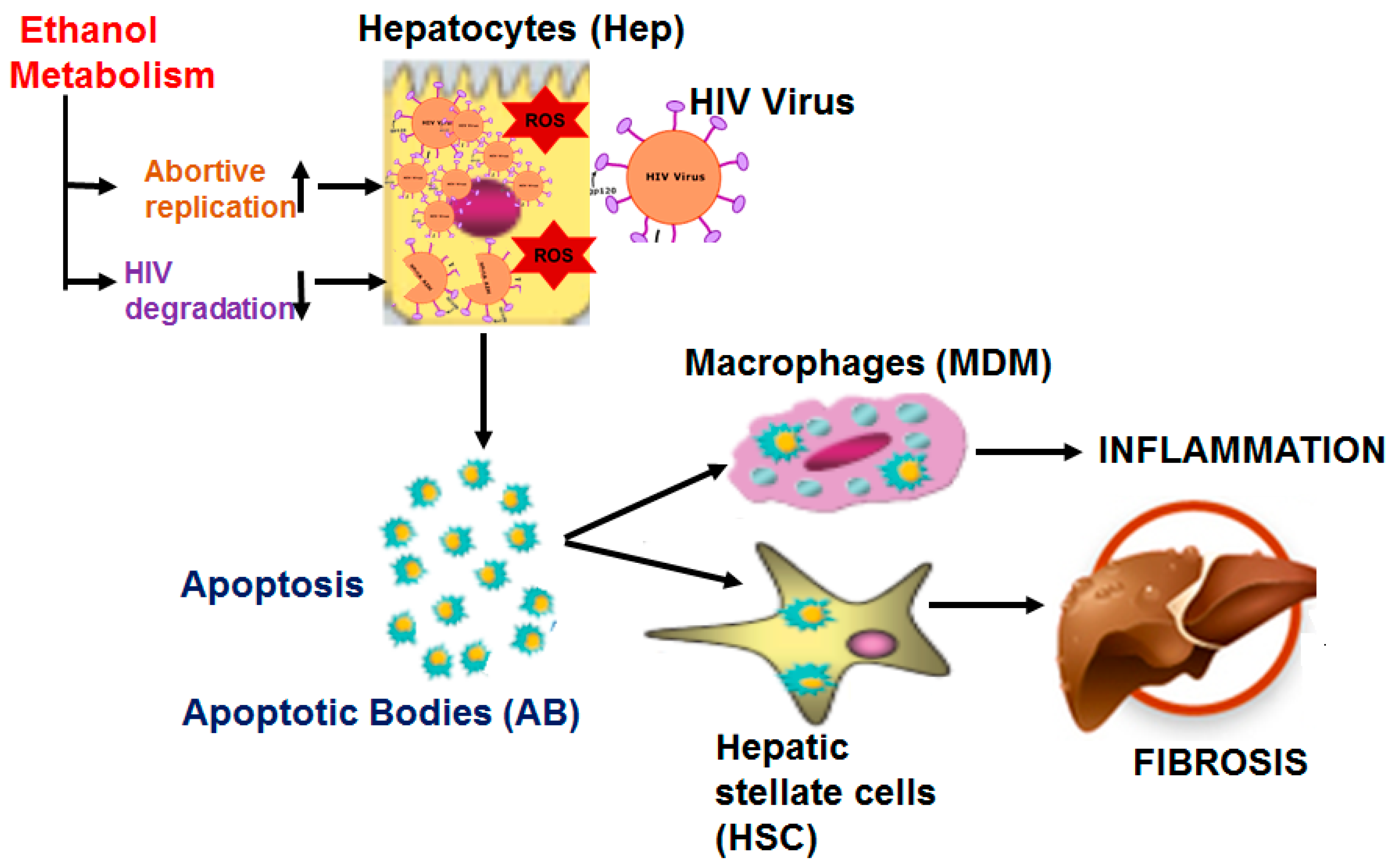
Alcohol Metabolism Potentiates HIV-Induced Hepatotoxicity: Contribution to End-Stage Liver Disease

 ,
,  ,
,  , , and
, , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Reagents and Media
2.2. Cells and Treatments
2.3. Human Monocyte-Derived Macrophages
2.4. Hepatic Stellate Cells (HSCs)
2.5. Apoptotic Body (AB) Generation and Treatment Macrophages and Hepatic Stellate Cells with Apoptotic Hepatocytes
2.6. RNA Isolation, Real-Time Polymerase Chain Reaction, and Western Blotting
2.7. Total HIV-1 DNA Quantification Using Semi-Nested Polymerase Chain Reaction
2.8. Integrated HIV-1 DNA Quantification Using Digital-Droplet PCR (ddPCR)
2.9. Activities of Proteasome and Cathepsins
2.10. Validation of In Vitro Results by In Vivo Studies
2.11. Statistical Analyses
3. Results
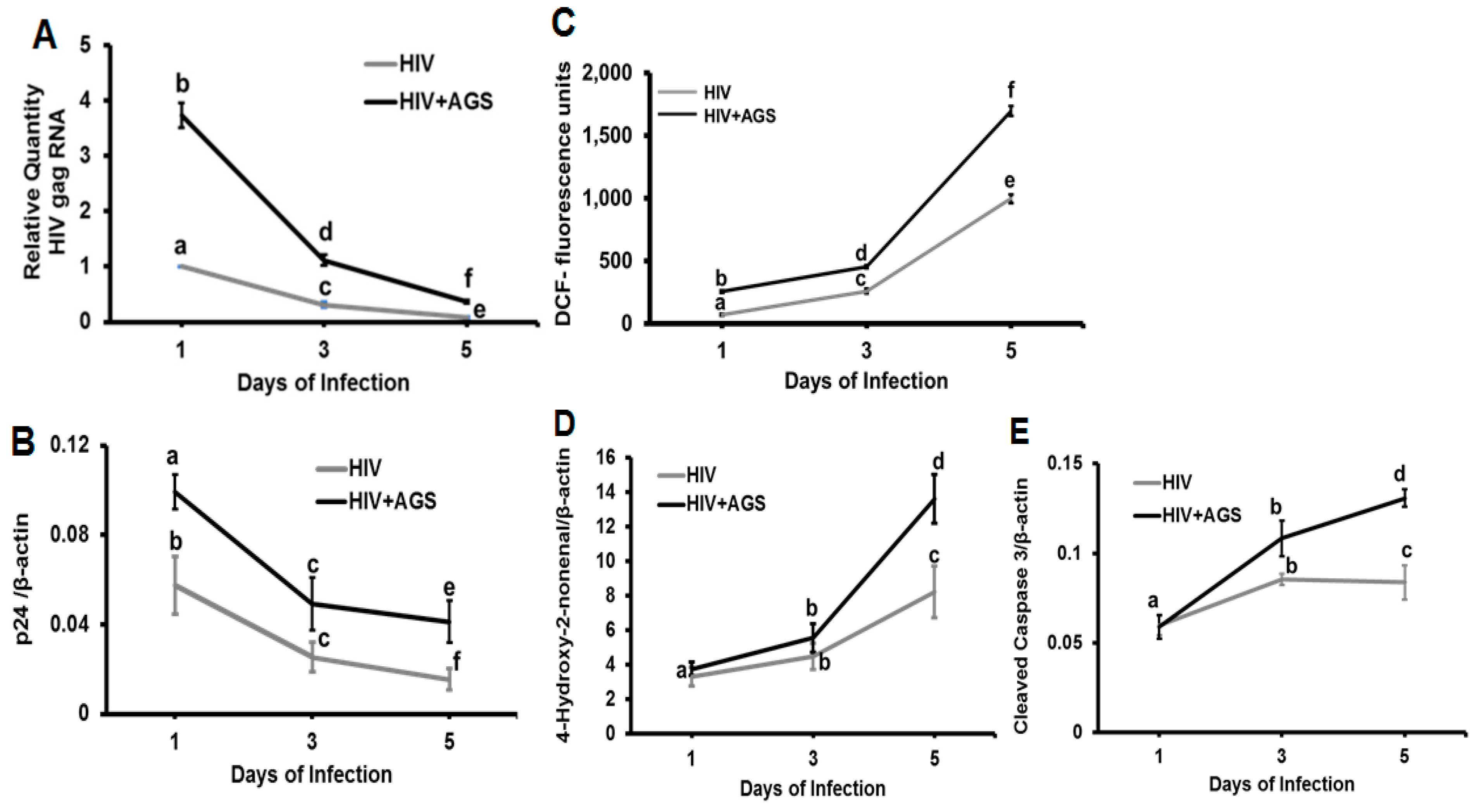
3.1. Ethanol Promotes Apoptosis in HIV-Infected Hepatocytes
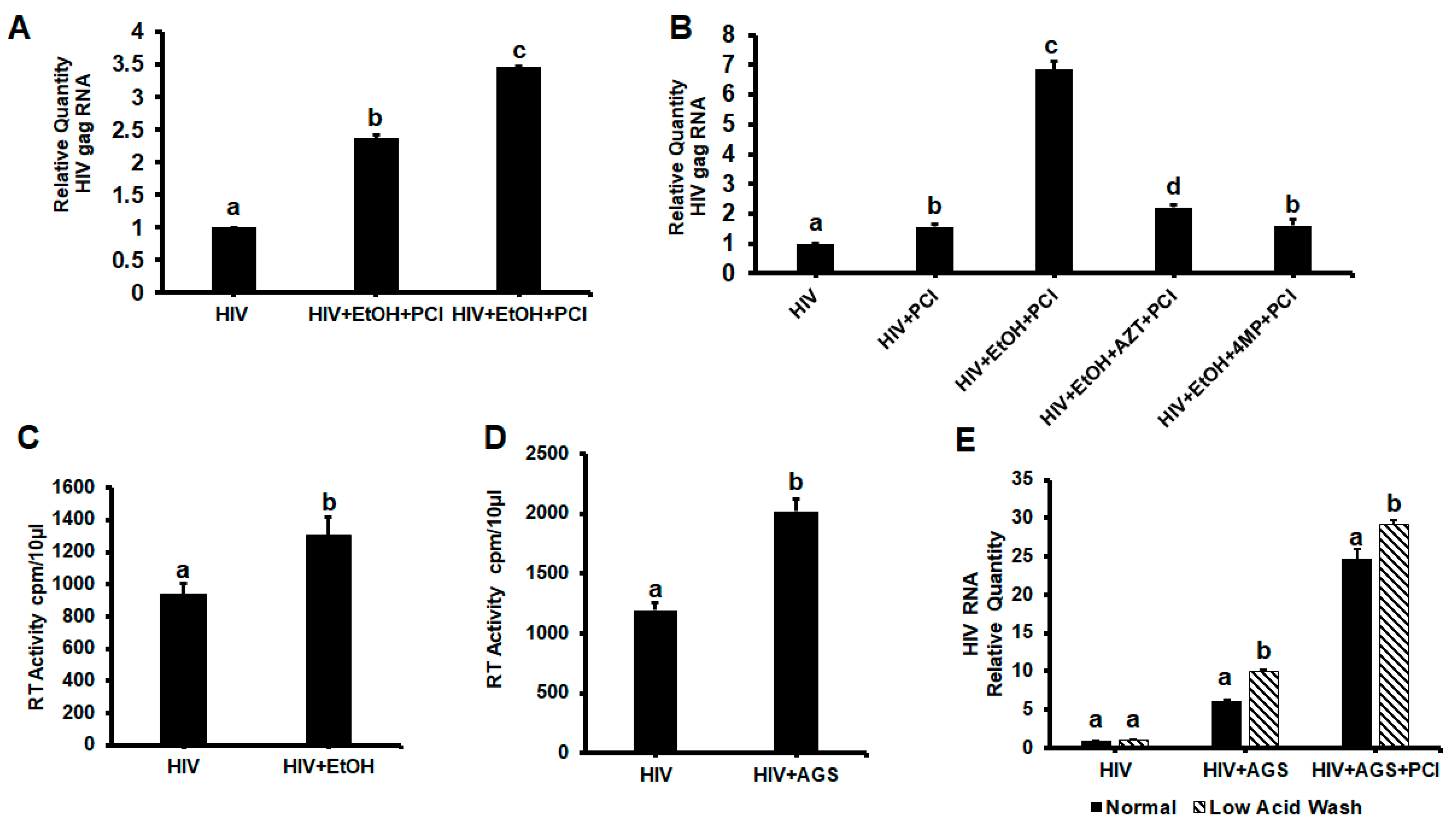
3.2. HIVgag Expression in Hepatocytes Exposed to Ethanol/AGS
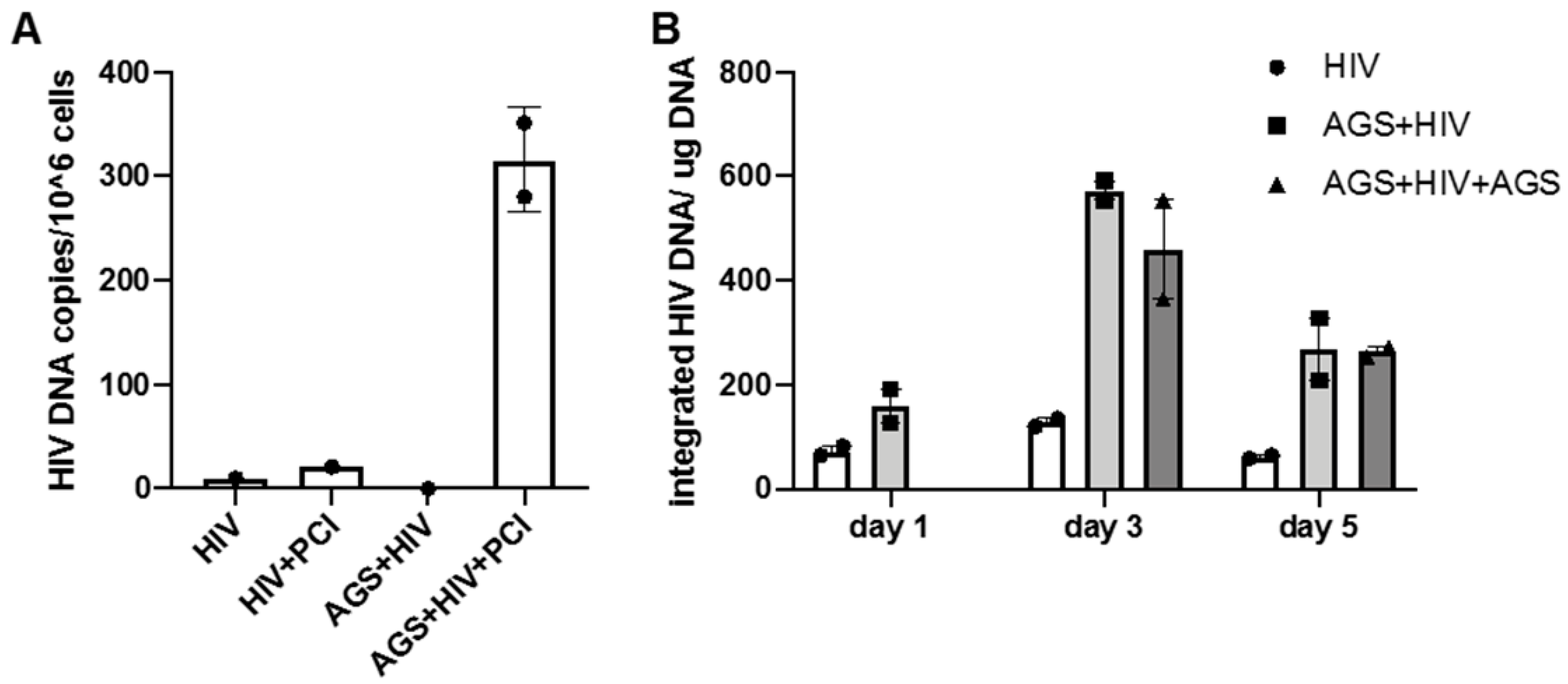
3.3. Effect of AGS on HIV DNA Levels in RLW Cells
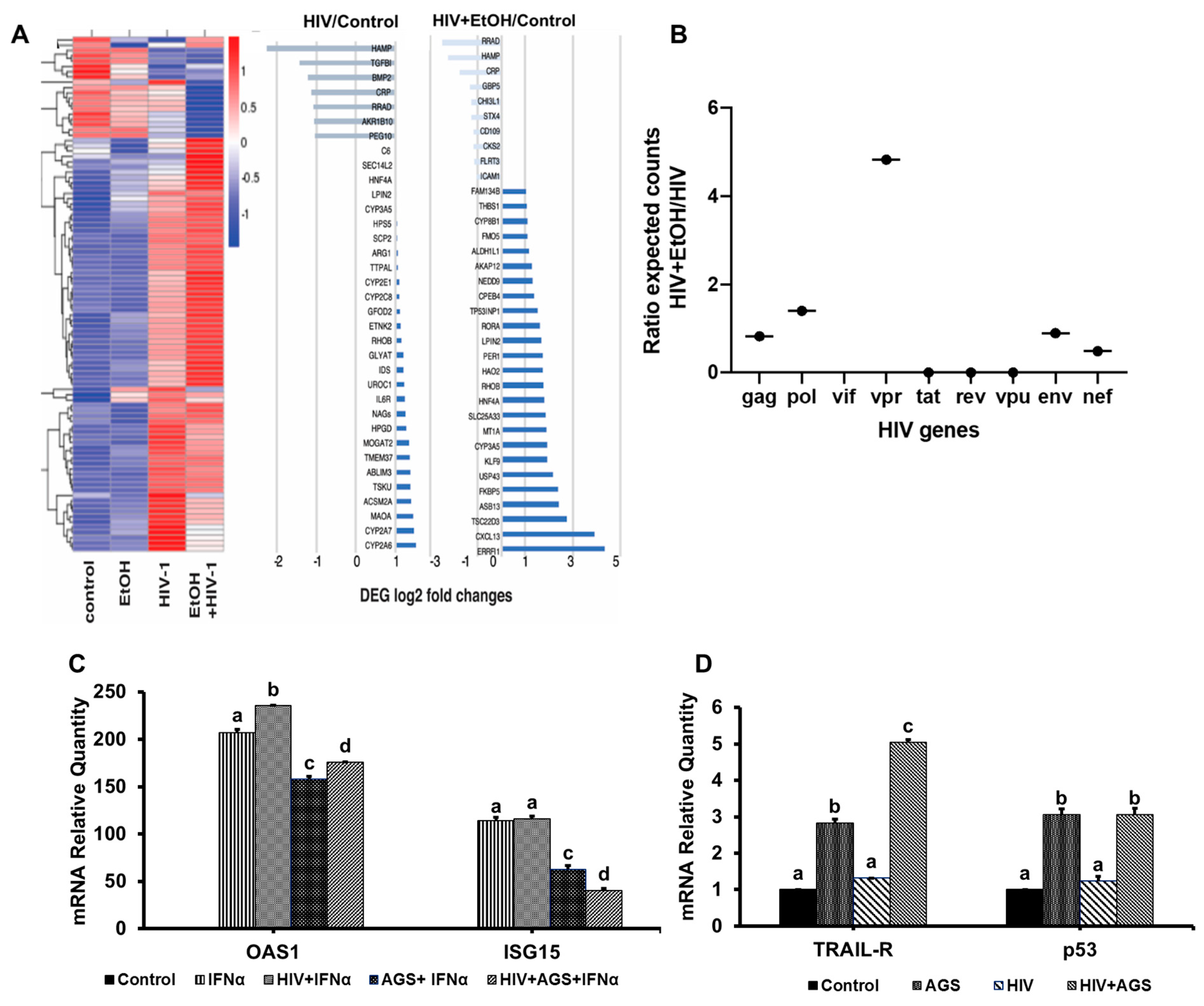
3.4. Kinetics of HIV Markers in RLW Cells in Response to AGS Treatment
3.5. Possible Mechanisms of Up-Regulation of HIV Expression by Ethanol Metabolites
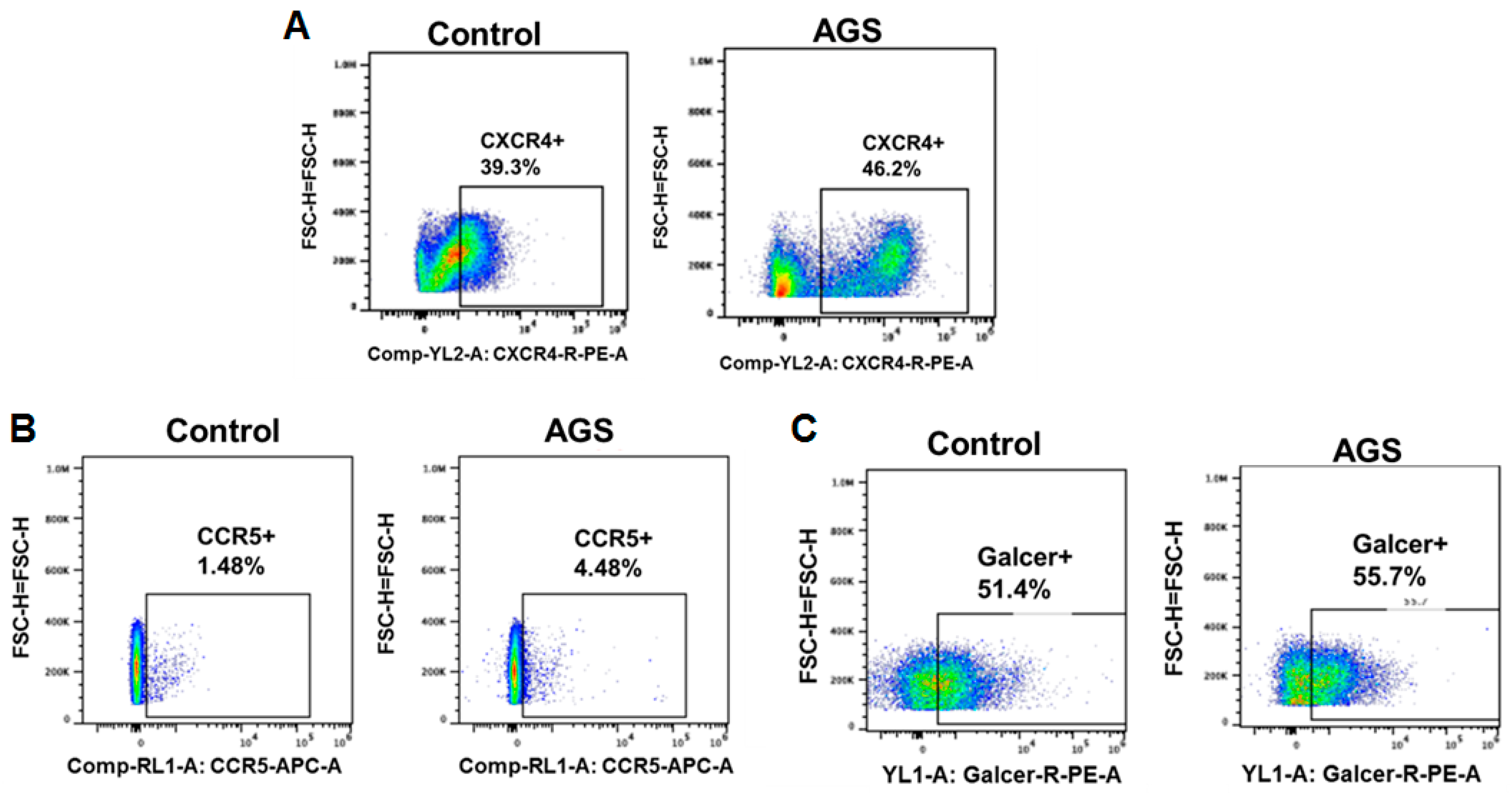
3.5.1. Receptors for Viral Entry
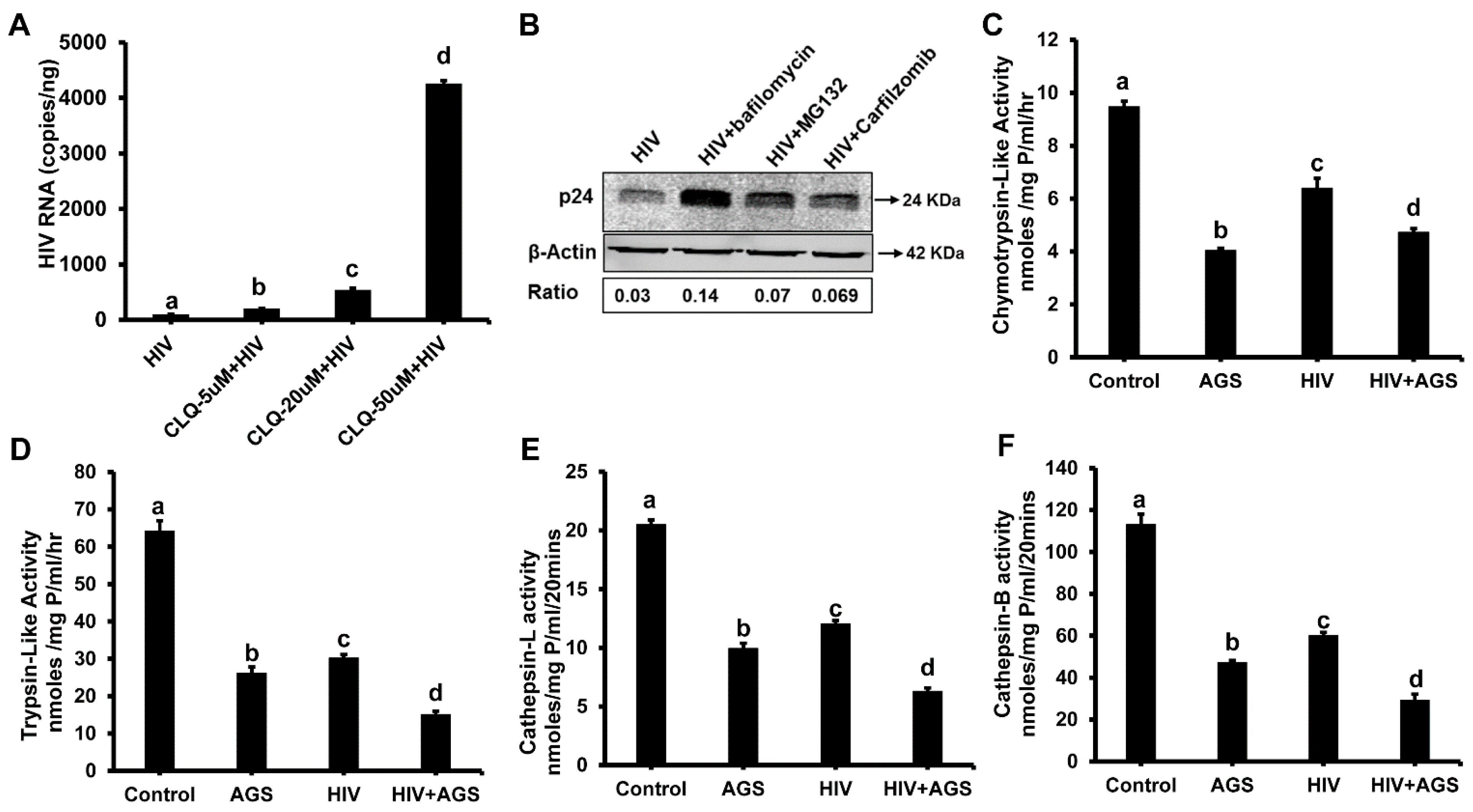
3.5.2. HIV-Accumulation in Hepatocytes Due to AGS-Suppressed Degradation
3.6. Ethanol Metabolism-Induced Gene Activation in HIV-Infected Hepatocytes
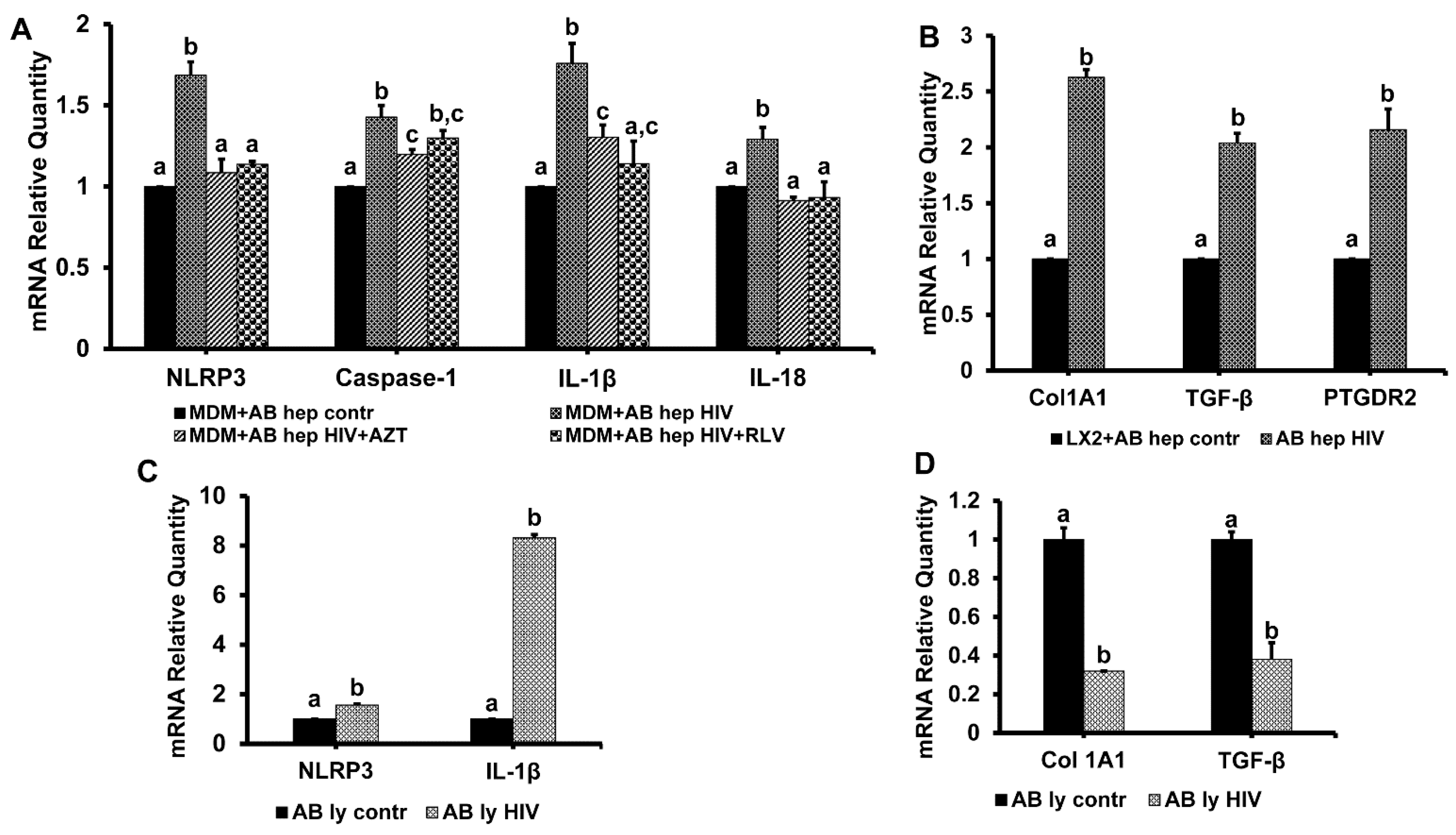
3.7. Hepatocyte-Derived Apoptotic Bodies (ABHep) and HIV Infection
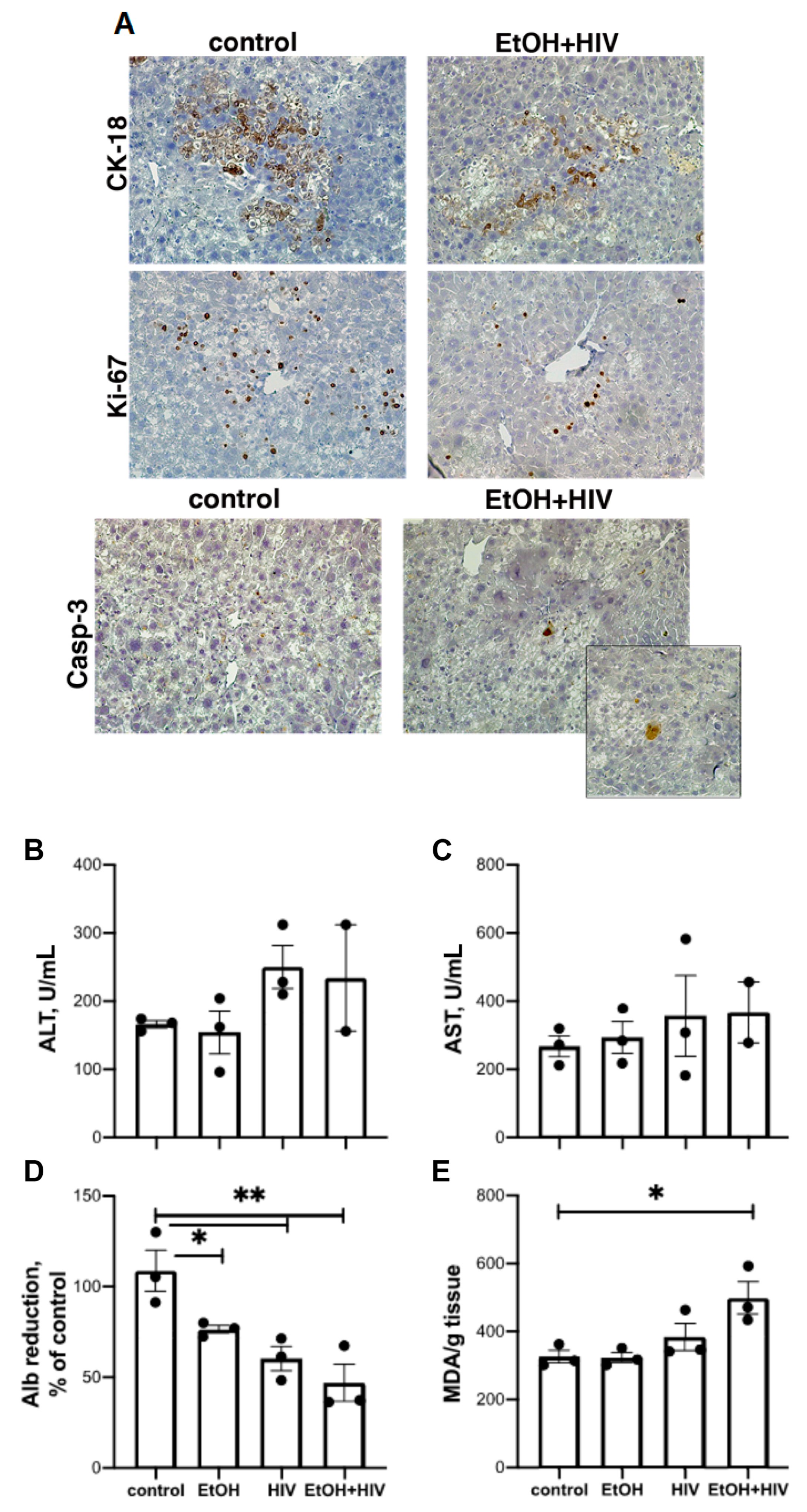
3.8. In Vivo Effects of HIV and Ethanol on Human Hepatocytes
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Debes, J.D.; Bohjanen, P.R.; Boonstra, A. Mechanisms of accelerated liver fibrosis progression during hiv infection. J. Clin. Transl. Hepatol. 2016, 4, 328–335. [Google Scholar] [PubMed]
