; Whitesell, L.; Santagata, S.; Zhang, J.; Liu, Q.; Gray, N.S.; Lindquist, S. Chaperones as thermodynamic sensors of drug-target interactions reveal kinase inhibitor specificities in living cells. Nat. Biotechnol. 2013, 31, 630–637. [Google Scholar] [CrossRef] [Green Version]
- Noguchi, M.; Ropars, V.; Roumestand, C.; Suizu, F. Proto-oncogene TCL1: More than just a coactivator for Akt. FASEB J. 2007, 21, 2273–2284. [Google Scholar] [CrossRef]
- Auguin, D.; Barthe, P.; Royer, C.; Stern, M.-H.; Noguchi, M.; Arold, S.T.; Roumestand, C. Structural basis for the co-activation of protein kinase B by T-cell leukemia-1 (TCL1) family proto-oncoproteins. J. Biol. Chem. 2004, 279, 35890–35902. [Google Scholar] [CrossRef]
- Lin, A.; Hu, Q.; Li, C.; Xing, Z.; Ma, G.; Wang, C.; Li, J.; Ye, Y.; Yao, J.; Liang, K.; et al. The LINK-A lncRNA interacts with PtdIns(3,4,5)P3 to hyperactivate AKT and confer resistance to AKT inhibitors. Nat. Cell Biol. 2017, 19, 238–251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, R.; D’Alessandro, L.A.; Kar, S.; Kramer, B.; She, B.; Kaschek, D.; Hahn, B.; Wrangborg, D.; Karlsson, J.; Kvarnström, M.; et al. Heterogeneous kinetics of AKT signaling in individual cells are accounted for by variable protein concentration. Front. Physiol. 2012, 3, 451. [Google Scholar] [CrossRef] [PubMed]
- Norris, D.M.; Yang, P.; Krycer, J.R.; Fazakerley, D.J.; James, D.E.; Burchfield, J.G. An improved Akt reporter reveals intra- and inter-cellular heterogeneity and oscillations in signal transduction. J. Cell Sci. 2017, 130, 2757–2766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruss, M.D.; Arias, E.B.; Lienhard, G.E.; Cartee, G.D. Increased phosphorylation of Akt substrate of 160 kDa (AS160) in rat skeletal muscle in response to insulin or contractile activity. Diabetes 2005, 54, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Galbaugh, T.; Cerrito, M.G.; Jose, C.C.; Cutler, M.L. EGF-induced activation of Akt results in mTOR-dependent p70S6 kinase phosphorylation and inhibition of HC11 cell lactogenic differentiation. BMC Cell Biol. 2006, 7, 34. [Google Scholar] [CrossRef] [PubMed]
- Kubota, H.; Noguchi, R.; Toyoshima, Y.; Ozaki, Y.-I.; Uda, S.; Watanabe, K.; Ogawa, W.; Kuroda, S. Temporal coding of insulin action through multiplexing of the AKT pathway. Mol. Cell 2012, 46, 820–832. [Google Scholar] [CrossRef] [PubMed]
- Kunkel, M.T.; Ni, Q.; Tsien, R.Y.; Zhang, J.; Newton, A.C. Spatio-temporal dynamics of protein kinase B/Akt signaling revealed by a genetically encoded fluorescent reporter. J. Biol. Chem. 2005, 280, 5581–5587. [Google Scholar] [CrossRef] [PubMed]
