The Integrity of α-β-α Sandwich Conformation Is Essential for a Novel Adjuvant TFPR1 to Maintain Its Adjuvanticity

Abstract
1. Introduction
2. Materials and Methods
2.1. Mice
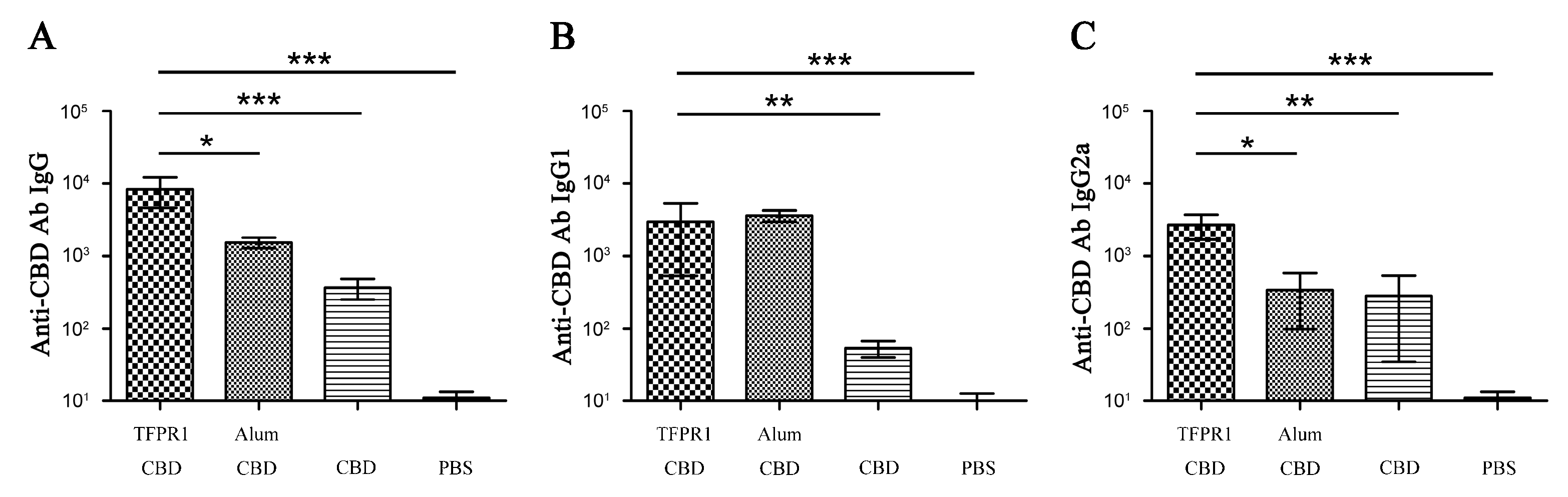
2.2. Evaluation of Adjuvanticity of TFPR1 for the Peptide Antigen HIV-1 CBD
2.3. Analysis and Identification of B Cell Epitopes on TFPR1
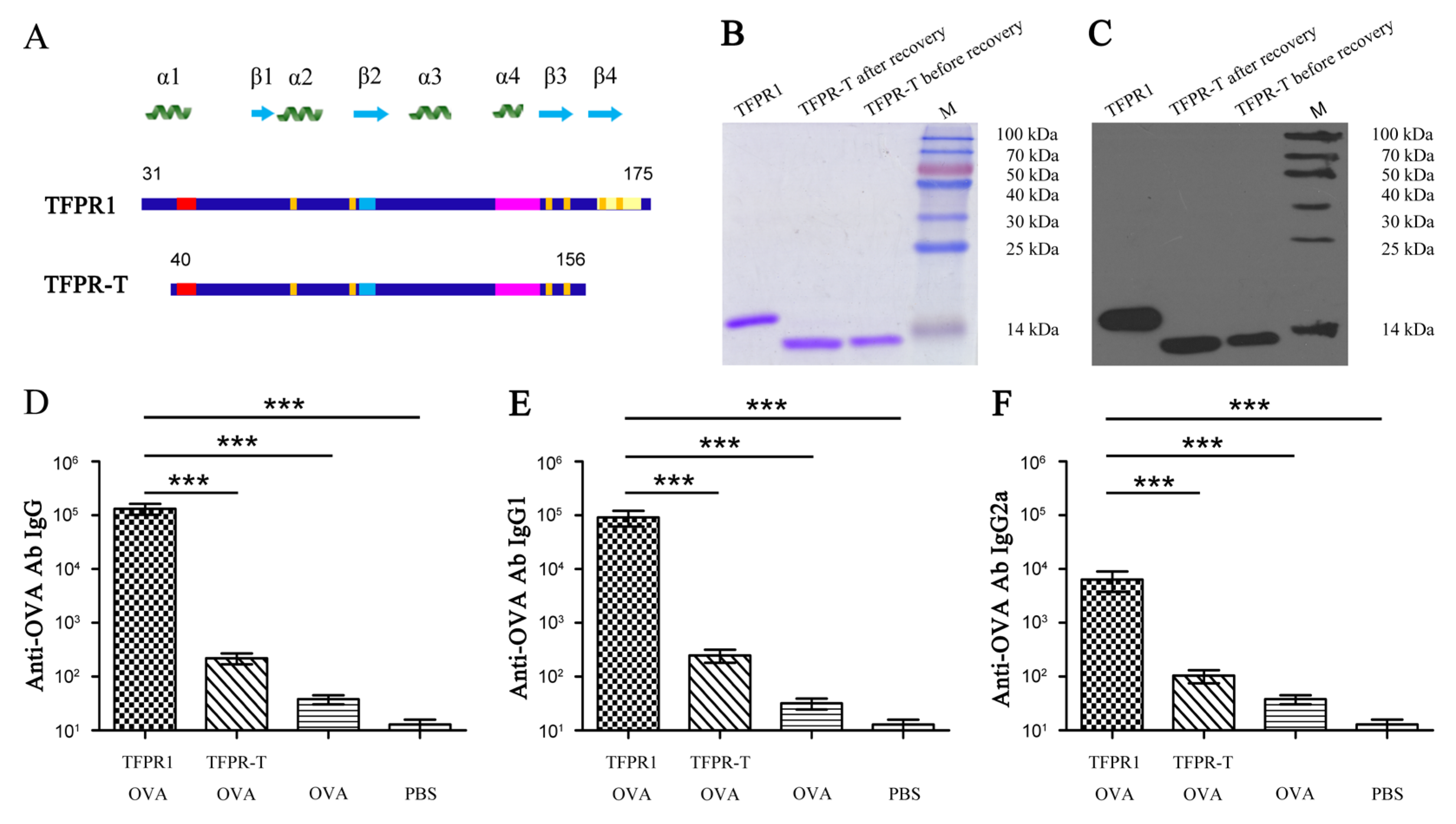
2.4. Expression, Purification, and Structure Prediction of Different Truncated Fragments of TFPR1
2.5. Adjuvanticity Evaluation of Each Truncated Fragment of TFPR1 Using Mouse Bone Marrow–Derived Dendritic Cells in Vitro
2.6. Adjuvanticity Evaluation of Each Truncated Fragment of TFPR1 Using the Model Antigen OVA In Vivo
2.7. Statistical Analysis
3. Results
3.1. TFPR1 Acts as an Effective Adjuvant for Peptide Antigen HIV-1 CBD
3.2. Prediction and Identification of Dominant B cell Epitopes on TFPR1
3.3. TFPR-T, the Core PR-1 Domain of TFPR1 in Absence of Partial α1-Helix and β4-fold, Has Little Adjuvant Activity In Vivo and In Vitro
3.4. Analysis, Design and Expression of Different Fragments Containing Either α1-Helix (TFPR-hd) or β4-Fold (TFPR-ta)
3.5. Both TFPR-Hd and TFPR-Ta Activate Dendritic Cells to Produce Cytokines Effectively
3.6. TFPR-Hd and TFPR-Ta Retain the Adjuvanticity of TFPR1 for the Model Antigen OVA
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Karch, C.P.; Burkhard, P. Vaccine technologies: From whole organisms to rationally designed protein assemblies. Biochem. Pharmacol. 2016, 120, 1–14. [Google Scholar] [CrossRef]
- Cunningham, A.L.; Garcon, N.; Leo, O.; Friedland, L.R.; Strugnell, R.; Laupeze, B.; Doherty, M.; Stern, P. Vaccine development: From concept to early clinical testing. Vaccine 2016, 34, 6655–6664. [Google Scholar] [CrossRef]
- Di Pasquale, A.; Preiss, S.; Tavares Da Silva, F.; Garcon, N. Vaccine Adjuvants: From 1920 to 2015 and Beyond. Vaccines 2015, 3, 320–343. [Google Scholar] [CrossRef] [PubMed]
- Cerofolini, L.; Giuntini, S.; Ravera, E.; Luchinat, C.; Berti, F.; Fragai, M. Structural characterization of a protein adsorbed on aluminum hydroxide adjuvant in vaccine formulation. Npj Vaccines 2019, 4, 20. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.; Shi, Y. Alum: An old dog with new tricks. Emerg. Microbes Infect. 2016, 5, e25. [Google Scholar] [CrossRef] [PubMed]
