Modulatory Mechanisms of the NLRP3 Inflammasomes in Diabetes



Abstract
1. Introduction
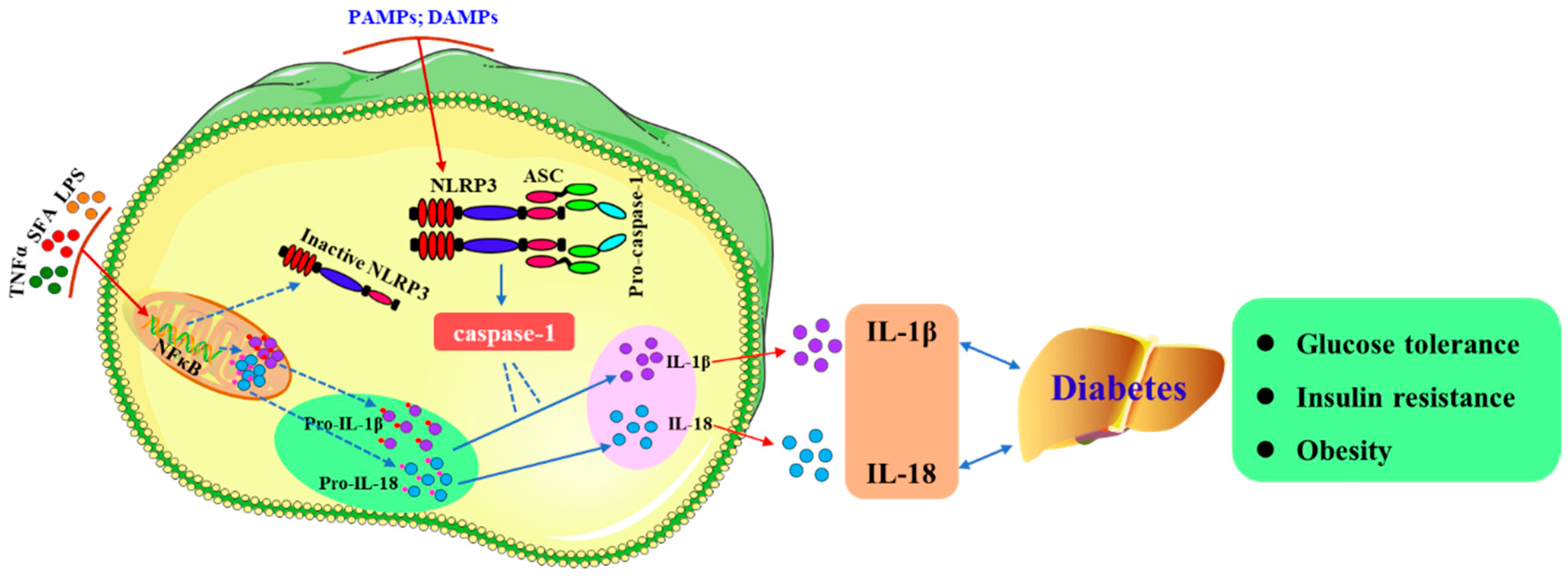
2. The NLRP3 Inflammasome
3. Metabolic Disease and Diabetes
4. NLRP3 and Diabetes
4.1. Development of Type 1 Diabetes Mellitus (T1DM) Is Regulated by NLRP3
4.2. The Development of Type 2 Diabetes Mellitus (T2DM) Is Regulated by NLRP3
5. NLRP3 and Adipose Tissue
6. NLRP3 and the Microbiome
7. Conclusions and Future Perspective
Author Contributions
Funding
Conflicts of Interest
References
- Cho, N.H.; Shaw, J.E.; Karuranga, S.; Huang, Y.; da Rocha Fernandes, J.D.; Ohlrogge, A.W.; Malanda, B. IDF Diabetes Atlas: Global estimates of diabetes prevalence for 2017 and projections for 2045. Diabetes Res. Clin. Pract. 2018, 138, 271–281. [Google Scholar] [CrossRef] [PubMed]
- Ashcroft, F.M.; Rorsman, P. Diabetes mellitus and the beta cell: The last ten years. Cell 2012, 148, 1160–1171. [Google Scholar] [CrossRef] [PubMed]
- Esser, N.; Legrand-Poels, S.; Piette, J.; Scheen, A.J.; Paquot, N. Inflammation as a link between obesity, metabolic syndrome and type 2 diabetes. Diabetes Res. Clin. Pract. 2014, 105, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Donath, M.Y.; Shoelson, S.E. Type 2 diabetes as an inflammatory disease. Nat. Rev. Immunol. 2011, 11, 98–107. [Google Scholar] [CrossRef] [PubMed]
- Mannering, S.I.; Di Carluccio, A.R.; Elso, C.M. Neoepitopes: A new take on beta cell autoimmunity in type 1 diabetes. Diabetologia 2019, 62, 351–356. [Google Scholar] [CrossRef] [PubMed]
- Nicolucci, A.; Maione, A.; Franciosi, M.; Amoretti, R.; Busetto, E.; Capani, F.; Bruttomesso, D.; Di Bartolo, P.; Girelli, A.; Leonetti, F.; et al. Quality of life and treatment satisfaction in adults with Type 1 diabetes: A comparison between continuous subcutaneous insulin infusion and multiple daily injections. Diabet. Med. 2008, 25, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Sharma, D.; Kanneganti, T.D. The cell biology of inflammasomes: Mechanisms of inflammasome activation and regulation. J. Cell Biol. 2016, 213, 617–629. [Google Scholar] [CrossRef]
- Hong, P.; Gu, R.N.; Li, F.X.; Xiong, X.X.; Liang, W.B.; You, Z.J.; Zhang, H.F. NLRP3 inflammasome as a potential treatment in ischemic stroke concomitant with diabetes. J. Neuroinflamm. 2019, 16, 121. [Google Scholar] [CrossRef]
- Hoseini, Z.; Sepahvand, F.; Rashidi, B.; Sahebkar, A.; Masoudifar, A.; Mirzaei, H. NLRP3 inflammasome: Its regulation and involvement in atherosclerosis. J. Cell. Physiol. 2018, 233, 2116–2132. [Google Scholar] [CrossRef]