- Chan, T.O.; Zhang, J.; Rodeck, U.; Pascal, J.M.; Armen, R.S.; Spring, M.; Dumitru, C.D.; Myers, V.; Li, X.; Cheung, J.Y.; et al. Resistance of Akt kinases to dephosphorylation through ATP-dependent conformational plasticity. Proc. Natl. Acad. Sci. USA 2011, 108, E1120–E1127. [Google Scholar] [CrossRef]
- Lin, K.; Lin, J.; Wu, W.-I.; Ballard, J.; Lee, B.B.; Gloor, S.L.; Vigers, G.P.A.; Morales, T.H.; Friedman, L.S.; Skelton, N.; et al. An ATP-site on-off switch that restricts phosphatase accessibility of Akt. Sci. Signal. 2012, 5, ra37. [Google Scholar] [CrossRef]
- Chan, T.O.; Zhang, J.; Tiegs, B.C.; Blumhof, B.; Yan, L.; Keny, N.; Penny, M.; Li, X.; Pascal, J.M.; Armen, R.S.; et al. Akt kinase C-terminal modifications control activation loop dephosphorylation and enhance insulin response. Biochem. J. 2015, 471, 37–51. [Google Scholar] [CrossRef] [Green Version]
- Han, E.K.-H.; Leverson, J.D.; McGonigal, T.; Shah, O.J.; Woods, K.W.; Hunter, T.; Giranda, V.L.; Luo, Y. Akt inhibitor A-443654 induces rapid Akt Ser-473 phosphorylation independent of mTORC1 inhibition. Oncogene 2007, 26, 5655–5661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okuzumi, T.; Fiedler, D.; Zhang, C.; Gray, D.C.; Aizenstein, B.; Hoffman, R.; Shokat, K.M. Inhibitor hijacking of Akt activation. Nat. Methods 2009, 5, 484–493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, T.; Furnari, F.; Newton, A.C. PHLPP: A Phosphatase that Directly Dephosphorylates Akt, Promotes Apoptosis, and Suppresses Tumor Growth. Mol. Cell 2005, 18, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Yang, H.; Liu, J.; Schmidt, M.D.; Gao, T. Scribble-mediated membrane targeting of PHLPP1 is required for its negative regulation of Akt. EMBO Rep. 2011, 12, 818–824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newton, A.C.; Trotman, L.C. Turning off AKT: PHLPP as a drug target. Annu. Rev. Pharmacol. Toxicol. 2014, 54, 537–558. [Google Scholar] [CrossRef] [PubMed]
- Zhande, R.; Zhang, W.; Zheng, Y.; Pendleton, E.; Li, Y.; Polakiewicz, R.D.; Sun, X.J. Dephosphorylation by default, a potential mechanism for regulation of insulin receptor substrate-1/2, Akt, and ERK1/2. J. Biol. Chem. 2006, 281, 39071–39080. [Google Scholar] [CrossRef] [PubMed]
- Kleiman, L.B.; Maiwald, T.; Conzelmann, H.; Lauffenburger, D.A.; Sorger, P.K. Rapid phospho-turnover by receptor tyrosine kinases impacts downstream signaling and drug binding. Mol. Cell 2011, 43, 723–737. [Google Scholar] [CrossRef]