- He, P.; Zou, Y.; Hu, Z. Advances in aluminum hydroxide-based adjuvant research and its mechanism. Hum. Vaccines Immunother. 2015, 11, 477–488. [Google Scholar] [CrossRef]
- HogenEsch, H.; O’Hagan, D.T.; Fox, C.B. Optimizing the utilization of aluminum adjuvants in vaccines: You might just get what you want. Npj Vaccines 2018, 3, 51. [Google Scholar] [CrossRef]
- Shah, R.R.; Hassett, K.J.; Brito, L.A. Overview of Vaccine Adjuvants: Introduction, History, and Current Status. Methods Mol. Biol. 2017, 1494, 1–13. [Google Scholar]
- Martinon, S.; Cisneros, A.; Villicana, S.; Hernandez-Miramontes, R.; Mixcoha, E.; Calderon-Vargas, P. Chemical and Immunological Characteristics of Aluminum-Based, Oil-Water Emulsion, and Bacterial-Origin Adjuvants. J. Immunol. Res. 2019, 2019, 3974127. [Google Scholar] [CrossRef]
- McKee, A.S.; Marrack, P. Old and new adjuvants. Curr. Opin. Immunol. 2017, 47, 44–51. [Google Scholar] [CrossRef]
- Sanina, N. Vaccine Adjuvants Derived from Marine Organisms. Biomolecules 2019, 9, 340. [Google Scholar] [CrossRef] [PubMed]
- Garcon, N.; Chomez, P.; Van Mechelen, M. GlaxoSmithKline Adjuvant Systems in vaccines: Concepts, achievements and perspectives. Expert Rev. Vaccines 2007, 6, 723–739. [Google Scholar] [CrossRef] [PubMed]
- Fensterheim, B.A.; Young, J.D.; Luan, L.; Kleinbard, R.R.; Stothers, C.L.; Patil, N.K.; McAtee-Pereira, A.G.; Guo, Y.; Trenary, I.; Hernandez, A.; et al. The TLR4 Agonist Monophosphoryl Lipid A Drives Broad Resistance to Infection via Dynamic Reprogramming of Macrophage Metabolism. J. Immunol. 2018, 200, 3777–3789. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Wang, Y.; Zhou, Y.; Kou, Z. Advances in novel vaccine adjuvants based on TLR4 agonists. Chin. J. Microbiol. Immunol. 2019, 39, 638–644. [Google Scholar]
- Hajam, I.A.; Dar, P.A.; Shahnawaz, I.; Jaume, J.C.; Lee, J.H. Bacterial flagellin-a potent immunomodulatory agent. Exp. Mol. Med. 2017, 49, e373. [Google Scholar] [CrossRef]
- Mizel, S.B.; Bates, J.T. Flagellin as an adjuvant: Cellular mechanisms and potential. J. Immunol. 2010, 185, 5677–5682. [Google Scholar] [CrossRef]
- Nikoofal-Sahlabadi, S.; Matbou Riahi, M.; Sadri, K.; Badiee, A.; Nikpoor, A.R.; Jaafari, M.R. Liposomal CpG-ODN: An in vitro and in vivo study on macrophage subtypes responses, biodistribution and subsequent therapeutic efficacy in mice models of cancers. Eur. J. Pharm. Sci.: Off. J. Eur. Fed. Pharm. Sci. 2018, 119, 159–170. [Google Scholar] [CrossRef]