- Bauernfeind, F.G.; Horvath, G.; Stutz, A.; Alnemri, E.S.; MacDonald, K.; Speert, D.; Fernandes-Alnemri, T.; Wu, J.; Monks, B.G.; Fitzgerald, K.A.; et al. Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J. Immunol. 2009, 183, 787–791. [Google Scholar] [CrossRef]
- Yang, Y.; Wang, H.; Kouadir, M.; Song, H.; Shi, F. Recent advances in the mechanisms of NLRP3 inflammasome activation and its inhibitors. Cell Death Dis. 2019, 10, 128. [Google Scholar] [CrossRef] [PubMed]
- Netea, M.G.; Nold-Petry, C.A.; Nold, M.F.; Joosten, L.A.; Opitz, B.; van der Meer, J.H.; van de Veerdonk, F.L.; Ferwerda, G.; Heinhuis, B.; Devesa, I.; et al. Differential requirement for the activation of the inflammasome for processing and release of IL-1beta in monocytes and macrophages. Blood 2009, 113, 2324–2335. [Google Scholar] [CrossRef] [PubMed]
- Gaidt, M.M.; Ebert, T.S.; Chauhan, D.; Schmidt, T.; Schmid-Burgk, J.L.; Rapino, F.; Robertson, A.A.; Cooper, M.A.; Graf, T.; Hornung, V. Human Monocytes Engage an Alternative Inflammasome Pathway. Immunity 2016, 44, 833–846. [Google Scholar] [CrossRef] [PubMed]
- Han, J.H.; Shin, H.; Rho, J.G.; Kim, J.E.; Son, D.H.; Yoon, J.; Lee, Y.J.; Park, J.H.; Song, B.J.; Choi, C.S.; et al. Peripheral cannabinoid 1 receptor blockade mitigates adipose tissue inflammation via NLRP3 inflammasome in mouse models of obesity. Diabetes Obes. Metab. 2018, 20, 2179–2189. [Google Scholar] [CrossRef]
- Wu, D.; Yan, Z.B.; Cheng, Y.G.; Zhong, M.W.; Liu, S.Z.; Zhang, G.Y.; Hu, S.Y. Deactivation of the NLRP3 inflammasome in infiltrating macrophages by duodenal-jejunal bypass surgery mediates improvement of beta cell function in type 2 diabetes. Metab. Clin. Exp. 2018, 81, 1–12. [Google Scholar] [CrossRef]
- Hu, C.; Ding, H.; Li, Y.; Pearson, J.A.; Zhang, X.; Flavell, R.A.; Wong, F.S.; Wen, L. NLRP3 deficiency protects from type 1 diabetes through the regulation of chemotaxis into the pancreatic islets. Proc. Natl. Acad. Sci. USA 2015, 112, 11318–11323. [Google Scholar] [CrossRef]
- Mathews, R.J.; Robinson, J.I.; Battellino, M.; Wong, C.; Taylor, J.C.; Eyre, S.; Churchman, S.M.; Wilson, A.G.; Isaacs, J.D.; Hyrich, K.; et al. Evidence of NLRP3-inflammasome activation in rheumatoid arthritis (RA); genetic variants within the NLRP3-inflammasome complex in relation to susceptibility to RA and response to anti-TNF treatment. Ann. Rheum. Dis. 2014, 73, 1202–1210. [Google Scholar] [CrossRef]