- Pascual-Pareja, J.F.; Caminoa, A.; Larrauri, C.; Gonzalez-Garcia, J.; Montes, M.L.; Diez, J.; Grande, M.; Arribas, J.R. Haart is associated with lower hepatic necroinflammatory activity in hiv-hepatitis c virus-coinfected patients with cd4 cell count of more than 350 cells/microl at the time of liver biopsy. AIDS 2009, 23, 971–975. [Google Scholar] [CrossRef] [PubMed]
- Mata-Marin, J.A.; Gaytan-Martinez, J.; Grados-Chavarria, B.H.; Fuentes-Allen, J.L.; Arroyo-Anduiza, C.I.; Alfaro-Mejia, A. Correlation between hiv viral load and aminotransferases as liver damage markers in hiv infected naive patients: A concordance cross-sectional study. Virol. J. 2009, 6, 181. [Google Scholar] [CrossRef] [PubMed]
- Penton, P.K.; Blackard, J.T. Analysis of hiv quasispecies suggests compartmentalization in the liver. AIDS Res. Hum. Retrovir. 2014, 30, 394–402. [Google Scholar] [CrossRef] [PubMed]
- Sterling, R.K.; Chiu, S.; Snider, K.; Nixon, D. The prevalence and risk factors for abnormal liver enzymes in hiv-positive patients without hepatitis B or C coinfections. Dig. Dis. Sci. 2008, 53, 1375–1382. [Google Scholar] [CrossRef]
- Kooij, K.W.; Wit, F.W.; van Zoest, R.A.; Schouten, J.; Kootstra, N.A.; van Vugt, M.; Prins, M.; Reiss, P.; van der Valk, M.; Group, A.G.C.S. Liver fibrosis in hiv-infected individuals on long-term antiretroviral therapy: Associated with immune activation, immunodeficiency and prior use of didanosine. AIDS 2016, 30, 1771–1780. [Google Scholar] [CrossRef]
- Kong, L.; Cardona Maya, W.; Moreno-Fernandez, M.E.; Ma, G.; Shata, M.T.; Sherman, K.E.; Chougnet, C.; Blackard, J.T. Low-level hiv infection of hepatocytes. Virol. J. 2012, 9, 157. [Google Scholar] [CrossRef]
- Pandrea, I.; Happel, K.I.; Amedee, A.M.; Bagby, G.J.; Nelson, S. Alcohol’s role in hiv transmission and disease progression. Alcohol. Res. Health 2010, 33, 203–218. [Google Scholar]
- Koziel, M.J.; Peters, M.G. Viral hepatitis in hiv infection. N. Engl. J. Med. 2007, 356, 1445–1454. [Google Scholar] [CrossRef]
