Phosphatases in Mitosis: Roles and Regulation



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. Serine/Threonine Phosphatases
1.1.1. Protein Phosphatase 1
1.1.2. Protein Phosphatase 2A
1.2. Tyrosine Phosphatases
CDC25 Phosphatases
2. Roles of Protein Phosphatases in Mitosis
2.1. Mitotic Entry: CDC25 Phosphatases Set up the Commitment
2.2. Sister Chromatid Cohesion: PP2A-B56 Holds Them Together
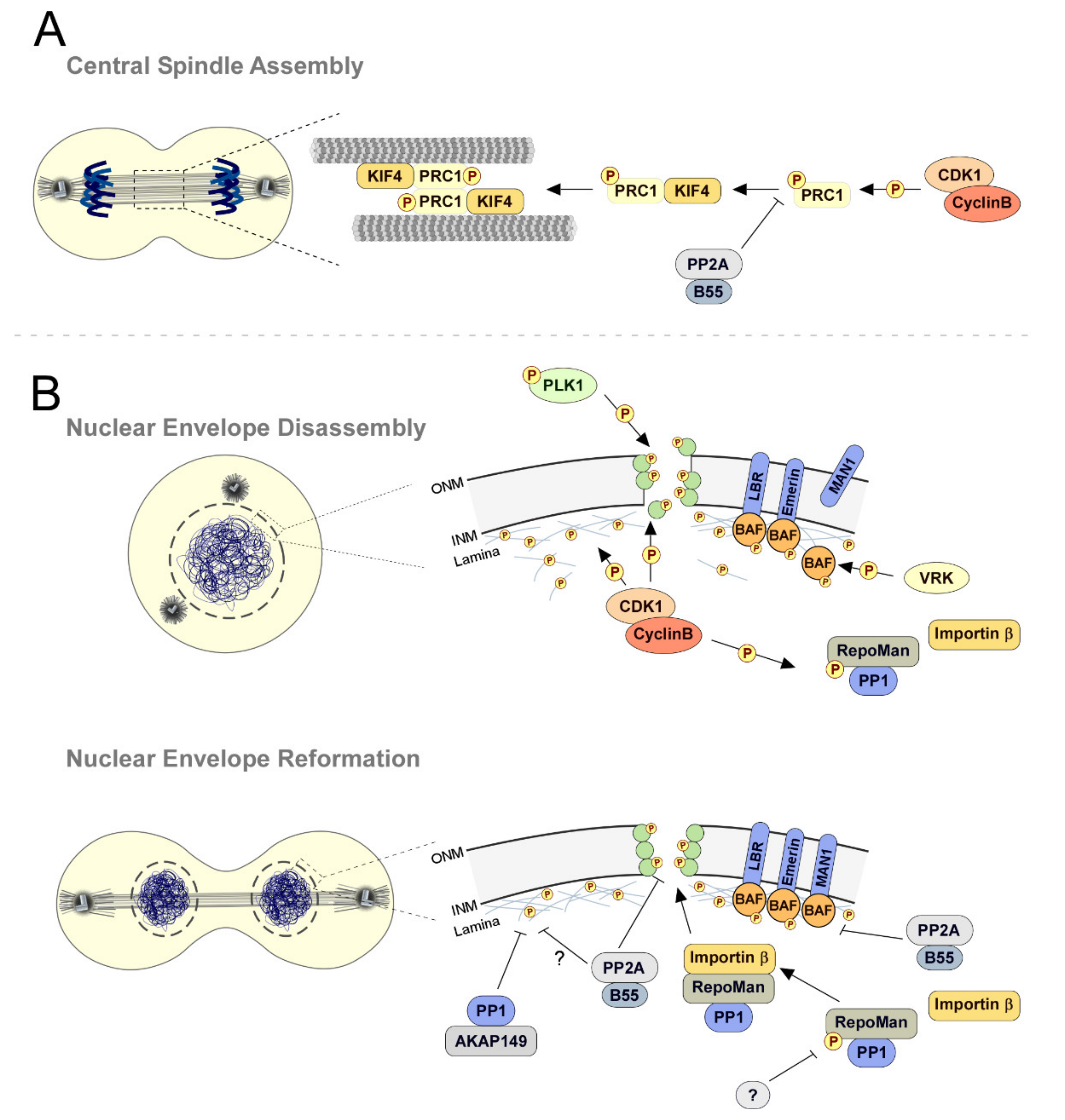
2.3. To Build a Spindle: A PP1, PP2A, PP4, and PP6 Groundwork
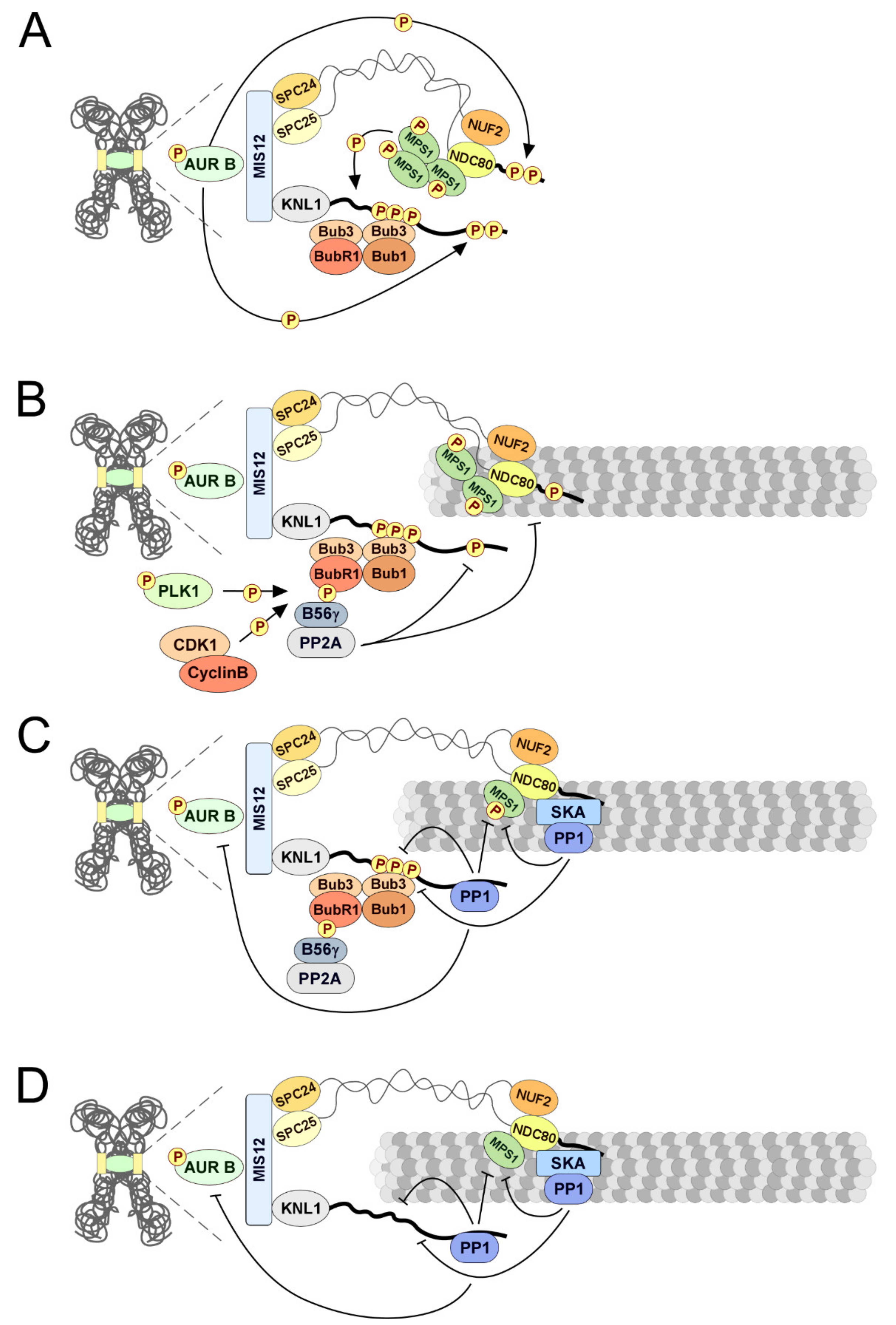
2.4. Kinetochore-Microtubule Attachments: PP1 and PP2A-B56 Keep Them Stable
2.5. Silencing the Spindle Assembly Checkpoint: PP1 and PP2A-B56 Shut It Up
2.6. Mitotic Exit: PP1 and PP2A-B55 Clear the Way
3. Spatiotemporal Control of Phosphatase Activities
4. Concluding Remarks and Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Olsen, J.V.; Blagoev, B.; Gnad, F.; Macek, B.; Kumar, C.; Mortensen, P.; Mann, M. Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell 2006, 127, 635–648. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.J.; Dixon, J.E.; Manning, G. Genomics and evolution of protein phosphatases. Sci. Signal. 2017, 10, eaag1796. [Google Scholar] [CrossRef] [PubMed]
- Manning, G.; Whyte, D.B.; Martinez, R.; Hunter, T.; Sudarsanam, S. The protein kinase complement of the human genome. Science 2002, 298, 1912–1934. [Google Scholar] [CrossRef] [PubMed]
- Andersen, J.N.; Mortensen, O.H.; Peters, G.H.; Drake, P.G.; Iversen, L.F.; Olsen, O.H.; Jansen, P.G.; Andersen, H.S.; Tonks, N.K.; Møller, N.P. Structural and evolutionary relationships among protein tyrosine phosphatase domains. Mol. Cell. Biol. 2001, 21, 7117–7136. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y. Serine/Threonine Phosphatases: Mechanism through Structure. Cell 2009, 139, 468–484. [Google Scholar] [CrossRef] [PubMed]
- Brautigan, D.L.; Shenolikar, S. Protein serine/threonine phosphatases: Keys to unlocking regulators and substrates. Annu. Rev. Biochem. 2018, 87, 921–964. [Google Scholar] [CrossRef] [PubMed]
- Mitić, N.; Miraula, M.; Selleck, C.; Hadler, K.S.; Uribe, E.; Pedroso, M.M.; Schenk, G. Catalytic mechanisms of metallohydrolases containing two metal ions. Adv. Protein Chem. Struct. Biol. 2014, 97, 49–81. [Google Scholar]
- Heroes, E.; Rip, J.; Beullens, M.; Van Meervelt, L.; De Gendt, S.; Bollen, M. Metals in the active site of native protein phosphatase-1. J. Inorg. Biochem. 2015, 149, 1–5. [Google Scholar] [CrossRef]
- Verbinnen, I.; Ferreira, M.; Bollen, M. Biogenesis and activity regulation of protein phosphatase 1. Biochem. Soc. Trans. 2017, 45, 89–99. [Google Scholar] [CrossRef]
- McNall, S.J.; Fischer, E.H. Phosphorylase phosphatase. Comparison of active forms using peptide substrates. J. Biol. Chem. 1988, 263, 1893–1897. [Google Scholar]
- Agostinis, P.; Goris, J.; Pinna, L.A.; Marchiori, F.; Perich, J.W.; Meyer, H.E.; Merlevede, W. Synthetic peptides as model substrates for the study of the specificity of the polycation-stimulated protein phosphatases. Eur. J. Biochem. 1990, 189, 235–241. [Google Scholar] [CrossRef] [PubMed]
- Agostinis, P.; Derua, R.; Sarno, S.; Goris, J.; Merlevede, W. Specificity of the polycation-stimulated (type-2A) and ATP, Mg-dependent (type-1) protein phosphatases toward substrates phosphorylated by P34cdc2 kinase. Eur. J. Biochem. 1992, 205, 241–248. [Google Scholar] [CrossRef] [PubMed]
- Imaoka, T.; Lynham, J.A.; Haslam, R.J. Purification and characterization of the 47,000-dalton protein phosphorylated during degranulation of human platelets. J. Biol. Chem. 1983, 258, 11404–11414. [Google Scholar] [PubMed]
- Mumby, M.C.; Russell, K.L.; Garrard, L.J.; Green, D.D. Cardiac contractile protein phosphatases. Purification of two enzyme forms and their characterization with subunit-specific antibodies. J. Biol. Chem. 1987, 262, 6257–6265. [Google Scholar] [PubMed]
- Xing, Y.; Xu, Y.; Chen, Y.; Jeffrey, P.D.; Chao, Y.; Lin, Z.; Li, Z.; Strack, S.; Stock, J.B.; Shi, Y. Structure of Protein Phosphatase 2A Core Enzyme Bound to Tumor-Inducing Toxins. Cell 2006, 127, 341–353. [Google Scholar] [CrossRef] [PubMed]
- Barr, F.A.; Elliott, P.R.; Gruneberg, U. Protein phosphatases and the regulation of mitosis. J. Cell Sci. 2011, 124, 2323–2334. [Google Scholar] [CrossRef] [PubMed]
- Ingebritsen, T.S.; Cohen, P. Protein phosphatases: Properties and role in cellular regulation. Science 1983, 221, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Okano, K.; Heng, H.; Trevisanato, S.; Tyers, M.; Varmuza, S. Genomic organization and functional analysis of the murine protein phosphatase 1c γ (Ppp1cc) gene. Genomic organization and functional analysis of the murine protein phosphatase 1c γ (Ppp1cc) gene. Genomics 1997, 45, 211–215. [Google Scholar] [CrossRef] [PubMed]
- Boens, S.; Szekér, K.; Van Eynde, A.; Bollen, M. Interactor-guided dephosphorylation by protein phosphatase-1. Methods Mol. Biol. 2013, 1053, 271–281. [Google Scholar]
- Sinha, N.; Pilder, S.; Vijayaraghavan, S. Significant expression levels of transgenic PPP1CC2 in testis and sperm are required to overcome the male infertility phenotype of Ppp1cc null mice. PLoS ONE 2012, 7, e47623. [Google Scholar] [CrossRef] [PubMed]
- Sinha, N.; Puri, P.; Nairn, A.C.; Vijayaraghavan, S. Selective ablation of Ppp1cc gene in testicular germ cells causes oligo-teratozoospermia and infertility in mice. Biol. Reprod. 2013, 89, 128. [Google Scholar] [CrossRef] [PubMed]
- Gibbons, J.A.; Kozubowski, L.; Tatchell, K.; Shenolikar, S. Expression of human protein phosphatase-1 in Saccharomyces cerevisiae highlights the role of phosphatase isoforms in regulating eukaryotic functions. J. Biol. Chem. 2007, 282, 21838–21847. [Google Scholar] [CrossRef] [PubMed]
- Alessi, D.R.; Street, A.J.; Cohen, P.; Cohen, P.T. Inhibitor-2 functions like a chaperone to fold three expressed isoforms of mammalian protein phosphatase-1 into a conformation with the specificity and regulatory properties of the native enzyme. Eur. J. Biochem. 1993, 213, 1055–1066. [Google Scholar] [CrossRef]
- Liu, R.; Correll, R.N.; Davis, J.; Vagnozzi, R.J.; York, A.J.; Sargent, M.A.; Nairn, A.C.; Molkentin, J.D. Cardiac-specific deletion of protein phosphatase 1β promotes increased myofilament protein phosphorylation and contractile alterations. J. Mol. Cell. Cardiol. 2015, 87, 204–213. [Google Scholar] [CrossRef] [PubMed]
- Egloff, M.P.; Cohen, P.T.; Reinemer, P.; Barford, D. Crystal structure of the catalytic subunit of human protein phosphatase 1 and its complex with tungstate. J. Mol. Biol. 1995, 254, 942–959. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, J.; Huang, H.B.; Kwon, Y.G.; Greengard, P.; Nairn, A.C.; Kuriyan, J. Three-dimensional structure of the catalytic subunit of protein serine/threonine phosphatase-1. Nature 1995, 376, 745–753. [Google Scholar] [CrossRef]
- Bollen, M.; Peti, W.; Ragusa, M.J.; Beullens, M. The extended PP1 toolkit: Designed to create specificity. Trends Biochem. Sci. 2010, 35, 450–458. [Google Scholar] [CrossRef]
- Heroes, E.; Lesage, B.; Görnemann, J.; Beullens, M.; Van Meervelt, L.; Bollen, M. The PP1 binding code: A molecular-lego strategy that governs specificity. FEBS J. 2013, 280, 584–595. [Google Scholar] [CrossRef]
- Roy, J.; Cyert, M.S. Cracking the phosphatase code: Docking interactions determine substrate specificity. Sci. Signal. 2009, 2, re9. [Google Scholar] [CrossRef]
- Marsh, J.A.; Dancheck, B.; Ragusa, M.J.; Allaire, M.; Forman-Kay, J.D.; Peti, W. Structural diversity in free and bound states of intrinsically disordered protein phosphatase 1 regulators. Structure 2010, 18, 1094–1103. [Google Scholar] [CrossRef]
- Davey, N.E.; Van Roey, K.; Weatheritt, R.J.; Toedt, G.; Uyar, B.; Altenberg, B.; Budd, A.; Diella, F.; Dinkel, H.; Gibson, T.J. Attributes of short linear motifs. Mol. Biosyst. 2012, 8, 268–281. [Google Scholar] [CrossRef] [PubMed]
- Davey, N.E.; Cyert, M.S.; Moses, A.M. Short linear motifs–ex nihilo evolution of protein regulation. Cell Commun. Signal. 2015, 13, 43. [Google Scholar] [CrossRef] [PubMed]
- Wakula, P.; Beullens, M.; Ceulemans, H.; Stalmans, W.; Bollen, M. Degeneracy and function of the ubiquitous RVXF-motif that mediates binding to protein phosphatase-1. J. Biol. Chem. 2003, 278, 18817–18823. [Google Scholar] [CrossRef] [PubMed]
- Meiselbach, H.; Sticht, H.; Enz, R. Structural analysis of the protein phosphatase 1 docking motif: Molecular description of binding specificities identifies interacting proteins. Chem. Biol. 2006, 13, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Egloff, M.P.; Johnson, D.F.; Moorhead, G.; Cohen, P.T.; Cohen, P.; Barford, D. Structural basis for the recognition of regulatory subunits by the catalytic subunit of protein phosphatase 1. EMBO J. 1997, 16, 1876–1887. [Google Scholar] [CrossRef] [PubMed]
- Hirano, K.; Phan, B.C.; Hartshorne, D.J. Interactions of the subunits of smooth muscle myosin phosphatase. J. Biol. Chem. 1997, 272, 3683–3688. [Google Scholar] [CrossRef] [PubMed]
- Ceulemans, H.; Stalmans, W.; Bollen, M. Regulator-driven functional diversification of protein phosphatase-1 in eukaryotic evolution. Bioessays 2002, 24, 371–381. [Google Scholar] [CrossRef]
- Terrak, M.; Kerff, F.; Langsetmo, K.; Tao, T.; Dominguez, R. Structural basis of protein phosphatase 1 regulation. Nature 2004, 429, 780–784. [Google Scholar] [CrossRef]
- Ragusa, M.J.; Dancheck, B.; Critton, D.A.; Nairn, A.C.; Page, R.; Peti, W. Spinophilin directs protein phosphatase 1 specificity by blocking substrate binding sites. Nat. Struct. Mol. Biol. 2010, 17, 459–464. [Google Scholar] [CrossRef]
- Choy, M.S.; Hieke, M.; Kumar, G.S.; Lewis, G.R.; Gonzalez-DeWhitt, K.R.; Kessler, R.P.; Stein, B.J.; Hessenberger, M.; Nairn, A.C.; Nairn Peti, W.; et al. Understanding the antagonism of retinoblastoma protein dephosphorylation by PNUTS provides insights into the PP1 regulatory code. Proc. Natl. Acad. Sci. USA 2014, 111, 4097–4102. [Google Scholar] [CrossRef]
- O’Connell, N.; Nichols, S.R.; Heroes, E.; Beullens, M.; Bollen, M.; Peti, W.; Page, R. The molecular basis for substrate specificity of the nuclear NIPP1: PP1 holoenzyme. Structure 2012, 20, 1746–1756. [Google Scholar] [CrossRef] [PubMed]
- Hurley, T.D.; Yang, J.; Zhang, L.; Goodwin, K.D.; Zou, Q.; Cortese, M.; Dunker, A.K.; DePaoli-Roach, A.A. Structural basis for regulation of protein phosphatase 1 by inhibitor-2. J. Biol. Chem. 2007, 282, 28874–28883. [Google Scholar] [CrossRef] [PubMed]
- Barford, D.; Das, A.K.; Egloff, M.P. The structure and mechanism of protein phosphatases: Insights into catalysis and regulation. Annu. Rev. Biophys. Biomol. Struct. 1998, 27, 133–164. [Google Scholar] [CrossRef] [PubMed]
- Eto, M.; Kirkbride, J.; Elliott, E.; Lo, S.H.; Brautigan, D.L. Association of the tensin N-terminal protein-tyrosine phosphatase domain with the α isoform of protein phosphatase-1 in focal adhesions. J. Biol. Chem. 2007, 282, 17806–17815. [Google Scholar] [CrossRef]
- Dumitru, A.M.G.; Rusin, S.F.; Clark, A.E.; Kettenbach, A.N.; Compton, D.A. Cyclin A/Cdk1 modulates Plk1 activity in prometaphase to regulate kinetochore-microtubule attachment stability. Elife 2017, 6, e29303. [Google Scholar] [CrossRef] [PubMed]
- Terry-Lorenzo, R.T.; Elliot, E.; Weiser, D.C.; Prickett, T.D.; Brautigan, D.L.; Shenolikar, S. Neurabins recruit protein phosphatase-1 and inhibitor-2 to the actin cytoskeleton. J. Biol. Chem. 2002, 277, 46535–46543. [Google Scholar] [CrossRef] [PubMed]
- Dancheck, B.; Ragusa, M.J.; Allaire, M.; Nairn, A.C.; Page, R.; Peti, W. Molecular Investigations of the Structure and Function of the Protein Phosphatase 1−Spinophilin−Inhibitor 2 Heterotrimeric Complex. Biochemistry 2011, 50, 1238–1246. [Google Scholar] [CrossRef]
- Arino, J.; Woon, C.W.; Brautigan, D.L.; Miller, T.B.; Johnson, G.L. Human liver phosphatase 2A: cDNA and amino acid sequence of two catalytic subunit isotypes. Proc. Natl. Acad. Sci. USA 1988, 85, 4252–4256. [Google Scholar] [CrossRef]
- Janssens, V.; Goris, J. Protein phosphatase 2A: A highly regulated family of serine/threonine phosphatases implicated in cell growth and signalling. Biochem. J. 2001, 353, 417–439. [Google Scholar] [CrossRef]
- Seshacharyulu, P.; Pandey, P.; Datta, K.; Batra, S.K. Phosphatase: PP2A structural importance, regulation and its aberrant expression in cancer. Cancer Lett. 2013, 335, 9–18. [Google Scholar] [CrossRef]
- Nasa, I.; Kettenbach, A.N. Coordination of Protein Kinase and Phosphoprotein Phosphatase Activities in Mitosis. Front. Cell Dev. Biol. 2018, 6, 30. [Google Scholar] [CrossRef] [PubMed]
- Groves, M.R.; Hanlon, N.; Turowski, P.; Hemmings, B.A.; Barford, D. The structure of the protein phosphatase 2A PR65/A subunit reveals the conformation of its 15 tandemly repeated HEAT motifs. Cell 1999, 96, 99–110. [Google Scholar] [CrossRef]
- Kremmer, E.; Ohst, K.; Kiefer, J.; Brewis, N.; Walter, G. Separation of PP2A core enzyme and holoenzyme with monoclonal antibodies against the regulatory A subunit: Abundant expression of both forms in cells. Mol. Cell. Biol. 1997, 17, 1692–1701. [Google Scholar] [CrossRef] [PubMed]
- Slupe, A.M.; Merrill, R.A.; Strack, S. Determinants for substrate specificity of protein phosphatase 2A. Enzyme Res. 2011, 2011, 398751. [Google Scholar] [CrossRef] [PubMed]
- Cho, U.S.; Xu, W. Crystal structure of a protein phosphatase 2A heterotrimeric holoenzyme. Nature 2006, 445, 53–57. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Xing, Y.; Chen, Y.; Chao, Y.; Lin, Z.; Fan, E.; Yu, J.W.; Strack, S.; Jeffrey, P.D.; Shi, Y. Structure of the protein phosphatase 2A holoenzyme. Cell 2006, 127, 1239–1251. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Chen, Y.; Zhang, P.; Jeffrey, P.D.; Shi, Y. Structure of a Protein Phosphatase 2A Holoenzyme: Insights into B55-Mediated Tau Dephosphorylation. Mol. Cell 2008, 31, 873–885. [Google Scholar] [CrossRef]
- Cundell, M.J.; Hutter, L.H.; Bastos, R.N.; Poser, E.; Holder, J.; Mohammed, S.; Novak, B.; Barr, F.A. A PP2A-B55 recognition signal controls substrate dephosphorylation kinetics during mitotic exit. J. Cell Biol. 2016, 214, 539–554. [Google Scholar] [CrossRef]
- Pereira, G.; Schiebel, E. Mitotic exit: Determining the PP2A dephosphorylation program. J. Cell Biol. 2016, 214, 499–501. [Google Scholar] [CrossRef]
- Pinna, L.A.; Donella, A.; Clari, G.; Moret, V. Preferential dephosphorylation of protein bound phosphorylthreonine and phosphorylserine residues by cytosol and mitochondrial “casein phosphatase”. Biochem. Biophys. Res. Commun. 1976, 70, 1308–1315. [Google Scholar] [CrossRef]
- Deana, A.D.; Marchiori, F.; Meggio, F.; Pinna, L.A. Dephosphorylation of synthetic phosphopeptides by protein phosphatase-T, a phosphothreonyl protein phosphatase. J. Biol. Chem. 1982, 257, 8565–8568. [Google Scholar] [PubMed]
- Deana, A.D.; Pinna, L.A. Identification of pseudo ‘phosphothreonyl-specific’ protein phosphatase T with a fraction of polycation-stimulated protein phosphatase 2A. Biochim. Biophys. Acta 1988, 968, 179–185. [Google Scholar] [CrossRef]
- Godfrey, M.; Touati, S.A.; Kataria, M.; Jones, A.; Snijders, A.P.; Uhlmann, F. PP2ACdc55 Phosphatase Imposes Ordered Cell-Cycle Phosphorylation by Opposing Threonine Phosphorylation. Mol. Cell 2017, 65, 393–402. [Google Scholar] [CrossRef] [PubMed]
- Hein, J.B.; Hertz, E.P.T.; Garvanska, D.H.; Kruse, T.; Nilsson, J. Distinct kinetics of serine and threonine dephosphorylation are essential for mitosis. Nat. Cell Biol. 2017, 19, 1433–1440. [Google Scholar] [CrossRef] [PubMed]
- Hertz, E.P.T.; Kruse, T.; Davey, N.E.; Lopéz-Méndez, B.; Sigurðsson, J.O.; Montoya, G.; Olsen, J.V.; Nilsson, J. A Conserved Motif Provides Binding Specificity to the PP2A-B56 Phosphatase. Mol. Cell 2016, 63, 686–695. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, Z.; Yu, T.; Yang, H.; Virshup, D.M.; Kops, G.J.; Lee, S.H.; Zhou, W.; Li, X.; Xu, W.; et al. Crystal structure of a PP2A B56-BubR1 complex and its implications for PP2A substrate recruitment and localization. Protein Cell 2016, 7, 516–526. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Bajaj, R.; Bollen, M.; Peti, W.; Page, R. Expanding the PP2A interactome by defining a B56-specific SLiM. Structure 2016, 24, 2174–2181. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.G.; Chen, H.; Guo, F.; Yadav, V.K.; Mcilwain, S.J.; Rowse, M.; Choudhary, A.; Lin, Z.; Li, Y.; Gu, T.; et al. PP2A-B′ holoenzyme substrate recognition, regulation and role in cytokinesis. Cell Discov. 2017, 3, 17027. [Google Scholar] [CrossRef]
