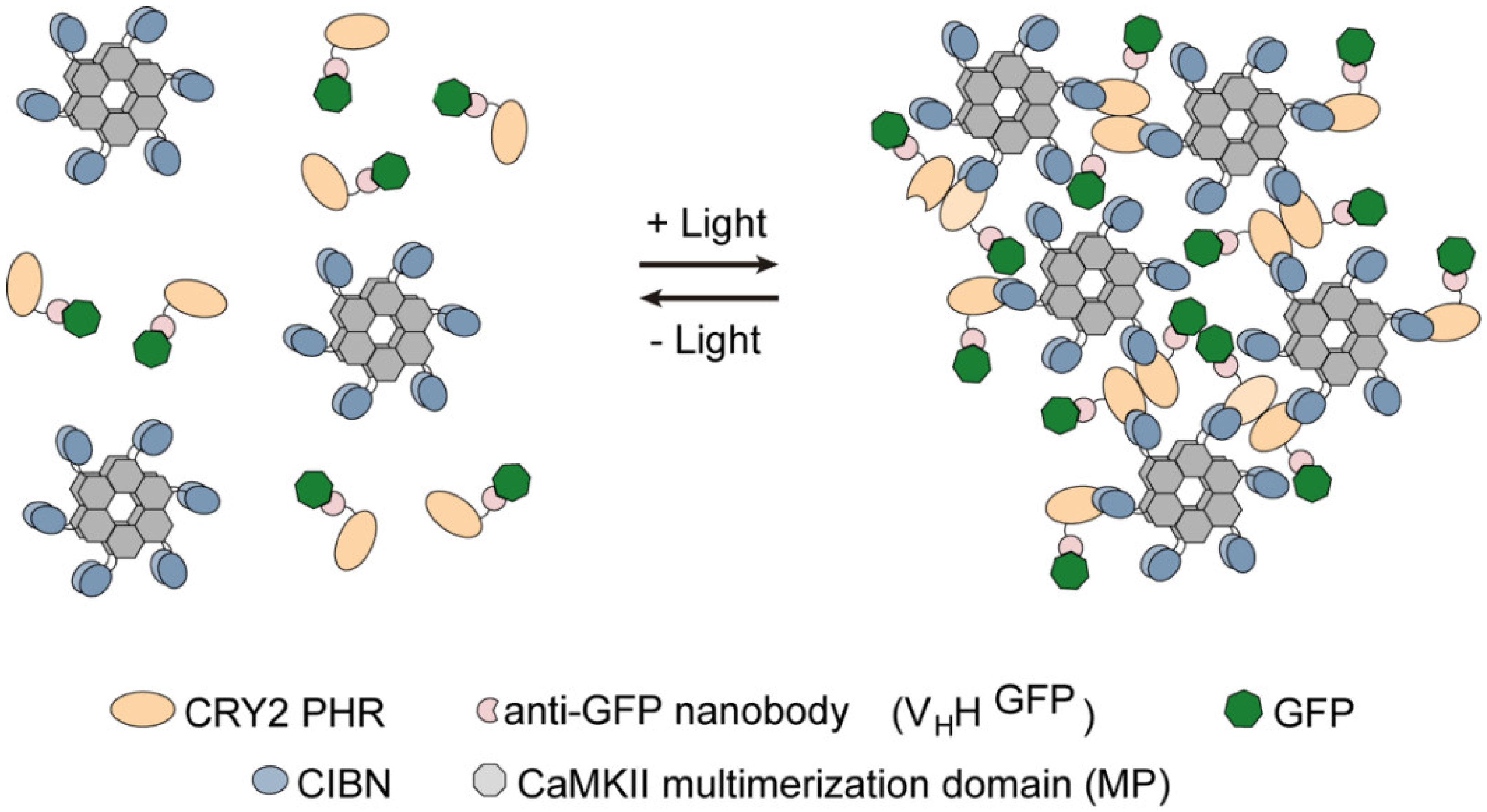
Light-Induced Protein Clustering for Optogenetic Interference and Protein Interaction Analysis in Drosophila S2 Cells


Abstract
:1. Introduction
2. Materials and Methods
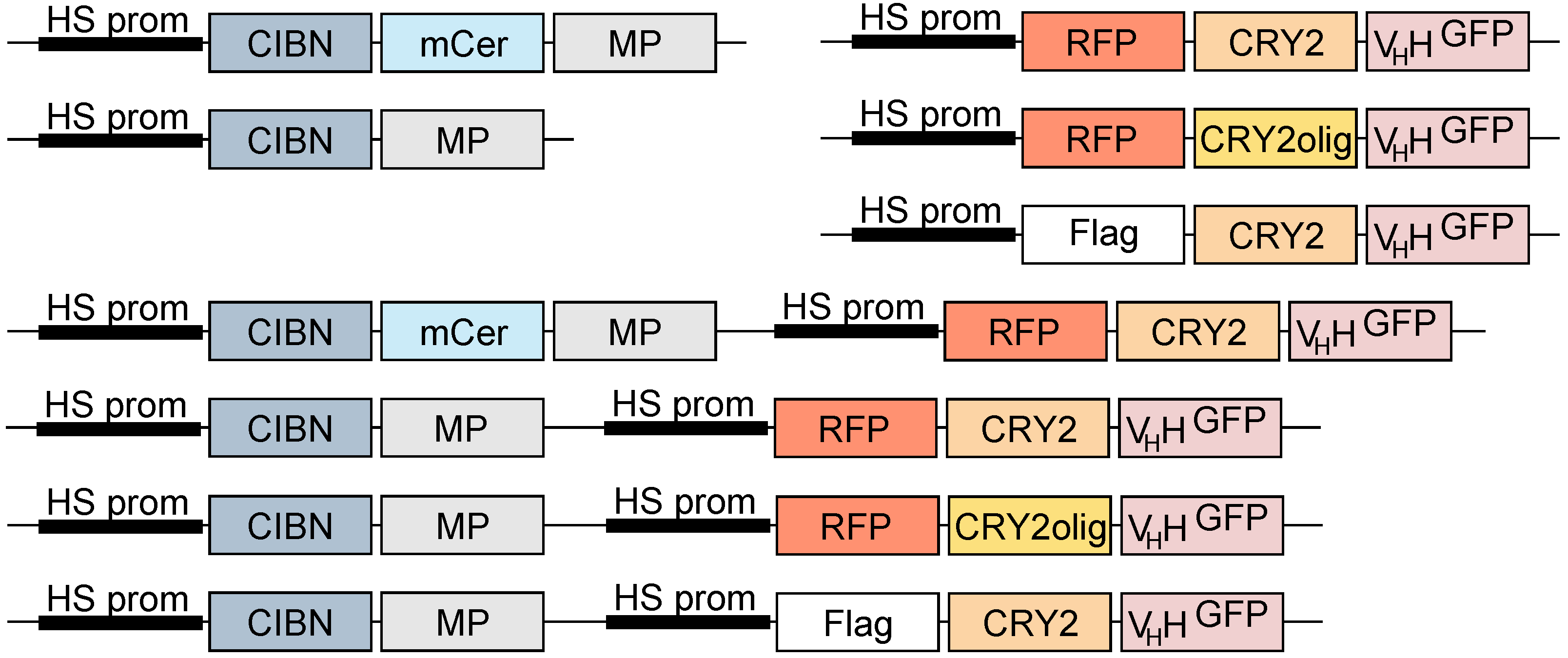
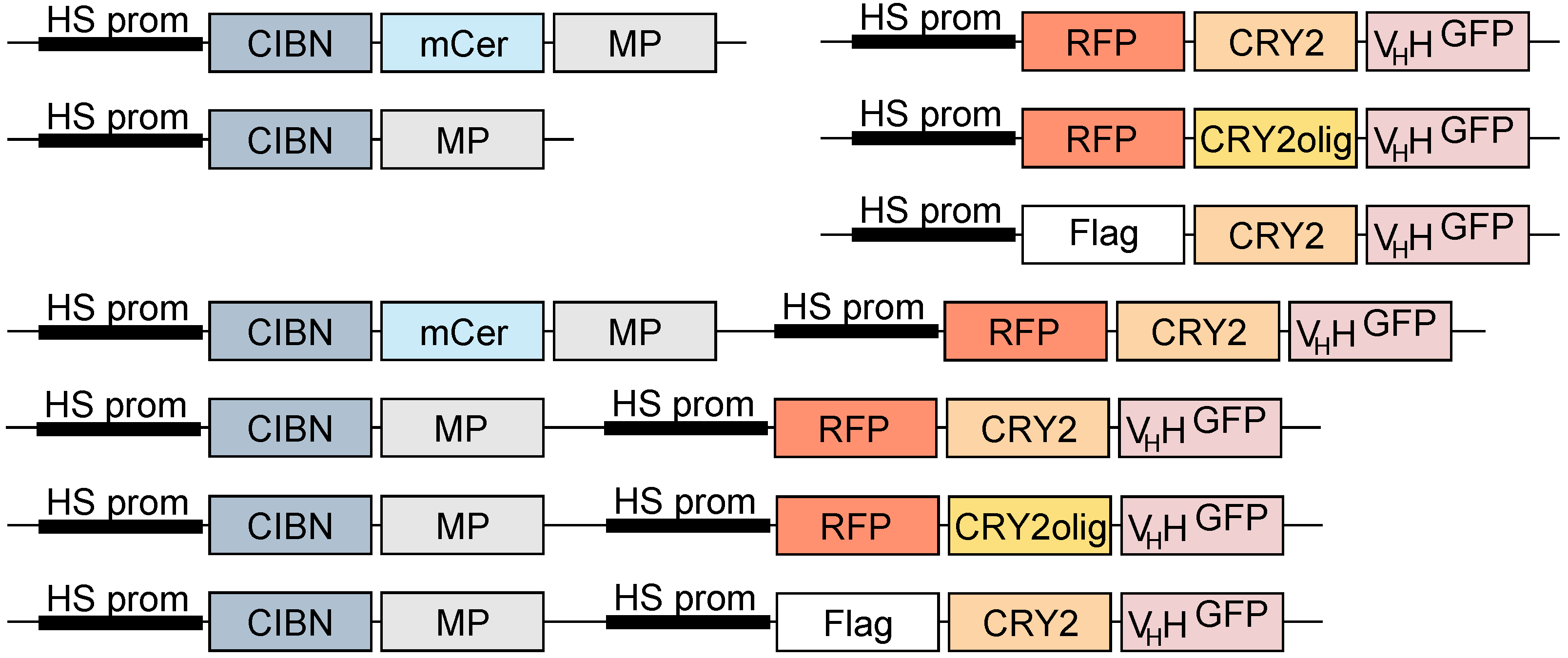
2.1. Molecular Biology
2.2. Schneider 2 Cell Culture, Transfection, and Imaging
2.3. Immunofluorescence
2.4. Image Analysis
3. Results and Discussion
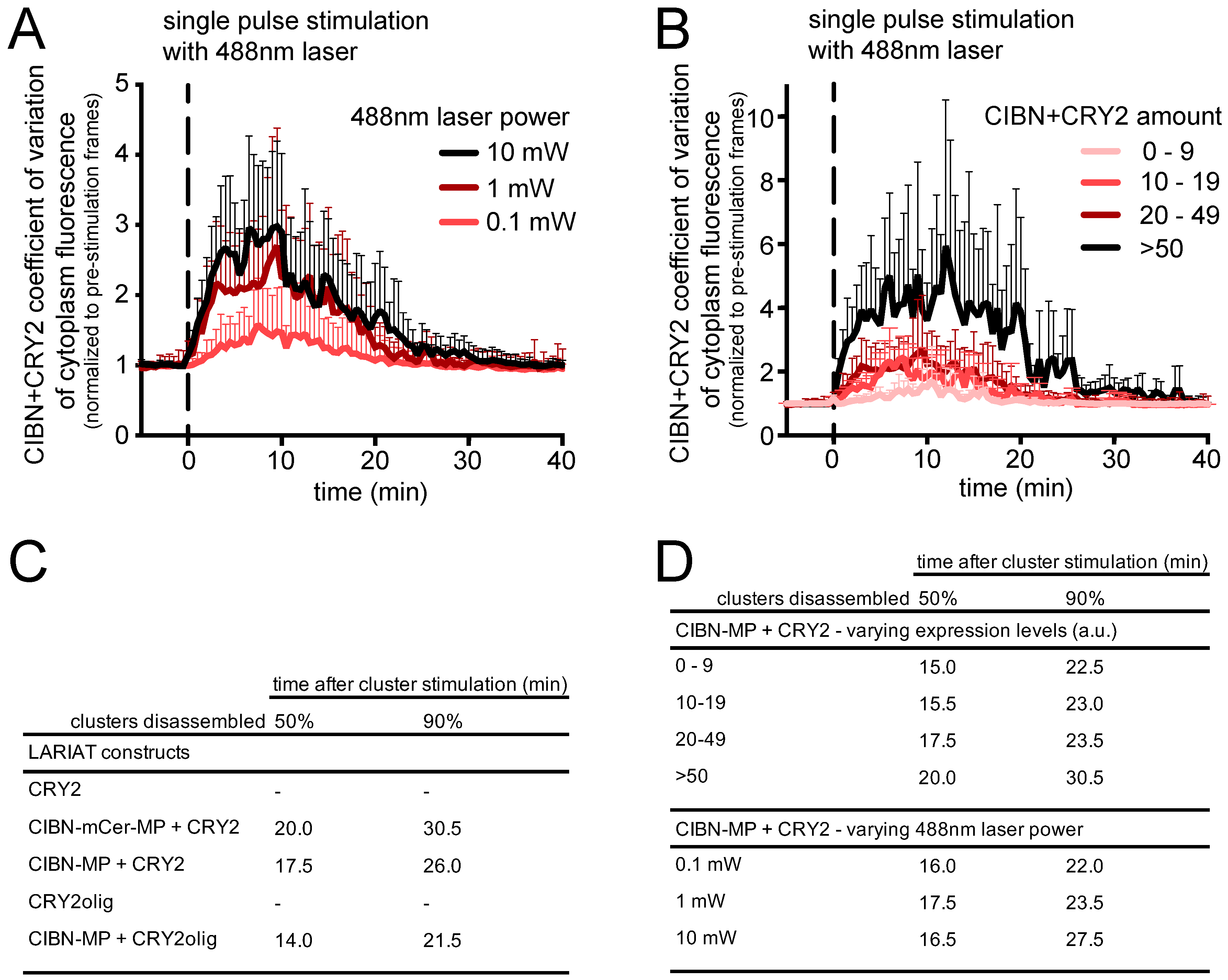
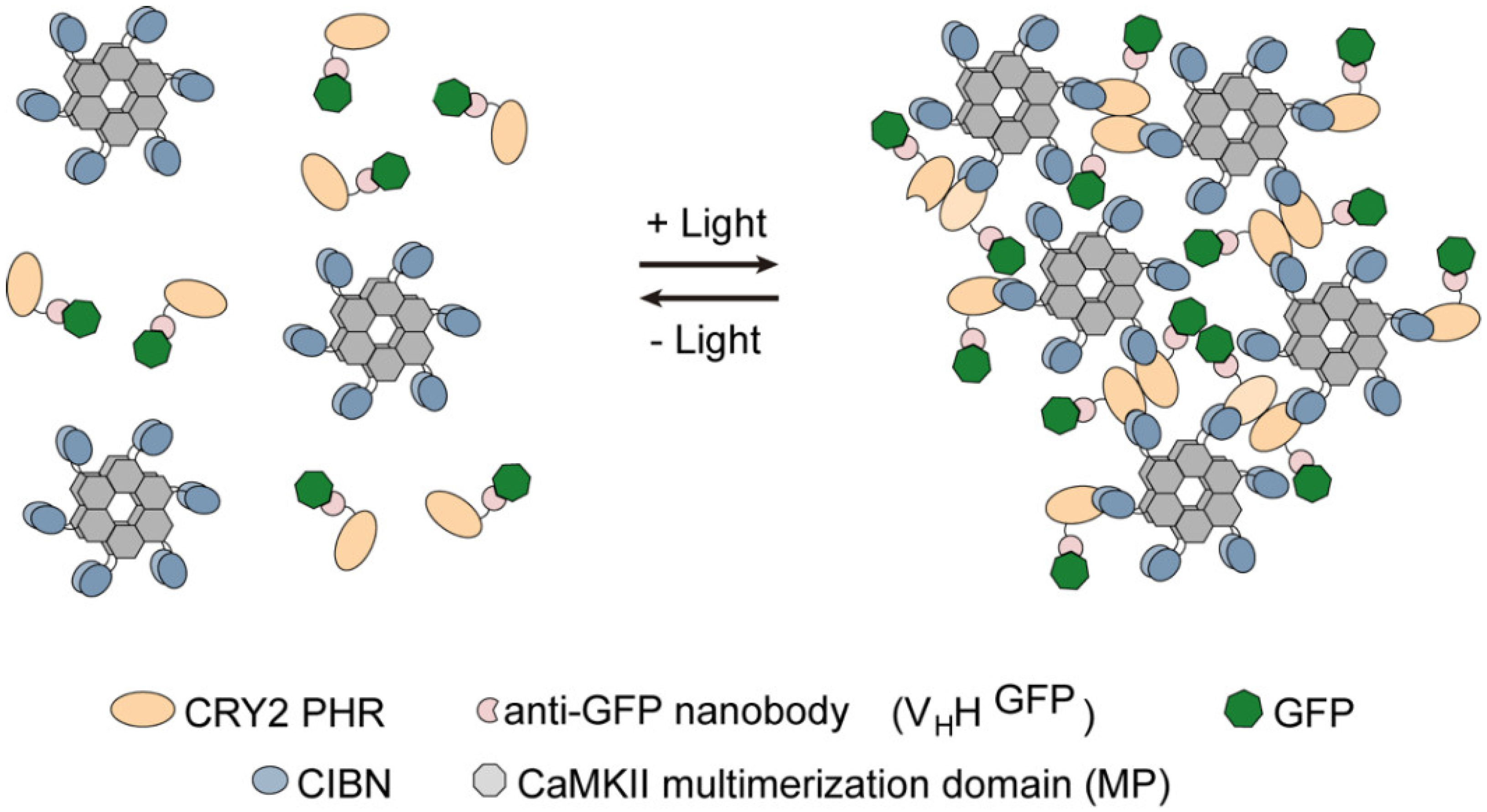
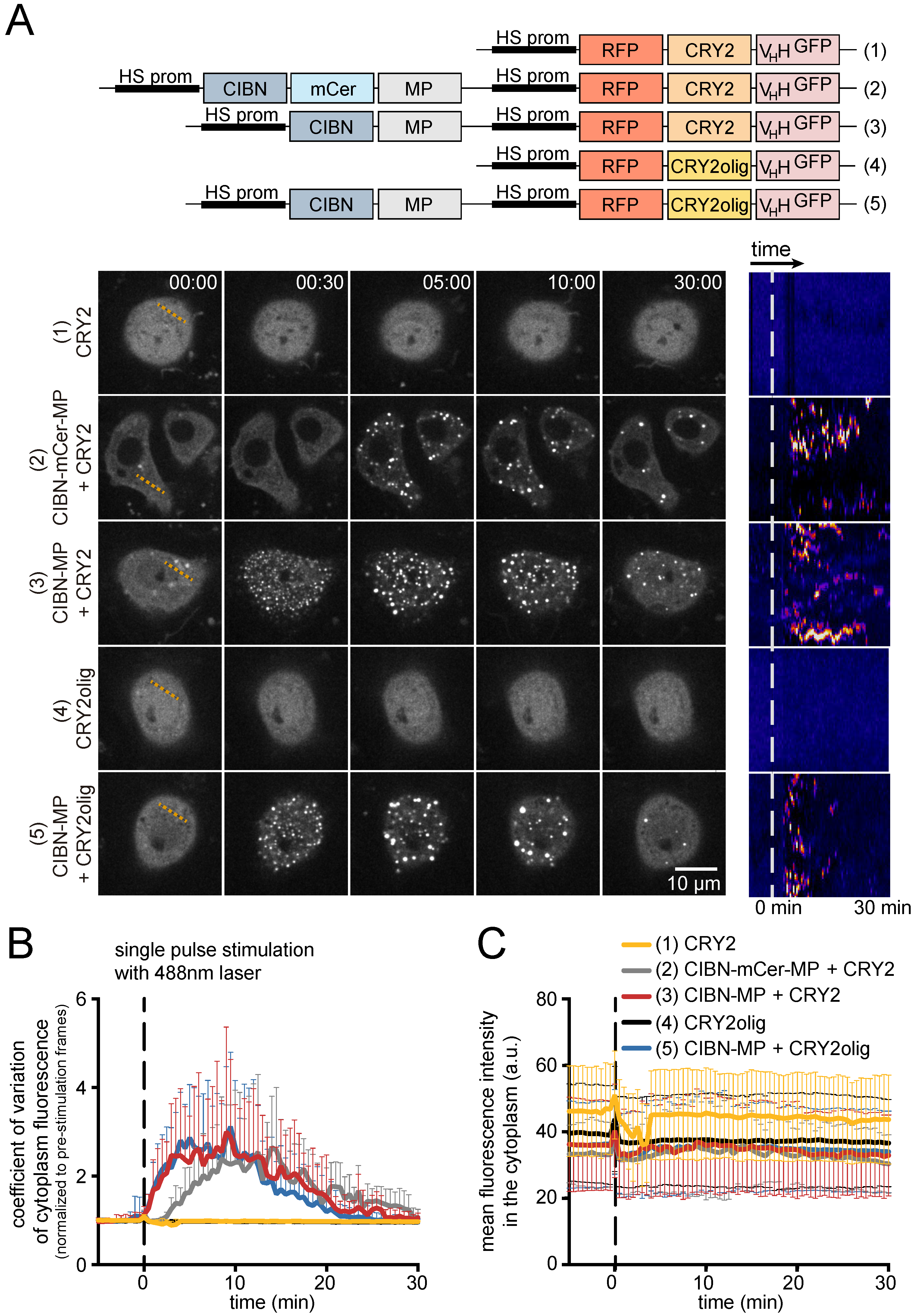
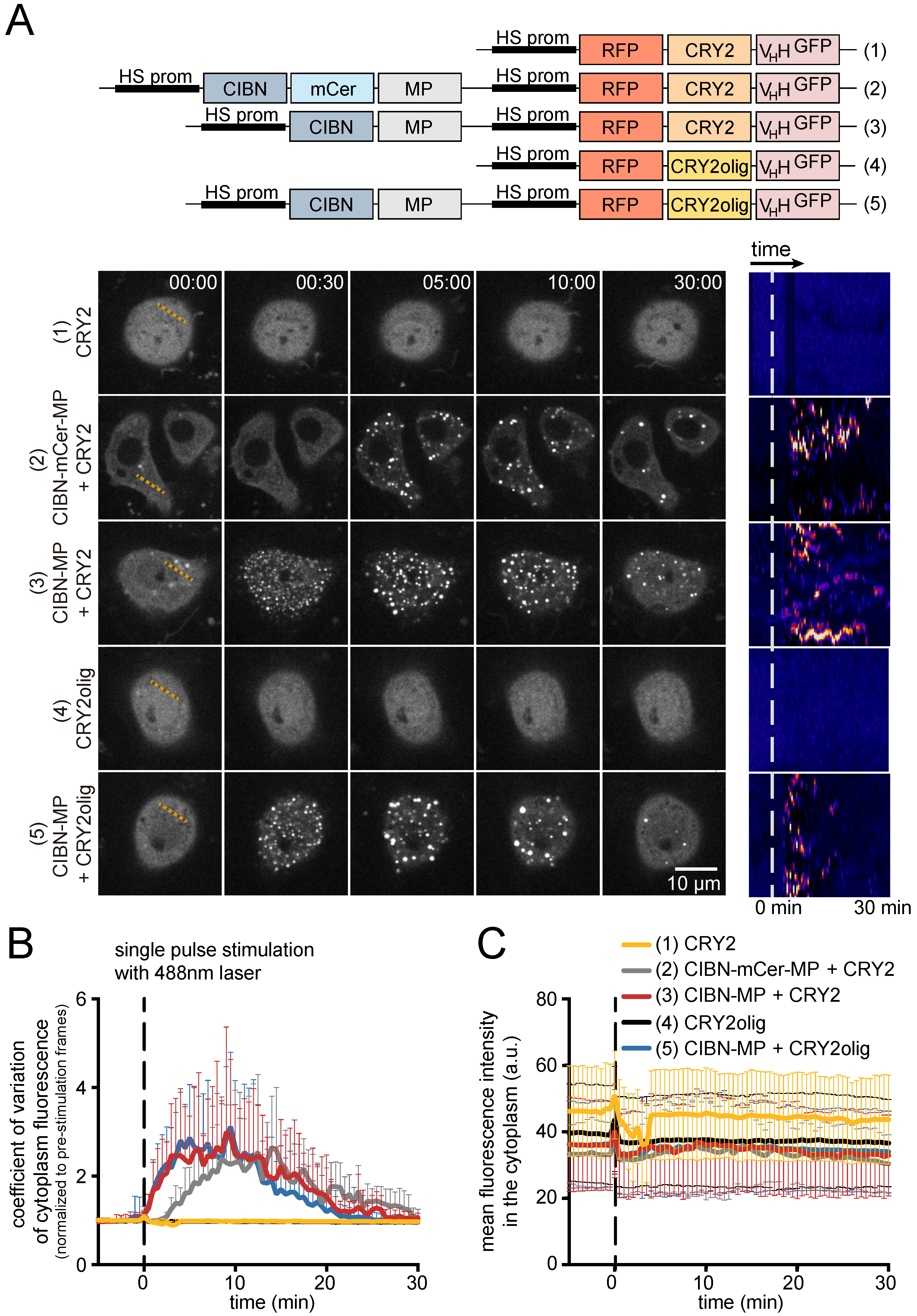
3.1. Generation and Characterization of Heat Shock-Inducible LARIAT Modules in S2 Cells
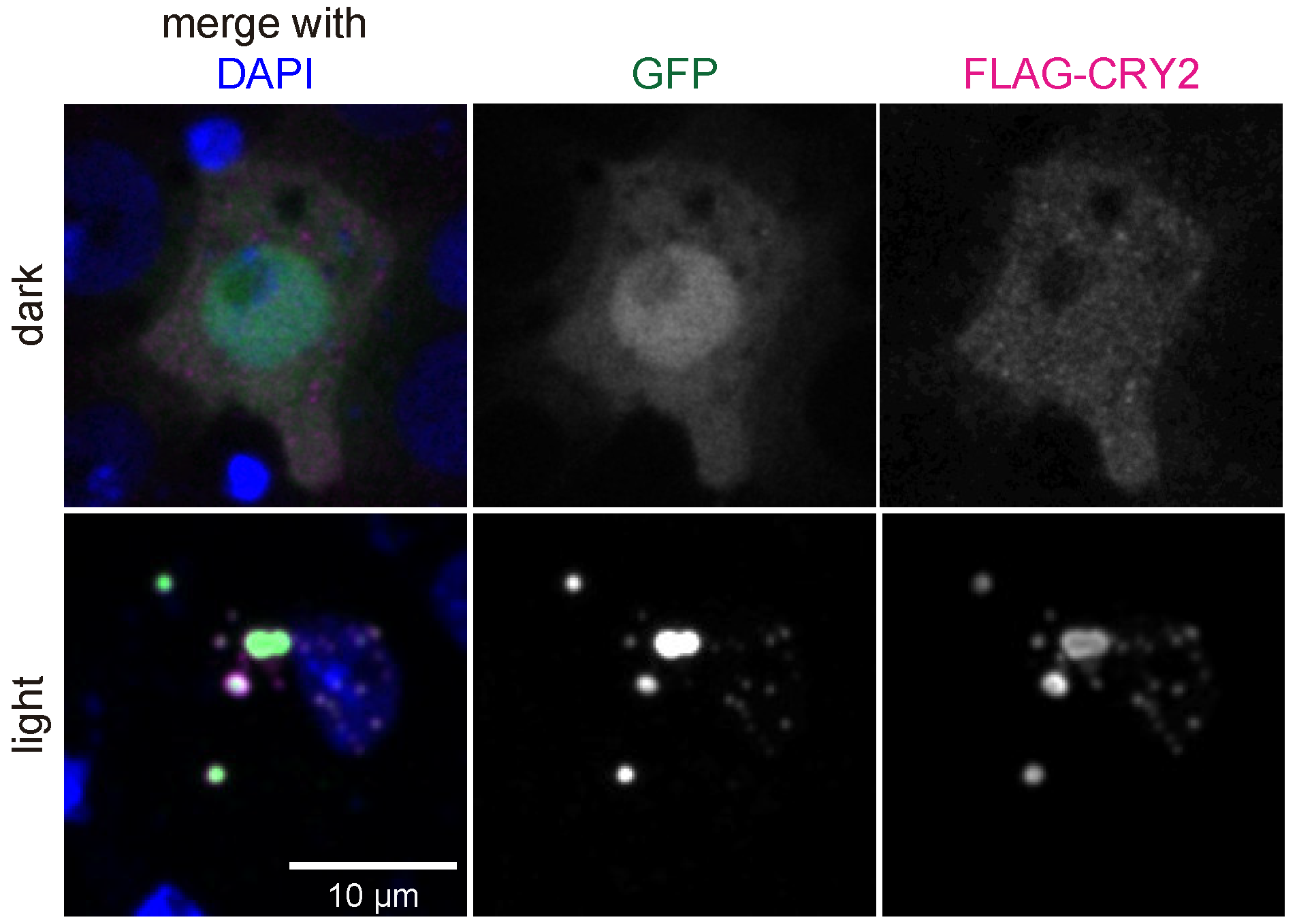
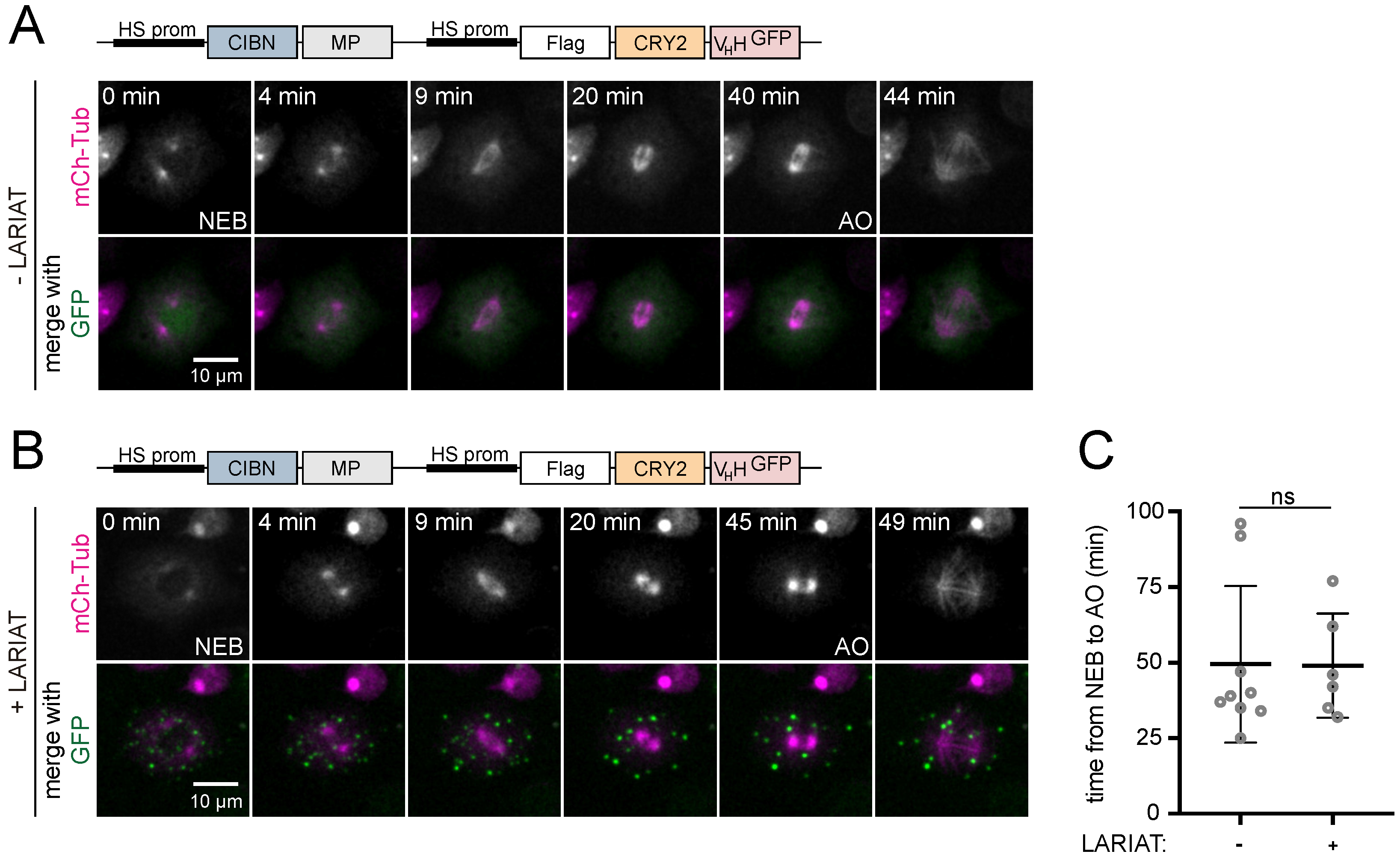
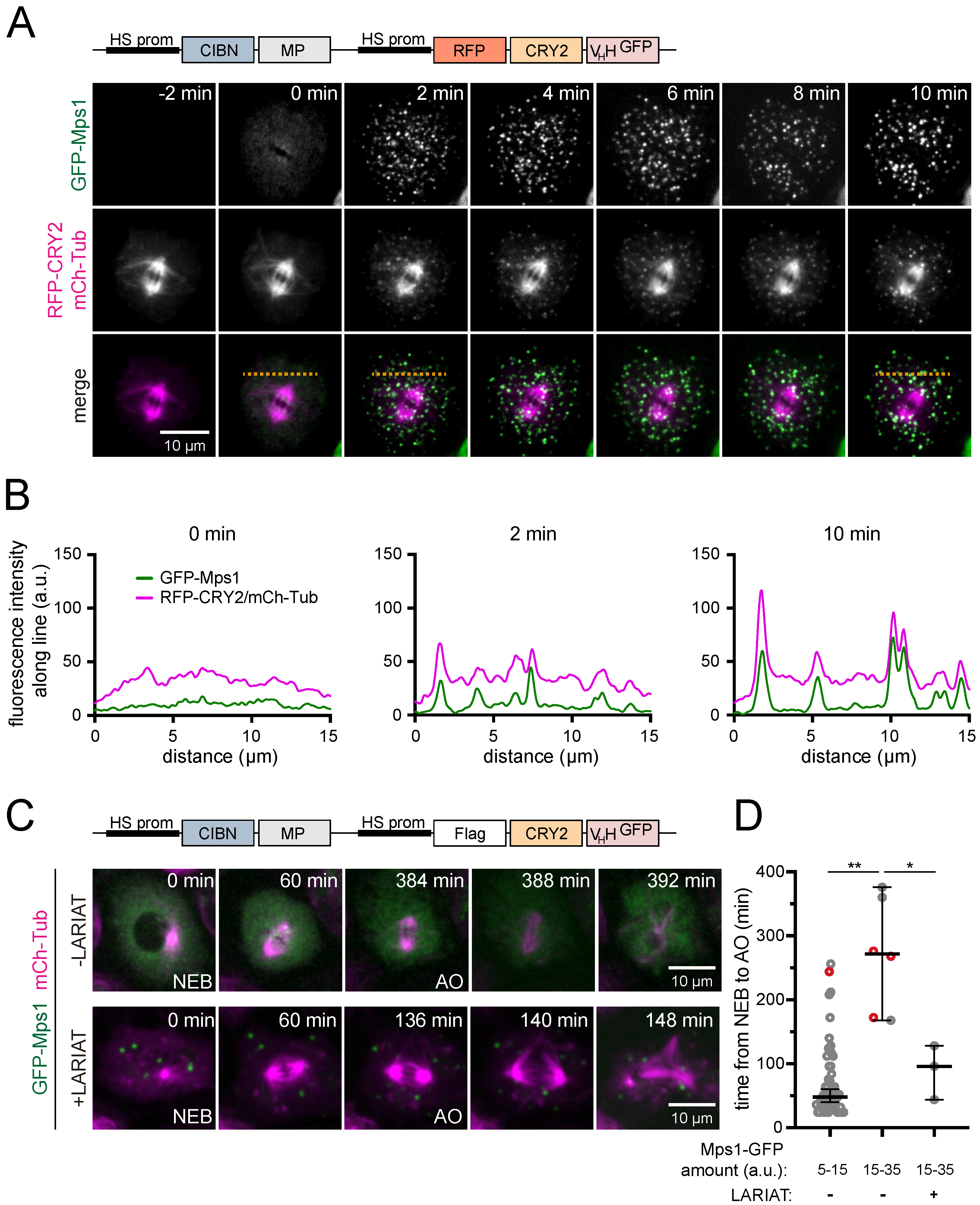
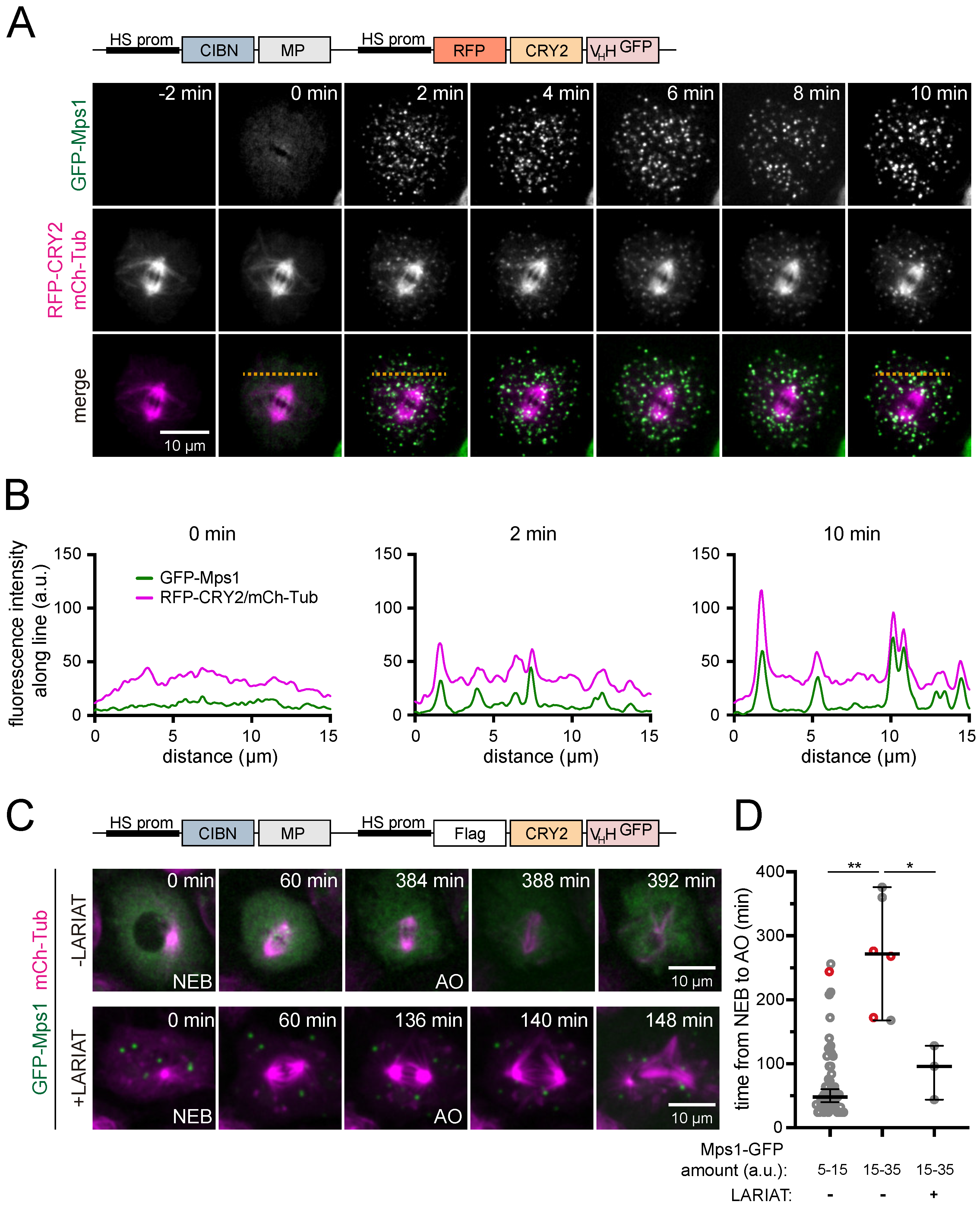
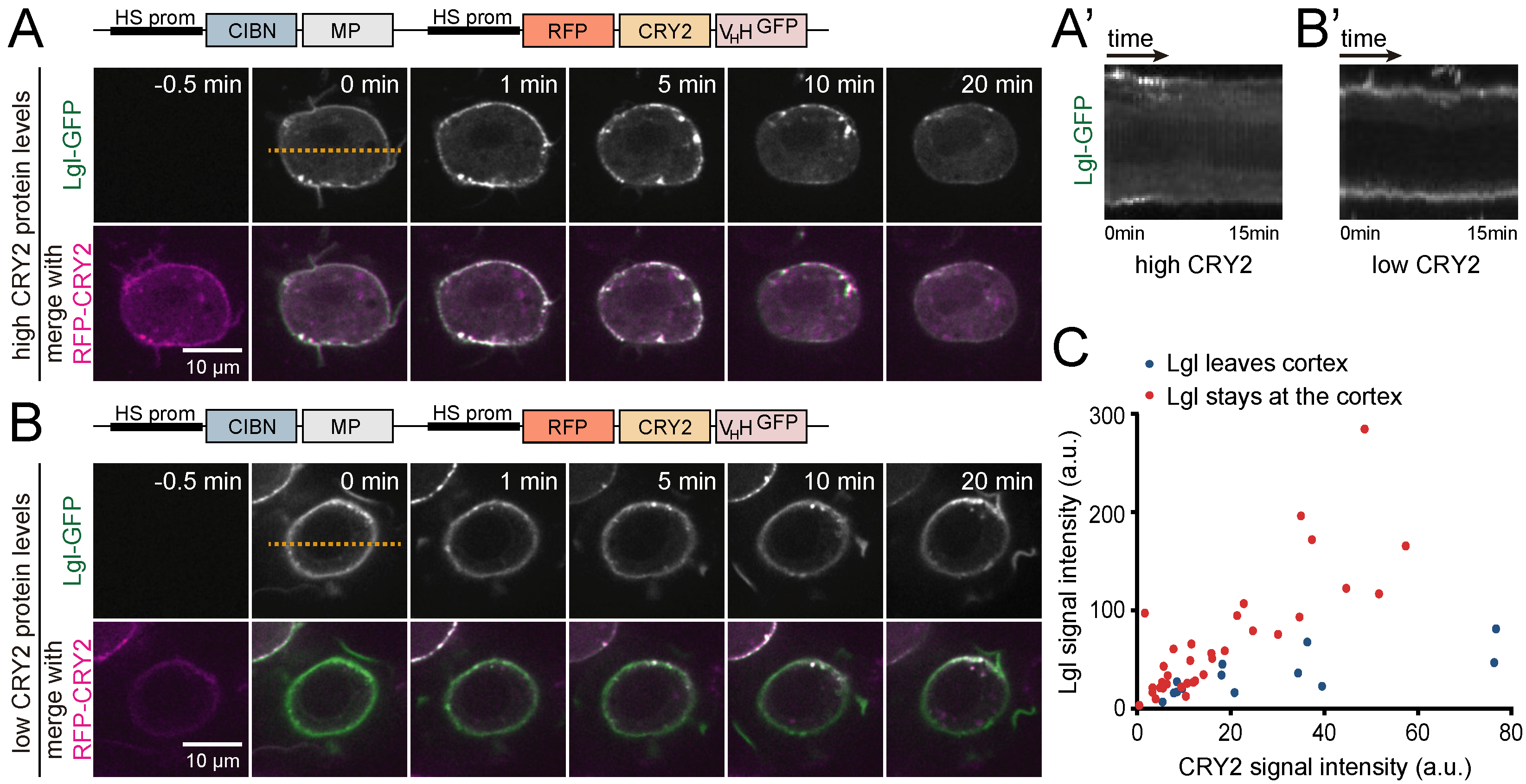
3.2. Interference with GFP-Tagged Proteins by Optogenetic Clustering
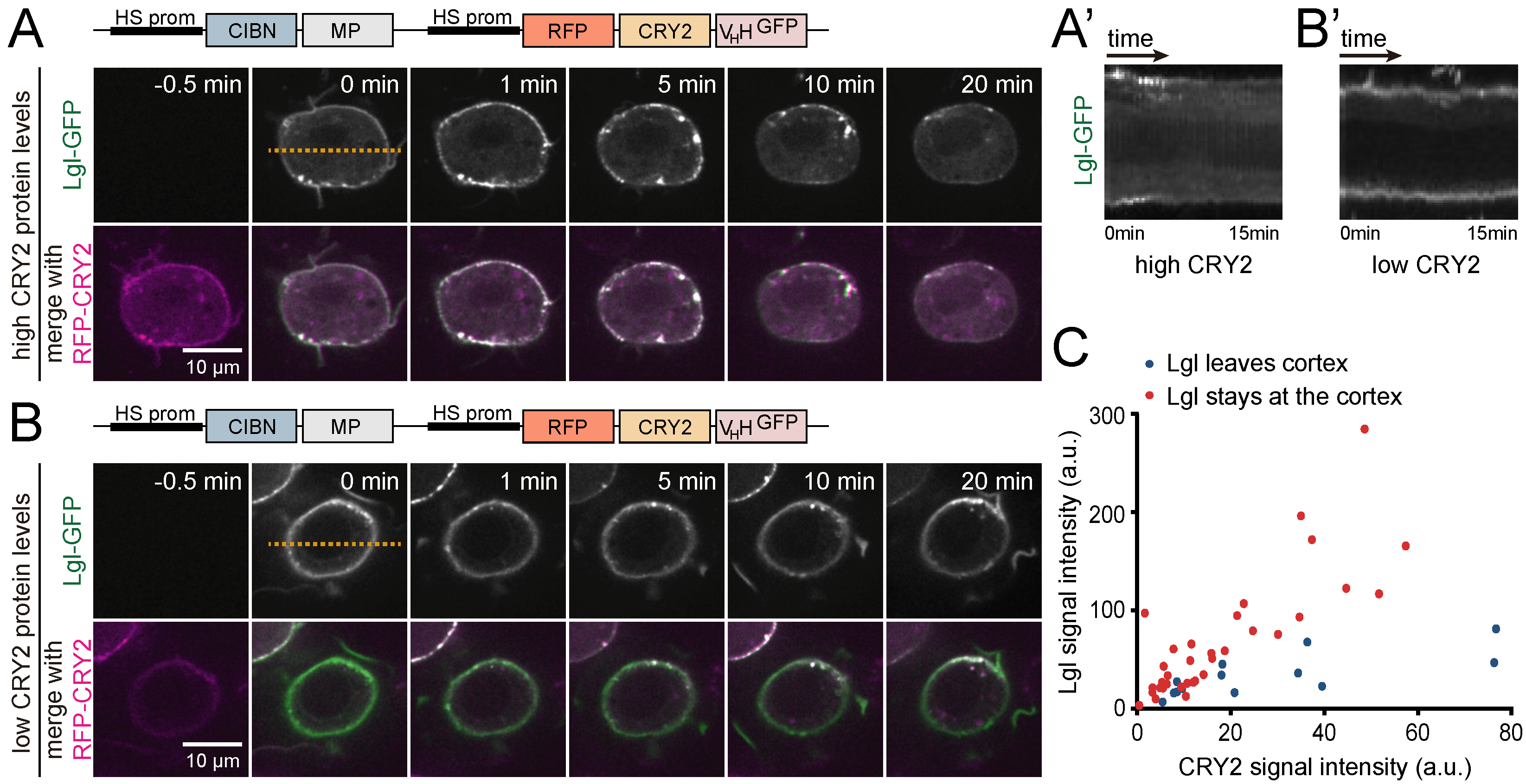
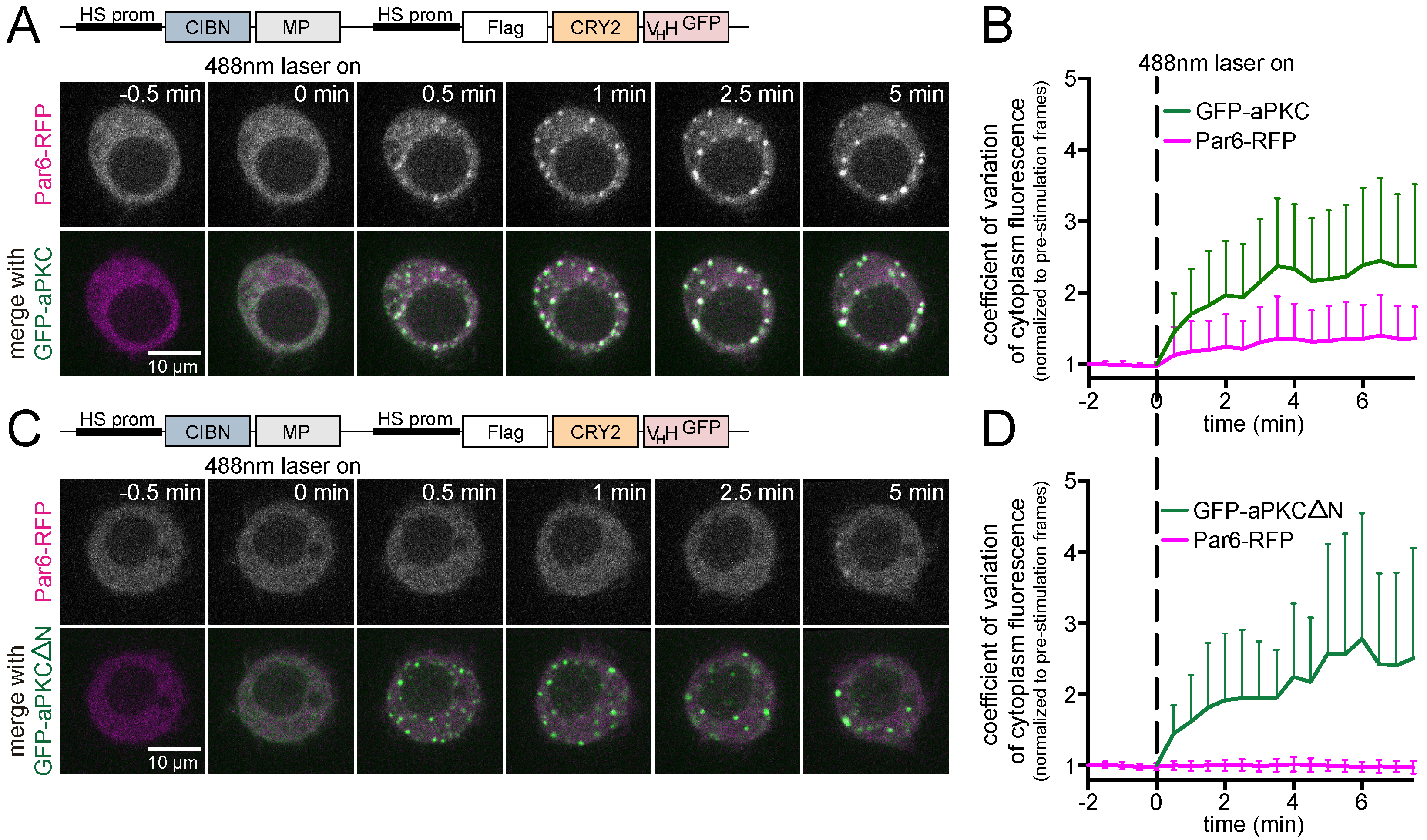
3.3. Analysis of Protein-Protein Interactions via Optogenetic Co-Clustering
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A
References
- Bettencourt-Dias, M.; Giet, R.; Sinka, R.; Mazumdar, A.; Lock, W.G.; Balloux, F.; Zafiropoulos, P.J.; Yamaguchi, S.; Winter, S.; Carthew, R.W.; et al. Genome-wide survey of protein kinases required for cell cycle progression. Nature 2004, 432, 980–987. [Google Scholar] [CrossRef]
- Goshima, G.; Wollman, R.; Goodwin, S.S.; Zhang, N.; Scholey, J.M.; Vale, R.D.; Stuurman, N. Genes required for mitotic spindle assembly in Drosophila S2 cells. Science 2007, 316, 417–421. [Google Scholar] [CrossRef] [PubMed]