- Barve, S.; Kapoor, R.; Moghe, A.; Ramirez, J.A.; Eaton, J.W.; Gobejishvili, L.; Joshi-Barve, S.; McClain, C.J. Focus on the liver: Alcohol use, highly active antiretroviral therapy, and liver disease in hiv-infected patients. Alcohol. Res. Health 2010, 33, 229–236. [Google Scholar]
- Chaudhry, A.A.; Sulkowski, M.S.; Chander, G.; Moore, R.D. Hazardous drinking is associated with an elevated aspartate aminotransferase to platelet ratio index in an urban hiv-infected clinical cohort. HIV Med. 2009, 10, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Wada, N.I.; Jacobson, L.P.; Margolick, J.B.; Breen, E.C.; Macatangay, B.; Penugonda, S.; Martinez-Maza, O.; Bream, J.H. The effect of haart-induced hiv suppression on circulating markers of inflammation and immune activation. AIDS 2015, 29, 463–471. [Google Scholar] [CrossRef] [PubMed]
- Schnabl, B.; Brenner, D.A. Interactions between the intestinal microbiome and liver diseases. Gastroenterology 2014, 146, 1513–1524. [Google Scholar] [CrossRef] [PubMed]
- Bansal, M.B.; Blackard, J.T. Effects of hiv on liver cell populations. In Hiv and Liver Disease; Sherman, K.E., Ed.; Springer: Berlin, Germany, 2012; pp. 81–90. [Google Scholar]
- Cao, Y.Z.; Dieterich, D.; Thomas, P.A.; Huang, Y.X.; Mirabile, M.; Ho, D.D. Identification and quantitation of hiv-1 in the liver of patients with aids. AIDS 1992, 6, 65–70. [Google Scholar] [CrossRef]
- Lin, W.; Weinberg, E.M.; Tai, A.W.; Peng, L.F.; Brockman, M.A.; Kim, K.A.; Kim, S.S.; Borges, C.B.; Shao, R.X.; Chung, R.T. Hiv increases hcv replication in a tgf-beta1-dependent manner. Gastroenterology 2008, 134, 803–811. [Google Scholar] [CrossRef]
- Nunez, M. Hepatotoxicity of antiretrovirals: Incidence, mechanisms and management. J. Hepatol. 2006, 44, S132–S139. [Google Scholar] [CrossRef]
- Qin, F.; Jiang, J.; Qin, C.; Huang, Y.; Liang, B.; Xu, Y.; Huang, J.; Xu, Z.; Ning, C.; Liao, Y.; et al. Liver damage in patients living with hiv on antiretroviral treatment with normal baseline liver function and without hbv/hcv infection: An 11-year retrospective cohort study in Guangxi, China. BMJ Open 2019, 9, e023140. [Google Scholar] [CrossRef]