- Vallardi, G.; Allan, L.A.; Crozier, L.; Saurin, A.T. A molecular explanation for PP2A-B56 isoform specificity during mitosis. bioRxiv 2018. [Google Scholar] [CrossRef]
- Tonks, N.K. Protein tyrosine phosphatases: From genes, to function, to disease. Nat. Rev. Mol. Cell. Biol. 2006, 7, 833–846. [Google Scholar] [CrossRef]
- Tiganis, T.; Bennett, A.M. Protein tyrosine phosphatase function: The substrate perspective. Biochem. J. 2007, 402, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Alonso, A.; Sasin, J.; Bottini, N.; Friedberg, I.; Friedberg, I.; Osterman, A.; Godzik, A.; Hunter, T.; Dixon, J.; Mustelin, T. Protein tyrosine phosphatases in the human genome. Cell 2004, 117, 699–711. [Google Scholar] [CrossRef] [PubMed]
- Andersen, J.N.; Jansen, P.G.; Echwald, S.M.; Mortensen, O.H.; Fukada, T.; Del Vecchio, R.; Tonks, N.K.; Møller, N.P. A genomic perspective on protein tyrosine phosphatases: Gene structure, pseudogenes, and genetic disease linkage. FASEB J. 2004, 18, 8–30. [Google Scholar] [CrossRef] [PubMed]
- Sacco, F.; Perfetto, L.; Castagnoli, L.; Cesareni, G. The human phosphatase interactome: An intricate family portrait. FEBS Lett. 2012, 586, 2732–2739. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Ryu, S.E. Structure and catalytic mechanism of human protein tyrosine phosphatome. BMB Rep. 2012, 45, 693–699. [Google Scholar] [CrossRef] [PubMed]
- Barford, D.; Flint, A.J.; Tonks, N.K. Crystal structure of human protein tyrosine phosphatase 1B. Science 1994, 263, 1397–1404. [Google Scholar] [CrossRef] [PubMed]
- Jia, Z.; Barford, D.; Flint, A.J.; Tonks, N.K. Structural basis for phosphotyrosine peptide recognition by protein tyrosine phosphatase 1B. Science 1995, 268, 1754–1758. [Google Scholar] [CrossRef] [PubMed]
- Yuvaniyama, J.; Denu, J.M.; Dixon, J.E.; Saper, M.A. Crystal structure of the dual specificity protein phosphatase VHR. Science 1996, 272, 1328–1331. [Google Scholar] [CrossRef]
- Wee, P.; Shi, H.; Jiang, J.; Wang, Y.; Wang, Z. EGF stimulates the activation of EGF receptors and the selective activation of major signaling pathways during mitosis. Cell Signal. 2015, 27, 638–651. [Google Scholar] [CrossRef]
- Jiang, Y.; Li, X.; Yang, W.; Hawke, D.H.; Zheng, Y.; Xia, Y.; Aldape, K.; Wei, C.; Guo, F.; Chen, Y.; et al. PKM2 regulates chromosome segregation and mitosis progression of tumor cells. Mol. Cell 2014, 53, 75–87. [Google Scholar] [CrossRef]
- Hitosugi, T.; Kang, S.; Vander Heiden, M.G.; Chung, T.W.; Elf, S.; Lythgoe, K.; Dong, S.; Lonial, S.; Wang, X.; Chen, G.Z.; et al. Tyrosine phosphorylation inhibits PKM2 to promote the Warburg effect and tumor growth. Sci. Signal. 2009, 2, ra73. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, S.; Hamasaki, M.; Yamamoto, T.; Ebisuya, M.; Sato, M.; Nishida, E.; Toyoshima, F. ABL1 regulates spindle orientation in adherent cells and mammalian skin. Nat. Commun. 2012, 3, 626. [Google Scholar] [CrossRef] [PubMed]
- Caron, D.; Byrne, D.P.; Thebault, P.; Soulet, D.; Landry, C.R.; Eyers, P.A.; Elowe, S. Mitotic phosphotyrosine network analysis reveals that tyrosine phosphorylation regulates Polo-like kinase 1 (PLK1). Sci. Signal. 2016, 9, rs14. [Google Scholar] [CrossRef] [PubMed]
- Queralt, E.; Uhlmann, F. Cdk-counteracting phosphatases unlock mitotic exit. Curr. Opin. Cell Biol. 2008, 20, 661–668. [Google Scholar] [CrossRef] [PubMed]
- Trinkle-Mulcahy, L.; Lamond, A.I. Mitotic phosphatases: No longer silent partners. Curr. Opin. Cell Biol. 2006, 18, 623–631. [Google Scholar] [CrossRef] [PubMed]
- Wurzenberger, C.; Gerlich, D.W. Phosphatases: Providing safe passage through mitotic exit. Nat. Rev. Mol. Cell. Biol. 2011, 12, 469–482. [Google Scholar] [CrossRef] [PubMed]
- Galaktionov, K.; Beach, D. Specific activation of cdc25 tyrosine phosphatases by B-type cyclins: Evidence for multiple roles of mitotic cyclins. Cell 1991, 67, 1181–1194. [Google Scholar] [CrossRef]
- Sadhu, K.; Reed, S.I.; Richardson, H.; Russell, P. Human homolog of fission yeast cdc25 mitotic inducer is predominantly expressed in G2. Proc. Natl. Acad. Sci. USA 1990, 87, 5139–5143. [Google Scholar] [CrossRef]
- Nagata, A.; Igarashi, M.; Jinno, S.; Suto, K.; Okayama, H. An additional homolog of the fission yeast cdc25+ gene occurs in humans and is highly expressed in some cancer cells. New Biol. 1991, 3, 959–968. [Google Scholar]
- Kristjánsdóttir, K.; Rudolph, J. Cdc25 phosphatases and cancer. Chem. Biol. 2004, 11, 1043–1051. [Google Scholar] [CrossRef]
- Rudolph, J. Cdc25 phosphatases: Structure, specificity, and mechanism. Biochemistry 2007, 46, 3595–3604. [Google Scholar] [CrossRef] [PubMed]
- Boutros, R.; Lobjois, V.; Ducommun, B. CDC25 phosphatases in cancer cells: Key players? Good targets? Nat. Rev. Cancer 2007, 7, 495–507. [Google Scholar] [CrossRef]
- Aressy, B.; Ducommun, B. Cell cycle control by the CDC25 phosphatases. Anticancer Agents Med. Chem. 2008, 8, 818–824. [Google Scholar] [CrossRef] [PubMed]
- Lavecchia, A.; Di Giovanni, C.; Novellino, E. CDC25 phosphatase inhibitors: An update. Mini Rev. Med. Chem. 2012, 12, 62–73. [Google Scholar] [CrossRef] [PubMed]
- Fauman, E.B.; Cogswell, J.P.; Lovejoy, B.; Rocque, W.J.; Holmes, W.; Montana, V.G.; Piwnica-Worms, H.; Rink, M.J.; Saper, M.A. Crystal structure of the catalytic domain of the human cell cycle control phosphatase, Cdc25A. Cell 1998, 93, 617–625. [Google Scholar] [CrossRef]
- Reynolds, R.A.; Yem, A.W.; Wolfe, C.L.; Deibel, M.R.Jr.; Chidester, C.G.; Watenpaugh, K.D. Crystal structure of the catalytic subunit of Cdc25B required for G2/M phase transition of the cell cycle. J. Mol. Biol. 1999, 293, 559–568. [Google Scholar] [CrossRef]
- Sohn, J.; Kristjánsdóttir, K.; Safi, A.; Parker, B.; Kiburz, B.; Rudolph, J. Remote hot spots mediate protein substrate recognition for the Cdc25 phosphatase. Proc. Natl. Acad. Sci. USA 2004, 101, 16437–16441. [Google Scholar] [CrossRef]
- Sohn, J.; Buhrman, G.; Rudolph, J. Kinetic and structural studies of specific protein-protein interactions in substrate catalysis by Cdc25B phosphatase. Biochemistry 2007, 46, 807–818. [Google Scholar] [CrossRef]
- Rudolph, J.; Epstein, D.M.; Parker, L.; Eckstein, J. Specificity of natural and artificial substrates for human Cdc25A. Anal. Biochem. 2001, 289, 43–51. [Google Scholar] [CrossRef]
- Wilborn, M.; Free, S.; Ban, A.; Rudolph, J. The C-terminal tail of the dual-specificity Cdc25B phosphatase mediates modular substrate recognition. Biochemistry 2001, 40, 14200–14206. [Google Scholar] [CrossRef]
- Denu, J.M.; Dixon, J.E. A catalytic mechanism for the dual-specific phosphatases. Proc. Natl. Acad. Sci. USA 1995, 92, 5910–5914. [Google Scholar] [CrossRef] [PubMed]
- Denu, J.M.; Stuckey, J.A.; Saper, M.A.; Dixon, J.E. Form and function in protein dephosphorylation. Cell 1996, 87, 361–364. [Google Scholar] [CrossRef]
- Chen, W.; Wilborn, M.; Rudolph, J. Dual-specific Cdc25B phosphatase: In search of the catalytic acid. Biochemistry 2000, 39, 10781–10789. [Google Scholar] [CrossRef] [PubMed]
- Rudolph, J. Catalytic mechanism of Cdc25. Biochemistry 2002, 41, 14613–14623. [Google Scholar] [CrossRef] [PubMed]
- McCain, D.F.; Catrina, I.E.; Hengge, A.C.; Zhang, Z.Y. The catalytic mechanism of Cdc25A phosphatase. J. Biol. Chem. 2002, 277, 11190–11200. [Google Scholar] [CrossRef] [PubMed]
- Sharma, K.; D’Souza, R.C.; Tyanova, S.; Schaab, C.; Wiśniewski, J.R.; Cox, J.; Mann, M. Ultradeep human phosphoproteome reveals a distinct regulatory nature of Tyr and Ser/Thr-based signaling. Cell Rep. 2014, 8, 1583–1594. [Google Scholar] [CrossRef] [PubMed]
- Kuilman, T.; Maiolica, A.; Godfrey, M.; Scheidel, N.; Aebersold, R.; Uhlmann, F. Identification of Cdk targets that control cytokinesis. EMBO J. 2015, 34, 81–96. [Google Scholar] [CrossRef]
- McCloy, R.A.; Parker, B.L.; Rogers, S.; Chaudhuri, R.; Gayevskiy, V.; Hoffman, N.J.; Ali, N.; Watkins, D.N.; Daly, R.J.; James, D.E.; et al. Global Phosphoproteomic Mapping of Early Mitotic Exit in Human Cells Identifies Novel Substrate Dephosphorylation Motifs. Mol. Cell Proteom. 2015, 14, 2194–2212. [Google Scholar] [CrossRef]
- Burgess, A.; Vuong, J.; Rogers, S.; Malumbres, M.; O’Donoghue, S.I. SnapShot: Phosphoregulation of Mitosis. Cell 2017, 169, 1358. [Google Scholar] [CrossRef]
- Pines, J.; Hunter, T. Isolation of a human cyclin cDNA: Evidence for cyclin mRNA and protein regulation in the cell cycle and for interaction with p34cdc2. Cell 1989, 58, 833–846. [Google Scholar] [CrossRef]
- Parker, L.L.; Piwnica-Worms, H. Inactivation of the p34cdc2-cyclin B complex by the human WEE1 tyrosine kinase. Science 1992, 257, 1955–1957. [Google Scholar] [CrossRef] [PubMed]
- Morgan, D.O. Principles of CDK regulation. Nature 1995, 374, 131–134. [Google Scholar] [CrossRef] [PubMed]
- Lindqvist, A.; Källström, H.; Lundgren, A.; Barsoum, E.; Rosenthal, C.K. Cdc25B cooperates with Cdc25A to induce mitosis but has a unique role in activating cyclin B1–Cdk1 at the centrosome. J. Cell Biol. 2005, 171, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Gabrielli, B.G.; De Souza, C.P.; Tonks, I.D.; Clark, J.M.; Hayward, N.K.; Ellem, K.A. Cytoplasmic accumulation of cdc25B phosphatase in mitosis triggers centrosomal microtubule nucleation in HeLa cells. J. Cell Sci. 1996, 109, 1081–1093. [Google Scholar] [PubMed]
- De Souza, C.P.; Ellem, K.A.; Gabrielli, B.G. Centrosomal and cytoplasmic Cdc2/cyclin B1 activation precedes nuclear mitotic events. Exp. Cell Res. 2000, 257, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Gabrielli, B.G.; Clark, J.M.; McCormack, A.K.; Ellem, K.A.O. Ultraviolet light-induced G2 phase cell cycle checkpoint blocks cdc25-dependent progression into mitosis. Oncogene 1997, 15, 749–758. [Google Scholar] [CrossRef] [PubMed]
- Molinari, M.; Mercurio, C.; Dominguez, J.; Goubin, F.; Draetta, G.F. Human Cdc25 A inactivation in response to S phase inhibition and its role in preventing premature mitosis. EMBO Rep. 2000, 1, 71–79. [Google Scholar] [CrossRef]
- Mailand, N.; Podtelejnikov, A.V.; Groth, A.; Mann, M.; Bartek, J.; Lukas, J. Regulation of G2/M events by Cdc25A through phosphorylation-dependent modulation of its stability. EMBO J. 2002, 21, 5911–5920. [Google Scholar] [CrossRef]
- Boutros, R.; Dozier, C.; Ducommun, B. The when and wheres of CDC25 phosphatases. Curr. Opin. Cell Biol. 2006, 18, 185–191. [Google Scholar] [CrossRef]
- Watanabe, N.; Arai, H.; Nishihara, Y.; Taniguchi, M.; Watanabe, N.; Hunter, T.; Osada, H. M-phase kinases induce phospho-dependent ubiquitination of somatic Wee1 by SCFβ-TrCP. Proc. Natl. Acad. Sci. USA 2004, 101, 4419–4424. [Google Scholar] [CrossRef]
- Booher, R.N.; Holman, P.S.; Fattaey, A. Human Myt1 is a cell cycle-regulated kinase that inhibits Cdc2 but not Cdk2 activity. J. Biol. Chem. 1997, 272, 22300–22306. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, H.; Toyoshima-Morimoto, F.; Taniguchi, E.; Nishida, E. Identification of a consensus motif for Plk (Polo-like kinase) phosphorylation reveals Myt1 as a Plk1 substrate. J. Biol. Chem. 2003, 278, 25227–25280. [Google Scholar] [CrossRef] [PubMed]
- O’Farrell, P.H. Triggering the all-or-nothing switch into mitosis. Trends Cell Biol. 2001, 11, 512–519. [Google Scholar] [CrossRef]
- Lindqvist, A.; Rodríguez-Bravo, V.; Medema, R.H. The decision to enter mitosis: Feedback and redundancy in the mitotic entry network. J. Cell Biol. 2009, 185, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Donzelli, M.; Squatrito, M.; Ganoth, D.; Hershko, A.; Pagano, M.; Draetta, G.F. Dual mode of degradation of Cdc25 A phosphatase. EMBO J. 2002, 21, 4875–4884. [Google Scholar] [CrossRef] [PubMed]
- Jinno, S.; Suto, K.; Nagata, A.; Igarashi, M.; Kanaoka, Y.; Nojima, H.; Okayama, H. Cdc25A is a novel phosphatase functioning early in the cell cycle. EMBO J. 1994, 13, 1549–1556. [Google Scholar] [CrossRef] [PubMed]
- Garner-Hamrick, P.A.; Fisher, C. Antisense phosphorothioate oligonucleotides specifically down-regulate cdc25B causing S-phase delay and persistent antiproliferative effects. Int. J. Cancer 1998, 76, 720–728. [Google Scholar] [CrossRef]
- Kieffer, I.; Lorenzo, C.; Dozier, C.; Schmitt, E.; Ducommun, B. Differential mitotic degradation of the CDC25B phosphatase variants. Oncogene 2007, 26, 7847–7858. [Google Scholar] [CrossRef]
- Baldin, V.; Cans, C.; Knibiehler, M.; Ducommun, B. Phosphorylation of human CDC25B phosphatase by CDK1-cyclin A triggers its proteasome-dependent degradation. J. Biol. Chem. 1997, 272, 32731–32734. [Google Scholar] [CrossRef]
- Kanemori, Y.; Uto, K.; Sagata, N. β-TrCP recognizes a previously undescribed nonphosphorylated destruction motif in Cdc25A and Cdc25B phosphatases. Proc. Natl. Acad. Sci. USA 2005, 102, 6279–6284. [Google Scholar] [CrossRef]
- Furnari, B.; Rhind, N.; Russell, P. Cdc25 mitotic inducer targeted by chk1 DNA damage checkpoint kinase. Science 1997, 277, 1495–1497. [Google Scholar] [CrossRef] [PubMed]
- Furnari, B.; Blasina, A.; Boddy, M.N.; McGowan, C.H.; Russell, P. Cdc25 inhibited in vivo and in vitro by checkpoint kinases Cds1 and Chk1. Mol. Biol. Cell 1999, 10, 833–845. [Google Scholar] [CrossRef] [PubMed]
- Kumagai, A.; Guo, Z.; Emami, K.H.; Wang, S.X.; Dunphy, W.G. The Xenopus Chk1 protein kinase mediates a caffeine-sensitive pathway of checkpoint control in cell-free extracts. J. Cell Biol. 1998, 142, 1559–1569. [Google Scholar] [CrossRef] [PubMed]
- Peng, C.Y.; Graves, P.R.; Ogg, S.; Thoma, R.S.; Byrnes, M.J., 3rd.; Wu, Z.; Stephenson, M.T.; Piwnica-Worms, H. C-TAK1 protein kinase phosphorylates human Cdc25C on serine 216 and promotes 14-3-3 protein binding. Cell Growth Differ. 1998, 9, 197–208. [Google Scholar] [PubMed]
- Zeng, Y.; Forbes, K.C.; Wu, Z.; Moreno, S.; Piwnica-Worms, H.; Enoch, T. Replication checkpoint requires phosphorylation of the phosphatase Cdc25 by Cds1 or Chk1. Nature 1998, 395, 507–510. [Google Scholar] [CrossRef] [PubMed]
- Duckworth, B.C.; Weaver, J.S.; Ruderman, J.V. G2 arrest in Xenopus oocytes depends on phosphorylation of Cdc25 by protein kinase A. Proc. Natl. Acad. Sci. USA 2002, 99, 16794–16799. [Google Scholar] [CrossRef] [PubMed]
- Peng, C.Y.; Graves, P.R.; Thoma, R.S.; Wu, Z.; Shaw, A.S.; Piwnica-Worms, H. Mitotic and G2 Checkpoint Control: Regulation of 14-3-3 Protein Binding by Phosphorylation of Cdc25C on Serine-216. Science 1997, 277, 1501–1505. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, Y.; Wong, C.; Thoma, R.S.; Richman, R.; Wu, Z.; Piwnica-Worms, H.; Elledge, S.J. Conservation of the Chk1 Checkpoint Pathway in Mammals: Linkage of DNA Damage to Cdk Regulation Through Cdc25. Science 1997, 277, 1497–1501. [Google Scholar] [CrossRef]
- Kumagai, A.; Yakowec, P.S.; Dunphy, W.G. 14-3-3 proteins act as negative regulators of the mitotic inducer Cdc25 in Xenopus egg extracts. Mol. Biol. Cell 1998, 9, 345–354. [Google Scholar] [CrossRef]
- Hermeking, H.; Benzinger, A. 14-3-3 proteins in cell cycle regulation. Semin. Cancer Biol. 2006, 16, 183–192. [Google Scholar] [CrossRef]
- Margolis, S.S.; Perry, J.A.; Weitzel, D.H.; Freel, C.D.; Yoshida, M.; Haystead, T.A.; Kornbluth, S. A role for PP1 in the Cdc2/Cyclin B-mediated positive feedback activation of Cdc25. Mol. Biol. Cell 2006, 17, 1779–1789. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.S.; Ryan, C.E.; Piwnica-Worms, H. Chk1 kinase negatively regulates mitotic function of Cdc25A phosphatase through 14-3-3 binding. Mol. Cell. Biol. 2003, 23, 7488–7497. [Google Scholar] [CrossRef]
- Dutertre, S.; Cazales, M.; Quaranta, M.; Froment, C.; Trabut, V.; Dozier, C.; Mirey, G.; Bouché, J.; Theis-Febvre, N.; Schmitt, E.; et al. Phosphorylation of CDC25B by Aurora-A at the centrosome contributes to the G2–M transition. J. Cell Sci. 2004, 117, 2523–2531. [Google Scholar] [CrossRef] [PubMed]
- Cazales, M.; Schmitt, E.; Montembault, E.; Dozier, C.; Prigent, C.; Ducommun, B. CDC25B phosphorylation by Aurora A occurs at the G2/M transition and is inhibited by DNA damage. Cell Cycle 2005, 4, 1233–1238. [Google Scholar] [CrossRef] [PubMed]
- Seki, A.; Coppinger, J.A.; Jang, C.Y.; Yates, J.R.; Fang, G. Bora and the kinase Aurora a cooperatively activate the kinase Plk1 and control mitotic entry. Science 2008, 320, 1655–1658. [Google Scholar] [CrossRef]
- Kumagai, A.; Dunphy, W.G. Purification and molecular cloning of Plx1, a Cdc25-regulatory kinase from Xenopus egg extracts. Science 1996, 237, 1377–1380. [Google Scholar] [CrossRef]