- Takeda, K.; Kaisho, T.; Akira, S. Toll-like receptors. Annu. Rev. Immunol. 2003, 21, 335–376. [Google Scholar] [CrossRef]
- Kumar, S.; Sunagar, R.; Gosselin, E. Bacterial Protein Toll-Like-Receptor Agonists: A Novel Perspective on Vaccine Adjuvants. Front. Immunol. 2019, 10, 1144. [Google Scholar] [CrossRef]
- Kwissa, M.; Kasturi, S.P.; Pulendran, B. The science of adjuvants. Expert Rev. Vaccines 2007, 6, 673–684. [Google Scholar] [CrossRef]
- Sunagar, K.; Johnson, W.E.; O’Brien, S.J.; Vasconcelos, V.; Antunes, A. Evolution of CRISPs associated with toxicoferan-reptilian venom and mammalian reproduction. Mol. Biol. Evol. 2012, 29, 1807–1822. [Google Scholar] [CrossRef] [PubMed]
- Darwiche, R.; Mene-Saffrane, L.; Gfeller, D.; Asojo, O.A.; Schneiter, R. The pathogen-related yeast protein Pry1, a member of the CAP protein superfamily, is a fatty acid-binding protein. J. Biol. Chem. 2017, 292, 8304–8314. [Google Scholar] [CrossRef] [PubMed]
- Breen, S.; Williams, S.J.; Outram, M.; Kobe, B.; Solomon, P.S. Emerging Insights into the Functions of Pathogenesis-Related Protein 1. Trends Plant Sci. 2017, 22, 871–879. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, G.M.; Roelants, K.; O’Bryan, M.K. The CAP superfamily: Cysteine-rich secretory proteins, antigen 5, and pathogenesis-related 1 proteins--roles in reproduction, cancer, and immune defense. Endocr. Rev. 2008, 29, 865–897. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Wang, Y.; Zhu, Q.; Ning, X.; Sun, W.; Zhou, Y.; Kou, Z. TFPR1 acts as a novel adjuvant by activating Dendritic cells and promoting its maturation. Mil. Med Sci. 2018, 42, 101–104. [Google Scholar]
- Sun, W.; Li, Q.; Ning, X.; Yang, Y.; Guo, J.; Zhu, Q.; Guo, Y.; Li, H.; Wang, Y.; Zhou, Y.; et al. TFPR1 acts as an immune regulator and an efficient adjuvant for proteins and peptides by activating immune cells, primarily through TLR2. Vaccine 2019. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Yang, Y.; Ning, X.; Guo, J.; Yu, H.; Kou, Z.; Zhou, Y. Production and Immunogenicity Analysis of Recombinant Snake Venom Protein TFPR1. Lett. Biotechnol. 2015, 26, 329–333. [Google Scholar]