- Cummins, N.W.; Badley, A.D. Mechanisms of hiv-associated lymphocyte apoptosis: 2010. Cell Death Dis. 2010, 1, e99. [Google Scholar] [CrossRef]
- Finkel, T.H.; Tudor-Williams, G.; Banda, N.K.; Cotton, M.F.; Curiel, T.; Monks, C.; Baba, T.W.; Ruprecht, R.M.; Kupfer, A. Apoptosis occurs predominantly in bystander cells and not in productively infected cells of hiv- and siv-infected lymph nodes. Nat. Med. 1995, 1, 129–134. [Google Scholar] [CrossRef]
- Gorantla, S.; Che, M.; Gendelman, H.E. Isolation, propagation, and hiv-1 infection of monocyte-derived macrophages and recovery of virus from brain and cerebrospinal fluid. Methods Mol. Biol. 2005, 304, 35–48. [Google Scholar]
- Godoy, P.; Hewitt, N.J.; Albrecht, U.; Andersen, M.E.; Ansari, N.; Bhattacharya, S.; Bode, J.G.; Bolleyn, J.; Borner, C.; Bottger, J.; et al. Recent advances in 2d and 3d in vitro systems using primary hepatocytes, alternative hepatocyte sources and non-parenchymal liver cells and their use in investigating mechanisms of hepatotoxicity, cell signaling and adme. Arch. Toxicol. 2013, 87, 1315–1530. [Google Scholar] [PubMed]
- Ganesan, M.; Dagur, R.S.; Makarov, E.; Poluektova, L.I.; Kidambi, S.; Osna, N.A. Matrix stiffness regulate apoptotic cell death in hiv-hcv co-infected hepatocytes: Importance for liver fibrosis progression. Biochem. Biophys. Res. Commun. 2018, 500, 717–722. [Google Scholar] [CrossRef] [PubMed]
- Ganesan, M.; Zhang, J.; Bronich, T.; Poluektova, L.I.; Donohue, T.M., Jr.; Tuma, D.J.; Kharbanda, K.K.; Osna, N.A. Acetaldehyde accelerates hcv-induced impairment of innate immunity by suppressing methylation reactions in liver cells. Am. J. Physiol. Gastrointest Liver Physiol. 2015, 309, G566–G577. [Google Scholar] [CrossRef] [PubMed]
- Ganesan, M.; Natarajan, S.K.; Zhang, J.; Mott, J.L.; Poluektova, L.I.; McVicker, B.L.; Kharbanda, K.K.; Tuma, D.J.; Osna, N.A. Role of apoptotic hepatocytes in hcv dissemination: Regulation by acetaldehyde. Am. J. Physiol. Gastrointest Liver Physiol. 2016, 310, G930–G940. [Google Scholar] [CrossRef]