- Qian, Y.W.; Erikson, E.; Maller, J.L. Purification and cloning of a protein kinase that phosphorylates and activates the polo-like kinase Plx1. Science 1998, 282, 1701–1704. [Google Scholar] [CrossRef]
- Karaïskou, A.; Jessus, C.; Brassac, T.; Ozon, R. Phosphatase 2A and polo kinase, two antagonistic regulators of cdc25 activation and MPF auto-amplification. J. Cell Sci. 1999, 112, 3747–3756. [Google Scholar]
- Roshak, A.K.; Capper, E.A.; Imburgia, C.; Fornwald, J.; Scott, G.; Marshall, L.A. The human polo-like kinase, PLK, regulates cdc2/cyclin B through phosphorylation and activation of the cdc25C phosphatase. Cell Signal. 2000, 12, 405–411. [Google Scholar] [CrossRef]
- van Vugt, M.A.; Brás, A.; Medema, R.H. Polo-like kinase-1 controls recovery from a G2 DNA damage-induced arrest in mammalian cells. Mol. Cell 2004, 15, 799–811. [Google Scholar] [CrossRef]
- Lobjois, V.; Froment, C.; Braud, E.; Grimal, F.; Burlet-Schiltz, O.; Ducommun, B.; Bouche, J.P. Study of the docking-dependent PLK1 phosphorylation of the CDC25B phosphatase. Biochem. Biophys. Res. Commun. 2011, 410, 87–90. [Google Scholar] [CrossRef] [PubMed]
- Gheghiani, L.; Loew, D.; Lombard, B.; Mansfeld, J.; Gavet, O. PLK1 activation in late G2 sets up commitment to mitosis. Cell Rep. 2017, 19, 2060–2073. [Google Scholar] [CrossRef] [PubMed]
- Qi, F.; Chen, Q.; Chen, H.; Yan, H.; Chen, B.; Xiang, X.; Liang, C.; Yi, Q.; Zhang, M.; Cheng, H.; et al. WAC Promotes Polo-like Kinase 1 Activation for Timely Mitotic Entry. Cell Rep. 2018, 24, 546–556. [Google Scholar] [CrossRef] [PubMed]
- Vigneron, S.; Sundermann, L.; Labbé, J.C.; Pintard, L.; Radulescu, O.; Castro, A.; Lorca, T. Cyclin A-Cdk1-dependent phosphorylation of Bora is the triggering factor promoting mitotic entry. Dev. Cell 2018, 45, 637–650. [Google Scholar] [CrossRef] [PubMed]
- Toyoshima-Morimoto, F.; Taniguchi, E.; Nishida, E. Plk1 promotes nuclear translocation of human Cdc25C during prophase. EMBO Rep. 2002, 3, 341–348. [Google Scholar] [CrossRef] [PubMed]
- Lobjois, V.; Jullien, D.; Bouché, J.P.; Ducommun, B. The polo-like kinase 1 regulates CDC25B-dependent mitosis entry. Biochim. Biophys. Acta 2009, 1793, 462–468. [Google Scholar] [CrossRef] [PubMed]
- Margolis, S.S.; Walsh, S.; Weiser, D.C.; Yoshida, M.; Shenolikar, S.; Kornbluth, S. PP1 control of M phase entry exerted through 14-3-3-regulated Cdc25 dephosphorylation. EMBO J. 2003, 22, 5734–5745. [Google Scholar] [CrossRef]
- Clarke, P.R.; Hoffmann, I.; Draetta, G.; Karsenti, E. Dephosphorylation of Cdc25-C by a type-2A protein phosphatase: Specific regulation during the cell cycle in Xenopus egg extracts. Mol. Biol. Cell 1993, 4, 397–411. [Google Scholar] [CrossRef]
- Mayer-Jaekel, R.E.; Ohkura, H.; Ferrigno, P.; Andjelkovic, N.; Shiomi, K.; Uemura, T.; Glover, D.M.; Hemmings, B.A. Drosophila mutants in the 55 kDa regulatory subunit of protein phosphatase 2A show strongly reduced ability to dephosphorylate substrates of p34cdc2. J. Cell Sci. 1994, 107, 2609–2616. [Google Scholar]
- Mochida, S.; Hunt, T. Calcineurin is required to release Xenopus egg extracts from meiotic M phase. Nature 2007, 449, 336–340. [Google Scholar] [CrossRef]
- Castilho, P.V.; Williams, B.C.; Mochida, S.; Zhao, Y.; Goldberg, M.L. The M Phase Kinase Greatwall (Gwl) Promotes Inactivation of PP2A/B55δ, a Phosphatase Directed Against CDK Phosphosites. Mol. Biol. Cell 2009, 20, 4777–4789. [Google Scholar] [CrossRef] [PubMed]
- Mochida, S.; Ikeo, S.; Gannon, J.; Hunt, T. Regulated activity of PP2A-B55δ is crucial for controlling entry into and exit from mitosis in Xenopus egg extracts. EMBO J. 2009, 28, 2777–2785. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, M.H.A.; Held, M.; Janssens, V.; Hutchins, J.R.A.; Hudecz, O.; Ivanova, E.; Goris, J.; Trinkle-Mulcahy, L.; Lamond, A.I.; Poser, I.; et al. Live-cell imaging RNAi screen identifies PP2A-B55α and importin-β1 as key mitotic exit regulators in human cells. Nat. Cell Biol. 2010, 12, 886–893. [Google Scholar] [CrossRef] [PubMed]
- Cundell, M.J.; Bastos, R.N.; Zhang, T.; Holder, J.; Gruneberg, U.; Novak, B.; Barr, F.A. The BEG (PP2A-B55/ENSA/Greatwall) pathway ensures cytokinesis follows chromosome separation. Mol. Cell 2013, 52, 393–405. [Google Scholar] [CrossRef] [PubMed]
- Gharbi-Ayachi, A.; Labbé, J.C.; Burgess, A.; Vigneron, S.; Strub, J.M.; Brioudes, E.; Van-Dorsselaer, A.; Castro, A.; Lorca, T. The Substrate of Greatwall Kinase, Arpp19, Controls Mitosis by Inhibiting Protein Phosphatase 2A. Science 2010, 330, 1673–1677. [Google Scholar] [CrossRef] [PubMed]
- Mochida, S.; Maslen, S.L.; Skehel, M.; Hunt, T. Greatwall Phosphorylates an Inhibitor of Protein Phosphatase 2A That Is Essential for Mitosis. Science 2010, 330, 1670–1673. [Google Scholar] [CrossRef] [PubMed]
- Archambault, V.; Zhao, X.; White-Cooper, H.; Carpenter, A.T.C.; Glover, D.M. Mutations in Drosophila Greatwall/Scant Reveal Its Roles in Mitosis and Meiosis and Interdependence with Polo Kinase. PLoS Genet. 2007, 3, e200. [Google Scholar] [CrossRef]
- Burgess, A.; Vigneron, S.; Brioudes, E.; Labbé, J.C.; Lorca, T.; Castro, A. Loss of human Greatwall results in G2 arrest and multiple mitotic defects due to deregulation of the cyclin B-Cdc2/PP2A balance. Proc. Natl. Acad. Sci. USA 2010, 107, 12564–12569. [Google Scholar] [CrossRef]
- Hara, M.; Abe, Y.; Tanaka, T.; Yamamoto, T.; Okumura, E.; Kishimoto, T. Greatwall kinase and cyclin B-Cdk1 are both critical constituents of M-phase-promoting factor. Nat. Commun. 2012, 3, 1059. [Google Scholar] [CrossRef]
- Lorca, T.; Bernis, C.; Vigneron, S.; Burgess, A.; Brioudes, E.; Labbé, J.C.; Castro, A. Constant regulation of both the MPF amplification loop and the Greatwall-PP2A pathway is required for metaphase II arrest and correct entry into the first embryonic cell cycle. J. Cell Sci. 2010, 123, 2281–2291. [Google Scholar] [CrossRef]
- Voets, E.; Wolthuis, R.M. MASTL is the human orthologue of Greatwall kinase that facilitates mitotic entry, anaphase and cytokinesis. Cell Cycle 2010, 9, 3591–3601. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Fleming, S.L.; Williams, B.; Williams, E.V.; Li, Z.; Somma, P.; Rieder, C.L.; Goldberg, M.L. Greatwall kinase: A nuclear protein required for proper chromosome condensation and mitotic progression in Drosophila. J. Cell Biol. 2004, 164, 487–492. [Google Scholar] [CrossRef] [PubMed]
- Álvarez-Fernández, M.; Sánchez-Martinez, R.; Sanz-Castillo, B.; Can, P.P.; Sanz-Flores, M.; Trakala, M.; Ruiz-Torres, M.; Lorca, T.; Castro, A.; Malumbres, M. Greatwall is essential to prevent mitotic collapse after nuclear envelope breakdown in mammals. Proc. Natl. Acad. Sci. USA 2013, 110, 17374–17379. [Google Scholar] [CrossRef] [PubMed]
- Diril, M.K.; Bisteau, X.; Kitagawa, M.; Caldez, M.J.; Wee, S.; Gunaratne, J.; Lee, S.H.; Kaldis, P. Loss of the Greatwall Kinase Weakens the Spindle Assembly Checkpoint. PLoS Genet. 2016, 12, e1006310. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Zhao, Y.; Li, Z.; Galas, S.; Goldberg, M.L. Greatwall kinase participates in the Cdc2 autoregulatory loop in Xenopus egg extracts. Mol. Cell 2006, 22, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Blake-Hodek, K.A.; Williams, B.C.; Zhao, Y.; Castilho, P.V.; Chen, W.; Mao, Y.; Yamamoto, T.M.; Goldberg, M.L. Determinants for activation of the atypical AGC kinase Greatwall during M phase entry. Mol. Cell. Biol. 2012, 32, 1337–1353. [Google Scholar] [CrossRef] [PubMed]
- Vigneron, S.; Gharbi-Ayachi, A.; Raymond, A.A.; Burgess, A.; Labbé, J.C.; Labesse, G.; Monsarrat, B.; Lorca, T.; Castro, A. Characterization of the mechanisms controlling Greatwall activity. Mol. Cell. Biol. 2011, 31, 2262–2275. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Haccard, O.; Wang, R.; Yu, J.; Kuang, J.; Jessus, C.; Goldberg, M.LL. Roles of Greatwall Kinase in the Regulation of Cdc25 Phosphatase. Mol. Biol. Cell 2008, 19, 1317–1327. [Google Scholar] [CrossRef] [PubMed]
- Peters, J.M.; Tedeschi, A.; Schmitz, J. The cohesin complex and its roles in chromosome biology. Genes Dev. 2008, 22, 3089–3114. [Google Scholar] [CrossRef]
- Nasmyth, K.; Haering, C.H. Cohesin: Its roles and mechanisms. Annu. Rev. Genet. 2009, 43, 525–558. [Google Scholar] [CrossRef]
- Haering, C.H.; Löwe, J.; Hochwagen, A.; Nasmyth, K. Molecular architecture of SMC proteins and the yeast cohesion complex. Mol. Cell 2002, 9, 773–788. [Google Scholar] [CrossRef]
- Gandhi, R.; Gillespie, P.J.; Hirano, T. Human WapI is a cohesin-binding protein that promotes sister-chromatid resolution in mitotic prophase. Curr. Biol. 2006, 16, 2406–2417. [Google Scholar] [CrossRef] [PubMed]
- Kueng, S.; Hegemann, B.; Peters, B.H.; Lipp, J.J.; Schleiffer, A.; Mechtler, K.; Peters, J.M. WapI controls the dynamic association of cohesion with chromatin. Cell 2006, 127, 955–967. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.L.; Roig, M.B.; Hu, B.; Beckouët, F.; Metson, J.; Nasmyth, K. Cohesin’s DNA exit gate is distinct from its entrance gate and is regulated by acetylation. Cell 2012, 150, 961–974. [Google Scholar] [CrossRef] [PubMed]
- Buheitel, J.; Stemmann, O. Prophase pathway-dependent removal of cohesin from human chromosomes requires opening of the Smc3–Scc1 gate. EMBO J. 2013, 32, 666–676. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, A.; Zakian, S.; Hu, X.W.; Singleton, M.R. Structural insights into the regulation of cohesion establishment by Wpl1. EMBO J. 2013, 32, 677–687. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, Z.; Zheng, G.; Song, J.; Borek, D.M.; Otwinowski, Z.; Brautigam, C.A.; Tomchick, D.R.; Rankin, S.; Yu, H. Structure of the human cohesin inhibitor Wapl. Proc. Natl. Acad. Sci. USA 2013, 110, 11355–11360. [Google Scholar] [CrossRef]
- Eichinger, C.S.; Kurze, A.; Oliveira, R.A.; Nasmyth, K. Disengaging the Smc3/kleisin interface releases cohesin from Drosophila chromosomes during interphase and mitosis. EMBO J. 2013, 32, 656–665. [Google Scholar] [CrossRef]
- Ben-Shahar, T.R.; Heeger, S.; Lehane, C.; East, P.; Flynn, H.; Skehel, M.; Uhlmann, F. Eco1-dependent cohesin acetylation during establishment of sister chromatid cohesion. Science 2008, 321, 563–566. [Google Scholar] [CrossRef]
- Rowland, B.D.; Roig, M.B.; Nishino, T.; Kurze, A.; Uluocak, P.; Mishra, A.; Beckouët, F.; Underwood, P.; Metson, J.; Imre, R.; et al. Building sister chromatid cohesion: Smc3 acetylation counteracts an antiestablishment activity. Mol. Cell 2009, 33, 763–774. [Google Scholar] [CrossRef]
- Sutani, T.; Kawaguchi, T.; Kanno, R.; Itoh, T.; Shirahige, K. Budding yeast Wpl1 (Rad61)-Pds5 complex counteracts sister chromatid cohesion-establishing reaction. Curr. Biol. 2009, 19, 492–497. [Google Scholar] [CrossRef] [PubMed]
- Ünal, E.; Heidinger-Pauli, J.M.; Kim, W.; Guacci, V.; Onn, I.; Gygi, S.P.; Koshland, D.E. A molecular determinant for the establishment of sister chromatid cohesion. Science 2008, 321, 566–569. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Shi, X.; Li, Y.; Kim, B.J.; Jia, J.; Huang, Z.; Yang, T.; Fu, X.; Jung, S.Y.; Wang, Y.; et al. Acetylation of Smc3 by Eco1 is required for S phase sister chromatid cohesion in both human and yeast. Mol. Cell 2008, 31, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Lafont, A.L.; Song, J.; Rankin, S. Sororin cooperates with the acetyltransferase Eco2 to ensure DNA replication-dependent sister chromatid cohesion. Proc. Natl. Acad. Sci. USA 2010, 107, 20364–20369. [Google Scholar] [CrossRef] [PubMed]
- Nishiyama, T.; Ladurner, R.; Schmitz, J.; Kreidl, E.; Schleiffer, A.; Bhaskara, V.; Bando, M.; Shirahige, K.; Hyman, A.A.; Mechtler, K.; et al. Sororin mediates sister chromatid cohesion by antagonizing WapI. Cell 2010, 143, 737–749. [Google Scholar] [CrossRef] [PubMed]
- Dreier, M.R.; Bekier, M.E., 2nd.; Taylor, W.R. Regulation of sororin by Cdk1-mediated phosphorylation. J. Cell Sci. 2011, 124, 2976–2987. [Google Scholar] [CrossRef] [PubMed]
- Giménez-Abián, J.F.; Sumara, I.; Hirota, T.; Hauf, S.; Gerlich, D.; de la Torre, C.; Ellenberg, J.; Peters, J.M. Regulation of sister chromatid cohesion between chromosome arms. Curr. Biol. 2004, 14, 1187–1193. [Google Scholar] [CrossRef] [PubMed]
- Hauf, S.; Roitinger, E.; Koch, B.; Dittrich, C.M.; Mechtler, K.; Peters, J.M. Dissociation of cohesin from chromosome arms and loss of arm cohesion during early mitosis depends on phosphorylation of SA2. PLoS Biol. 2005, 3, e69. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Rankin, S.; Yu, H. Phosphorylation-enabled binding of SGO1-PP2A to cohesin protects sororin and centromeric cohesion during mitosis. Nat. Cell Biol. 2013, 15, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Losada, A.; Hirano, M.; Hirano, T. Cohesin release is required for sister chromatid resolution, but not for condensin-mediated compaction, at the onset of mitosis. Genes Dev. 2002, 16, 3004–3016. [Google Scholar] [CrossRef] [PubMed]
- Nishiyama, T.; Sykora, M.M.; Huis, P.J.; Mechtler, K.; Peters, J.M. Aurora B and Cdk1 mediate WapI activation and release of acetylated cohesin from chromosomes by phosphorylating Sororin. Proc. Natl. Acad. Sci. USA 2013, 110, 13404–13409. [Google Scholar] [CrossRef] [PubMed]
- Sumara, I.; Vorlaufer, E.; Stukenberg, P.T.; Kelm, O.; Redemann, N.; Nigg, E.A.; Peters, J.M. The dissociation of cohesin from chromosomes in prophase is regulated by Polo-like kinase. Mol. Cell 2002, 9, 515–525. [Google Scholar] [CrossRef]
- Zhang, N.; Panigrahi, A.K.; Mao, Q.; Pati, D. Interaction of Sororin protein with Polo-like kinase 1 mediates resolution of chromosomal arm cohesion. J. Biol. Chem. 2011, 286, 41826–41837. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.H.; Mayer, B.; Stemmann, O.; Nigg, E.A. Centromere DNA decatenation depends on cohesin removal and is required for mammalian cell division. J. Cell Sci. 2010, 123, 806–813. [Google Scholar] [CrossRef] [PubMed]
- Deardorff, M.A.; Wilde, J.J.; Albrecht, M.; Dickinson, E.; Tennstedt, S.; Braunholz, D.; Mönnich, M.; Yan, Y.; Xu, W.; Gil-Rodríguez, M.C.; et al. RAD21 mutations cause a human cohesinopathy. Am. J. Hum. Genet. 2012, 90, 1014–1027. [Google Scholar] [CrossRef] [PubMed]
- Haarhuis, J.H.; Elbatsh, A.M.O.; van den Broek, B.; Camps, D.; Erkan, H.; Jalink, K.; Medema, R.H.; Rowland, B.D. WAPL-mediated removal of cohesin protects against segregation errors and aneuploidy. Curr. Biol. 2013, 23, 2071–2077. [Google Scholar] [CrossRef] [PubMed]
- Tedeschi, A.; Wutz, G.; Huet, S.; Jaritz, M.; Wuensche, A.; Schirghuber, E.; Davidson, I.F.; Tang, W.; Cisneros, D.A.; Bhaskara, V.; et al. WapI is an essential regulator of chromatin structure and chromosome segregation. Nature 2013, 501, 564–568. [Google Scholar] [CrossRef]
- Nicklas, R.B. How cells get the right chromosomes. Science 1997, 275, 632–637. [Google Scholar] [CrossRef]
- Tanaka, T.; Fuchs, J.; Loidl, J.; Nasmyth, K. Cohesin ensures bipolar attachment of microtubules to sister centromeres and resists their precocious separation. Nat. Cell Biol. 2000, 2, 492–499. [Google Scholar] [CrossRef]
- Haarhuis, J.H.I.; Elbatsh, A.M.O.; Rowland, B.D. Cohesin and its regulation: On the logic of X-shaped chromosomes. Dev. Cell 2014, 31, 7–18. [Google Scholar] [CrossRef]
- Morales, C.; Losada, A. Establishing and dissolving cohesion during the vertebrate cell cycle. Curr. Opin. Cell Biol. 2018, 52, 51–57. [Google Scholar] [CrossRef] [PubMed]
- McGuinness, B.E.; Hirota, T.; Kudo, N.R.; Peters, J.M.; Masmyth, K. Shugoshin prevents dissociation of cohesin from centromeres during mitosis in vertebrate cells. PLoS Biol. 2005, 3, e86. [Google Scholar] [CrossRef] [PubMed]
- Kitajima, T.S.; Sakuno, T.; Ishiguro, K.; Iemura, S.; Natsume, T.; Kawashima, S.A.; Watanabe, Y. Shugoshin collaborates with protein phosphatase 2A to protect cohesin. Nature 2006, 441, 46–52. [Google Scholar] [CrossRef] [PubMed]
- Riedel, C.G.; Katis, V.L.; Katou, Y.; Mori, S.; Itoh, T.; Helmhart, W.; Gálová, M.; Petronczki, M.; Gregan, J.; Cetin, B.; et al. Protein phosphatase 2A protects centromeric sister chromatid cohesion during meiosis I. Nature 2006, 441, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Shu, H.; Qi, W.; Mahmood, N.A.; Mumby, M.C.; Yu, H. PP2A is required for centromeric localization of Sgo1 and proper chromosome segregation. Dev. Cell 2006, 10, 575–585. [Google Scholar] [CrossRef] [PubMed]
- Rivera, T.; Losada, A. Shugoshin regulates cohesion by driving relocalization of PP2A in Xenopus extracts. Chromosoma 2009, 118, 223–233. [Google Scholar] [CrossRef] [PubMed]