- Walle, L.V.; Van Opdenbosch, N.; Jacques, P.; Fossoul, A.; Verheugen, E.; Vogel, P.; Beyaert, R.; Elewaut, D.; Kanneganti, T.D.; van Loo, G.; et al. Negative regulation of the NLRP3 inflammasome by A20 protects against arthritis. Nature 2014, 512, 69–73. [Google Scholar] [CrossRef]
- Wang, B.R.; Shi, J.Q.; Ge, N.N.; Ou, Z.; Tian, Y.Y.; Jiang, T.; Zhou, J.S.; Xu, J.; Zhang, Y.D. PM2.5 exposure aggravates oligomeric amyloid beta-induced neuronal injury and promotes NLRP3 inflammasome activation in an in vitro model of Alzheimer’s disease. J. Neuroinflamm. 2018, 15, 132. [Google Scholar] [CrossRef]
- Szabo, G.; Petrasek, J. Inflammasome activation and function in liver disease. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 387–400. [Google Scholar] [CrossRef]
- Savage, C.D.; Lopez-Castejon, G.; Denes, A.; Brough, D. NLRP3-Inflammasome Activating DAMPs Stimulate an Inflammatory Response in Glia in the Absence of Priming Which Contributes to Brain Inflammation after Injury. Front. Immunol. 2012, 3, 288. [Google Scholar] [CrossRef] [PubMed]
- Munoz-Planillo, R.; Kuffa, P.; Martinez-Colon, G.; Smith, B.L.; Rajendiran, T.M.; Nunez, G. K(+) efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity 2013, 38, 1142–1153. [Google Scholar] [CrossRef] [PubMed]
- Coll, R.C.; Robertson, A.A.; Chae, J.J.; Higgins, S.C.; Munoz-Planillo, R.; Inserra, M.C.; Vetter, I.; Dungan, L.S.; Monks, B.G.; Stutz, A.; et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat. Med. 2015, 21, 248–255. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Lu, Z.; Luo, Y.; Liu, Y.; Cao, Z.; Shen, S.; Li, H.; Liu, J.; Chen, K.; Chen, Z.; et al. Targeting of NLRP3 inflammasome with gene editing for the amelioration of inflammatory diseases. Nat. Commun. 2018, 9, 4092. [Google Scholar] [CrossRef] [PubMed]
- Tang, T.; Lang, X.; Xu, C.; Wang, X.; Gong, T.; Yang, Y.; Cui, J.; Bai, L.; Wang, J.; Jiang, W.; et al. CLICs-dependent chloride efflux is an essential and proximal upstream event for NLRP3 inflammasome activation. Nat. Commun. 2017, 8, 202. [Google Scholar] [CrossRef] [PubMed]
- Daniels, M.J.; Rivers-Auty, J.; Schilling, T.; Spencer, N.G.; Watremez, W.; Fasolino, V.; Booth, S.J.; White, C.S.; Baldwin, A.G.; Freeman, S.; et al. Fenamate NSAIDs inhibit the NLRP3 inflammasome and protect against Alzheimer’s disease in rodent models. Nat. Commun. 2016, 7, 12504. [Google Scholar] [CrossRef] [PubMed]
- Sorbara, M.T.; Girardin, S.E. Mitochondrial ROS fuel the inflammasome. Cell Res. 2011, 21, 558–560. [Google Scholar] [CrossRef] [PubMed]