- Rogers, S.L.; Rogers, G.C. Culture of Drosophila S2 cells and their use for RNAi-mediated loss-of-function studies and immunofluorescence microscopy. Nat. Protoc. 2008, 3, 606–611. [Google Scholar] [CrossRef] [PubMed]
- Moutinho-Pereira, S.; Matos, I.; Maiato, H. Drosophila S2 cells as a model system to investigate mitotic spindle dynamics, architecture, and function. Methods Cell. Biol. 2010, 97, 243–257. [Google Scholar] [CrossRef] [PubMed]
- Heigwer, F.; Port, F.; Boutros, M. RNA Interference (RNAi) Screening in Drosophila. Genetics 2018, 208, 853–874. [Google Scholar] [CrossRef] [PubMed]
- Johnston, C.A.; Hirono, K.; Prehoda, K.E.; Doe, C.Q. Identification of an Aurora-A/PinsLINKER/Dlg spindle orientation pathway using induced cell polarity in S2 cells. Cell 2009, 138, 1150–1163. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, S.A.; D’Ambrosio, M.V.; Vale, R.D. Induction of focal adhesions and motility in Drosophila S2 cells. Mol. Biol. Cell 2014, 25, 3861–3869. [Google Scholar] [CrossRef]
- Carvalho, C.A.; Moreira, S.; Ventura, G.; Sunkel, C.E.; Morais-de-Sa, E. Aurora A triggers Lgl cortical release during symmetric division to control planar spindle orientation. Curr. Biol. 2015, 25, 53–60. [Google Scholar] [CrossRef]
- Guglielmi, G.; Falk, H.J.; De Renzis, S. Optogenetic Control of Protein Function: From Intracellular Processes to Tissue Morphogenesis. Trends Cell Biol. 2016, 26, 864–874. [Google Scholar] [CrossRef]
- Liu, Q.; Tucker, C.L. Engineering genetically-encoded tools for optogenetic control of protein activity. Curr. Opin. Chem. Biol. 2017, 40, 17–23. [Google Scholar] [CrossRef]