- Tanno, Y.; Kitajima, T.S.; Honda, T.; Ando, Y.; Ishiguro, K.; Watanabe, Y. Phosphorylation of mammalian Sgo2 by Aurora B recruits PP2A and MCAK to centromeres. Genes Dev. 2010, 24, 2169–2179. [Google Scholar] [CrossRef]
- Xu, Z.; Cetin, B.; Anger, M.; Cho, U.S.; Helmhart, W.; Nasmyth, K.; Xu, W. Structure and function of the PP2A-shugoshin interaction. Mol. Cell 2009, 35, 426–441. [Google Scholar] [CrossRef]
- Gutiérrez-Caballero, C.; Cebollero, L.R.; Pendás, A.M. Shugoshins: From protectors of cohesion to versatile adaptors at the centromere. Trends Genet. 2012, 28, 351–360. [Google Scholar] [CrossRef]
- Huang, H.; Feng, J.; Famulski, J.; Rattner, J.B.; Liu, S.T.; Kao, G.D.; Muschel, R.; Chan, G.K.; Yen, T.J. Tripin/hSgo2 recruits MCAK to the inner centromere to correct defective kinetochore attachments. J. Cell Biol. 2007, 177, 413–424. [Google Scholar] [CrossRef]
- Kitajima, T.S.; Hauf, S.; Ohsugi, M.; Yamamoto, T.; Watanabe, Y. Human Bub1 defines the persistent cohesion site along the mitotic chromosome by affecting Shugoshin localization. Curr. Biol. 2005, 15, 353–359. [Google Scholar] [CrossRef] [PubMed]
- Llano, E.; Gómez, R.; Gutiérrez-Caballero, C.; Herrán, Y.; Sánchez-Martín, M.; Vázquez-Quiñones, L.; Hernández, T.; de Alava, E.; Cuadrado, A.; Barbero, J.L.; et al. Shugoshin-2 is essential for the completion of meiosis but not for mitotic cell division in mice. Genes Dev. 2008, 22, 2400–2413. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Kitajima, T.S.; Tanno, Y.; Yoshida, K.; Morita, T.; Miyano, T.; Miyake, M.; Watanabe, Y. Unified mode of centromeric protection by shugoshin in mammalian oocytes and somatic cells. Nat. Cell Biol. 2008, 10, 42–52. [Google Scholar] [CrossRef] [PubMed]
- Wittmann, T.; Hyman, A.; Desai, A. The spindle: A dynamic assembly of microtubules and motors. Nat. Cell Biol. 2001, 3, E28–E34. [Google Scholar] [CrossRef] [PubMed]
- Mennella, V.; Agard, D.A.; Huang, B.; Pelletier, L. Amorphous no more: Subdiffraction view of the pericentriolar material architecture. Trends Cell Biol. 2014, 24, 188–197. [Google Scholar] [CrossRef] [PubMed]
- Woodruff, J.B.; Wueseke, O.; Hyman, A.A. Pericentriolar material structure and dynamics. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2014, 369, 20130459. [Google Scholar] [CrossRef] [PubMed]
- Conduit, P.T.; Wainman, A.; Raff, J.W. Centrosome function and assembly in animal cells. Nat. Rev. Mol. Cell Biol. 2015, 16, 611–624. [Google Scholar] [CrossRef]
- Stearns, T.; Kirschner, M. In vitro reconstitution of centrosome assembly and function: The central role of γ-tubulin. Cell 1994, 76, 623–637. [Google Scholar] [CrossRef]
- Sunkel, C.E.; Gomes, R.; Sampaio, P.; Perdigão, J.; González, C. γ-tubulin is required for the structure and function of the microtubule organizing centre in Drosophila neuroblasts. EMBO J. 1995, 14, 28–36. [Google Scholar] [CrossRef]
- Lattao, R.; Kovács, L.; Glover, D.M. The Centrioles, Centrosomes, Basal Bodies, and Cilia of Drosophila melanogaster. Genetics 2017, 206, 33–53. [Google Scholar] [CrossRef]
- Sumiyoshi, E.; Sugimoto, A.; Yamamoto, M. Protein phosphatase 4 is required for centrosome maturation in mitosis and sperm meiosis in C. elegans. J. Cell Sci. 2002, 115, 1403–1410. [Google Scholar] [PubMed]
- Martin-Granados, C.; Philp, A.; Oxenham, S.K.; Prescott, A.R.; Cohen, P.T. Depletion of protein phosphatase 4 in human cells reveals essential roles in centrosome maturation, cell migration and the regulation of Rho GTPases. Int. J. Biochem. Cell Biol. 2008, 40, 2315–2332. [Google Scholar] [CrossRef] [PubMed]
- Toyo-oka, K.; Mori, D.; Yano, Y.; Shiota, M.; Iwao, H.; Goto, H.; Inagaki, M.; Hiraiwa, N.; Muramatsu, M.; Wynshaw-Boris, A.; et al. Protein phosphatase 4 catalytic subunit regulates Cdk1 activity and microtubule organization via NDEL1 dephosphorylation. J. Cell Biol. 2008, 180, 1133–1147. [Google Scholar] [CrossRef] [PubMed]
- McNally, F.J.; Vale, R.D. Identification of katanin, an ATPase that severs and disassembles stable microtubules. Cell 1993, 75, 419–429. [Google Scholar] [CrossRef]
- McNally, F. Capturing a ring of samurai. Nat. Cell Biol. 2000, 2, E4–E7. [Google Scholar] [CrossRef] [PubMed]
- Toyo-Oka, K.; Sasaki, S.; Yanon, Y.; Mori, D.; Kobayashi, T.; Toyoshima, Y.Y.; Tokuoka, S.M.; Ishii, S.; Shimizu, T.; Muramatsu, M.; et al. Recruitment of katanin p60 by phosphorylated NDEL1, an LIS1 interacting protein, is essential for mitotic cell division and neuronal migration. Hum. Mol. Genet. 2005, 14, 3113–3128. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Gomes, J.E.; Birmingham, C.L.; Pintard, L.; Sugimoto, A.; Mains, P.E. The role of protein phosphatase 4 in regulating microtubule severing in the Caenorhabditis elegans embryo. Genetics 2009, 181, 933–943. [Google Scholar] [CrossRef]
- Gomes, J.E.; Tavernier, N.; Richaudeau, B.; Formstecher, E.; Boulin, T.; Mains, P.E.; Dumont, J.; Pintard, L. Microtubule severing by the katanin complex is activated by PPFR-1-dependent MEI-1 dephosphorylation. J. Cell Biol. 2013, 202, 431–439. [Google Scholar] [CrossRef]
- Srayko, M.; Kaya, A.; Stamford, J.; Hyman, A.A. Identification and characterization of factors required for microtubule growth and nucleation in the early C. elegans embryo. Dev. Cell 2005, 9, 223–226. [Google Scholar] [CrossRef]
- Thawani, A.; Kadzik, R.S.; Petry, S. XMAP215 is a microtubule nucleation factor that functions synergistically with the γ-tubulin ring complex. Nat. Cell Biol. 2018, 20, 575–585. [Google Scholar] [CrossRef]
- Kinoshita, K.; Noetzel, T.L.; Pelletier, L.; Mechtler, K.; Drechsel, D.N.; Schwager, A.; Lee, M.; Raff, J.W.; Hyman, A.A. Aurora A phosphorylation of TACC3/maskin is required for centrosome-dependent microtubule assembly in mitosis. J. Cell Biol. 2005, 170, 1047–1055. [Google Scholar] [CrossRef] [PubMed]
- Schlaitz, A.L.; Srayko, M.; Dammermann, A.; Quintin, S.; Wielsch, N.; MacLeod, I.; de Robillard, Q.; Zinke, A.; Yates, J.R., 3rd.; Müller-Reichert, T.; et al. The C. elegans RSA complex localizes protein phosphatase 2A to centrosomes and regulates mitotic spindle assembly. Cell 2007, 128, 115–127. [Google Scholar] [CrossRef] [PubMed]
- Kufer, T.A.; Silljé, H.H.; Körner, R.; Gruss, O.J.; Meraldi, P.; Nigg, E.A. Human TPX2 is required for targeting Aurora-A kinase to the spindle. J. Cell Biol. 2002, 158, 617–623. [Google Scholar] [CrossRef] [PubMed]
- Ozlü, N.; Srayko, M.; Kinoshita, K.; Habermann, B.; O’toole, E.T.; Müller-Reichert, T.; Schmalz, N.; Desai, A.; Hyman, A.A. An essential function of the C. elegans ortholog of TPX2 is to localize activated aurora A kinase to mitotic spindles. Dev. Cell 2005, 9, 237–248. [Google Scholar] [CrossRef] [PubMed]
- Wittmann, T.; Wilm, M.; Karsenti, E.; Vernos, I. TPX2, A novel xenopus MAP involved in spindle pole organization. J. Cell Biol. 2000, 149, 1405–1418. [Google Scholar] [CrossRef] [PubMed]
- Gruss, O.J.; Carazo-Salas, R.E.; Schatz, C.A.; Guarguaglini, G.; Kast, J.; Wilm, M.; Le Bot, N.; Vernos, I.; Karsenti, E.; Mattaj, I.W. Ran induces spindle assembly by reversing the inhibitory effect of importin α on TPX2 activity. Cell 2001, 104, 83–93. [Google Scholar] [CrossRef]
- Meunier, S.; Vernos, I. Acentrosomal Microtubule Assembly in Mitosis: The Where, When, and How. Trends Cell Biol. 2016, 26, 80–87. [Google Scholar] [CrossRef]
- Kalab, P.; Heald, R. The RanGTP gradient—A GPS for the mitotic spindle. J. Cell Sci. 2008, 121, 1577–1586. [Google Scholar] [CrossRef]
- Cavazza, T.; Vernos, I. The RanGTP Pathway: From Nucleo-Cytoplasmic Transport to Spindle Assembly and Beyond. Front Cell Dev. Biol. 2016, 3, 82. [Google Scholar] [CrossRef]
- Bayliss, R.; Sardon, T.; Vernos, I.; Conti, E. Structural basis of Aurora-A activation by TPX2 at the mitotic spindle. Mol. Cell 2003, 12, 851–862. [Google Scholar] [CrossRef]
- Eyers, P.A.; Erikson, E.; Chen, L.G.; Maller, J.L. A novel mechanism for activation of the protein kinase Aurora A. Curr. Biol. 2003, 13, 691–697. [Google Scholar] [CrossRef]
- Tsai, M.Y.; Wiese, C.; Cao, K.; Martin, O.; Donovan, P.; Ruderman, J.; Prigent, C.; Zheng, Y. A Ran signalling pathway mediated by the mitotic kinase Aurora A in spindle assembly. Nat. Cell Biol. 2003, 5, 242–248. [Google Scholar] [CrossRef] [PubMed]
- Groen, A.C.; Cameron, L.A.; Coughlin, M.; Miyamoto, D.T.; Mitchison, T.J.; Ohi, R. XRHAMM functions in ran-dependent microtubule nucleation and pole formation during anastral spindle assembly. Curr. Biol. 2004, 14, 1801–1811. [Google Scholar] [CrossRef] [PubMed]
- Scrofani, J.; Sardon, T.; Meunier, S.; Vernos, I. Microtubule nucleation in mitosis by a RanGTP-dependent protein complex. Curr. Biol. 2015, 25, 131–140. [Google Scholar] [CrossRef]
- Pinoyl, R.; Scrofani, J.; Vernos, I. The role of NEDD1 phosphorylation by Aurora A in chromosomal microtubule nucleation and spindle function. Curr. Biol. 2013, 23, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Zeng, K.; Bastos, R.N.; Barr, F.A.; Gruneberg, U. Protein phosphatase 6 regulates mitotic spindle formation by controlling the T-loop phosphorylation state of Aurora A bound to its activator TPX2. J. Cell Biol. 2010, 191, 1315–1332. [Google Scholar] [CrossRef] [PubMed]
- Zorba, A.; Buosi, V.; Kutter, S.; Kern, N.; Pontiggia, F.; Cho, Y.J.; Kern, D. Molecular mechanism of Aurora A kinase autophosphorylation and its allosteric activation by TPX2. Elife 2014, 3, e02667. [Google Scholar] [CrossRef]
- Cyphers, S.; Ruff, E.F.; Behr, J.M.; Chodera, J.D.; Levinson, N.M. A water-mediated allosteric network governs activation of Aurora kinase A. Nat. Chem. Biol. 2017, 13, 402–408. [Google Scholar] [CrossRef]
- Ruff, E.F.; Muretta, J.M.; Thompson, A.R.; Lake, E.W.; Cyphers, S.; Albanese, S.K.; Hanson, S.M.; Behr, J.M.; Thomas, D.D.; Chodera, J.D.; et al. A dynamic mechanism for allosteric activation of Aurora kinase A by activation loop phosphorylation. eLife 2018, 7, e32766. [Google Scholar] [CrossRef]
- Andrésson, T.; Ruderman, J.V. The kinase Eg2 is a component of the Xenopus oocyte progesterone-activated signaling pathway. EMBO J. 1998, 17, 5627–5637. [Google Scholar] [CrossRef]
- Walter, A.O.; Seghezzi, W.; Korver, W.; Sheung, J.; Lees, E. The mitotic serine/threonine kinase Aurora2/AIK is regulated by phosphorylation and degradation. Oncogene 2000, 19, 4906–4916. [Google Scholar] [CrossRef] [PubMed]
- Rusin, S.F.; Schlosser, K.A.; Adamo, M.E.; Kettenbach, A.N. Quantitative phosphoproteomics reveals new roles for the protein phosphatase PP6 in mitotic cells. Sci. Signal. 2015, 8, rs12. [Google Scholar] [CrossRef] [PubMed]
- Kettenbach, A.N.; Schlosser, K.A.; Lyons, S.P.; Nasa, I.; Gui, J.; Adamo, M.E.; Gerber, S.A. Global assessment of its network dynamics reveals that the kinase Plk1 inhibits the phosphatase PP6 to promote Aurora A activity. Sci. Signal. 2018, 11, eaaq1441. [Google Scholar] [CrossRef] [PubMed]
- Dodson, C.A.; Bayliss, R. Activation of Aurora-A kinase by protein partner binding and phosphorylation are independent and synergistic. J. Biol. Cell 2012, 287, 1150–1157. [Google Scholar] [CrossRef] [PubMed]
- Joukov, V.; De Nicolo, A. Aurora-PLK1 cascades as key signaling modules in the regulation of mitosis. Sci. Signal. 2018, 11, eaar4195. [Google Scholar] [CrossRef] [PubMed]
- Tournebize, R.; Popov, A.; Kinoshita, K.; Ashford, A.J.; Rybina, S.; Pozniakovsky, A.; Mayer, T.U.; Walczak, C.E.; Karsenti, E.; Hyman, A.A. Control of microtubule dynamics by the antagonistic activities of XMAP215 and XKCM1 in Xenopus egg extracts. Nat. Cell Biol. 2000, 2, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.J.; Gergely, F.; Jeffers, K.; Peak-Chew, S.Y.; Raff, J.W. Msps/XMAP215 interacts with the centrosomal protein D-TACC to regulate microtubule behavior. Nat. Cell Biol. 2001, 3, 643–649. [Google Scholar] [CrossRef]
- Gergely, F. Centrosomal TACCtics. Bioessays 2002, 24, 915–925. [Google Scholar] [CrossRef]
- Giet, R.; McLean, D.; Descamps, S.; Lee, M.J.; Raff, J.W.; Prigent, C.; Glover, D.M. Drosophila Aurora A kinase is required to localize D-TACC to centrosomes and to regulate astral microtubules. J. Cell Biol. 2002, 156, 437–451. [Google Scholar] [CrossRef]
- Gergely, F.; Draviam, V.M.; Raff, J.W. The ch-TOG/XMAP215 protein is essential for spindle pole organization in human somatic cells. Genes Dev. 2003, 17, 336–341. [Google Scholar] [CrossRef]
- Peset, I.; Seiler, J.; Sardon, T.; Bejarano, L.A.; Rybina, S.; Vernos, I. Function and regulation of Maskin, a TACC family protein, in microtubule growth during mitosis. J. Cell Biol. 2005, 170, 1057–1066. [Google Scholar] [CrossRef] [PubMed]
- Barros, T.P.; Kinoshita, K.; Hyman, A.A.; Raff, J.W. Aurora A activates D-TACC-Msps complexes exclusively at centrosomes to stabilize centrosomal microtubules. J. Cell Biol. 2005, 170, 1039–1046. [Google Scholar] [CrossRef]
- LeRoy, P.J.; Hunter, J.J.; Hoar, K.M.; Burke, K.E.; Shinde, V.; Ruan, J.; Bowman, D.; Galvin, K.; Ecsedy, J.A. Localization of human TACC3 to mitotic spindles is mediated by phosphorylation on Ser558 by Aurora A: A novel pharmacodynamic method for measuring Aurora A activity. Cancer Res. 2007, 67, 5362–5370. [Google Scholar] [CrossRef] [PubMed]
- Peset, I.; Vernos, I. The TACC proteins: TACC-ling microtubule dynamics and centrosome function. Trends Cell Biol. 2008, 18, 379–388. [Google Scholar] [CrossRef] [PubMed]
- Burgess, S.G.; Mukherjee, M.; Sabir, S.; Joseph, N.; Gutiérrez-Caballero, C.; Richards, M.W.; Huguenin-Dezot, N.; Chin, J.W.; Kennedy, E.J.; Pfuhl, M.; et al. Mitotic spindle association of TACC3 requires Aurora-A-dependent stabilization of a cryptic α-helix. EMBO J. 2018, 37, e97902. [Google Scholar] [CrossRef]
- Tanenbaum, M.E.; Medema, R.H. Mechanisms of centrosome separation and bipolar spindle assembly. Dev. Cell 2010, 19, 797–806. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Paweletz, N.; Schroeter, D. Cdc2-independent induction of premature mitosis by okadaic acid in HeLa cells. Exp. Cell Res. 1998, 242, 1–9. [Google Scholar] [CrossRef]
- Meraldi, P.; Nigg, E.A. Centrosome cohesion is regulated by a balance of kinase and phosphatase activities. J. Cell Sci. 2001, 114, 3749–3757. [Google Scholar]
- Agircan, F.G.; Schiebel, E.; Mardin, B.R. Separate to operate: Control of centrosome positioning and separation. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2014, 369, 20130461. [Google Scholar] [CrossRef]
- Wang, G.; Jiang, Q.; Zhang, C. The role of mitotic kinases in coupling the centrosome cycle with the assembly of the mitotic spindle. J. Cell Sci. 2014, 127, 4111–4122. [Google Scholar] [CrossRef]
- Fry, A.M.; Mayor, T.; Meraldi, P.; Stierhof, Y.D.; Tanaka, K.; Nigg, E.A. C-Nap1, a novel centrosomal coiled-coil protein and candidate substrate of the cell cycle regulated protein kinase Nek2. Cell Biol. 1998, 141, 1563–1574. [Google Scholar] [CrossRef]
- Yang, J.; Liu, X.; Yue, G.; Adamian, M.; Bulgakov, O.; Li, T. Rootletin, a novel coiled-coil protein, is a structural component of the ciliary rootlet. J. Cell Biol. 2002, 159, 431–440. [Google Scholar] [CrossRef] [PubMed]
- Bahe, S.; Stierhof, Y.D.; Wilkinson, C.J.; Leiss, F.; Nigg, E.A. Rootletin forms centriole-associated filaments and functions in centrosome cohesion. J. Cell Biol. 2005, 171, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Graser, S.; Stierhof, Y.D.; Nigg, E.A. Cep68 and Cep215 (Cdk5rap2) are required for centrosome cohesion. J. Cell Sci. 2007, 120, 4321–4331. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Adamian, M.; Li, T. Rootletin interacts with C-Nap1 and may function as a physical linker between the pair of centrioles/basal bodies in cells. Mol. Biol. Cell 2006, 17, 1033–1040. [Google Scholar] [CrossRef] [PubMed]
- Vlijm, R.; Li, X.; Panic, M.; Rüthnick, D.; Hata, S.; Herrmannsdörfer, F.; Kuner, T.; Heilemann, M.; Engelhardt, J.; Hell, S.W.; et al. STED nanoscopy of the centrosome linker reveals a CEP68-organized, periodic rootletin network anchored to a C-Nap1 ring at centrioles. Proc. Natl. Acad. Sci. USA 2018, 115, E2246–E2253. [Google Scholar] [CrossRef] [PubMed]
- Hardy, T.; Lee, M.; Hames, R.S.; Prosser, S.L.; Cheary, D.M.; Samant, M.D.; Schultz, F.; Baxter, J.E.; Rhee, K.; Fry, A.M. Multisite phosphorylation of C-Nap1 releases it from Cep135 to trigger centrosome disjunction. J. Cell Sci. 2014, 127, 2493–2506. [Google Scholar] [CrossRef]
- Faragher, A.J.; Fry, A.M. Nek2A kinase stimulates centrosome disjunction and is required for formation of bipolar mitotic spindles. Mol. Biol. Cell 2003, 14, 2876–2889. [Google Scholar] [CrossRef]