- Luo, Y.; Smith, R.A.; Guan, R.; Liu, X.; Klinghofer, V.; Shen, J.; Hutchins, C.; Richardson, P.; Holzman, T.; Rosenberg, S.H.; et al. Pseudosubstrate peptides inhibit Akt and induce cell growth inhibition. Biochemistry 2004, 43, 1254–1263. [Google Scholar] [CrossRef]
- Lindhurst, M.J.; Sapp, J.C.; Teer, J.K.; Johnston, J.J.; Finn, E.M.; Peters, K.; Turner, J.; Cannons, J.L.; Bick, D.; Blakemore, L.; et al. A mosaic activating mutation in AKT1 associated with the Proteus syndrome. N. Engl. J. Med. 2011, 365, 611–619. [Google Scholar] [CrossRef]
- Jansen, L.A.; Mirzaa, G.M.; Ishak, G.E.; O’Roak, B.J.; Hiatt, J.B.; Roden, W.H.; Gunter, S.A.; Christian, S.L.; Collins, S.; Adams, C.; et al. PI3K/AKT pathway mutations cause a spectrum of brain malformations from megalencephaly to focal cortical dysplasia. Brain 2015, 138, 1613–1628. [Google Scholar] [CrossRef] [Green Version]
- Bellacosa, A.; Testa, J.R.; Staal, S.P.; Tsichlis, P.N. A retroviral oncogene, akt, encoding a serine-threonine kinase containing an SH2-like region. Science 1991, 254, 274–277. [Google Scholar] [CrossRef] [PubMed]
- Yi, K.H.; Lauring, J. Recurrent AKT mutations in human cancers: Functional consequences and effects on drug sensitivity. Oncotarget 2015, 7, 4241–4251. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.-L.; Wang, Z.-G.; Hu, Y.; Xin, Y.; Singaram, I.; Gorai, S.; Zhou, X.; Shim, Y.; Min, J.-H.; Gong, L.-W.; et al. Quantitative Lipid Imaging Reveals a New Signaling Function of Phosphatidylinositol-3,4-Bisphophate: Isoform- and Site-Specific Activation of Akt. Mol. Cell 2018, 71, 1092–1104.e5. [Google Scholar] [CrossRef] [PubMed]
- Ivetac, I.; Gurung, R.; Hakim, S.; Horan, K.A.; Sheffield, D.A.; Binge, L.C.; Majerus, P.W.; Tiganis, T.; Mitchell, C.A. Regulation of PI(3)K/Akt signalling and cellular transformation by inositol polyphosphate 4-phosphatase-1. EMBO Rep. 2009, 10, 487–493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braccini, L.; Ciraolo, E.; Campa, C.C.; Perino, A.; Longo, D.L.; Tibolla, G.; Pregnolato, M.; Cao, Y.; Tassone, B.; Damilano, F.; et al. PI3K-C2γ is a Rab5 effector selectively controlling endosomal Akt2 activation downstream of insulin signalling. Nat. Commun. 2015, 6, 7400. [Google Scholar] [CrossRef] [PubMed]
- Su, X.; Lodhi, I.J.; Saltiel, A.R.; Stahl, P.D. Insulin-stimulated Interaction between insulin receptor substrate 1 and p85alpha and activation of protein kinase B/Akt require Rab5. J. Biol. Chem. 2006, 281, 27982–27990. [Google Scholar] [CrossRef] [PubMed]