- Lee, S.; Park, H.; Kyung, T.; Kim, N.Y.; Kim, S.; Kim, J.; Heo, W.D. Reversible protein inactivation by optogenetic trapping in cells. Nat. Methods 2014, 11, 633–636. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, M.J.; Hughes, R.M.; Peteya, L.A.; Schwartz, J.W.; Ehlers, M.D.; Tucker, C.L. Rapid blue-light-mediated induction of protein interactions in living cells. Nat. Methods 2010, 7, 973–975. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, M.K.; Kim, C.Y.; Kim, J.M.; Park, B.O.; Lee, S.; Park, H.; Heo, W.D. Optogenetic oligomerization of Rab GTPases regulates intracellular membrane trafficking. Nat. Chem. Biol. 2016, 12, 431–436. [Google Scholar] [CrossRef] [PubMed]
- Qin, X.; Park, B.O.; Liu, J.; Chen, B.; Choesmel-Cadamuro, V.; Belguise, K.; Heo, W.D.; Wang, X. Cell-matrix adhesion and cell-cell adhesion differentially control basal myosin oscillation and Drosophila egg chamber elongation. Nat. Commun. 2017, 8, 14708. [Google Scholar] [CrossRef] [PubMed]
- Qin, X.; Hannezo, E.; Mangeat, T.; Liu, C.; Majumder, P.; Liu, J.; Choesmel-Cadamuro, V.; McDonald, J.A.; Liu, Y.; Yi, B.; et al. A biochemical network controlling basal myosin oscillation. Nat. Commun. 2018, 9, 1210. [Google Scholar] [CrossRef] [PubMed]
- Betschinger, J.; Mechtler, K.; Knoblich, J.A. The Par complex directs asymmetric cell division by phosphorylating the cytoskeletal protein Lgl. Nature 2003, 422, 326–330. [Google Scholar] [CrossRef] [PubMed]
- Peng, C.Y.; Manning, L.; Albertson, R.; Doe, C.Q. The tumour-suppressor genes lgl and dlg regulate basal protein targeting in Drosophila neuroblasts. Nature 2000, 408, 596–600. [Google Scholar] [CrossRef]
- Hao, Y.; Du, Q.; Chen, X.; Zheng, Z.; Balsbaugh, J.L.; Maitra, S.; Shabanowitz, J.; Hunt, D.F.; Macara, I.G. Par3 controls epithelial spindle orientation by aPKC-mediated phosphorylation of apical Pins. Curr. Biol. 2010, 20, 1809–1818. [Google Scholar] [CrossRef]
- Guilgur, L.G.; Prudencio, P.; Ferreira, T.; Pimenta-Marques, A.R.; Martinho, R.G. Drosophila aPKC is required for mitotic spindle orientation during symmetric division of epithelial cells. Development 2012, 139, 503–513. [Google Scholar] [CrossRef]