- Bahmanyar, S.; Kaplan, D.D.; Deluca, J.G.; Giddings, T.H., Jr.; O’Toole, T.E.; Winey, M.; Salmon, E.D.; Casey, P.J.; Nelson, W.J.; Barth, A.I. beta-Catenin is a Nek2 substrate involved in centrosome separation. Genes Dev. 2008, 22, 91–105. [Google Scholar] [CrossRef]
- He, R.; Huang, N.; Bao, Y.; Zhou, H.; Teng, J.; Chen, J. LRRC45 is a centrosome linker component required for centrosome cohesion. Cell Rep. 2013, 4, 1100–1107. [Google Scholar] [CrossRef]
- Zhang, Y.; Foreman, O.; Wigle, D.A.; Kosari, F.; Vasmatzis, G.; Salisbury, J.L.; van Deursen, J.; Galardy, P.J. USP44 regulates centrosome positioning to prevent aneuploidy and suppress tumorigenesis. J. Clin. Investig. 2012, 122, 4362–4374. [Google Scholar] [CrossRef] [PubMed]
- Nam, H.J.; van Deursen, J.M. Cyclin B2 and p53 control proper timing of centrosome separation. Nat. Cell Biol. 2014, 16, 538–549. [Google Scholar] [CrossRef] [PubMed]
- Decarreau, J.; Wagenbach, M.; Lynch, E.; Halpern, A.R.; Vaughan, J.C.; Kollman, J.; Wordeman, L. The tetrameric kinesin Kif25 supresses pre-mitotic centrosome separation to establish proper spindle orientation. Nat. Cell Biol. 2017, 19, 384–390. [Google Scholar] [CrossRef]
- Fry, A.M.; Arnaud, L.; Nigg, E.A. Activity of the human centrosomal kinase, Nek2, depends on an unusual leucine zipper dimerization motif. J. Biol. Chem. 1999, 274, 16304–16310. [Google Scholar] [CrossRef] [PubMed]
- Rellos, P.; Ivins, F.J.; Baxter, J.E.; Pike, A.; Nott, T.J.; Parkinson, D.M.; Das, S.; Howell, S.; Fedorov, O.; Shen, Q.Y.; et al. Structure and regulation of the human Nek2 centrosomal kinase. J. Biol. Chem. 2007, 282, 6833–6842. [Google Scholar] [CrossRef] [PubMed]
- Croasdale, R.; Ivins, F.J.; Muskett, F.; Daviter, T.; Scott, D.J.; Hardy, T.; Smerdon, S.J.; Fry, A.M.; Pfuhl, M. An undecided coiled coil: The leucine zipper of Nek2 kinase exhibits atypical conformational exchange dynamics. J. Biol. Chem. 2011, 286, 27537–27547. [Google Scholar] [CrossRef] [PubMed]
- Mardin, B.R.; Lange, C.; Baxter, J.E.; Hardy, T.; Scholz, S.R.; Fry, A.M.; Schiebel, E. Components of the Hippo pathway cooperate with Nek2 kinase to regulate centrosome disjunction. Nat. Cell Biol. 2010, 12, 1166–1176. [Google Scholar] [CrossRef]
- Helps, N.R.; Luo, X.; Barker, H.M.; Cohen, P.T. NIMA-related kinase 2 (Nek2), a cell-cycle-regulated protein kinase localized to centrosomes, is complexed to protein phosphatase 1. Biochem. J. 2000, 349, 509–518. [Google Scholar] [CrossRef]
- Mardin, B.R.; Agircan, F.G.; Lange, C.; Schiebel, E. Plk1 controls the Nek2A-PP1γ antagonism in centrosome disjunction. Curr. Biol. 2011, 21, 1145–1151. [Google Scholar] [CrossRef]
- Eto, M.; Elliot, E.; Prickett, T.D.; Brautigan, D.L. Inhibitor-2 regulates protein phosphatase-1 complexed with NimA-related kinase to induce centrosome separation. J. Biol. Chem. 2002, 277, 44013–44020. [Google Scholar] [CrossRef]
- Mi, J.; Guo, C.; Brautigan, D.L.; Larner, J.M. Protein phosphatase-1alpha regulates centrosome splitting through Nek2. Cancer Res. 2007, 67, 1082–1089. [Google Scholar] [CrossRef] [PubMed]
- Hames, R.S.; Wattam, S.L.; Yamano, H.; Bacchieri, R.; Fry, A.M. APC/C-mediated destruction of the centrosomal kinase Nek2A occurs in early mitosis and depends upon a cyclin A-type D-box. EMBO J. 2001, 20, 7117–7127. [Google Scholar] [CrossRef] [PubMed]
- Magidson, V.; O’Connell, C.B.; Lončarek, J.; Paul, R.; Mogilner, A.; Khodjakov, A. The spatial arrangement of chromosomes during prometaphase facilitates spindle assembly. Cell 2011, 146, 555–567. [Google Scholar] [CrossRef] [PubMed]
- Silkworth, W.T.; Nardi, I.K.; Paul, R.; Mogilner, A.; Cimini, D. Timing of centrosome separation is important for accurate chromosome segregation. Mol. Biol. Cell 2012, 23, 401–411. [Google Scholar] [CrossRef] [PubMed]
- Kaseda, K.; McAinsh, A.D.; Cross, R.A. Dual pathway spindle assembly increases both the speed and the fidelity of mitosis. Biol. Open 2012, 1, 12–18. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Jüschke, C.; Esk, C.; Hirotsune, S.; Knoblich, J.A. The phosphatase PP4c controls spindle orientation to maintain proliferative symmetric divisions in the developing neocortex. Neuron 2013, 79, 254–265. [Google Scholar] [CrossRef] [PubMed]
- Siller, K.H.; Doe, C.Q. Spindle orientation during asymmetric cell division. Nat. Cell Biol. 2009, 11, 365–374. [Google Scholar] [CrossRef] [PubMed]
- di Pietro, F.; Echard, A.; Morin, X. Regulation of mitotic spindle orientation: An integrated view. EMBO Rep. 2016, 17, 1106–1130. [Google Scholar] [CrossRef] [PubMed]
- Seldin, L.; Macara, I. Epithelial spindle orientation diversities and uncertainties: Recent developments and lingering questions. F1000Research 2017, 6, 984. [Google Scholar] [CrossRef] [PubMed]
- Bergstralh, D.T.; Dawney, N.S.; St. Johnston, D. Spindle orientation: A question of complex positioning. Development 2017, 144, 1137–1145. [Google Scholar] [CrossRef] [PubMed]
- Nguyen-Ngoc, T.; Afshar, K.; Gönczy, P. Coupling of cortical dynein and G α proteins mediates spindle positioning in Caenorhabditis elegans. Nat. Cell Biol. 2007, 9, 1294–1302. [Google Scholar] [CrossRef]
- Kotak, S.; Busso, C.; Gönczy, P. Cortical dynein is critical for proper spindle positioning in human cells. J. Cell Biol. 2012, 199, 97–110. [Google Scholar] [CrossRef] [PubMed]
- Laan, L.; Pavin, N.; Husson, J.; Romet-Lemonne, G.; van Duijn, M.; López, M.P.; Vale, R.D.; Jülicher, F.; Reck-Peterson, S.L.; Dogterom, M. Cortical dynein controls microtubule dynamics to generate pulling forces that position microtubule asters. Cell 2012, 148, 502–514. [Google Scholar] [CrossRef] [PubMed]
- Du, Q.; Macara, I.G. Mammalian Pins is a conformational switch that links NuMA to heterotrimeric G proteins. Cell 2004, 119, 503–516. [Google Scholar] [CrossRef] [PubMed]
- Gallini, S.; Carminati, M.; De Mattia, F.; Pirovano, L.; Martini, E.; Oldani, A.; Asteriti, I.A.; Guarguaglini, G.; Mapelli, M. NuMA Phosphorylation by Aurora-A Orchestrates Spindle Orientation. Curr. Biol. 2016, 26, 458–469. [Google Scholar] [CrossRef] [PubMed]
- Kotak, S.; Afshar, K.; Busso, C.; Gönczy, P. Aurora A kinase regulates proper spindle positioning in C. elegans and human cells. J. Cell Sci. 2016, 129, 3015–3025. [Google Scholar] [CrossRef] [PubMed]
- Kotak, S.; Busso, C.; Gönczy, P. NuMA phosphorylation by CDK1 couples mitotic progression with cortical dynein function. EMBO J. 2013, 32, 2517–2529. [Google Scholar] [CrossRef]
- Sana, S.; Keshri, R.; Rajeevan, A.; Kapoor, S.; Kotak, S. Plk1 regulates spindle orientation by phosphorylating NuMA in human cells. Life Sci. Alliance 2018, 1, e201800223. [Google Scholar] [CrossRef]
- Zheng, Z.; Wan, Q.; Meixiong, G.; Du, Q. Cell cycle-regulated membrane binding of NuMA contributes to efficient anaphase chromosome separation. Mol. Biol. Cell 2014, 25, 606–619. [Google Scholar] [CrossRef]
- Lee, B.H.; Schwager, F.; Meraldi, P.; Gotta, M. p37/UBXN2B regulates spindle orientation by limiting cortical NuMA recruitment via PP1/Repo-Man. J. Cell Biol. 2018, 217, 483–493. [Google Scholar] [CrossRef]
- Afshar, K.; Werner, M.E.; Tse, Y.C.; Glotzer, M.; Gönczy, P. Regulation of cortical contractility and spindle positioning by the protein phosphatase 6 PPH-6 in one-cell stage C. elegans embryos. Development 2010, 137, 237–247. [Google Scholar] [CrossRef] [PubMed]
- Musacchio, A.; Desai, A. A molecular view of kinetochore assembly and function. Biology 2017, 6, 5. [Google Scholar] [CrossRef] [PubMed]
- Cimini, D.; Howell, B.; Maddox, P.; Khodjakov, A.; Degrassi, F.; Salmon, E.D. Merotelic Kinetochore Orientation Is a Major Mechanism of Aneuploidy in Mitotic Mammalian Tissue Cells. J. Cell Biol. 2001, 153, 517–527. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Mukae, N.; Dewar, H.; Breugel, M.; James, E.K.; Prescott, A.R.; Anthony, A.; Tanaka, T.U. Molecular mechanisms of kinetochore capture by spindle microtubules. Nature 2005, 434, 987–994. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, T.M.; Lampson, M.A.; Hergert, P.; Cameron, L.; Cimini, D.; Salmon, E.D.; McEwen, B.F.; Khodjakov, A. Chromosomes Can Congress to the Metaphase Plate Before Biorientation. Science 2006, 311, 388–391. [Google Scholar] [CrossRef] [PubMed]
- Thompson, S.L.; Compton, D.A. Chromosome missegregation in human cells arises through specific types of kinetochore–microtubule attachment errors. Proc. Natl Acad. Sci. USA 2011, 108, 17974–17978. [Google Scholar] [CrossRef]
- Foley, E.A.; Kapoor, T.M. Microtubule attachment and spindle assembly checkpoint signalling at the kinetochore. Nat. Rev. Mol. Cell Biol. 2013, 14, 25–37. [Google Scholar] [CrossRef]
- Barisic, M.; Aguiar, P.; Geley, S.; Maiato, H. Kinetochore motors drive congression of peripheral polar chromosomes by overcoming random arm-ejection forces. Nat. Cell Biol. 2014, 16, 1249–1256. [Google Scholar] [CrossRef]
- Bakhoum, S.F.; Thompson, S.L.; Manning, A.L.; Compton, D.A. Genome stability is ensured by temporal control of kinetochore-microtubule dynamics. Nat. Cell Biol. 2009, 11, 27–35. [Google Scholar] [CrossRef]
- Bakhoum, S.F.; Genovese, G.; Compton, D.A. Deviant Kinetochore Microtubule Dynamics Underlie Chromosomal Instability. Curr. Biol. 2009, 19, 1937–1942. [Google Scholar] [CrossRef]
- Zaytsev, A.V.; Grishchuk, E.L. Basic mechanism for biorientation of mitotic chromosomes is provided by the kinetochore geometry and indiscriminate turnover of kinetochore microtubules. Mol. Biol. Cell 2015, 26, 3985–3998. [Google Scholar] [CrossRef] [PubMed]
- Cheeseman, I.M.; Chappie, J.S.; Wilson-Kubalek, E.M.; Desai, A. The Conserved KMN Network Constitutes the Core Microtubule-Binding Site of the Kinetochore. Cell 2006, 127, 983–997. [Google Scholar] [CrossRef] [PubMed]
- Wei, R.R.; Al-Bassam, J.; Harrison, S.C. The Ndc80/HEC1 complex is a contact point for kinetochore-microtubule attachment. Nat. Struct. Mol. Biol. 2006, 14, 54–59. [Google Scholar] [CrossRef] [PubMed]
- Ciferri, C.; Pasqualato, S.; Screpanti, E.; Varetti, G.; Santaguida, S.; Reis, G.; Maiolica, A.; Polka, J.; DeLuca, J.G.; de Wulf, P.; et al. Implications for Kinetochore-Microtubule Attachment from the Structure of an Engineered Ndc80 Complex. Cell 2008, 133, 427–439. [Google Scholar] [CrossRef] [PubMed]
- Alushin, G.M.; Ramey, V.H.; Pasqualato, S.; Ball, D.A.; Grigorieff, N.; Musacchio, A.; Nogales, E. The Ndc80 kinetochore complex forms oligomeric arrays along microtubules. Nature 2011, 467, 805–810. [Google Scholar] [CrossRef] [PubMed]
- DeLuca, J.G.; Gall, W.E.; Ciferri, C.; Cimini, D.; Musacchio, A.; Salmon, E.D. Kinetochore Microtubule Dynamics and Attachment Stability Are Regulated by Hec1. Cell 2006, 127, 969–982. [Google Scholar] [CrossRef] [PubMed]
- Guimarães, G.J.; Dong, Y.; McEwen, B.F.; DeLuca, J.G. Kinetochore-Microtubule Attachment Relies on the Disordered N-Terminal Tail Domain of Hec1. Curr. Biol. 2008, 18, 1778–1784. [Google Scholar] [CrossRef]
- Miller, S.A.; Johnson, M.L.; Stukenberg, P.T. Kinetochore Attachments Require an Interaction between Unstructured Tails on Microtubules and Ndc80Hec1. Curr. Biol. 2008, 18, 1785–1791. [Google Scholar] [CrossRef]
- Zaytsev, A.V.; Sundin, L.J.R.; DeLuca, K.F.; Grishchuk, E.L.; DeLuca, J.G. Accurate phosphoregulation of kinetochore–microtubule affinity requires unconstrained molecular interactions. J. Cell Biol. 2014, 206, 45–59. [Google Scholar] [CrossRef]
- Zaytsev, A.V.; Mick, J.E.; Maslennikov, E.; Nikashin, B.; DeLuca, J.G.; Grishchuk, E.L. Multisite phosphorylation of the NDC80 complex gradually tunes its microtubule-binding affinity. Mol. Biol. Cell 2015, 26, 1829–1844. [Google Scholar] [CrossRef]
- Tanaka, T.U.; Rachidi, N.; Janke, C.; Pereira, G.; Galova, M.; Schiebel, E.; Stark, M.J.R.; Nasmyth, K. Evidence that the Ipl1-Sli15 (Aurora Kinase-INCENP) Complex Promotes Chromosome Bi-orientation by Altering Kinetochore-Spindle Pole Connections. Cell 2002, 108, 317–329. [Google Scholar] [CrossRef]
- Liu, D.; Vader, G.; Vromans, M.J.M.; Lampson, M.A.; Lens, S.M.A. Sensing Chromosome Bi-Orientation by Spatial Separation of Aurora B Kinase from Kinetochore Substrates. Science 2009, 323, 1350–1353. [Google Scholar] [CrossRef] [PubMed]
- Krenn, V.; Musacchio, A. The Aurora B kinase in chromosome bi-orientation and spindle checkpoint signaling. Front. Oncol. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Lampson, M.A.; Cheeseman, I.M. Sensing centromere tension: Aurora B and the regulation of kinetochore function. Trends Cell Biol. 2011, 21, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Bishop, J.D.; Schumacher, J.M. Phosphorylation of the Carboxyl Terminus of Inner Centromere Protein (INCENP) by the Aurora B Kinase Stimulates Aurora B Kinase Activity. J. Biol. Chem. 2002, 277, 27577–27580. [Google Scholar] [CrossRef] [PubMed]
- Honda, R.; Korner, R.; Nigg, E.A. Exploring the functional interactions between Aurora B, INCENP, and survivin in mitosis. Mol. Biol. Cell 2003, 14, 3325–3341. [Google Scholar] [CrossRef] [PubMed]
- Yasui, Y.; Urano, T.; Kawajiri, A.; Nagata, K.; Tatsuka, M.; Saya, H.; Furukawa, K.; Takahashi, T.; Izawa, I.; Inagaki, M. Autophosphorylation of a newly identified site of Aurora-B is indispensable for cytokinesis. J. Biol. Chem. 2004, 279, 12997–13003. [Google Scholar] [CrossRef]
- Sessa, F.; Mapelli, M.; Ciferri, C.; Tarricone, C.; Areces, L.B.; Schneider, T.R.; Stukenberg, P.T.; Musacchio, A. Mechanism of Aurora B Activation by INCENP and Inhibition by Hesperadin. Mol. Cell 2005, 18, 379–391. [Google Scholar] [CrossRef]
- Kelly, A.E.; Sampath, S.C.; Maniar, T.A.; Woo, E.M.; Chait, B.T.; Funabiki, H. Chromosomal Enrichment and Activation of the Aurora B Pathway Are Coupled to Spatially Regulate Spindle Assembly. Dev. Cell 2007, 12, 31–43. [Google Scholar] [CrossRef]
- Wang, E.; Ballister, E.R.; Lampson, M.A. Aurora B dynamics at centromeres create a diffusion-based phosphorylation gradient. J. Cell Biol. 2011, 194, 539–549. [Google Scholar] [CrossRef]
- Zaytsev, A.V.; Segura-Peña, D.; Godzi, M.; Calderon, A.; Ballister, E.R.; Stamatov, R.; Mayo, A.M.; Peterson, L.; Black, B.E.; Ataullakhanov, F.I.; et al. Bistability of a coupled Aurora B kinase-phosphatase system in cell division. Elife 2016, 5, e10644. [Google Scholar] [CrossRef] [PubMed]
- Ruppert, F.G.; Samejima, K.; Platani, M.; Molina, O.; Kimura, H.; Jeyaprakash, A.A.; Ohta, S.; Earnshaw, W.C. HP1α targets the chromosomal passenger complex for activation at heterochromatin before mitotic entry. EMBO J. 2018, e97677. [Google Scholar] [CrossRef] [PubMed]
- Maresca, T.J.; Salmon, E.D. Intrakinetochore stretch is associated with changes in kinetochore phosphorylation and spindle assembly checkpoint activity. J. Cell Biol. 2009, 184, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Uchida, K.S.; Takagaki, K.; Kumada, K.; Hirayama, Y.; Noda, T.; Hirota, T. Kinetochore stretching inactivates the spindle assembly checkpoint. J. Cell Biol. 2009, 184, 383–390. [Google Scholar] [CrossRef] [PubMed]
- Wan, X.; O’Quinn, R.P.; Pierce, H.L.; Joglekar, A.P.; Gall, W.E.; DeLuca, J.G.; Carroll, C.W.; Liu, S.; Yen, T.J.; McEwen, B.F.; et al. Protein Architecture of the Human Kinetochore Microtubule Attachment Site. Cell 2009, 137, 672–684. [Google Scholar] [CrossRef] [PubMed]
- Maresca, T.J.; Salmon, E.D. Welcome to a new kind of tension: Translating kinetochore mechanics into a wait-anaphase signal. J. Cell Sci. 2010, 123, 825–835. [Google Scholar] [CrossRef] [PubMed]
- Lampson, M.A.; Renduchitala, K.; Khodjakov, A.; Kapoor, T.M. Correcting improper chromosome–spindle attachments during cell division. Nat. Cell Biol. 2004, 6, 232–237. [Google Scholar] [CrossRef]
- Pinsky, B.A.; Kung, C.; Shokat, K.M.; Biggins, S. The Ipl1-Aurora protein kinase activates the spindle checkpoint by creating unattached kinetochores. Nat. Cell Biol. 2006, 8, 78–83. [Google Scholar] [CrossRef]
- Welburn, J.P.; Vleugel, M.; Liu, D.; Yates, J.R.; Lampson, M.A.; Fukagawa, T.; Cheeseman, I.M. Aurora B phosphorylates spatially distinct targets to differentially regulate the kinetochore-microtubule interface. Mol. Cell 2010, 38, 383–392. [Google Scholar] [CrossRef]
- Shrestha, R.L.; Conti, D.; Tamura, N.; Braun, D.; Ramalingam, R.A.; Cieslinski, K.; Ries, J.; Draviam, V.M. Aurora-B kinase pathway controls the lateral to end-on conversion of kinetochore-microtubule attachments in human cells. Nat. Commun. 2017, 8, 150. [Google Scholar] [CrossRef]