- Hafner-Bratkovic, I.; Bencina, M.; Fitzgerald, K.A.; Golenbock, D.; Jerala, R. NLRP3 inflammasome activation in macrophage cell lines by prion protein fibrils as the source of IL-1beta and neuronal toxicity. Cell. Mol. Life Sci. 2012, 69, 4215–4228. [Google Scholar] [CrossRef]
- Dempsey, C.; Rubio Araiz, A.; Bryson, K.J.; Finucane, O.; Larkin, C.; Mills, E.L.; Robertson, A.A.B.; Cooper, M.A.; O’Neill, L.A.J.; Lynch, M.A. Inhibiting the NLRP3 inflammasome with MCC950 promotes non-phlogistic clearance of amyloid-beta and cognitive function in APP/PS1 mice. Brain Behav. Immun. 2017, 61, 306–316. [Google Scholar] [CrossRef]
- Kayagaki, N.; Stowe, I.B.; Lee, B.L.; O’Rourke, K.; Anderson, K.; Warming, S.; Cuellar, T.; Haley, B.; Roose-Girma, M.; Phung, Q.T.; et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 2015, 526, 666–671. [Google Scholar] [CrossRef]
- Baker, P.J.; Boucher, D.; Bierschenk, D.; Tebartz, C.; Whitney, P.G.; D’Silva, D.B.; Tanzer, M.C.; Monteleone, M.; Robertson, A.A.; Cooper, M.A.; et al. NLRP3 inflammasome activation downstream of cytoplasmic LPS recognition by both caspase-4 and caspase-5. Eur. J. Immunol. 2015, 45, 2918–2926. [Google Scholar] [CrossRef] [PubMed]
- Zu, Y.; Wan, L.J.; Cui, S.Y.; Gong, Y.P.; Li, C.L. The mitochondrial Na(+)/Ca(2+) exchanger may reduce high glucose-induced oxidative stress and nucleotide-binding oligomerization domain receptor 3 inflammasome activation in endothelial cells. J. Geriatr. Cardiol. JGC 2015, 12, 270–278. [Google Scholar] [CrossRef] [PubMed]
- Pillon, N.J.; Chan, K.L.; Zhang, S.; Mejdani, M.; Jacobson, M.R.; Ducos, A.; Bilan, P.J.; Niu, W.; Klip, A. Saturated fatty acids activate caspase-4/5 in human monocytes, triggering IL-1beta and IL-18 release. Am. J. Physiol. Endocrinol. Metab. 2016, 311, E825–E835. [Google Scholar] [CrossRef] [PubMed]
- Ralston, J.C.; Lyons, C.L.; Kennedy, E.B.; Kirwan, A.M.; Roche, H.M. Fatty Acids and NLRP3 Inflammasome-Mediated Inflammation in Metabolic Tissues. Annu. Rev. Nutr. 2017, 37, 77–102. [Google Scholar] [CrossRef]
- Ahima, R.S.; Lazar, M.A. The health risk of obesity--better metrics imperative. Science 2013, 341, 856–858. [Google Scholar] [CrossRef]
- Kopelman, P.G. Obesity as a medical problem. Nature 2000, 404, 635–643. [Google Scholar] [CrossRef]
- Khadka, R.; Tian, W.; Hao, X.; Koirala, R. Risk factor, early diagnosis and overall survival on outcome of association between pancreatic cancer and diabetes mellitus: Changes and advances, a review. Int. J. Surg. 2018, 52, 342–346. [Google Scholar] [CrossRef]
- Chari, S.T.; Leibson, C.L.; Rabe, K.G.; Ransom, J.; de Andrade, M.; Petersen, G.M. Probability of pancreatic cancer following diabetes: A population-based study. Gastroenterology 2005, 129, 504–511. [Google Scholar] [CrossRef]