- Ganesan, M.; Poluektova, L.Y.; Enweluzo, C.; Kharbanda, K.K.; Osna, N.A. Hepatitis c virus-infected apoptotic hepatocytes program macrophages and hepatic stellate cells for liver inflammation and fibrosis development: Role of ethanol as a second hit. Biomolecules 2018, 8, 113. [Google Scholar] [CrossRef]
- Ganesan, M.; Tikhanovich, I.; Vangimalla, S.S.; Dagur, R.S.; Wang, W.; Poluektova, L.I.; Sun, Y.; Mercer, D.F.; Tuma, D.; Weinman, S.A.; et al. Demethylase jmjd6 as a new regulator of interferon signaling: Effects of hcv and ethanol metabolism. Cell. Mol. Gastroenterol. Hepatol. 2018, 5, 101–112. [Google Scholar] [CrossRef]
- Gendelman, H.E.; Orenstein, J.M.; Martin, M.A.; Ferrua, C.; Mitra, R.; Phipps, T.; Wahl, L.A.; Lane, H.C.; Fauci, A.S.; Burke, D.S.; et al. Efficient isolation and propagation of human immunodeficiency virus on recombinant colony-stimulating factor 1-treated monocytes. J. Exp. Med. 1988, 167, 1428–1441. [Google Scholar] [CrossRef]
- Dagur, R.S.; Wang, W.; Cheng, Y.; Makarov, E.; Ganesan, M.; Suemizu, H.; Gebhart, C.L.; Gorantla, S.; Osna, N.; Poluektova, L.Y. Human hepatocytes depletion in the presence of hiv-1 infection in dual reconstituted humanized mice. Biol. Open 2018, 7, bio029785. [Google Scholar] [CrossRef]
- Dagur, R.S.; Branch-Woods, A.; Mathews, S.; Joshi, P.S.; Quadros, R.M.; Harms, D.W.; Cheng, Y.; Miles, S.M.; Pirruccello, S.J.; Gurumurthy, C.B.; et al. Human-like nsg mouse glycoproteins sialylation pattern changes the phenotype of human lymphocytes and sensitivity to hiv-1 infection. BMC Immunol. 2019, 20, 2. [Google Scholar] [CrossRef]
- Ganesan, M.; Krutik, V.M.; Makarov, E.; Mathews, S.; Kharbanda, K.K.; Poluektova, L.Y.; Casey, C.A.; Osna, N.A. Acetaldehyde suppresses the display of hbv-mhc class i complexes on hbv-expressing hepatocytes. Am. J. Physiol. Gastrointest Liver Physiol. 2019, 317, G127–G140. [Google Scholar] [CrossRef]