- Li, Y.; Yu, W.; Liang, Y.; Zhu, X. Kinetochore dynein generates a poleward pulling force to facilitate congression and full chromosome alignment. Cell Res. 2007, 17, 701–712. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Tulu, U.S.; Wadsworth, P.; Rieder, C.L. Kinetochore dynein is required for chromosome motion and congression independent of the spindle checkpoint. Curr. Biol. 2007, 17, 973–980. [Google Scholar] [CrossRef] [PubMed]
- Vorozhko, V.V.; Emanuele, M.J.; Kallio, M.J.; Stukenberg, P.T.; Gorbsky, G.J. Multiple mechanisms of chromosome movement in vertebrate cells mediated through the Ndc80 complex and dynein/dynactin. Chromosoma 2008, 117, 169–179. [Google Scholar] [CrossRef] [PubMed]
- Monda, J.K.; Cheeseman, I.M. The kinetochore–microtubule interface at a glance. J. Cell Sci. 2018, 131, jcs214577. [Google Scholar] [CrossRef] [PubMed]
- Cai, S.; O’connell, C.B.; Khodjakov, A.; Walczak, C.E. Chromosome congression in the absence of kinetochore fibres. Nat. Cell Biol. 2009, 11, 832–838. [Google Scholar] [CrossRef] [PubMed]
- Barisic, M.; Sousa, R.S.; Tripathy, S.K.; Magiera, M.M.; Zaytsev, A.V.; Pereira, A.L.; Janke, C.; Grishchuk, E.L.; Maiato, H. Microtubule detyrosination guides chromosomes during mitosis. Science 2015, 348, 799–803. [Google Scholar] [CrossRef] [PubMed]
- Iemura, K.; Tanaka, K. Chromokinesin Kid and kinetochore kinesin CENP-E differentially support chromosome congression without end-on attachment to microtubules. Nat. Commun. 2015, 6, 6447. [Google Scholar] [CrossRef]
- Barisic, M.; Maiato, H. The tubulin code: A navigation system for chromosomes during mitosis. Trends Cell Biol. 2016, 26, 766–775. [Google Scholar] [CrossRef]
- Itoh, G.; Ikeda, M.; Iemura, K.; Amin, M.A.; Kuriyama, S.; Tanaka, M.; Mizuno, N.; Osakada, H.; Haraguchi, T.; Tanaka, K. Lateral attachment of kinetochores to microtubules is enriched in prometaphase rosette and facilitates chromosome alignment and bi-orientation establishment. Sci. Rep. 2018, 8, 3888. [Google Scholar] [CrossRef]
- Lombillo, V.A.; Nislow, C.; Yen, T.J.; Gelfand, V.I.; McIntosh, J.R. Antibodies to the kinesin motor domain and CENP-E inhibit microtubule depolymerization-dependent motion of chromosomes in vitro. J. Cell Biol. 1995, 128, 107–115. [Google Scholar] [CrossRef]
- Gudimchuk, N.; Vitre, B.; Kim, Y.; Kiyatkin, A.; Cleveland, D.W.; Ataullakhanov, F.I.; Grishchuk, E.L. Kinetochore kinesin CENP-E is a processive bi-directional tracker of dynamic microtubule tips. Nat. Cell Biol. 2013, 15, 1079–1088. [Google Scholar] [CrossRef] [PubMed]
- Shrestha, R.L.; Draviam, V.M. Lateral to end-on conversion of chromosome-microtubule attachment requires kinesins CENP-E and MCAK. Curr. Biol. 2013, 23, 1514–1526. [Google Scholar] [CrossRef] [PubMed]
- Suijkerbuijk, S.J.; Vleugel, M.; Teixeira, A.; Kops, G.J. Integration of kinase and phosphatase activities by BUBR1 ensures formation of stable kinetochore-microtubule attachments. Dev. Cell 2012, 23, 745–755. [Google Scholar] [CrossRef] [PubMed]
- Kruse, T.; Zhang, G.; Larsen, M.S.Y.; Lischetti, T.; Streicher, W.; Nielsen, T.K.; Bjørn, S.P.; Nilsson, J. Direct binding between BubR1 and B56-PP2A phosphatase complexes regulate mitotic progression. J. Cell Sci. 2013, 126, 1086–1092. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Raetz, E.A.; Kitagawa, M.; Virshup, D.M.; Lee, S.H. BUBR1 recruits PP2A via the B56 family of targeting subunits to promote chromosome congression. Biol. Open 2013, 2, 479–486. [Google Scholar] [CrossRef] [PubMed]
- Elowe, S.; Hümmer, S.; Uldschmid, A.; Li, X.; Nigg, E.A. Tension-sensitive Plk1 phosphorylation on BubR1 regulates the stability of kinetochore–microtubule interactions. Genes Dev. 2007, 21, 2205–2219. [Google Scholar] [CrossRef]
- Huang, H.; Hittle, J.; Zappacosta, F.; Annan, R.S.; Hershko, A.; Yen, T.J. Phosphorylation sites in BubR1 that regulate kinetochore attachment, tension, and mitotic exit. J. Cell Biol. 2008, 183, 667–680. [Google Scholar] [CrossRef]
- Rivera, T.; Ghenoiu, C.; Rodríguez-Corsino, M.; Mochida, S.; Funabiki, H.; Losada, A. Xenopus Shugoshin 2 regulates the spindle assembly pathway mediated by the chromosomal passenger complex. EMBO J. 2012, 31, 1467–1479. [Google Scholar] [CrossRef]
- Nijenhuis, W.; Vallardi, G.; Teixeira, A.; Kops, G.J.P.L.; Saurin, A.T. Negative feedback at kinetochores underlies a responsive spindle checkpoint signal. Nat. Cell Biol. 2014, 16, 1257–1264. [Google Scholar] [CrossRef]
- Meppelink, A.; Kabeche, L.; Vromans, M.J.; Compton, D.A.; Lens, S.M. Shugoshin-1 balances Aurora B kinase activity via PP2A to promote chromosome bi-orientation. Cell Rep. 2015, 11, 508–515. [Google Scholar] [CrossRef]
- Foley, E.A.; Maldonado, M.; Kapoor, T.M. Formation of stable attachments between kinetochores and microtubules depends on the B56-PP2A phosphatase. Nat. Cell Biol. 2011, 13, 1265–1271. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Vleugel, M.; Backer, C.B.; Hori, T.; Fukagawa, T.; Cheeseman, I.M.; Lampson, M.A. Regulated targeting of protein phosphatase 1 to the outer kinetochore by KNL1 opposes Aurora B kinase. J. Cell Biol. 2010, 188, 809–820. [Google Scholar] [CrossRef] [PubMed]
- Vallardi, G.; Cordeiro, M.H.; Saurin, A.T. A kinase-phosphatase network that regulates kinetochore-microtubule attachments and the SAC. Prog. Mol. Subcell. Biol. 2017, 56, 457–484. [Google Scholar] [PubMed]
- Saurin, A.T. Kinase and phosphatase cross-talk at the kinetochore. Front. Cell Dev. Biol. 2018, 6, 62. [Google Scholar] [CrossRef] [PubMed]
- Posch, M.; Khoudoli, G.A.; Swift, S.; King, E.M.; DeLuca, J.G.; Swedlow, J.R. Sds22 regulates aurora B activity and microtubule–kinetochore interactions at mitosis. J. Cell Biol. 2010, 131, 61–74. [Google Scholar] [CrossRef] [PubMed]
- Wurzenberger, C.; Held, M.; Lampson, M.A.; Poser, I.; Hyman, A.A.; Gerlich, D.W. Sds22 and Repo-Man stabilize chromosome segregation by counteracting Aurora B on anaphase kinetochores. J. Cell Biol. 2012, 198, 173–183. [Google Scholar] [CrossRef]
- Murnion, M.E.; Adams, R.R.; Callister, D.M.; Allis, C.D.; Earnshaw, W.C.; Swedlow, J.R. Chromatin-associated protein phosphatase 1 regulates aurora-B and histone H3 phosphorylation. J. Biol. Chem. 2001, 276, 26656–26665. [Google Scholar] [CrossRef]
- Rosasco-Nitcher, S.E.; Lan, W.; Khorasanizadeh, S.; Stukenberg, P.T. Centromeric Aurora-B activation requires TD-60, microtubules, and substrate priming phosphorylation. Science 2008, 319, 469–472. [Google Scholar] [CrossRef]
- Cheerambathur, D.K.; Prevo, B.; Hattersley, N.; Lewellyn, L.; Corbett, K.D.; Oegema, K.; Desai, A. Dephosphorylation of the Ndc80 tail stabilizes kinetochore-microtubule attachments via the Ska complex. Dev. Cell 2017, 41, 424–437. [Google Scholar] [CrossRef]
- Janczyk, P.Ł.; Skorupka, K.A.; Tooley, J.G.; Matson, D.R.; Kestner, C.A.; West, T.; Pornillos, O.; Stukenberg, P.T. Mechanism of Ska recruitment by Ndc80 complexes to kinetochores. Dev. Cell 2017, 41, 438–449. [Google Scholar] [CrossRef]
- Schmidt, J.C.; Arthanari, H.; Boeszoermenyi, A.; Dashkevich, N.M.; Wilson-Kubalek, E.M.; Monnier, N.; Markus, M.; Oberer, M.; Milligan, R.A.; Bathe, M.; et al. The kinetochore-bound Ska1 complex tracks depolymerizing microtubules and binds to curved protofilaments. Dev. Cell 2012, 23, 968–980. [Google Scholar] [CrossRef] [PubMed]
- Hanisch, A.; Silljé, H.H.; Nigg, E.A. Timely anaphase onset requires a novel spindle and kinetochore complex comprising Ska1 and Ska2. EMBO J. 2006, 25, 5504–5515. [Google Scholar] [CrossRef]
- Gaitanos, T.N.; Santamaria, A.; Jeyaprakash, A.A.; Wang, B.; Conti, E.; Nigg, E.A. Stable kinetochore–microtubule interactions depend on the Ska complex and its new component Ska3/C13Orf3. EMBO J. 2009, 28, 1442–1452. [Google Scholar] [CrossRef] [PubMed]
- Raaijmakers, J.A.; Tanenbaum, M.E.; Maia, A.F.; Medema, R.H. RAMA1 is a novel kinetochore protein involved in kinetochore-microtubule attachment. J. Cell Sci. 2009, 122, 2436–2445. [Google Scholar] [CrossRef] [PubMed]
- Welburn, J.P.; Grishchuk, E.L.; Backer, C.B.; Wilson-Kubalek, E.M.; Yates III, J.R.; Cheeseman, I.M. The human kinetochore Ska1 complex facilitates microtubule depolymerization-coupled motility. Dev. Cell 2009, 16, 374–385. [Google Scholar] [CrossRef] [PubMed]
- Jeyaprakash, A.A.; Santamaria, A.; Jayachandran, U.; Chan, Y.W.; Benda, C.; Nigg, E.A.; Conti, E. Structural and functional organization of the Ska complex, a key component of the kinetochore-microtubule interface. Mol. Cell 2012, 46, 274–286. [Google Scholar] [CrossRef] [PubMed]
- Monda, J.K.; Whitney, I.P.; Tarasovetc, E.V.; Wilson-Kubalek, E.; Milligan, R.A.; Grishchuk, E.L.; Cheeseman, I.M. Microtubule Tip Tracking by the Spindle and Kinetochore Protein Ska1 Requires Diverse Tubulin-Interacting Surfaces. Curr. Biol. 2017, 27, 3666–3675. [Google Scholar] [CrossRef]
- Auckland, P.; Clarke, N.I.; Royle, S.J.; McAinsh, A.D. Congressing kinetochores progressively load Ska complexes to prevent force-dependent detachment. J. Cell Biol. 2017, 216, 1623–1638. [Google Scholar] [CrossRef]
- Helgeson, L.A.; Zelter, A.; Riffle, M.; MacCoss, M.J.; Asbury, C.L.; Davis, T.N. Human Ska complex and Ndc80 complex interact to form a load-bearing assembly that strengthens kinetochore–microtubule attachments. Proc. Nat. Acad. Sci. USA 2018, 115, 2740–2745. [Google Scholar] [CrossRef]
- Chan, Y.W.; Jeyaprakash, A.A.; Nigg, E.A.; Santamaria, A. Aurora B controls kinetochore–microtubule attachments by inhibiting Ska complex–KMN network interaction. J. Cell Biol. 2012, 196, 563–571. [Google Scholar] [CrossRef]
- Abad, M.A.; Medina, B.; Santamaria, A.; Zou, J.; Plasberg-Hill, C.; Madhumalar, A.; Jayachandran, U.; Redli, P.M.; Rappsilber, J.; Nigg, E.A.; et al. Structural basis for microtubule recognition by the human kinetochore Ska complex. Nat. Commun. 2014, 5, 2964. [Google Scholar] [CrossRef] [PubMed]
- Redli, P.M.; Gasic, I.; Meraldi, P.; Nigg, E.A.; Santamaria, A. The Ska complex promotes Aurora B activity to ensure chromosome biorientation. J. Cell Biol. 2016, 215, 77–93. [Google Scholar] [CrossRef] [PubMed]
- Sivakumar, S.; Janczyk, P.Ł.; Qu, Q.; Brautigam, C.A.; Stukenberg, P.T.; Yu, H.; Gorbsky, G.J. The human SKA complex drives the metaphase-anaphase cell cycle transition by recruiting protein phosphatase 1 to kinetochores. Elife 2016, 5, e12902. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, A.; Gupta, A.; Long, S.K.; Evans, R.; Badger, B.L.; Salmon, E.D.; Biggings, S.; Bloom, K. A Kinesin-5, Cin8, Recruits Protein Phosphatase 1 to Kinetochores and Regulates Chromosome Segregation. Curr. Biol. 2018, 28, 2697–2704. [Google Scholar] [CrossRef] [PubMed]
- De Wever, V.; Nasa, I.; Chamousset, D.; Lloyd, D.; Nimick, M.; Xu, H.; Trinkle-Mulcahy, L.; Moorhead, G.B. The human mitotic kinesin KIF18A binds protein phosphatase 1 (PP1) through a highly conserved docking motif. Biochem. Biophys. Res. Commun. 2014, 453, 432–437. [Google Scholar] [CrossRef] [PubMed]
- Häfner, J.; Mayr, M.I.; Möckel, M.M.; Mayer, T.U. Pre-anaphase chromosome oscillations are regulated by the antagonistic activities of Cdk1 and PP1 on Kif18A. Nat. Commun. 2014, 5, 4397. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Holland, A.J.; Lan, W.; Cleveland, D.W. Aurora kinases and protein phosphatase 1 mediate chromosome congression through regulation of CENP-E. Cell 2010, 142, 444–455. [Google Scholar] [CrossRef]
- Kim, H.; Stumpff, J.K. Kif18A promotes Hec1 dephosphorylation to coordinate chromosome alignment with kinetochore microtubule attachment. bioRxiv 2018. [Google Scholar] [CrossRef]
- Totsukawa, G.; Yamakita, Y.; Yamashiro, S.; Hosoya, H.; Hartshorne, D.J.; Matsumura, F. Activation of myosin phosphatase targeting subunit by mitosis-specific phosphorylation. J. Cell Biol. 1999, 144, 735–744. [Google Scholar] [CrossRef]
- Yamashiro, S.; Yamakita, Y.; Totsukawa, G.; Goto, H.; Kaibuchi, K.; Ito, M.; Hartshorne, D.J.; Matsumura, F. Myosin phosphatase-targeting subunit 1 regulates mitosis by antagonizing polo-like kinase 1. Dev. Cell 2008, 14, 787–797. [Google Scholar] [CrossRef]
- Kabeche, L.; Compton, D.A. Cyclin A regulates kinetochore microtubules to promote faithful chromosome segregation. Nature 2013, 502, 110–113. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Liu, X.S.; Yang, X.; Wang, Y.; Wang, Y.; Turner, J.R.; Liu, X. Phosphorylation of CLIP-170 by Plk1 and CK2 promotes timely formation of kinetochore–microtubule attachments. EMBO J. 2010, 29, 2953–2965. [Google Scholar] [CrossRef] [PubMed]
- Bader, J.R.; Kasuboski, J.M.; Winding, M.; Vaughan, P.S.; Hinchcliffe, E.H.; Vaughan, K.T. Polo-like kinase 1 is required for recruitment of dynein to kinetochores during mitosis. J. Biol. Chem. 2011, jbc-M111. [Google Scholar]
- Hood, E.A.; Kettenbach, A.N.; Gerber, S.A.; Compton, D.A. Plk1 regulates the kinesin-13 protein Kif2b to promote faithful chromosome segregation. Mol. Biol. Cell 2012, 23, 2264–2274. [Google Scholar] [CrossRef]
- Maia, A.R.; Garcia, Z.; Kabeche, L.; Barisic, M.; Maffini, S.; Macedo-Ribeiro, S.; Cheeseman, I.M.; Compton, D.A.; Kaverina, I.; Maiato, H. Cdk1 and Plk1 mediate a CLASP2 phospho-switch that stabilizes kinetochore–microtubule attachments. J. Cell Biol. 2012, 199, 285–301. [Google Scholar] [CrossRef] [PubMed]
- Kakeno, M.; Matsuzawa, K.; Matsui, T.; Akita, H.; Sugiyama, I.; Ishidate, F.; Nakano, A.; Takashima, S.; Goto, H.; Inagaki, M.; et al. Plk1 phosphorylates CLIP-170 and regulates its binding to microtubules for chromosome alignment. Cell Struct Funct. 2014, 39, 45–59. [Google Scholar] [CrossRef]
- Shao, H.; Huang, Y.; Zhang, L.; Yuan, K.; Chu, Y.; Dou, Z.; Jin, C.; Garcia-Barrio, M.; Liu, X.; Yao, X. Spatiotemporal dynamics of Aurora B-PLK1-MCAK signaling axis orchestrates kinetochore bi-orientation and faithful chromosome segregation. Sci. Rep. 2015, 5, 12204. [Google Scholar] [CrossRef] [PubMed]
- Nishino, M.; Kurasawa, Y.; Evans, R.; Lin, S.H.; Brinkley, B.R.; Yu-Lee, L.Y. NudC is required for Plk1 targeting to the kinetochore and chromosome congression. Curr. Biol. 2006, 16, 1414–1421. [Google Scholar] [CrossRef] [PubMed]
- Amin, M.A.; Itoh, G.; Iemura, K.; Ikeda, M.; Tanaka, K. CLIP-170 is required to recruit PLK1 to kinetochores during early mitosis for chromosome alignment. J. Cell Sci. 2014, jcs-150755. [Google Scholar]
- Ehlen, A.; Martin, C.; Julien, M.; Miron, S.; Theillet, F.X.; Boucherit, V.; Duchambon, P.; Marjou, A.E.; Justin, S.Z.; Carreira, A. Proper chromosome alignment depends on BRCA2 phosphorylation by PLK1. bioRxiv 2018. [Google Scholar] [CrossRef]
- Lénárt, P.; Petronczki, M.; Steegmaier, M.; Di Fiore, B.; Lipp, J.J.; Hoffmann, M.; Rettig, W.J.; Kraut, N.; Peter, J.M. The small-molecule inhibitor BI 2536 reveals novel insights into mitotic roles of polo-like kinase 1. Curr. Biol. 2007, 17, 304–315. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Davydenko, O.; Lampson, M.A. Polo-like kinase-1 regulates kinetochore–microtubule dynamics and spindle checkpoint silencing. J. Cell Biol. 2012, 198, 491–499. [Google Scholar] [CrossRef] [PubMed]
- Moutinho-Santos, T.; Conde, C.; Sunkel, C.E. POLO ensures chromosome bi-orientation by preventing and correcting erroneous chromosome–spindle attachments. J. Cell Sci. 2012, 125, 576–583. [Google Scholar] [CrossRef] [PubMed]
- Lera, R.F.; Potts, G.K.; Suzuki, A.; Johnson, J.M.; Salmon, E.D.; Coon, J.J.; Burkard, M.E. Decoding Polo-like kinase 1 signaling along the kinetochore–centromere axis. Nat. Chem. Biol. 2016, 12, 411–418. [Google Scholar] [CrossRef] [PubMed]
- Eiteneuer, A.; Seiler, J.; Weith, M.; Beullens, M.; Lesage, B.; Krenn, V.; Musacchio, A.; Bollen, M.; Meyer, H. Inhibitor-3 ensures bipolar mitotic spindle attachment by limiting association of SDS22 with kinetochore-bound protein phosphatase-1. EMBO J. 2014, 33, 2704–2720. [Google Scholar] [CrossRef] [PubMed]
- Weith, M.; Seiler, J.; van den Boom, J.; Kracht, M.; Hülsmann, J.; Primorac, I.; Garcia, J.P.; Kaschani, F.; Kaiser, M.; Musacchio, A.; et al. Ubiquitin-independent disassembly by a p97 AAA-ATPase complex drives PP1 holoenzyme formation. Mol. Cell 2018, 72, 766–777. [Google Scholar] [CrossRef] [PubMed]
- Lesage, B.; Beullens, M.; Pedelini, L.; Garcia-Gimeno, M.A.; Waelkens, E.; Sanz, P.; Bollen, M. A complex of catalytically inactive protein phosphatase-1 sandwiched between Sds22 and inhibitor-3. Biochemistry 2007, 46, 8909–8919. [Google Scholar] [CrossRef]
- Cheng, Y.L.; Chen, R.H. Assembly and quality control of protein phosphatase 1 holoenzyme involve Cdc48-Shp1 chaperone. J. Cell Sci. 2015, 128, 1180–1192. [Google Scholar] [CrossRef]