- Posor, Y.; Eichhorn-Gruenig, M.; Puchkov, D.; Schöneberg, J.; Ullrich, A.; Lampe, A.; Müller, R.; Zarbakhsh, S.; Gulluni, F.; Hirsch, E.; et al. Spatiotemporal control of endocytosis by phosphatidylinositol-3,4-bisphosphate. Nature 2013, 499, 233–237. [Google Scholar] [CrossRef] [PubMed]
- Marat, A.L.; Wallroth, A.; Lo, W.-T.; Müller, R.; Norata, G.D.; Falasca, M.; Schultz, C.; Haucke, V. mTORC1 activity repression by late endosomal phosphatidylinositol 3,4-bisphosphate. Science 2017, 356, 968–972. [Google Scholar] [CrossRef]
- Wang, H.; Lo, W.-T.; Vujičić Žagar, A.; Gulluni, F.; Lehmann, M.; Scapozza, L.; Haucke, V.; Vadas, O. Autoregulation of Class II Alpha PI3K Activity by Its Lipid-Binding PX-C2 Domain Module. Mol. Cell 2018, 71, 343–351.e4. [Google Scholar] [CrossRef]
- Schenck, A.; Goto-Silva, L.; Collinet, C.; Rhinn, M.; Giner, A.; Habermann, B.; Brand, M.; Zerial, M. The endosomal protein Appl1 mediates Akt substrate specificity and cell survival in vertebrate development. Cell 2008, 133, 486–497. [Google Scholar] [CrossRef]
- Nazarewicz, R.R.; Salazar, G.; Patrushev, N.; San Martin, A.; Hilenski, L.; Xiong, S.; Alexander, R.W. Early endosomal antigen 1 (EEA1) is an obligate scaffold for angiotensin II-induced, PKC-alpha-dependent Akt activation in endosomes. J. Biol. Chem. 2011, 286, 2886–2895. [Google Scholar] [CrossRef] [PubMed]
- Chaturvedi, A.; Martz, R.; Dorward, D.; Waisberg, M.; Pierce, S.K. Endocytosed BCRs sequentially regulate MAPK and Akt signaling pathways from intracellular compartments. Nat. Immunol. 2011, 12, 1119–1126. [Google Scholar] [CrossRef] [Green Version]
- Zoncu, R.; Perera, R.M.; Balkin, D.M.; Pirruccello, M.; Toomre, D.; De Camilli, P. A phosphoinositide switch controls the maturation and signaling properties of APPL endosomes. Cell 2009, 136, 1110–1121. [Google Scholar] [CrossRef] [PubMed]
- Naguib, A.; Bencze, G.; Cho, H.; Zheng, W.; Tocilj, A.; Elkayam, E.; Faehnle, C.R.; Jaber, N.; Pratt, C.P.; Chen, M.; et al. PTEN functions by recruitment to cytoplasmic vesicles. Mol. Cell 2015, 58, 255–268. [Google Scholar] [CrossRef] [PubMed]
- Malek, M.; Kielkowska, A.; Chessa, T.; Anderson, K.E.; Barneda, D.; Pir, P.; Nakanishi, H.; Eguchi, S.; Koizumi, A.; Sasaki, J.; et al. PTEN Regulates PI(3,4)P2 Signaling Downstream of Class I PI3K. Mol. Cell 2017, 68, 566–580.e10. [Google Scholar] [CrossRef] [PubMed]
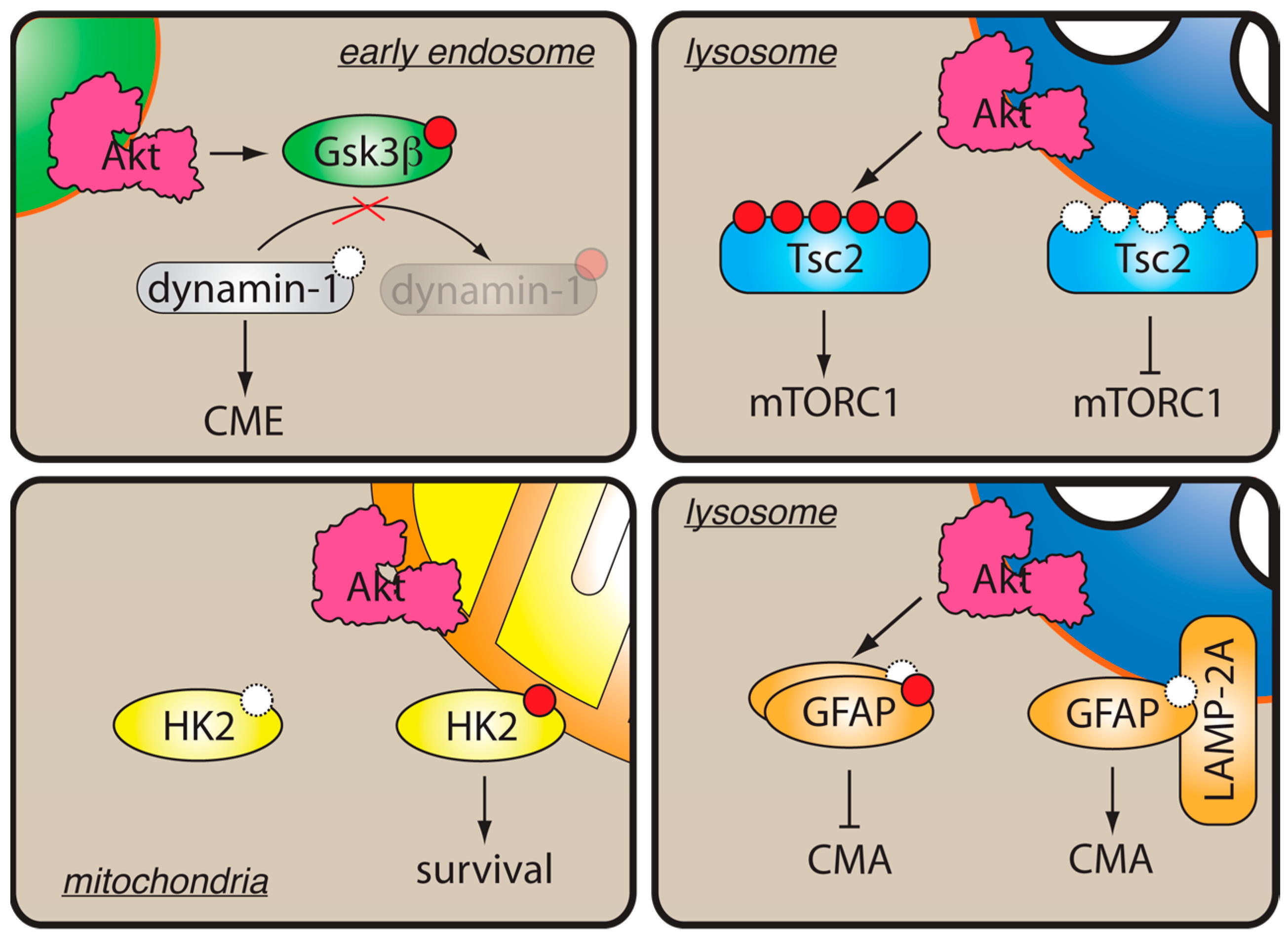