- Bell, G.P.; Fletcher, G.C.; Brain, R.; Thompson, B.J. Aurora kinases phosphorylate Lgl to induce mitotic spindle orientation in Drosophila epithelia. Curr. Biol. 2015, 25, 61–68. [Google Scholar] [CrossRef]
- Graybill, C.; Prehoda, K.E. Ordered multisite phosphorylation of lethal giant larvae by atypical protein kinase C. Biochemistry 2014, 53, 4931–4937. [Google Scholar] [CrossRef] [PubMed]
- Moreira, S.; Osswald, M.; Ventura, G.; Goncalves, M.; Sunkel, C.E.; Morais-de-Sa, E. PP1-Mediated Dephosphorylation of Lgl Controls Apical-basal Polarity. Cell Rep. 2019, 26, 293–301. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Wen, A.; Shen, B.; Lu, J.; Huang, Y.; Chang, Y. FastCloning: A highly simplified, purification-free, sequence- and ligation-independent PCR cloning method. BMC Biotechnol. 2011, 11, 92. [Google Scholar] [CrossRef] [PubMed]
- Quan, J.; Tian, J. Circular polymerase extension cloning for high-throughput cloning of complex and combinatorial DNA libraries. Nat. Protoc. 2011, 6, 242–251. [Google Scholar] [CrossRef] [PubMed]
- Conde, C.; Osswald, M.; Barbosa, J.; Moutinho-Santos, T.; Pinheiro, D.; Guimaraes, S.; Matos, I.; Maiato, H.; Sunkel, C.E. Drosophila Polo regulates the spindle assembly checkpoint through Mps1-dependent BubR1 phosphorylation. EMBO J. 2013, 32, 1761–1777. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef]
- Taslimi, A.; Vrana, J.D.; Chen, D.; Borinskaya, S.; Mayer, B.J.; Kennedy, M.J.; Tucker, C.L. An optimized optogenetic clustering tool for probing protein interaction and function. Nat. Commun. 2014, 5, 4925. [Google Scholar] [CrossRef]