- London, N.; Biggins, S. Signalling dynamics in the spindle checkpoint response. Nat. Rev. Mol. Cell. Biol. 2014, 15, 736–747. [Google Scholar] [CrossRef]
- Musacchio, A. The Molecular Biology of Spindle Assembly Checkpoint Signaling Dynamics. Curr. Biol. 2015, 25, 1002–1018. [Google Scholar] [CrossRef]
- Joglekar, A.P. A Cell Biological Perspective on Past, Present and Future Investigations of the Spindle Assembly Checkpoint. Biology 2016, 5, 44. [Google Scholar] [CrossRef] [PubMed]
- Corbett, K.D. Molecular Mechanisms of Spindle Assembly Checkpoint Activation and Silencing. Prog. Mol. Subcell. Biol. 2017, 56, 429–455. [Google Scholar] [PubMed]
- Michel, L.S.; Liberal, V.; Chatterjee, A.; Kirchwegger, R.; Pasche, B.; Gerald, W.; Dobles, M.; Sorger, P.K.; Murty, V.V.; Benezra, R. MAD2 haplo-insufficiency causes premature anaphase and chromosome instability in mammalian cells. Nature 2001, 409, 355–359. [Google Scholar] [CrossRef] [PubMed]
- Dai, W.; Wang, Q.; Liu, T.; Swamy, M.; Fang, Y.; Xie, S.; Mahmood, R.; Yang, Y.M.; Xu, M.; Rao, C.V. Slippage of mitotic arrest and enhanced tumor development in mice with BubR1 haploinsufficiency. Cancer Res. 2004, 64, 440–445. [Google Scholar] [CrossRef] [PubMed]
- Iwanaga, Y.; Chi, Y.H.; Miyazato, A.; Sheleg, S.; Haller, K.; Peloponese, J.M. Jr.; Li, Y.; Ward, J.M.; Benezra, R.; Jeang, K.T. Heterozygous deletion of mitotic arrest-deficient protein 1 (MAD1) increases the incidence of tumors in mice. Cancer Res. 2007, 67, 160–166. [Google Scholar] [CrossRef] [PubMed]
- Schvartzman, J.M.; Sotillo, R.; Benezra, R. Mitotic chromosomal instability and cancer: Mouse modelling of the human disease. Nat. Rev. Cancer 2010, 10, 102–115. [Google Scholar] [CrossRef] [PubMed]
- Gorbsky, G.J. Cohesion fatigue. Curr. Biol. 2013, 23, 986–988. [Google Scholar] [CrossRef]
- Rieder, C.L.; Cole, R.W.; Khodjakov, A.; Sluder, G. The checkpoint delaying anaphase in response to chromosome monoorientation is mediated by an inhibitory signal produced by unattached kinetochores. J. Cell Biol. 1995, 130, 941–948. [Google Scholar] [CrossRef]
- Dick, A.E.; Gerlich, D.W. Kinetic framework of spindle assembly checkpoint signalling. Nat. Cell Biol. 2013, 15, 1370–1377. [Google Scholar] [CrossRef]
- Liu, X.; Winey, M. The MPS1 family of protein kinases. Annu. Rev. Biochem. 2012, 81, 561–585. [Google Scholar] [CrossRef]
- Pachis, S.T.; Kops, G.J.P.L. Leader of the SAC: Molecular mechanisms of Mps1/TTK regulation in mitosis. Open Biol. 2018, 8, 180109. [Google Scholar] [CrossRef] [PubMed]
- London, N.; Ceto, S.; Ranish, J.A.; Biggins, S. Phosphoregulation of Spc105 by Mps1 and PP1 regulates Bub1 localization to kinetochores. Curr. Biol. 2012, 22, 900–906. [Google Scholar] [CrossRef] [PubMed]
- Shepperd, L.A.; Meadows, J.C.; Sochaj, A.M.; Lancaster, T.C.; Zou, J.; Buttrick, G.J.; Rappsilber, J.; Hardwick, K.G.; Millar, J.B. Phosphodependent recruitment of Bub1 and Bub3 to Spc7/KNL1 by Mph1 kinase maintains the spindle checkpoint. Curr. Biol. 2012, 22, 891–899. [Google Scholar] [CrossRef] [PubMed]
- Yamagishi, Y.; Yang, C.H.; Tanno, Y.; Watanabe, Y. MPS1/Mph1 phosphorylates the kinetochore protein KNL1/Spc7 to recruit SAC components. Nat. Cell. Biol. 2012, 14, 746–752. [Google Scholar] [CrossRef] [PubMed]
- Primorac, I.; Weir, J.R.; Chiroli, E.; Gross, F.; Hoffmann, I.; van Gerwen, S.; Ciliberto, A.; Musacchio, A. Bub3 reads phosphorylated MELT repeats to promote spindle assembly checkpoint signaling. Elife 2013, 2, e01030. [Google Scholar] [CrossRef] [PubMed]
- Vleugel, M.; Omerzu, M.; Groenewold, V.; Hadders, M.A.; Lens, S.M.A.; Kops, G.J.P.L. Sequential multisite phospho-regulation of KNL1-BUB3 interfaces at mitotic kinetochores. Mol. Cell 2015, 57, 824–835. [Google Scholar] [CrossRef] [PubMed]
- Overlack, K.; Primorac, I.; Vleugel, M.; Krenn, V.; Maffini, S.; Hoffmann, I.; Kops, G.J.; Musacchio, A. A molecular basis for the differential roles of Bub1 and BubR1 in the spindle assembly checkpoint. Elife 2015, 4, e05269. [Google Scholar] [CrossRef]
- London, N.; Biggins, S. Mad1 kinetochore recruitment by Mps1-mediated phosphorylation of Bub1 signals the spindle checkpoint. Genes Dev. 2014, 28, 140–152. [Google Scholar] [CrossRef]
- Mora-Santos, M.D.; Hervas-Aguilar, A.; Sewart, K.; Lancaster, T.C.; Meadows, J.C.; Millar, J.B. Bub3-Bub1 Binding to Spc7/KNL1 Toggles the Spindle Checkpoint Switch by Licensing the Interaction of Bub1 with Mad1-Mad2. Curr. Biol. 2016, 26, 2642–2650. [Google Scholar] [CrossRef]
- Faesen, A.C.; Thanasoula, M.; Maffini, S.; Breit, C.; Müller, F.; van Gerwen, S.; Bange, T.; Musacchio, A. Basis of catalytic assembly of the mitotic checkpoint complex. Nature 2017, 542, 498–502. [Google Scholar] [CrossRef]
- Ji, Z.; Gao, H.; Jia, L.; Li, B.; Yu, H. A sequential multi-target Mps1 phosphorylation cascade promotes spindle checkpoint signaling. Elife 2017, 6, e22513. [Google Scholar] [CrossRef]
- Qian, J.; García-Gimeno, M.A.; Beullens, M.; Manzione, M.G.; Van der Hoeven, G.; Igual, J.C.; Heredia, M.; Sanz, P.; Gelens, L.; Bollen, M. An Attachment-Independent Biochemical Timer of the Spindle Assembly Checkpoint. Mol. Cell 2017, 68, 715–730. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Kruse, T.; López-Méndez, B.; Sylvestersen, K.B.; Garvanska, D.H.; Schopper, S.; Nielsen, M.L.; Nilsson, J. Bub1 positions Mad1 close to KNL1 MELT repeats to promote checkpoint signalling. Nat. Commun. 2017, 8, 15822. [Google Scholar] [CrossRef] [PubMed]
- Ciliberto, A.; Hauf, S. Micromanaging checkpoint proteins. Elife 2017, 6, e25001. [Google Scholar] [CrossRef] [PubMed]
- Hiruma, Y.; Sacristan, C.; Pachis, S.T.; Adamopoulos, A.; Kuijt, T.; Ubbink, M.; von Castelmur, E.; Perrakis, A.; Kops, G.J. CELL DIVISION CYCLE. Competition between MPS1 and microtubules at kinetochores regulates spindle checkpoint signaling. Science 2015, 348, 1264–1267. [Google Scholar] [CrossRef] [PubMed]
- Ji, Z.; Gao, H.; Yu, H. CELL DIVISION CYCLE. Kinetochore attachment sensed by competitive Mps1 and microtubule binding to Ndc80C. Science 2015, 348, 1260–1264. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.; Chen, Y.; Zhao, Y.; Yu, H. Autophosphorylation-dependent activation of human Mps1 is required for the spindle checkpoint. Proc. Natl. Acad. Sci. USA 2007, 104, 20232–20237. [Google Scholar] [CrossRef] [PubMed]
- Mattison, C.P.; Old, W.M.; Steiner, E.; Huneycutt, B.J.; Resing, K.A.; Ahn, N.G.; Winey, M. Mps1 activation loop autophosphorylation enhances kinase activity. J. Biol. Chem. 2007, 282, 30553–30561. [Google Scholar] [CrossRef]
- Jelluma, N.; Brenkman, A.B.; McLeod, I.; Yates, J.R., 3rd.; Cleveland, D.W.; Medema, R.H.; Kops, G.J. Chromosomal instability by inefficient Mps1 auto-activation due to a weakened mitotic checkpoint and lagging chromosomes. PLoS ONE 2008, 3, e2415. [Google Scholar] [CrossRef]
- Tyler, R.K.; Chu, M.L.; Johnson, H.; McKenzie, E.A.; Gaskell, S.J.; Eyers, P.A. Phosphoregulation of human Mps1 kinase. Biochem. J. 2009, 417, 173–181. [Google Scholar] [CrossRef]
- Wang, W.; Yang, Y.; Gao, Y.; Xu, Q.; Wang, F.; Zhu, S.; Old, W.; Resing, K.; Ahn, N.; Lei, M.; et al. Structural and mechanistic insights into Mps1 kinase activation. J. Cell. Mol. Med. 2009, 13, 1679–1694. [Google Scholar] [CrossRef] [PubMed]
- Hewitt, L.; Tighe, A.; Santaguida, S.; White, A.M.; Jones, C.D.; Musacchio, A.; Green, S.; Taylor, S.S. Sustained Mps1 activity is required in mitosis to recruit O-Mad2 to the Mad1-C-Mad2 core complex. J. Cell Biol. 2010, 190, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Santaguida, S.; Vernieri, C.; Villa, F.; Ciliberto, A.; Musacchio, A. Evidence that Aurora B is implicated in spindle checkpoint signalling independently of errorcorrection. EMBO J. 2011, 30, 1508–1519. [Google Scholar] [CrossRef] [PubMed]
- Saurin, A.T.; van der Waal, M.S.; Medema, R.H.; Lens, S.M.; Kops, G.J. Aurora B potentiates Mps1 activation to ensure rapid checkpoint establishment at the onset of mitosis. Nat. Commun. 2011, 2, 316. [Google Scholar] [CrossRef] [PubMed]
- Morin, V.; Prieto, S.; Melines, S.; Hem, S.; Rossignol, M.; Lorca, T.; Espeut, J.; Morin, N.; Abrieu, A. CDK-dependent potentiation of MPS1 kinase activity is essential to the mitotic checkpoint. Curr. Biol. 2012, 22, 289–295. [Google Scholar] [CrossRef] [PubMed]
- Nijenhuis, W.; von Castelmur, E.; Littler, D.; De Marco, V.; Tromer, E.; Vleugel, M.; van Osch, M.H.; Snel, B.; Perrakis, A.; Kops, G.J. A TPR domain-containing N-terminal module of MPS1 is required for its kinetochore localization by Aurora B. J. Cell Biol. 2013, 201, 217–231. [Google Scholar] [CrossRef] [PubMed]
- Conde, C.; Osswald, M.; Barbosa, J.; Moutinho-Santos, T.; Pinheiro, D.; Guimarães, S.; Matos, I.; Maiato, H.; Sunkel, C.E. Drosophila Polo regulates the spindle assembly checkpoint through Mps1-dependent BubR1phosphorylation. EMBO J. 2013, 32, 1761–1777. [Google Scholar] [CrossRef]
- von Schubert, C.; Cubizolles, F.; Bracher, J.M.; Sliedrecht, T.; Kops, G.J.P.L.; Nigg, E.A. Plk1 and Mps1 Cooperatively Regulate the Spindle Assembly Checkpoint in Human Cells. Cell Rep. 2015, 12, 66–78. [Google Scholar] [CrossRef]
- O’Connor, A.; Maffini, S.; Rainey, M.D.; Kaczmarczyk, A.; Gaboriau, D.; Musacchio, A.; Santocanale, C. Requirement for PLK1 kinase activity in the maintenance of a robust spindle assembly checkpoint. Biol. Open 2016, 5, 11–19. [Google Scholar] [CrossRef]
- Ikeda, M.; Tanaka, K. Plk1 bound to Bub1 contributes to spindle assembly checkpoint activity during mitosis. Sci Rep. 2017, 7, 8794. [Google Scholar] [CrossRef]
- Jelluma, N.; Brenkman, A.B.; van den Broek, N.J.; Cruijsen, C.W.; van Osch, M.H.; Lens, S.M.; Medema, R.H.; Kops, G.J. Mps1 phosphorylates Borealin to control Aurora B activity and chromosome alignment. Cell 2008, 132, 233–246. [Google Scholar] [CrossRef]
- van der Waal, M.S.; Saurin, A.T.; Vromans, M.J.; Vleugel, M.; Wurzenberger, C.; Gerlich, D.W.; Medema, R.H.; Kops, G.J.; Lens, S.M. Mps1 promotes rapid centromere accumulation of Aurora B. EMBO Rep. 2012, 13, 847–854. [Google Scholar] [CrossRef] [PubMed]
- Moura, M.; Osswald, M.; Leça, N.; Barbosa, J.; Pereira, A.J.; Maiato, H.; Sunkel, C.E.; Conde, C. Protein Phosphatase 1 inactivates Mps1 to ensure efficient Spindle Assembly Checkpoint silencing. Elife 2017, 6, e25366. [Google Scholar] [CrossRef] [PubMed]
- Sassoon, I.; Severin, F.F.; Andrews, P.D.; Taba, M.R.; Kaplan, K.B.; Ashford, A.J.; Stark, M.J.; Sorger, P.K.; Hyman, A.A. Regulation of Saccharomyces cerevisiae kinetochores by the type 1 phosphatase Glc7p. Genes Dev. 1999, 13, 545–555. [Google Scholar] [CrossRef] [PubMed]
- Bloecher, A.; Tatchell, K. Defects in Saccharomyces cerevisiae protein phosphatase type I activate the spindle/kinetochore checkpoint. Genes Dev. 1999, 13, 517–522. [Google Scholar] [CrossRef] [PubMed]
- Pinsky, B.A.; Nelson, C.R.; Biggins, S. Protein phosphatase 1 regulates exit from the spindle checkpoint in budding yeast. Curr. Biol. 2009, 19, 1182–1187. [Google Scholar] [CrossRef] [PubMed]
- Vanoosthuyse, V.; Hardwick, K.G. A novel protein phosphatase 1-dependent spindle checkpoint silencing mechanism. Curr. Biol. 2009, 19, 1176–1181. [Google Scholar] [CrossRef]
- Meadows, J.C.; Shepperd, L.A.; Vanoosthuyse, V.; Lancaster, T.C.; Sochaj, A.M.; Buttrick, G.J.; Hardwick, K.G.; Millar, J.B. Spindle checkpoint silencing requires association of PP1 to both Spc7 and kinesin-8 motors. Dev. Cell 2011, 20, 739–750. [Google Scholar] [CrossRef]
- Rosenberg, J.S.; Cross, F.R.; Funabiki, H. KNL1/Spc105 recruits PP1 to silence the spindle assembly checkpoint. Curr. Biol. 2011, 21, 942–947. [Google Scholar] [CrossRef]
- Espeut, J.; Cheerambathur, D.K.; Krenning, L.; Oegema, K.; Desai, A. Microtubule binding by KNL-1 contributes to spindle checkpoint silencing at the kinetochore. J. Cell Biol. 2012, 196, 469–482. [Google Scholar] [CrossRef]
- Zhang, G.; Lischetti, T.; Nilsson, J. A minimal number of MELT repeats supports all the functions of KNL1 in chromosome segregation. J. Cell Sci. 2014, 127, 871–884. [Google Scholar] [CrossRef] [PubMed]
- Tang, N.H.; Toda, T. Alp7/TACC recruits kinesin-8-PP1 to the Ndc80 kinetochore protein for timely mitotic progression and chromosome movement. J. Cell Sci. 2015, 128, 354–363. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Kelstrup, C.D.; Hu, X.W.; Kaas Hansen, M.J.; Singleton, M.R.; Olsen, J.V.; Nilsson, J. The Ndc80 internal loop is required for recruitment of the Ska complex to establish end-on microtubule attachment to kinetochores. J. Cell Sci. 2012, 125, 3243–3253. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Sivakumar, S.; Chen, Y.; Gao, H.; Yang, L.; Yuan, Z.; Yu, H.; Liu, H. Ska3 Phosphorylated by Cdk1 Binds Ndc80 and Recruits Ska to Kinetochores to Promote Mitotic Progression. Curr. Biol. 2017, 27, 1477–1484. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Chen, Y.; Yang, L.; Liu, H. Multitasking Ska in Chromosome Segregation: Its Distinct Pools Might Specify Various Functions. Bioessays 2018, 40, 1700176. [Google Scholar] [CrossRef] [PubMed]
- Bajaj, R.; Bollen, M.; Peti, W.; Page, R. KNL1 Binding to PP1 and Microtubules Is Mutually Exclusive. Structure 2018, 26, 1327–1336. [Google Scholar] [CrossRef]
- Espert, A.; Uluocak, P.; Bastos, R.N.; Mangat, D.; Graab, P.; Gruneberg, U. PP2A-B56 opposes Mps1 phosphorylation of Knl1 and thereby promotes spindle assemblycheckpoint silencing. J. Cell Biol. 2014, 206, 833–842. [Google Scholar] [CrossRef]
- Jia, L.; Li, B.; Yu, H. The Bub1-Plk1 kinase complex promotes spindle checkpoint signalling through Cdc20phosphorylation. Nat. Commun. 2016, 7, 10818. [Google Scholar] [CrossRef]
- Craney, A.; Kelly, A.; Jia, L.; Fedrigo, I.; Yu, H.; Rape, M. Control of APC/C-dependent ubiquitin chain elongation by reversible phosphorylation. Proc. Natl. Acad. Sci. USA 2016, 113, 1540–1545. [Google Scholar] [CrossRef]
- Labit, H.; Fujimitsu, K.; Bayin, N.S.; Takaki, T.; Gannon, J.; Yamano, H. Dephosphorylation of Cdc20 is required for its C-box-dependent activation of the APC/C. EMBO J. 2012, 31, 3351–3362. [Google Scholar] [CrossRef]
- Kim, T.; Lara-Gonzalez, P.; Prevo, B.; Meitinger, F.; Cheerambathur, D.K.; Oegema, K.; Desai, A. Kinetochores accelerate or delay APC/C activation by directing Cdc20 to opposing fates. Genes Dev. 2017, 31, 1089–1094. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Rodriguez-Bravo, V.; Kim, H.; Datta, S.; Foley, E.A. The PP2AB56 phosphatase promotes the association of Cdc20 with APC/C in mitosis. J. Cell Sci. 2017, 130, 1760–1771. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, J. Protein phosphatases in the regulation of mitosis. J. Cell Biol. 2018, 218, 395–409. [Google Scholar] [CrossRef] [PubMed]
- Neef, R.; Gruneberg, U.; Kopajtich, R.; Li, X.; Nigg, E.A.; Sillje, H.; Barr, F.A. Choice of Plk1 docking partners during mitosis and cytokinesis is controlled by the activation state of Cdk1. Nat. Cell Biol. 2007, 9, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Petronczki, M.; Glotzer, M.; Kraut, N.; Peters, J.M. Polo-like kinase 1 triggers the initiation of cytokinesis in human cells by promoting recruitment of the Rho GEF Ect2 to the central spindle. Dev. Cell 2007, 12, 713–725. [Google Scholar] [CrossRef] [PubMed]
- Santamaria, A.; Neef, R.; Eberspächer, U.; Eis, K.; Husemann, M.; Mumberg, D.; Prechtl, S.; Schulze, V.; Siemeister, G.; Wortmann, L.; et al. Use of the novel Plk1 inhibitor ZK-thiazolidinone to elucidate functions of Plk1 in early and latestages of mitosis. Mol. Biol. Cell 2007, 18, 4024–4036. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, B.A.; Takaki, T.; Petronczki, M.; Glotzer, M. Polo-like kinase 1 directs assembly of the HsCyk-4 RhoGAP/Ect2 RhoGEF complex to initiatecleavage furrow formation. PLoS Biol. 2009, 7, e1000110. [Google Scholar] [CrossRef] [PubMed]
- Walczak, C.E.; Shaw, S.L. A MAP for bundling microtubules. Cell 2010, 142, 364–367. [Google Scholar] [CrossRef]
- Jiang, W.; Jimenez, G.; Wells, N.J.; Hope, T.J.; Wahl, G.M.; Hunter, T.; Fukunaga, R. PRC1: A human mitotic spindle-associated CDK substrate protein required for cytokinesis. Mol. Cell 1998, 2, 877–885. [Google Scholar] [CrossRef]
- Mollinari, C.; Kleman, J.P.; Jiang, W.; Schoehn, G.; Hunter, T.; Margolis, R.L. PRC1 is a microtubule binding and bundling protein essential to maintain the mitotic spindle midzone. J. Cell Biol. 2002, 157, 1175–1186. [Google Scholar] [CrossRef]