- Hovanessian, A.G.; Briand, J.P.; Said, E.A.; Svab, J.; Ferris, S.; Dali, H.; Muller, S.; Desgranges, C.; Krust, B. The caveolin-1 binding domain of HIV-1 glycoprotein gp41 is an efficient B cell epitope vaccine candidate against virus infection. Immunity 2004, 21, 617–627. [Google Scholar] [CrossRef]
- Rey-Cuille, M.A.; Svab, J.; Benferhat, R.; Krust, B.; Briand, J.P.; Muller, S.; Hovanessian, A.G. HIV-1 neutralizing antibodies elicited by the candidate CBD1 epitope vaccine react with the conserved caveolin-1 binding motif of viral glycoprotein gp41. J. Pharm. Pharmacol. 2006, 58, 759–767. [Google Scholar] [CrossRef]
- Guo, J.; Yang, Y.; Xiao, W.; Sun, W.; Yu, H.; Du, L.; Lustigman, S.; Jiang, S.; Kou, Z.; Zhou, Y. A truncated fragment of Ov-ASP-1 consisting of the core pathogenesis-related-1 (PR-1) domain maintains adjuvanticity as the full-length protein. Vaccine 2015, 33, 1974–1980. [Google Scholar] [CrossRef]
- Micsonai, A.; Wien, F.; Kernya, L.; Lee, Y.H.; Goto, Y.; Refregiers, M.; Kardos, J. Accurate secondary structure prediction and fold recognition for circular dichroism spectroscopy. Proc. Natl. Acad. Sci. USA 2015, 112, E3095–E3103. [Google Scholar] [CrossRef] [PubMed]
- Greenfield, N.J. Using circular dichroism spectra to estimate protein secondary structure. Nat Protoc 2006, 1, 2876–2890. [Google Scholar] [CrossRef] [PubMed]
- Basak, S.K.; Harui, A.; Stolina, M.; Sharma, S.; Mitani, K.; Dubinett, S.M.; Roth, M.D. Increased dendritic cell number and function following continuous in vivo infusion of granulocyte macrophage-colony-stimulating factor and interleukin-4. Blood 2002, 99, 2869–2879. [Google Scholar] [CrossRef] [PubMed]
- Faulin, T.; Kazuma, S.M.; Tripodi, G.L.; Cavalcante, M.F.; Wakasuqui, F.; Oliveira, C.L.P.; Degenhardt, M.F.S.; Michaloski, J.; Giordano, R.J.; Ketelhuth, D.F.J.; et al. Proinflammatory Action of a New Electronegative Low-Density Lipoprotein Epitope. Biomolecules 2019, 9, 386. [Google Scholar] [CrossRef] [PubMed]
- Reed, S.G.; Orr, M.T.; Fox, C.B. Key roles of adjuvants in modern vaccines. Nat. Med. 2013, 19, 1597–1608. [Google Scholar] [CrossRef] [PubMed]
- Nabel, G.J. Designing tomorrow’s vaccines. New Engl. J. Med. 2013, 368, 551–560. [Google Scholar] [CrossRef]
- Montomoli, E.; Piccirella, S.; Khadang, B.; Mennitto, E.; Camerini, R.; De Rosa, A. Current adjuvants and new perspectives in vaccine formulation. Expert Rev. Vaccines 2011, 10, 1053–1061. [Google Scholar] [CrossRef]