- Park, H.; Kim, N.Y.; Lee, S.; Kim, N.; Kim, J.; Heo, W.D. Optogenetic protein clustering through fluorescent protein tagging and extension of CRY2. Nat. Commun. 2017, 8, 30. [Google Scholar] [CrossRef]
- Ormo, M.; Cubitt, A.B.; Kallio, K.; Gross, L.A.; Tsien, R.Y.; Remington, S.J. Crystal structure of the Aequorea victoria green fluorescent protein. Science 1996, 273, 1392–1395. [Google Scholar] [CrossRef]
- Bugaj, L.J.; Choksi, A.T.; Mesuda, C.K.; Kane, R.S.; Schaffer, D.V. Optogenetic protein clustering and signaling activation in mammalian cells. Nat. Methods 2013, 10, 249–252. [Google Scholar] [CrossRef]
- Duan, L.; Hope, J.; Ong, Q.; Lou, H.Y.; Kim, N.; McCarthy, C.; Acero, V.; Lin, M.Z.; Cui, B. Understanding CRY2 interactions for optical control of intracellular signaling. Nat. Commun. 2017, 8, 547. [Google Scholar] [CrossRef] [PubMed]
- Taslimi, A.; Zoltowski, B.; Miranda, J.G.; Pathak, G.P.; Hughes, R.M.; Tucker, C.L. Optimized second-generation CRY2-CIB dimerizers and photoactivatable Cre recombinase. Nat. Chem. Biol. 2016, 12, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Weiss, E.; Winey, M. The Saccharomyces cerevisiae spindle pole body duplication gene MPS1 is part of a mitotic checkpoint. J. Cell Biol. 1996, 132, 111–123. [Google Scholar] [CrossRef] [PubMed]
- Pachis, S.T.; Kops, G. Leader of the SAC: Molecular mechanisms of Mps1/TTK regulation in mitosis. Open Biol. 2018, 8. [Google Scholar] [CrossRef] [PubMed]
- Abrieu, A.; Magnaghi-Jaulin, L.; Kahana, J.A.; Peter, M.; Castro, A.; Vigneron, S.; Lorca, T.; Cleveland, D.W.; Labbe, J.C. Mps1 is a kinetochore-associated kinase essential for the vertebrate mitotic checkpoint. Cell 2001, 106, 83–93. [Google Scholar] [CrossRef]
- Stucke, V.M.; Sillje, H.H.; Arnaud, L.; Nigg, E.A. Human Mps1 kinase is required for the spindle assembly checkpoint but not for centrosome duplication. EMBO J. 2002, 21, 1723–1732. [Google Scholar] [CrossRef] [PubMed]