- Mishima, M.; Pavicic, V.; Grüneberg, U.; Nigg, E.A.; Glotzer, M. Cell cycle regulation of central spindle assembly. Nature 2004, 430, 908–913. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Lau, E.; Schwarzenbacher, R.; Bossy-Wetzel, E.; Jiang, W. Spatiotemporal control of spindle midzone formation by PRC1 in human cells. Proc. Natl. Acad. Sci. USA 2006, 103, 6196–6201. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Jiang, W. Cell cycle-dependent translocation of PRC1 on the spindle by Kif4 is essential for midzone formation and cytokinesis. Proc. Natl. Acad. Sci. USA 2005, 102, 343–348. [Google Scholar] [CrossRef] [PubMed]
- Bieling, P.; Telley, I.A.; Surrey, T. A minimal midzone protein module controls formation and length of antiparallel microtubule overlaps. Cell 2010, 142, 420–432. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, R.; Wilson-Kubalek, E.M.; Arthur, C.P.; Bick, M.J.; Campbell, E.A.; Darst, S.A.; Milligan, R.A.; Kapoor, T.M. Insights into antiparallel microtubule crosslinking by PRC1, a conserved nonmotor microtubule binding protein. Cell 2010, 142, 433–443. [Google Scholar] [CrossRef] [PubMed]
- de Castro, I.J.; Gokhan, E.; Vagnarelli, P. Resetting a functional G1 nucleus after mitosis. Chromosoma 2016, 125, 607–619. [Google Scholar] [CrossRef]
- Schellhaus, A.K.; De Magistris, P.; Antonin, W. Nuclear Reformation at the End of Mitosis. J. Mol. Biol. 2016, 428, 1962–1985. [Google Scholar] [CrossRef]
- LaJoie, D.; Ullman, K.S. Coordinated events of nuclear assembly. Curr. Opin. Cell Biol. 2017, 46, 39–45. [Google Scholar] [CrossRef]
- Peter, M.; Nakagawa, J.; Dorée, M.; Labbé, J.C.; Nigg, E.A. In vitro disassembly of the nuclear lamina and M phase-specific phosphorylation of lamins by cdc2 kinase. Cell 1990, 61, 591–602. [Google Scholar] [CrossRef]
- Heald, R.; McKeon, F. Mutations of phosphorylation sites in lamin A that prevent nuclear lamina disassembly in mitosis. Cell 1990, 61, 579–589. [Google Scholar] [CrossRef]
- Macaulay, C.; Meier, E.; Forbes, D.J. Differential mitotic phosphorylation of proteins of the nuclear pore complex. J. Biol. Chem. 1995, 270, 254–262. [Google Scholar] [CrossRef] [PubMed]
- Favreau, C.; Worman, H.J.; Wozniak, R.W.; Frappier, T.; Courvalin, J.C. Cell cycle-dependent phosphorylation of nucleoporins and nuclear pore membrane protein Gp210. Biochemistry 1996, 35, 8035–8044. [Google Scholar] [CrossRef] [PubMed]
- Mansfeld, J.; Güttinger, S.; Hawryluk-Gara, L.A.; Panté, N.; Mall, M.; Galy, V.; Haselmann, U.; Mühlhäusser, P.; Wozniak, R.W.; Mattaj, I.W.; et al. The conserved transmembrane nucleoporin NDC1 is required for nuclear pore complex assembly in vertebrate cells. Mol. Cell 2006, 22, 93–103. [Google Scholar] [CrossRef] [PubMed]
- Glavy, J.S.; Krutchinsky, A.N.; Cristea, I.M.; Berke, I.C.; Boehmer, T.; Blobel, G.; Chait, B.T. Cell-cycle-dependent phosphorylation of the nuclear pore Nup107-160 subcomplex. Proc. Natl. Acad. Sci. USA 2007, 104, 3811–3816. [Google Scholar] [CrossRef] [PubMed]
- Laurell, E.; Beck, K.; Krupina, K.; Theerthagiri, G.; Bodenmiller, B.; Horvath, P.; Aebersold, R.; Antonin, W.; Kutay, U. Phosphorylation of Nup98 by multiple kinases is crucial for NPC disassembly during mitotic entry. Cell 2011, 144, 539–550. [Google Scholar] [CrossRef] [PubMed]
- Linder, M.I.; Köhler, M.; Boersema, P.; Weberruss, M.; Wandke, C.; Marino, J.; Ashiono, C.; Picotti, P.; Antonin, W.; Kutay, U. Mitotic Disassembly of Nuclear Pore Complexes Involves CDK1- and PLK1-MediatedPhosphorylation of Key Interconnecting Nucleoporins. Dev. Cell 2017, 43, 141–156. [Google Scholar] [CrossRef] [PubMed]
- de Castro, I.J.; Gil, R.S.; Ligammari, L.; Di Giacinto, M.L.; Vagnarelli, P. CDK1 and PLK1 coordinate the disassembly and reassembly of the nuclear envelope in vertebrate mitosis. Oncotarget 2017, 9, 7763–7773. [Google Scholar] [CrossRef] [PubMed]
- Martino, L.; Morchoisne-Bolhy, S.; Cheerambathur, D.K.; Van Hove, L.; Dumont, J.; Joly, N.; Desai, A.; Doye, V.; Pintard, L. Channel Nucleoporins Recruit PLK-1 to Nuclear Pore Complexes to Direct Nuclear Envelope Breakdown in C. elegans. Dev. Cell 2017, 43, 157–171. [Google Scholar] [CrossRef]
- Nichols, R.J.; Wiebe, M.S.; Traktman, P. The vaccinia-related kinases phosphorylate the N’ terminus of BAF, regulating its interaction with DNA and its retention in the nucleus. Mol. Biol. Cell 2006, 17, 2451–2464. [Google Scholar] [CrossRef]
- Margalit, A.; Brachner, A.; Gotzmann, J.; Foisner, R.; Gruenbaum, Y. Barrier-to-autointegration factor—A BAF fling little protein. Trends Cell Biol. 2007, 17, 202–208. [Google Scholar] [CrossRef]
- Gorjánácz, M.; Klerkx, E.P.; Galy, V.; Santarella, R.; López-Iglesias, C.; Askjaer, P.; Mattaj, I.W. Caenorhabditis elegans BAF-1 and its kinase VRK-1 participate directly in post-mitotic nuclear envelope assembly. EMBO J. 2007, 26, 132–143. [Google Scholar] [CrossRef] [PubMed]
- Asencio, C.; Davidson, I.F.; Santarella-Mellwig, R.; Ly-Hartig, T.B.; Mall, M.; Wallenfang, M.R.; Mattaj, I.W.; Gorjánácz, M. Coordination of kinase and phosphatase activities by Lem4 enables nuclear envelope reassembly during mitosis. Cell 2012, 150, 122–135. [Google Scholar] [CrossRef] [PubMed]
- Mehsen, H.; Boudreau, V.; Garrido, D.; Bourouh, M.; Larouche, M.; Maddox, P.S.; Swan, A.; Archambault, V. PP2A-B55 promotes nuclear envelope reformation after mitosis in Drosophila. J. Cell Biol. 2018, 217, 4106–4123. [Google Scholar] [CrossRef] [PubMed]
- Thompson, L.J.; Bollen, M.; Fields, A.P. Identification of protein phosphatase 1 as a mitotic lamin phosphatase. J. Biol. Chem. 1997, 272, 29693–29697. [Google Scholar] [CrossRef] [PubMed]
- Steen, R.L.; Martins, S.B.; Tasken, K.; Collas, P. Recruitment of protein phosphatase 1 to the nuclear envelope by A-kinase anchoring protein AKAP149 is a prerequisite for nuclear lamina assembly. J. Cell Biol. 2000, 150, 1251–1262. [Google Scholar] [CrossRef] [PubMed]
- Steen, R.L.; Beullens, M.; Landsverk, H.B.; Bollen, M.; Collas, P. AKAP149 is a novel PP1 specifier required to maintain nuclear envelope integrity in G1 phase. J. Cell Sci. 2003, 116, 2237–2246. [Google Scholar] [CrossRef] [PubMed]
- Güttinger, S.; Laurell, E.; Kutay, U. Orchestrating nuclear envelope disassembly and reassembly during mitosis. Nat. Rev. Mol. Cell Biol. 2009, 10, 178–191. [Google Scholar] [CrossRef] [PubMed]
- Vagnarelli, P.; Ribeiro, S.; Sennels, L.; Sanchez-Pulido, L.; de Lima Alves, F.; Verheyen, T.; Kelly, D.A.; Ponting, C.P.; Rappsilber, J.; Earnshaw, W.C. Repo-Man coordinates chromosomal reorganization with nuclear envelope reassembly during mitotic exit. Dev. Cell 2011, 21, 328–342. [Google Scholar] [CrossRef]
- Ungricht, R.; Kutay, U. Mechanisms and functions of nuclear envelope remodelling. Nat. Rev. Mol. Cell Biol. 2017, 18, 229–245. [Google Scholar] [CrossRef]
- Zhang, C.; Hutchins, J.R.; Mühlhäusser, P.; Kutay, U.; Clarke, P.R. Role of importin-beta in the control of nuclear envelope assembly by Ran. Curr. Biol. 2002, 12, 498–502. [Google Scholar] [CrossRef]
- Harel, A.; Chan, R.C.; Lachish-Zalait, A.; Zimmerman, E.; Elbaum, M.; Forbes, D.J. Importin beta negatively regulates nuclear membrane fusion and nuclear pore complex assembly. Mol. Biol. Cell 2003, 14, 4387–4396. [Google Scholar] [CrossRef] [PubMed]
- Walther, T.C.; Askjaer, P.; Gentzel, M.; Habermann, A.; Griffiths, G.; Wilm, M.; Mattaj, I.W.; Hetzer, M. RanGTP mediates nuclear pore complex assembly. Nature 2003, 424, 689–694. [Google Scholar] [CrossRef] [PubMed]
- Clarke, P.R.; Zhang, C. Spatial and temporal coordination of mitosis by Ran GTPase. Nat. Rev. Mol. Cell Biol. 2008, 9, 464–477. [Google Scholar] [CrossRef] [PubMed]
- Rotem, A.; Gruber, R.; Shorer, H.; Shaulov, L.; Klein, E.; Harel, A. Importin beta regulates the seeding of chromatin with initiation sites for nuclear pore assembly. Mol. Biol. Cell 2009, 20, 4031–4042. [Google Scholar] [CrossRef] [PubMed]
- Hoelz, A.; Debler, E.W.; Blobel, G. The structure of the nuclear pore complex. Annu. Rev. Biochem. 2011, 80, 613–643. [Google Scholar] [CrossRef]
- Fernandez, A.G.; Piano, F. MEL-28 is downstream of the Ran cycle and is required for nuclear-envelope function and chromatin maintenance. Curr. Biol. 2006, 16, 1757–1763. [Google Scholar] [CrossRef] [PubMed]
- Galy, V.; Askjaer, P.; Franz, C.; López-Iglesias, C.; Mattaj, I.W. MEL-28, a novel nuclear-envelope and kinetochore protein essential for zygotic nuclear-envelopeassembly in C. elegans. Curr. Biol. 2006, 16, 1748–1756. [Google Scholar] [CrossRef] [PubMed]
- Rasala, B.A.; Orjalo, A.V.; Shen, Z.; Briggs, S.; Forbes, D.J. ELYS is a dual nucleoporin/kinetochore protein required for nuclear pore assembly and proper cell division. Proc. Natl. Acad. Sci. USA 2006, 103, 17801–17806. [Google Scholar] [CrossRef] [PubMed]
- Franz, C.; Walczak, R.; Yavuz, S.; Santarella, R.; Gentzel, M.; Askjaer, P.; Galy, V.; Hetzer, M.; Mattaj, I.W.; Antonin, W. MEL-28/ELYS is required for the recruitment of nucleoporins to chromatin and postmitotic nuclear pore complex assembly. EMBO Rep. 2007, 8, 165–172. [Google Scholar] [CrossRef]
- Gillespie, P.J.; Khoudoli, G.A.; Stewart, G.; Swedlow, J.R.; Blow, J.J. ELYS/MEL-28 chromatin association coordinates nuclear pore complex assembly and replication licensing. Curr. Biol. 2007, 17, 1657–1662. [Google Scholar] [CrossRef]
- Hattersley, N.; Cheerambathur, D.; Moyle, M.; Stefanutti, M.; Richardson, A.; Lee, K.Y.; Dumont, J.; Oegema, K.; Desai, A. A Nucleoporin Docks Protein Phosphatase 1 to Direct Meiotic Chromosome Segregation and Nuclear Assembly. Dev. Cell 2016, 38, 463–477. [Google Scholar] [CrossRef] [PubMed]
- Antonin, W.; Neumann, H. Chromosome condensation and decondensation during mitosis. Curr. Opin. Cell. Biol. 2016, 40, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Trinkle-Mulcahy, L.; Andersen, J.; Lam, Y.W.; Moorhead, G.; Mann, M.; Lamond, A.I. Repo-Man recruits PP1 gamma to chromatin and is essential for cell viability. J. Cell. Biol. 2006, 172, 679–692. [Google Scholar] [CrossRef] [PubMed]
- Vagnarelli, P.; Hudson, D.F.; Ribeiro, S.A.; Trinkle-Mulcahy, L.; Spence, J.M.; Lai, F.; Farr, C.J.; Lamond, A.I.; Earnshaw, W.C. Condensin and Repo-Man-PP1 co-operate in the regulation of chromosome architecture during mitosis. Nat. Cell. Biol. 2006, 8, 1133–1142. [Google Scholar] [CrossRef] [PubMed]
- Qian, J.; Beullens, M.; Lesage, B.; Bollen, M. Aurora B defines its own chromosomal targeting by opposing the recruitment of the phosphatase scaffold Repo-Man. Curr. Biol. 2013, 23, 1136–1143. [Google Scholar] [CrossRef] [PubMed]
- Qian, J.; Lesage, B.; Beullens, M.; Van Eynde, A.; Bollen, M. PP1/Repo-man dephosphorylates mitotic histone H3 at T3 and regulates chromosomal aurora B targeting. Curr. Biol. 2011, 21, 766–773. [Google Scholar] [CrossRef]
- Qian, J.; Beullens, M.; Huang, J.; De Munter, S.; Lesage, B.; Bollen, M. Cdk1 orders mitotic events through coordination of a chromosome-associated phosphatase switch. Nat. Commun. 2015, 6, 10215. [Google Scholar] [CrossRef]
- Fischle, W.; Tseng, B.S.; Dormann, H.L.; Ueberheide, B.M.; Garcia, B.A.; Shabanowitz, J.; Hunt, D.F.; Funabiki, H.; Allis, C.D. Regulation of HP1-chromatin binding by histone H3 methylation and phosphorylation. Nature 2005, 438, 1116–1122. [Google Scholar] [CrossRef]
- Booth, D.G.; Takagi, M.; Sanchez-Pulido, L.; Petfalski, E.; Vargiu, G.; Samejima, K.; Imamoto, N.; Ponting, C.P.; Tollervey, D.; Earnshaw, W.C.; et al. Ki-67 is a PP1-interacting protein that organises the mitotic chromosome periphery. Elife 2014, 3, e01641. [Google Scholar] [CrossRef]
- Kumar, G.S.; Gokhan, E.; De Munter, S.; Bollen, M.; Vagnarelli, P.; Peti, W.; Page, R. The Ki-67 and RepoMan mitotic phosphatases assemble via an identical, yet novel mechanism. Elife 2016, 5, e16539. [Google Scholar] [CrossRef]
- Landsverk, H.B.; Mora-Bermúdez, F.; Landsverk, O.J.; Hasvold, G.; Naderi, S.; Bakke, O.; Ellenberg, J.; Collas, P.; Syljuåsen, R.G.; Küntziger, T. The protein phosphatase 1 regulator PNUTS is a new component of the DNA damage response. EMBO Rep. 2010, 11, 868–875. [Google Scholar] [CrossRef] [PubMed]
- Williams, B.C.; Filter, J.J.; Blake-Hodek, K.A.; Wadzinski, B.E.; Fuda, N.J.; Shalloway, D.; Goldberg, M.L. Greatwall-phosphorylated Endosulfine is both an inhibitor and a substrate of PP2A-B55 heterotrimers. Elife 2014, 3, e01695. [Google Scholar] [CrossRef] [PubMed]
- Heim, A.; Konietzny, A.; Mayer, T.U. Protein phosphatase 1 is essential for Greatwall inactivation at mitotic exit. EMBO Rep. 2015, 16, 1501–1510. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Vigneron, S.; Robert, P.; Strub, J.M.; Cianferani, S.; Castro, A.; Lorca, T. Greatwall dephosphorylation and inactivation upon mitotic exit is triggered by PP1. J. Cell Sci. 2016, 129, 1329–1339. [Google Scholar] [CrossRef] [PubMed]
- Rogers, S.; Fey, D.; McCloy, R.A.; Parker, B.L.; Mitchell, N.J.; Payne, R.J.; Daly, R.J.; James, D.E.; Caldon, C.E.; Watkins, D.N.; et al. PP1 initiates the dephosphorylation of MASTL, triggering mitotic exit and bistability in human cells. J. Cell Sci. 2016, 129, 1340–1354. [Google Scholar] [CrossRef] [PubMed]
- Ren, D.; Fisher, L.A.; Zhao, J.; Wang, L.; Williams, B.C.; Goldberg, M.L.; Peng, A. Cell cycle-dependent regulation of Greatwall kinase by protein phosphatase 1 and regulatory subunit 3B. J. Biol. Chem. 2017, 292, 10026–10034. [Google Scholar] [CrossRef] [PubMed]
- Dohadwala, M.; e Silva, E.D.C.; Hall, F.L.; Williams, R.T.; Carbonaro-Hall, D.A.; Nairn, A.C.; Greengard, P.; Berndt, N. Phosphorylation and inactivation of protein phosphatase 1 by cyclin-dependent kinases. Proc. Natl. Acad. Sci. USA 1994, 91, 6408–6412. [Google Scholar] [CrossRef]
- Kwon, Y.G.; Lee, S.Y.; Choi, Y.; Greengard, P.; Nairn, A.C. Cell cycle-dependent phosphorylation of mammalian protein phosphatase 1 by cdc2 kinase. Proc. Natl. Acad. Sci. USA 1997, 94, 2168–2173. [Google Scholar] [CrossRef]
- Grallert, A.; Boke, E.; Hagting, A.; Hodgson, B.; Connolly, Y.; Griffiths, J.R.; Smith, D.L.; Pines, J.; Hagan, I.M. A PP1–PP2A phosphatase relay controls mitotic progression. Nature 2015, 517, 94–98. [Google Scholar] [CrossRef]
- Wu, J.Q.; Guo, J.Y.; Tang, W.; Yang, C.S.; Freel, C.D.; Chen, C.; Nairn, A.C.; Kornbluth, S. PP1-mediated dephosphorylation of phosphoproteins at mitotic exit is controlled by inhibitor-1 and PP1 phosphorylation. Nat. Cell Biol. 2009, 11, 644–651. [Google Scholar] [CrossRef]
- Olsen, J.V.; Vermeulen, M.; Santamaria, A.; Kumar, C.; Miller, M.L.; Jensen, L.J.; Gnad, F.; Cox, J.; Jensen, T.S.; Nigg, E.A.; et al. Quantitative phosphoproteomics reveals widespread full phosphorylation site occupancy during mitosis. Sci. Signal. 2010, 3, ra3. [Google Scholar] [CrossRef] [PubMed]
- Nasa, I.; Rusin, S.F.; Kettenbach, A.N.; Moorhead, G.B. Aurora B opposes PP1 function in mitosis by phosphorylating the conserved PP1-binding RVxF motif in PP1 regulatory proteins. Sci. Signal. 2018, 11, eaai8669. [Google Scholar] [CrossRef] [PubMed]
- Hengeveld, R.C.; Vromans, M.J.; Vleugel, M.; Hadders, M.A.; Lens, S.M. Inner centromere localization of the CPC maintains centromere cohesion and allows mitotic checkpoint silencing. Nat. Commun. 2017, 8, 15542. [Google Scholar] [CrossRef] [PubMed]
- Porter, I.M.; Schleicher, K.; Porter, M.; Swedlow, J.R. Bod1 regulates protein phosphatase 2A at mitotic kinetochores. Nat. Commun. 2013, 4, 2677. [Google Scholar] [CrossRef] [PubMed]
- Schleicher, K.; Porter, M.; ten Have, S.; Sundaramoorthy, R.; Porter, I.M.; Swedlow, J.R. The Ndc80 complex targets Bod1 to human mitotic kinetochores. Open Biol. 2017, 7, 170099. [Google Scholar] [CrossRef] [PubMed]
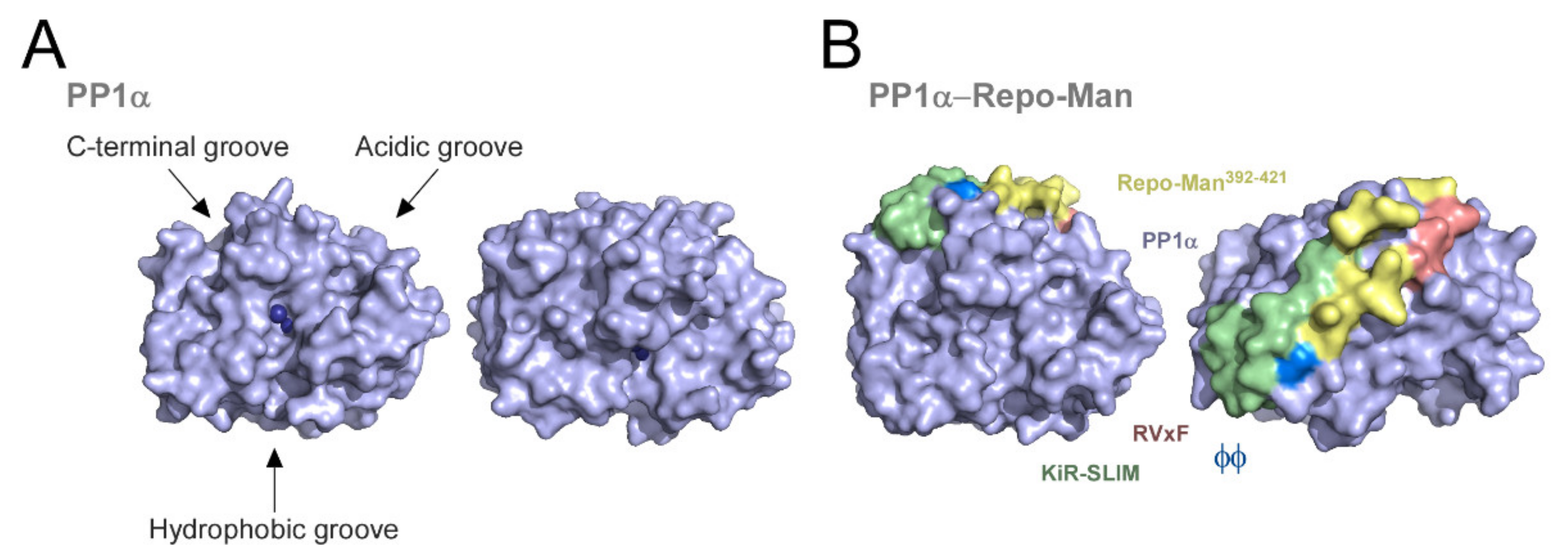
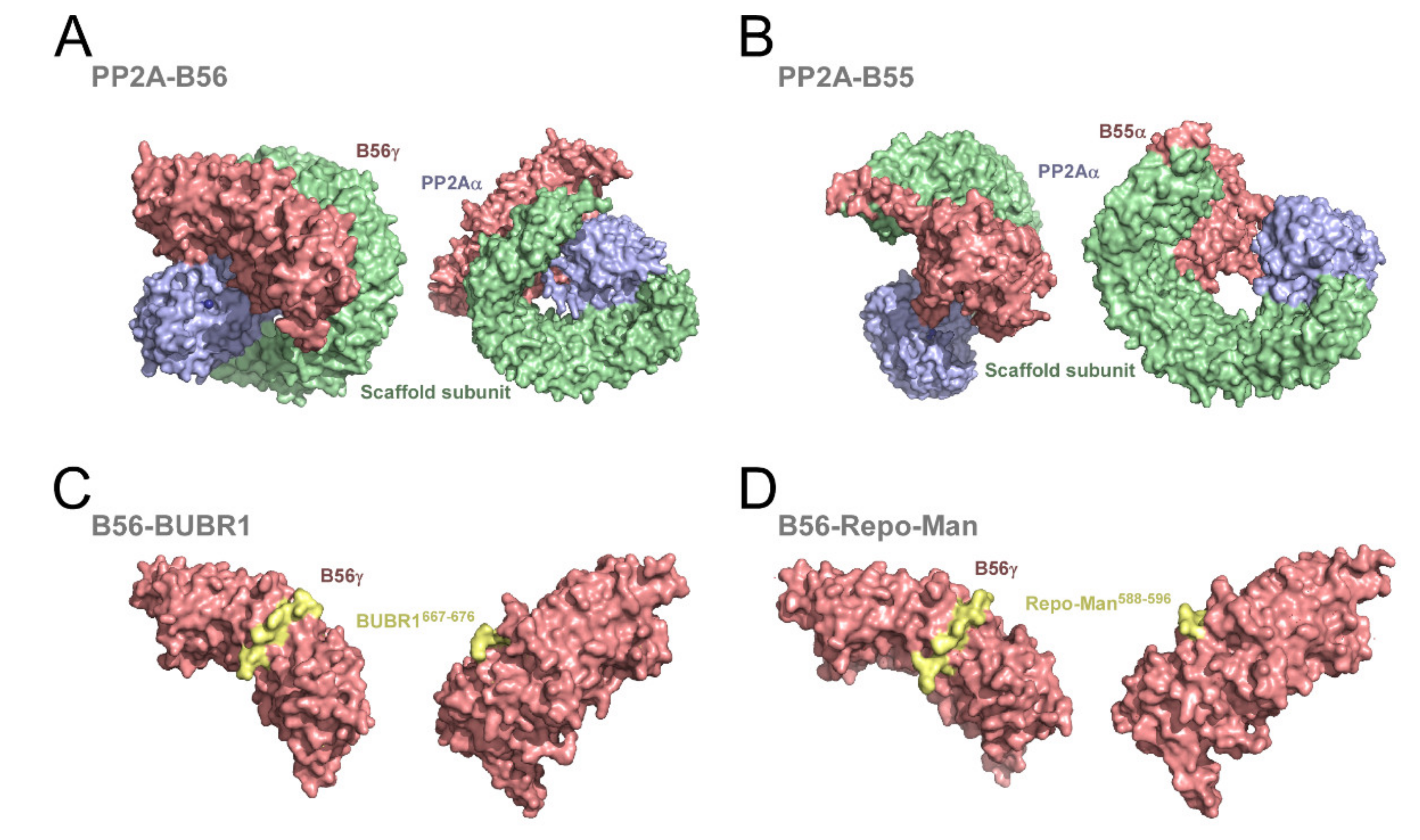
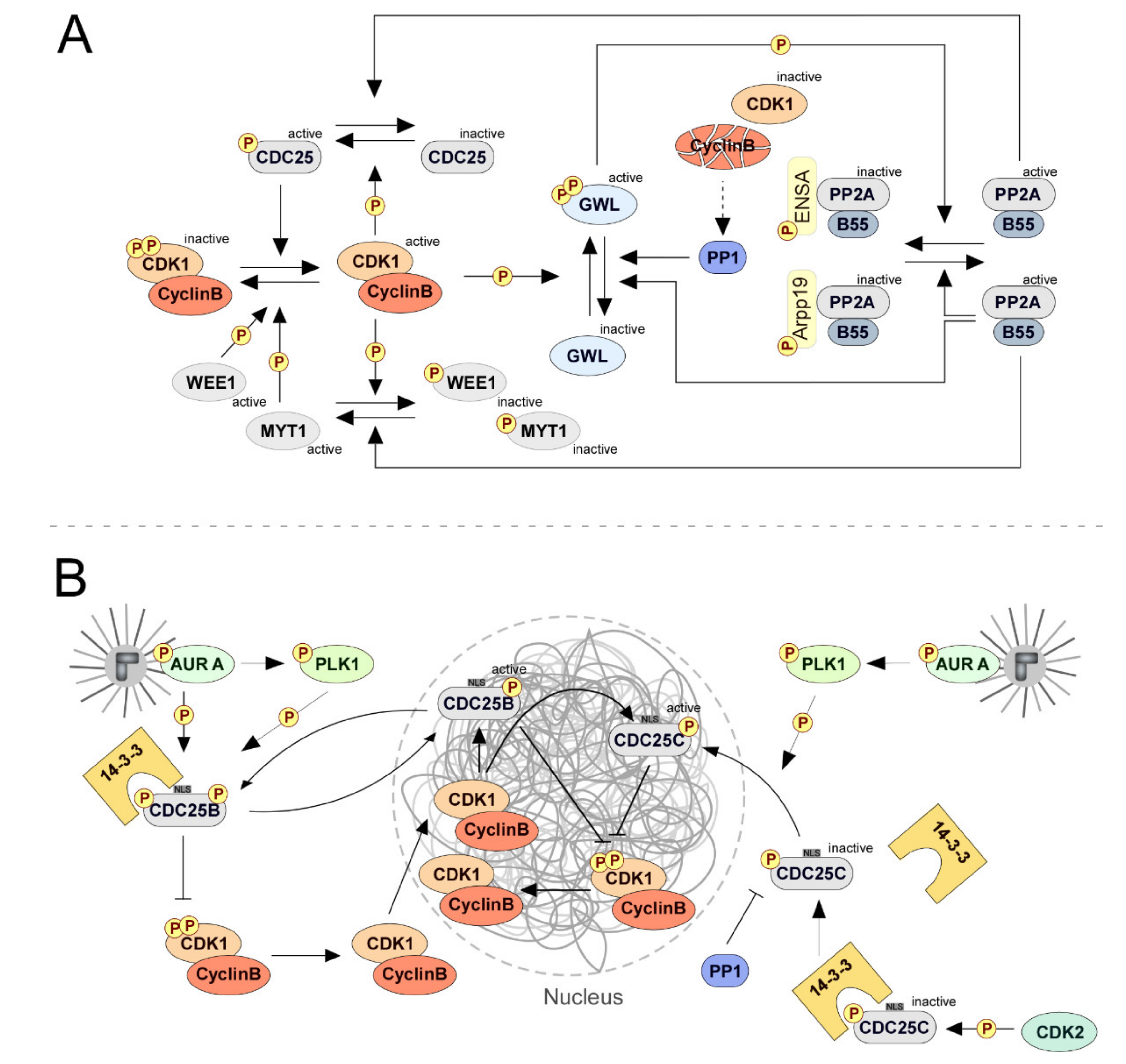
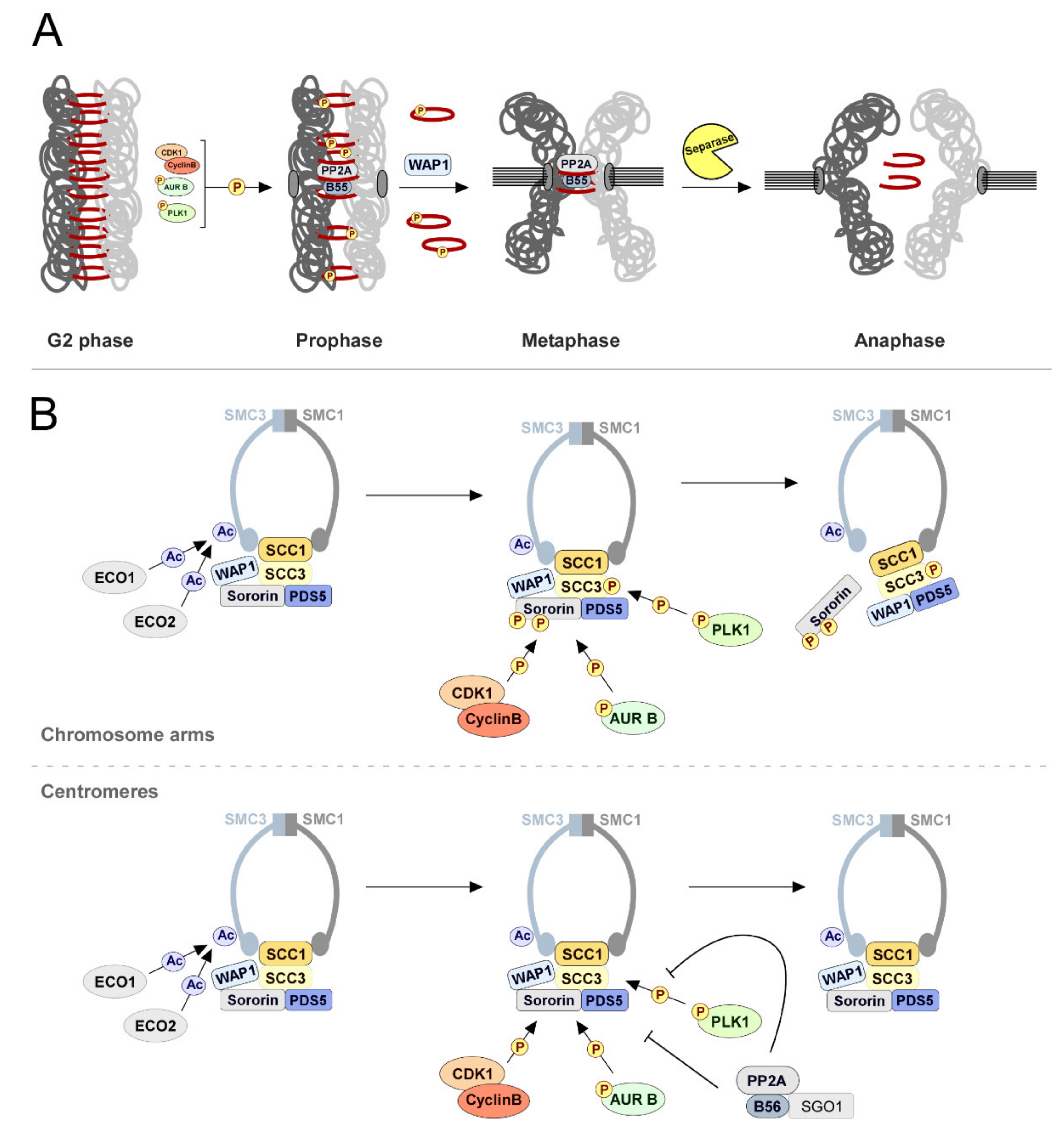
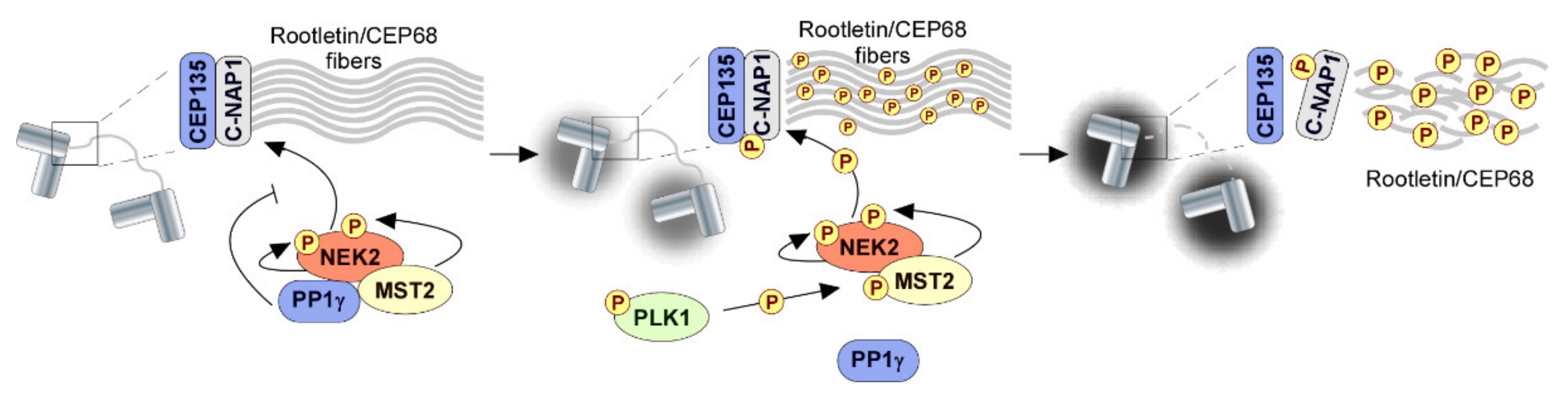
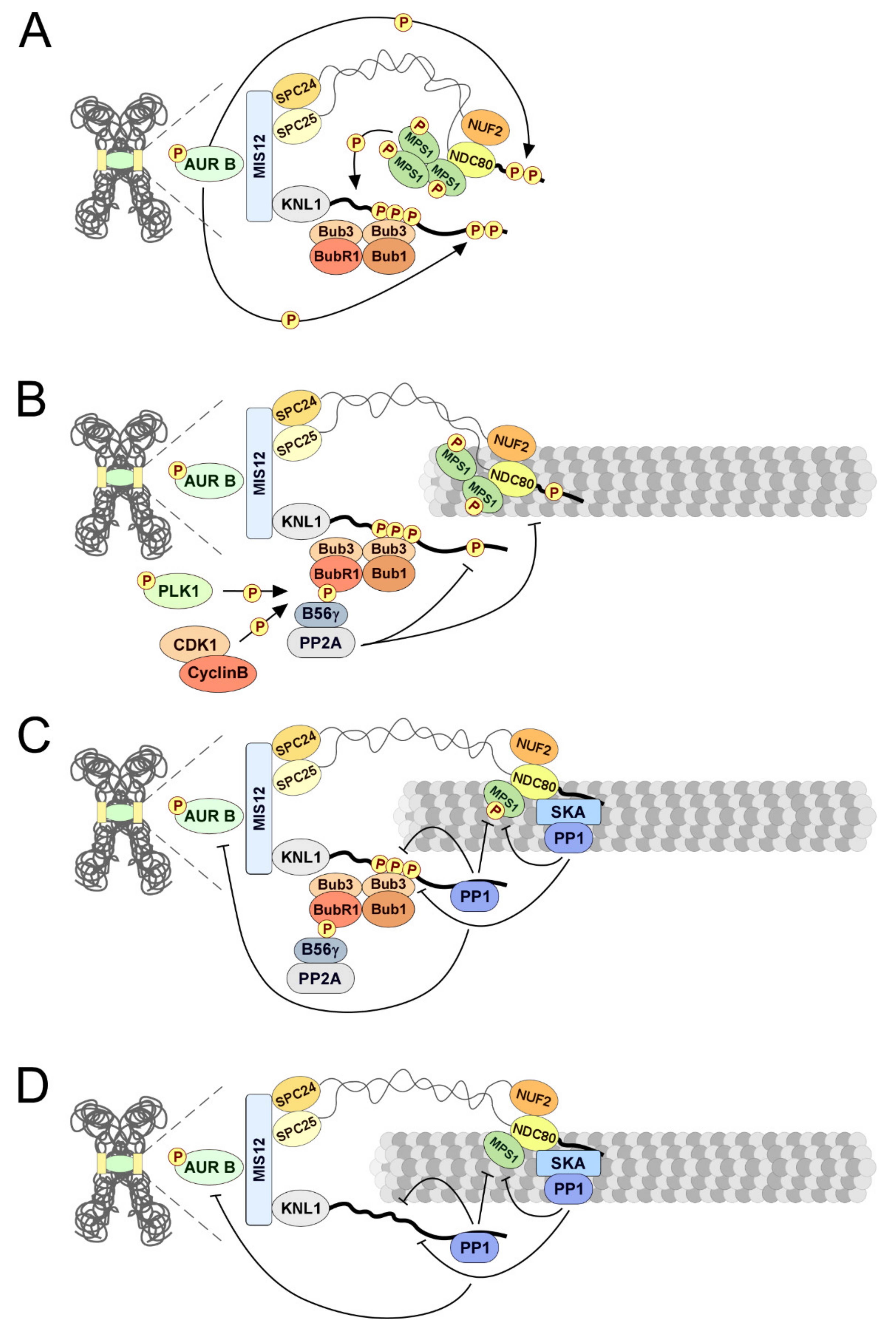
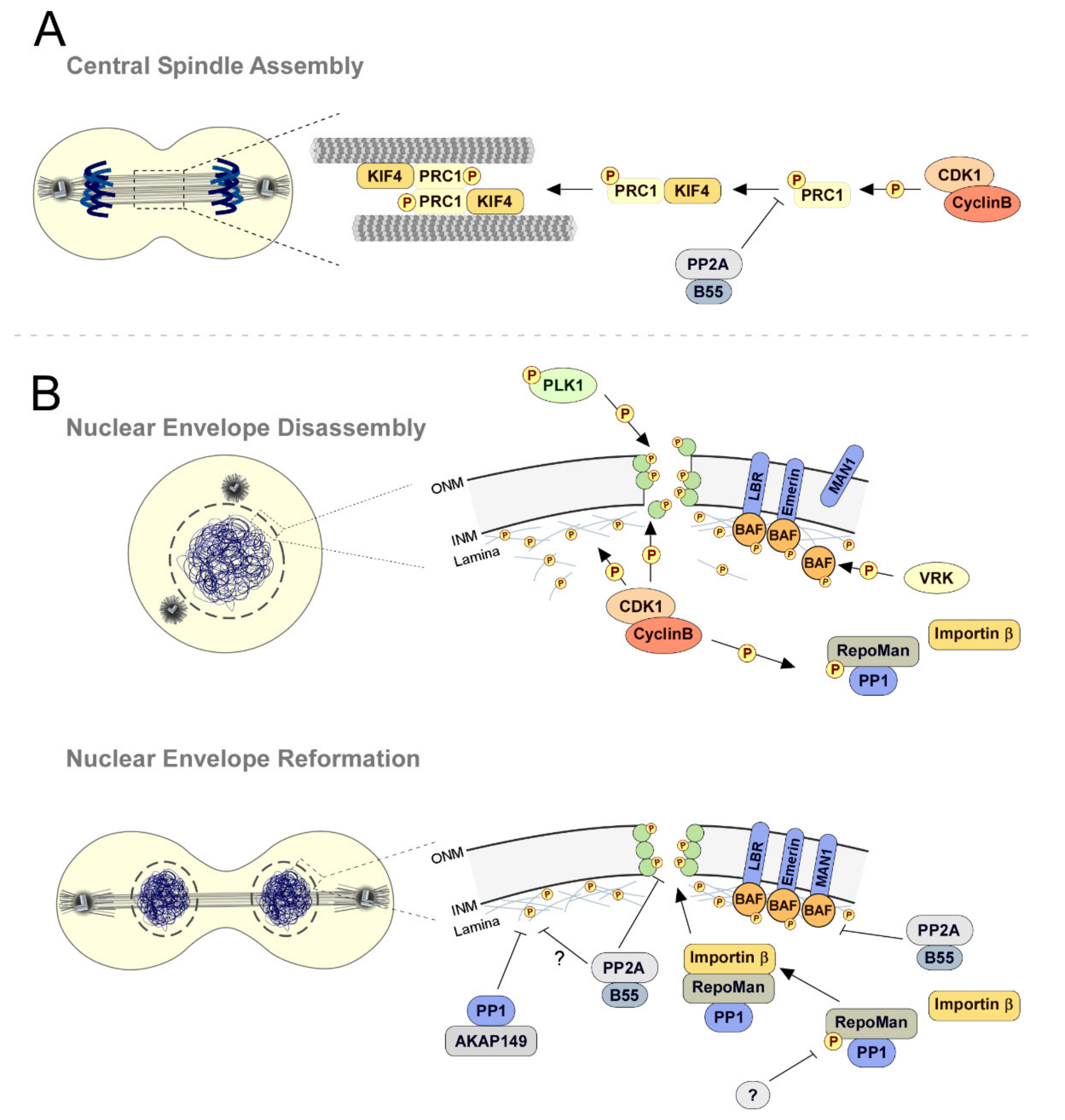
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moura, M.; Conde, C. Phosphatases in Mitosis: Roles and Regulation. Biomolecules 2019, 9, 55. https://doi.org/10.3390/biom9020055
Moura M, Conde C. Phosphatases in Mitosis: Roles and Regulation. Biomolecules. 2019; 9(2):55. https://doi.org/10.3390/biom9020055
Chicago/Turabian StyleMoura, Margarida, and Carlos Conde. 2019. "Phosphatases in Mitosis: Roles and Regulation" Biomolecules 9, no. 2: 55. https://doi.org/10.3390/biom9020055
APA StyleMoura, M., & Conde, C. (2019). Phosphatases in Mitosis: Roles and Regulation. Biomolecules, 9(2), 55. https://doi.org/10.3390/biom9020055