- Thomes, P.G.; Ehlers, R.A.; Trambly, C.S.; Clemens, D.L.; Fox, H.S.; Tuma, D.J.; Donohue, T.M. Multilevel regulation of autophagosome content by ethanol oxidation in hepg2 cells. Autophagy 2013, 9, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Bertola, A.; Mathews, S.; Ki, S.H.; Wang, H.; Gao, B. Mouse model of chronic and binge ethanol feeding (the niaaa model). Nat. Protoc. 2013, 8, 627–637. [Google Scholar] [CrossRef] [PubMed]
- Kameyama, S.; Horie, M.; Kikuchi, T.; Omura, T.; Tadokoro, A.; Takeuchi, T.; Nakase, I.; Sugiura, Y.; Futaki, S. Acid wash in determining cellular uptake of fab/cell-permeating peptide conjugates. Biopolymers 2007, 88, 98–107. [Google Scholar] [CrossRef] [PubMed]
- Xiao, P.; Usami, O.; Suzuki, Y.; Ling, H.; Shimizu, N.; Hoshino, H.; Zhuang, M.; Ashino, Y.; Gu, H.; Hattori, T. Characterization of a cd4-independent clinical hiv-1 that can efficiently infect human hepatocytes through chemokine (c-x-c motif) receptor 4. AIDS 2008, 22, 1749–1757. [Google Scholar] [CrossRef] [PubMed]
- Donohue, T.M., Jr.; Osna, N.A. Intracellular proteolytic systems in alcohol-induced tissue injury. Alcohol. Res. Health 2003, 27, 317–324. [Google Scholar]
- Jiang, J.X.; Mikami, K.; Venugopal, S.; Li, Y.; Torok, N.J. Apoptotic body engulfment by hepatic stellate cells promotes their survival by the jak/stat and akt/nf-kappab-dependent pathways. J. Hepatol. 2009, 51, 139–148. [Google Scholar] [CrossRef]
- Zhang, L.; Dailey, P.J.; Gettie, A.; Blanchard, J.; Ho, D.D. The liver is a major organ for clearing simian immunodeficiency virus in rhesus monkeys. J. Virol. 2002, 76, 5271–5273. [Google Scholar] [CrossRef]
- Hu, S.; Ghabril, M.; Amet, T.; Hu, N.; Byrd, D.; Yang, K.; Vuppalanchi, R.; Saxena, R.; Desai, M.; Lan, J.; et al. Hiv-1 coinfection profoundly alters intrahepatic chemokine but not inflammatory cytokine profiles in hcv-infected subjects. PLoS ONE 2014, 9, e86964. [Google Scholar] [CrossRef]