- Jelluma, N.; Dansen, T.B.; Sliedrecht, T.; Kwiatkowski, N.P.; Kops, G.J. Release of Mps1 from kinetochores is crucial for timely anaphase onset. J. Cell Biol. 2010, 191, 281–290. [Google Scholar] [CrossRef]
- Hiruma, Y.; Sacristan, C.; Pachis, S.T.; Adamopoulos, A.; Kuijt, T.; Ubbink, M.; von Castelmur, E.; Perrakis, A.; Kops, G.J. Competition between Mps1 and microtubules at kinetochores regulates spindle checkpoint signaling. Science 2015, 348, 1264–1267. [Google Scholar] [CrossRef]
- Ji, Z.; Gao, H.; Yu, H. Kinetochore attachment sensed by competitive Mps1 and microtubule binding to Ndc80C. Science 2015, 348, 1260–1264. [Google Scholar] [CrossRef]
- Moura, M.; Osswald, M.; Leca, N.; Barbosa, J.; Pereira, A.J.; Maiato, H.; Sunkel, C.E.; Conde, C. Protein Phosphatase 1 inactivates Mps1 to ensure efficient Spindle Assembly Checkpoint silencing. eLife 2017, 6. [Google Scholar] [CrossRef]
- Ohshiro, T.; Yagami, T.; Zhang, C.; Matsuzaki, F. Role of cortical tumour-suppressor proteins in asymmetric division of Drosophila neuroblast. Nature 2000, 408, 593–596. [Google Scholar] [CrossRef] [PubMed]
- Wirtz-Peitz, F.; Nishimura, T.; Knoblich, J.A. Linking cell cycle to asymmetric division: Aurora-A phosphorylates the Par complex to regulate Numb localization. Cell 2008, 135, 161–173. [Google Scholar] [CrossRef] [PubMed]
- Dong, W.; Zhang, X.; Liu, W.; Chen, Y.J.; Huang, J.; Austin, E.; Celotto, A.M.; Jiang, W.Z.; Palladino, M.J.; Jiang, Y.; et al. A conserved polybasic domain mediates plasma membrane targeting of Lgl and its regulation by hypoxia. J. Cell Biol. 2015, 211, 273–286. [Google Scholar] [CrossRef] [PubMed]