- World Health Organization. Global Report on Diabetes; World Health Organization: Geneva, Switzerland, 2016; Volume 2016. [Google Scholar]
- Chu, Y.; Rosso, L.G.; Huang, P.; Wang, Z.; Xu, Y.; Yao, X.; Bao, M.; Yan, J.; Song, H.; Wang, G. Liver Med23 ablation improves glucose and lipid metabolism through modulating FOXO1 activity. Cell Res. 2014, 24, 1250–1265. [Google Scholar] [CrossRef]
- Bandyopadhyay, G.K.; Lu, M.; Avolio, E.; Siddiqui, J.A.; Gayen, J.R.; Wollam, J.; Vu, C.U.; Chi, N.W.; O’Connor, D.T.; Mahata, S.K. Pancreastatin-dependent inflammatory signaling mediates obesity-induced insulin resistance. Diabetes 2015, 64, 104–116. [Google Scholar] [CrossRef]
- Palomer, X.; Salvado, L.; Barroso, E.; Vazquez-Carrera, M. An overview of the crosstalk between inflammatory processes and metabolic dysregulation during diabetic cardiomyopathy. Int. J. Cardiol. 2013, 168, 3160–3172. [Google Scholar] [CrossRef] [PubMed]
- Burcelin, R. Gut microbiota and immune crosstalk in metabolic disease. Mol. Metab. 2016, 5, 771–781. [Google Scholar] [CrossRef] [PubMed]
- Rajilic-Stojanovic, M.; de Vos, W.M. The first 1000 cultured species of the human gastrointestinal microbiota. FEMS Microbiol. Rev. 2014, 38, 996–1047. [Google Scholar] [CrossRef] [PubMed]
- Chu, H.; Mazmanian, S.K. Innate immune recognition of the microbiota promotes host-microbial symbiosis. Nat. Immunol. 2013, 14, 668–675. [Google Scholar] [CrossRef] [PubMed]
- Bassols, J.; Ortega, F.J.; Moreno-Navarrete, J.M.; Peral, B.; Ricart, W.; Fernandez-Real, J.M. Study of the proinflammatory role of human differentiated omental adipocytes. J. Cell. Biochem. 2009, 107, 1107–1117. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, F.H.; Tremaroli, V.; Nookaew, I.; Bergstrom, G.; Behre, C.J.; Fagerberg, B.; Nielsen, J.; Backhed, F. Gut metagenome in European women with normal, impaired and diabetic glucose control. Nature 2013, 498, 99–103. [Google Scholar] [CrossRef]
- Hotamisligil, G.S.; Shargill, N.S.; Spiegelman, B.M. Adipose expression of tumor necrosis factor-alpha: Direct role in obesity-linked insulin resistance. Science 1993, 259, 87–91. [Google Scholar] [CrossRef]
- Denou, E.; Lolmede, K.; Garidou, L.; Pomie, C.; Chabo, C.; Lau, T.C.; Fullerton, M.D.; Nigro, G.; Zakaroff-Girard, A.; Luche, E.; et al. Defective NOD2 peptidoglycan sensing promotes diet-induced inflammation, dysbiosis, and insulin resistance. EMBO Mol. Med. 2015, 7, 259–274. [Google Scholar] [CrossRef]