- Babu, C.K.; Suwansrinon, K.; Bren, G.D.; Badley, A.D.; Rizza, S.A. Hiv induces trail sensitivity in hepatocytes. PLoS ONE 2009, 4, e4623. [Google Scholar] [CrossRef]
- Vlahakis, S.R.; Villasis-Keever, A.; Gomez, T.S.; Bren, G.D.; Paya, C.V. Human immunodeficiency virus-induced apoptosis of human hepatocytes via cxcr4. J. Infect. Dis. 2003, 188, 1455–1460. [Google Scholar] [CrossRef]
- Balasubramanian, A.; Ganju, R.K.; Groopman, J.E. Signal transducer and activator of transcription factor 1 mediates apoptosis induced by hepatitis c virus and hiv envelope proteins in hepatocytes. J. Infect. Dis 2006, 194, 670–681. [Google Scholar] [CrossRef] [PubMed]
- Fromentin, R.; Tardif, M.R.; Tremblay, M.J. Inefficient fusion due to a lack of attachment receptor/co-receptor restricts productive human immunodeficiency virus type 1 infection in human hepatoma huh7.5 cells. J. Gen. Virol 2011, 92, 587–597. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Ao, Z.; Chen, L.; Kobinger, G.; Peng, J.; Yao, X. The cellular antiviral protein apobec3g interacts with hiv-1 reverse transcriptase and inhibits its function during viral replication. J. Virol. 2012, 86, 3777–3786. [Google Scholar] [CrossRef] [PubMed]
- Park, I.W.; Fan, Y.; Luo, X.; Ryou, M.G.; Liu, J.; Green, L.; He, J.J. Hiv-1 nef is transferred from expressing t cells to hepatocytic cells through conduits and enhances hcv replication. PLoS ONE 2014, 9, e99545. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Picado, J.; Zurakowski, R.; Buzon, M.J.; Stevenson, M. Episomal hiv-1 DNA and its relationship to other markers of hiv-1 persistence. Retrovirology 2018, 15, 15. [Google Scholar] [CrossRef] [PubMed]
- Kolchinsky, P.; Mirzabekov, T.; Farzan, M.; Kiprilov, E.; Cayabyab, M.; Mooney, L.J.; Choe, H.; Sodroski, J. Adaptation of a ccr5-using, primary human immunodeficiency virus type 1 isolate for cd4-independent replication. J. Virol. 1999, 73, 8120–8126. [Google Scholar]
- Westervelt, P.; Gendelman, H.E.; Ratner, L. Identification of a determinant within the human immunodeficiency virus 1 surface envelope glycoprotein critical for productive infection of primary monocytes. Proc. Natl. Acad. Sci. USA 1991, 88, 3097–3101. [Google Scholar] [CrossRef]