- Bailey, M.J.; Prehoda, K.E. Establishment of Par-Polarized Cortical Domains via Phosphoregulated Membrane Motifs. Dev. Cell 2015, 35, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Strand, D.; Jakobs, R.; Merdes, G.; Neumann, B.; Kalmes, A.; Heid, H.W.; Husmann, I.; Mechler, B.M. The Drosophila lethal(2)giant larvae tumor suppressor protein forms homo-oligomers and is associated with nonmuscle myosin II heavy chain. J. Cell Biol. 1994, 127, 1361–1373. [Google Scholar] [CrossRef] [PubMed]
- Kunzelmann, S.; Bottcher, R.; Schmidts, I.; Forstemann, K. A Comprehensive Toolbox for Genome Editing in Cultured Drosophila melanogaster Cells. G3 (Bethesda) 2016, 6, 1777–1785. [Google Scholar] [CrossRef] [PubMed]
- Joberty, G.; Petersen, C.; Gao, L.; Macara, I.G. The cell-polarity protein Par6 links Par3 and atypical protein kinase C to Cdc42. Nat. Cell Biol. 2000, 2, 531–539. [Google Scholar] [CrossRef]
- Suzuki, A.; Yamanaka, T.; Hirose, T.; Manabe, N.; Mizuno, K.; Shimizu, M.; Akimoto, K.; Izumi, Y.; Ohnishi, T.; Ohno, S. Atypical protein kinase C is involved in the evolutionarily conserved par protein complex and plays a critical role in establishing epithelia-specific junctional structures. J. Cell Biol. 2001, 152, 1183–1196. [Google Scholar] [CrossRef]
- Hirano, Y.; Yoshinaga, S.; Takeya, R.; Suzuki, N.N.; Horiuchi, M.; Kohjima, M.; Sumimoto, H.; Inagaki, F. Structure of a cell polarity regulator, a complex between atypical PKC and Par6 PB1 domains. J. Biol. Chem. 2005, 280, 9653–9661. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Purpose | Primer Name | Primer Sequence (5′–3′) |
|---|---|---|
| pENTR insertion | CIBN-mCer-MP Fw | GCTGCTCCATTTACAATGAATGGAGCTATAGGAG |
| CIBN-mCer-MP Rv | AGCGCGTCCACCTTTTCAATGGGGCAGGACG | |
| CRY2-VHH Fw | GCTGCTCCATTTACAATGGACAAAAAGACCATC | |
| CRY2-VHH Rv | AGCGCGTCCACCTTTTTAGCTGGAGACGGTGAC | |