- He, Y.; Barker, S.J.; MacDonald, A.J.; Yu, Y.; Cao, L.; Li, J.; Parhar, R.; Heck, S.; Hartmann, S.; Golenbock, D.T.; et al. Recombinant Ov-ASP-1, a Th1-biased protein adjuvant derived from the helminth Onchocerca volvulus, can directly bind and activate antigen-presenting cells. J. Immunol. 2009, 182, 4005–4016. [Google Scholar] [CrossRef]
- Cui, B.; Liu, X.; Fang, Y.; Zhou, P.; Zhang, Y.; Wang, Y. Flagellin as a vaccine adjuvant. Expert Rev. Vaccines 2018, 17, 335–349. [Google Scholar] [CrossRef]
- Biedma, M.E.; Cayet, D.; Tabareau, J.; Rossi, A.H.; Ivicak-Kocjan, K.; Moreno, G.; Errea, A.; Soulard, D.; Parisi, G.; Jerala, R.; et al. Recombinant flagellins with deletions in domains D1, D2, and D3: Characterization as novel immunoadjuvants. Vaccine 2019, 37, 652–663. [Google Scholar] [CrossRef]
- Wang, J.; Tricoche, N.; Du, L.; Hunter, M.; Zhan, B.; Goud, G.; Didier, E.S.; Liu, J.; Lu, L.; Marx, P.A.; et al. The adjuvanticity of an O. volvulus-derived rOv-ASP-1 protein in mice using sequential vaccinations and in non-human primates. PLoS ONE 2012, 7, e37019. [Google Scholar] [CrossRef] [PubMed]
- Onda, M. Reducing the immunogenicity of protein therapeutics. Curr. Drug Targets 2009, 10, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Sethu, S.; Govindappa, K.; Alhaidari, M.; Pirmohamed, M.; Park, K.; Sathish, J. Immunogenicity to biologics: Mechanisms, prediction and reduction. Arch. Immunol. Et Ther. Exp. 2012, 60, 331–344. [Google Scholar] [CrossRef] [PubMed]
- Arnold-Schrauf, C.; Berod, L.; Sparwasser, T. Dendritic cell specific targeting of MyD88 signalling pathways in vivo. Eur. J. Immunol. 2015, 45, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Benko, S.; Magyarics, Z.; Szabo, A.; Rajnavolgyi, E. Dendritic cell subtypes as primary targets of vaccines: The emerging role and cross-talk of pattern recognition receptors. Biol. Chem. 2008, 389, 469–485. [Google Scholar] [CrossRef] [PubMed]
- Levitz, S.M.; Golenbock, D.T. Beyond empiricism: Informing vaccine development through innate immunity research. Cell 2012, 148, 1284–1292. [Google Scholar] [CrossRef] [PubMed]
- Wculek, S.K.; Cueto, F.J.; Mujal, A.M.; Melero, I.; Krummel, M.F.; Sancho, D. Dendritic cells in cancer immunology and immunotherapy. Nat. Rev. Immunol. 2019. [Google Scholar] [CrossRef]