- Reis, C.R.; Chen, P.-H.; Srinivasan, S.; Aguet, F.; Mettlen, M.; Schmid, S.L. Crosstalk between Akt/GSK3β signaling and dynamin-1 regulates clathrin-mediated endocytosis. EMBO J. 2015, 34, 2132–2146. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.-H.; Bendris, N.; Hsiao, Y.-J.; Reis, C.R.; Mettlen, M.; Chen, H.-Y.; Yu, S.-L.; Schmid, S.L. Crosstalk between CLCb/Dyn1-Mediated Adaptive Clathrin-Mediated Endocytosis and Epidermal Growth Factor Receptor Signaling Increases Metastasis. Dev. Cell 2017, 40, 278–288.e5. [Google Scholar] [CrossRef] [Green Version]
- Schmid, S.L. Reciprocal regulation of signaling and endocytosis: Implications for the evolving cancer cell. J. Cell Biol. 2017, 216, 2623–2632. [Google Scholar] [CrossRef]
- Menon, S.; Dibble, C.C.; Talbott, G.; Hoxhaj, G.; Valvezan, A.J.; Takahashi, H.; Cantley, L.C.; Manning, B.D. Spatial control of the TSC complex integrates insulin and nutrient regulation of mTORC1 at the lysosome. Cell 2014, 156, 771–785. [Google Scholar] [CrossRef]
- Demetriades, C.; Doumpas, N.; Teleman, A.A. Regulation of TORC1 in response to amino acid starvation via lysosomal recruitment of TSC2. Cell 2014, 156, 786–799. [Google Scholar] [CrossRef]
- Roberts, D.J.; Miyamoto, S. Hexokinase II integrates energy metabolism and cellular protection: Akting on mitochondria and TORCing to autophagy. Cell Death Differ. 2015, 22, 248–257. [Google Scholar] [CrossRef] [PubMed]
- Maiuri, T.; Ho, J.; Stambolic, V. Regulation of adipocyte differentiation by distinct subcellular pools of protein kinase B (PKB/Akt). J. Biol. Chem. 2010, 285, 15038–15047. [Google Scholar] [CrossRef] [PubMed]
- Beg, M.; Abdullah, N.; Thowfeik, F.S.; Altorki, N.K.; McGraw, T.E. Distinct Akt phosphorylation states are required for insulin regulated Glut4 and Glut1-mediated glucose uptake. Elife 2017, 6, e26896. [Google Scholar] [CrossRef] [PubMed]
- Arias, E.; Koga, H.; Diaz, A.; Mocholi, E.; Patel, B.; Cuervo, A.M. Lysosomal mTORC2/PHLPP1/Akt Regulate Chaperone-Mediated Autophagy. Mol. Cell 2015, 59, 270–284. [Google Scholar] [CrossRef] [PubMed]
© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yudushkin, I. Getting the Akt Together: Guiding Intracellular Akt Activity by PI3K. Biomolecules 2019, 9, 67. https://doi.org/10.3390/biom9020067
Yudushkin I. Getting the Akt Together: Guiding Intracellular Akt Activity by PI3K. Biomolecules. 2019; 9(2):67. https://doi.org/10.3390/biom9020067
Chicago/Turabian StyleYudushkin, Ivan. 2019. "Getting the Akt Together: Guiding Intracellular Akt Activity by PI3K" Biomolecules 9, no. 2: 67. https://doi.org/10.3390/biom9020067
APA StyleYudushkin, I. (2019). Getting the Akt Together: Guiding Intracellular Akt Activity by PI3K. Biomolecules, 9(2), 67. https://doi.org/10.3390/biom9020067