- Chao, X.; Wang, S.; Zhao, K.; Li, Y.; Williams, J.A.; Li, T.; Chavan, H.; Krishnamurthy, P.; He, X.C.; Li, L.; et al. Impaired tfeb-mediated lysosome biogenesis and autophagy promote chronic ethanol-induced liver injury and steatosis in mice. Gastroenterology 2018, 155, 865–879. [Google Scholar] [CrossRef]
- Osna, N.A.; Donohue, T.M., Jr. Cyp2e1-catalyzed alcohol metabolism: Role of oxidant generation in interferon signaling, antigen presentation and autophagy. In Cytochrome P450 2E1: Its Role in Disease and Drug Metabolism; Springer: Dordrecht, The Netherlands, 2013; Volume 67, pp. 177–197. [Google Scholar]










© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ganesan, M.; New-Aaron, M.; Dagur, R.S.; Makarov, E.; Wang, W.; Kharbanda, K.K.; Kidambi, S.; Poluektova, L.Y.; Osna, N.A. Alcohol Metabolism Potentiates HIV-Induced Hepatotoxicity: Contribution to End-Stage Liver Disease. Biomolecules 2019, 9, 851. https://doi.org/10.3390/biom9120851
Ganesan M, New-Aaron M, Dagur RS, Makarov E, Wang W, Kharbanda KK, Kidambi S, Poluektova LY, Osna NA. Alcohol Metabolism Potentiates HIV-Induced Hepatotoxicity: Contribution to End-Stage Liver Disease. Biomolecules. 2019; 9(12):851. https://doi.org/10.3390/biom9120851
Chicago/Turabian StyleGanesan, Murali, Moses New-Aaron, Raghubendra Singh Dagur, Edward Makarov, Weimin Wang, Kusum K. Kharbanda, Srivatsan Kidambi, Larisa Y. Poluektova, and Natalia A. Osna. 2019. "Alcohol Metabolism Potentiates HIV-Induced Hepatotoxicity: Contribution to End-Stage Liver Disease" Biomolecules 9, no. 12: 851. https://doi.org/10.3390/biom9120851
APA StyleGanesan, M., New-Aaron, M., Dagur, R. S., Makarov, E., Wang, W., Kharbanda, K. K., Kidambi, S., Poluektova, L. Y., & Osna, N. A. (2019). Alcohol Metabolism Potentiates HIV-Induced Hepatotoxicity: Contribution to End-Stage Liver Disease. Biomolecules, 9(12), 851. https://doi.org/10.3390/biom9120851