- Worbs, T.; Hammerschmidt, S.I.; Forster, R. Dendritic cell migration in health and disease. Nat. Reviews. Immunol. 2017, 17, 30–48. [Google Scholar] [CrossRef]
- Scheiblhofer, S.; Laimer, J.; Machado, Y.; Weiss, R.; Thalhamer, J. Influence of protein fold stability on immunogenicity and its implications for vaccine design. Expert Rev. Vaccines 2017, 16, 479–489. [Google Scholar] [CrossRef]
- Ohkuri, T.; Nagatomo, S.; Oda, K.; So, T.; Imoto, T.; Ueda, T. A protein’s conformational stability is an immunologically dominant factor: Evidence that free-energy barriers for protein unfolding limit the immunogenicity of foreign proteins. J. Immunol. 2010, 185, 4199–4205. [Google Scholar] [CrossRef]
- McLaughlin, R.N., Jr.; Poelwijk, F.J.; Raman, A.; Gosal, W.S.; Ranganathan, R. The spatial architecture of protein function and adaptation. Nature 2012, 491, 138–142. [Google Scholar] [CrossRef] [PubMed]
- Andrade, F.D.; Forato, L.A.; Bernardes Filho, R.; Colnago, L.A. Quantification of protein secondary structure by (13)C solid-state NMR. Anal. Bioanal. Chem. 2016, 408, 3875–3879. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Shi, X.; Thompson, F.; Weber, W.S.; Mou, Q.; Yarger, J.L. Protein secondary structure of Green Lynx spider dragline silk investigated by solid-state NMR and X-ray diffraction. Int. J. Biol. Macromol. 2015, 81, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Nagata, S.; Pastan, I. Removal of B cell epitopes as a practical approach for reducing the immunogenicity of foreign protein-based therapeutics. Adv. Drug Deliv. Rev. 2009, 61, 977–985. [Google Scholar] [CrossRef] [PubMed]
- Mazor, R.; King, E.M.; Pastan, I. Strategies to Reduce the Immunogenicity of Recombinant Immunotoxins. Am. J. Pathol. 2018, 188, 1736–1743. [Google Scholar] [CrossRef]
- Mazor, R.; Onda, M.; Pastan, I. Immunogenicity of therapeutic recombinant immunotoxins. Immunol. Rev. 2016, 270, 152–164. [Google Scholar] [CrossRef]
- Onda, M.; Beers, R.; Xiang, L.; Nagata, S.; Wang, Q.C.; Pastan, I. An immunotoxin with greatly reduced immunogenicity by identification and removal of B cell epitopes. Proc. Natl. Acad. Sci. USA 2008, 105, 11311–11316. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Start | End | Amino Acid Sequence | Length (Aa) | Score | Secondary Structures # |
|---|---|---|---|---|---|---|