| aPKC Fw | GCTGCTCCATTTACAATGCAGAAAATGCCCTC | |
| aPKC/aPKCΔN Rv | AGCGCGTCCACCTTTGACGCAATCCTCCAGAG | |
| aPKC ΔN Fw | GCTGCTCCATTTACAATGAAGCTGTTGGTGCACAAG | |
| Par6 Fw | GCTGCTCCATTTACAATGTCGAAGAACAAGATAAACAC | |
| Par6 Rv | AGCGCGTCCACCTTTCAAATGCAGCACTCCATC | |
| pENTR Fw | AAAGGTGGACGCGCTGACCCAGCTTTCTT | |
| pENTR Rv | TGTAAATGGAGCAGCCGCGGAGC | |
| mutagenesis | CRY2olig E490G Fw | GATCTCTCGCACTCGGGGCGCCCAGATTATG |
| CRY2olig E490G Rv | CGAGTGCGAGAGATCGCTTTAG | |
| Gateway AfeI Fw | GAGTCAGTGAGCGAGCACGTGGAAGAGCGCTCAATACGC | |
| Gateway AfeI Rv | CTCGCTCACTGACTCGCTGCGCTC | |
| CPEC | Gateway CIBN Fw | TCCCGTTTGCGGCATTTTGCCTTC |
| Gateway CIBN Rv | GGCGCTCAATAAGGGCGACACGGA | |
| Gateway CRY2 Fw | CCCTTATTGAGCGCCCAATACGCAAAC | |
| Gateway CRY2 Rv | AATGCCGCAAACGGGATCCAGACATGATAAG |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Osswald, M.; Santos, A.F.; Morais-de-Sá, E. Light-Induced Protein Clustering for Optogenetic Interference and Protein Interaction Analysis in Drosophila S2 Cells. Biomolecules 2019, 9, 61. https://doi.org/10.3390/biom9020061
Osswald M, Santos AF, Morais-de-Sá E. Light-Induced Protein Clustering for Optogenetic Interference and Protein Interaction Analysis in Drosophila S2 Cells. Biomolecules. 2019; 9(2):61. https://doi.org/10.3390/biom9020061
Chicago/Turabian StyleOsswald, Mariana, A. Filipa Santos, and Eurico Morais-de-Sá. 2019. "Light-Induced Protein Clustering for Optogenetic Interference and Protein Interaction Analysis in Drosophila S2 Cells" Biomolecules 9, no. 2: 61. https://doi.org/10.3390/biom9020061
APA StyleOsswald, M., Santos, A. F., & Morais-de-Sá, E. (2019). Light-Induced Protein Clustering for Optogenetic Interference and Protein Interaction Analysis in Drosophila S2 Cells. Biomolecules, 9(2), 61. https://doi.org/10.3390/biom9020061