| 1 | 31 | 35 | PEIQN | 5 | 0.882 | α1-helix |
| 2 | 45 | 53 | RRSVNPTAS | 9 | 1.06 | α1-helix |
| 3 | 61 | 70 | YPEAAANAER | 10 | 1.287 | β1-fold, α2-helix |
| 4 | 78 | 86 | SHSSRDSRV | 9 | 1.07 | / |
| 5 | 101 | 105 | YPAKW | 5 | 0.741 | α3-helix |
| 6 | 114 | 132 | GEYKDFKYGVGAVPSDAVI | 19 | 1.033 | α3-helix |
| 7 | 145 | 147 | RAG | 3 | 0.246 | β3-fold |
| 8 | 149 | 157 | AAAYCPSSK | 9 | 0.649 | β3-fold |
| 9 | 170 | 174 | GNIIG | 5 | 0.283 | / |
| Peptide | Start | End | Amino Acid Sequence | Length (Aa) | Predicted B Epitope # | Secondary Structures |
|---|---|---|---|---|---|---|
| P1 | 31 | 48 | PEIQNEIIDLHNSLRRSV | 18 | high(#1+2) | α1-helix * |
| P2 | 41 | 55 | HNSLRRSVNPTASNM | 15 | high(#2) | α1-helix |
| P3 | 57 | 71 | KMEWYPEAAANAERW | 15 | high(#3) | β1-fold *, α2-helix |
| P4 | 89 | 103 | GIKCGENIYMATYPA | 15 | N.P.(#5Δ) | β2-fold * |
| P5 | 94 | 108 | ENIYMATYPAKWTDI | 15 | intermediate(#5) | β2-fold, α3-helix |
| P6 | 103 | 117 | AKWTDIIHAWHGEYK | 15 | intermediate(#5+6) | α3-helix * |
| P7 | 132 | 146 | IGHYTQIVWYKSYRA | 15 | N.P | α4-helix *, β3-fold |
| P8 | 138 | 153 | IVWYKSYRAGCAAAYC | 16 | low(#7) | β3-fold* |
| P9 | 158 | 172 | YSYFYVCQYCPAGNI | 15 | low(#9Δ) | β4-fold* |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Q.; Ning, X.; Wang, Y.; Zhu, Q.; Guo, Y.; Li, H.; Zhou, Y.; Kou, Z. The Integrity of α-β-α Sandwich Conformation Is Essential for a Novel Adjuvant TFPR1 to Maintain Its Adjuvanticity. Biomolecules 2019, 9, 869. https://doi.org/10.3390/biom9120869
Li Q, Ning X, Wang Y, Zhu Q, Guo Y, Li H, Zhou Y, Kou Z. The Integrity of α-β-α Sandwich Conformation Is Essential for a Novel Adjuvant TFPR1 to Maintain Its Adjuvanticity. Biomolecules. 2019; 9(12):869. https://doi.org/10.3390/biom9120869
Chicago/Turabian StyleLi, Qiao, Xiuzhe Ning, Yuepeng Wang, Qing Zhu, Yan Guo, Hao Li, Yusen Zhou, and Zhihua Kou. 2019. "The Integrity of α-β-α Sandwich Conformation Is Essential for a Novel Adjuvant TFPR1 to Maintain Its Adjuvanticity" Biomolecules 9, no. 12: 869. https://doi.org/10.3390/biom9120869
APA StyleLi, Q., Ning, X., Wang, Y., Zhu, Q., Guo, Y., Li, H., Zhou, Y., & Kou, Z. (2019). The Integrity of α-β-α Sandwich Conformation Is Essential for a Novel Adjuvant TFPR1 to Maintain Its Adjuvanticity. Biomolecules, 9(12), 869. https://doi.org/10.3390/biom9120869