- Cani, P.D.; Amar, J.; Iglesias, M.A.; Poggi, M.; Knauf, C.; Bastelica, D.; Neyrinck, A.M.; Fava, F.; Tuohy, K.M.; Chabo, C.; et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 2007, 56, 1761–1772. [Google Scholar] [CrossRef]
- Lassenius, M.I.; Pietilainen, K.H.; Kaartinen, K.; Pussinen, P.J.; Syrjanen, J.; Forsblom, C.; Porsti, I.; Rissanen, A.; Kaprio, J.; Mustonen, J.; et al. Bacterial endotoxin activity in human serum is associated with dyslipidemia, insulin resistance, obesity, and chronic inflammation. Diabetes Care 2011, 34, 1809–1815. [Google Scholar] [CrossRef]
- Meijnikman, A.S.; Gerdes, V.E.; Nieuwdorp, M.; Herrema, H. Evaluating Causality of Gut Microbiota in Obesity and Diabetes in Humans. Endocr. Rev. 2018, 39, 133–153. [Google Scholar] [CrossRef] [PubMed]
- Cao, M.; Wang, P.; Sun, C.; He, W.; Wang, F. Amelioration of IFN-gamma and TNF-alpha-induced intestinal epithelial barrier dysfunction by berberine via suppression of MLCK-MLC phosphorylation signaling pathway. PLoS ONE 2013, 8, e61944. [Google Scholar] [CrossRef]
- American Diabetes Association. Diagnosis and classification of diabetes mellitus. Diabetes Care 2013, 36 (Suppl. 1), S67–S74. [Google Scholar] [CrossRef]
- Atkinson, M.A. The pathogenesis and natural history of type 1 diabetes. Cold Spring Harb. Perspect. Med. 2012, 2. [Google Scholar] [CrossRef]
- Esser, N.; L’Homme, L.; De Roover, A.; Kohnen, L.; Scheen, A.J.; Moutschen, M.; Piette, J.; Legrand-Poels, S.; Paquot, N. Obesity phenotype is related to NLRP3 inflammasome activity and immunological profile of visceral adipose tissue. Diabetologia 2013, 56, 2487–2497. [Google Scholar] [CrossRef]
- Jourdan, T.; Godlewski, G.; Cinar, R.; Bertola, A.; Szanda, G.; Liu, J.; Tam, J.; Han, T.; Mukhopadhyay, B.; Skarulis, M.C.; et al. Activation of the Nlrp3 inflammasome in infiltrating macrophages by endocannabinoids mediates beta cell loss in type 2 diabetes. Nat. Med. 2013, 19, 1132–1140. [Google Scholar] [CrossRef]
- Miura, K.; Kodama, Y.; Inokuchi, S.; Schnabl, B.; Aoyama, T.; Ohnishi, H.; Olefsky, J.M.; Brenner, D.A.; Seki, E. Toll-like receptor 9 promotes steatohepatitis by induction of interleukin-1beta in mice. Gastroenterology 2010, 139, 323–334. [Google Scholar] [CrossRef]
- Bradshaw, E.M.; Raddassi, K.; Elyaman, W.; Orban, T.; Gottlieb, P.A.; Kent, S.C.; Hafler, D.A. Monocytes from patients with type 1 diabetes spontaneously secrete proinflammatory cytokines inducing Th17 cells. J. Immunol. 2009, 183, 4432–4439. [Google Scholar] [CrossRef]
- Dogan, Y.; Akarsu, S.; Ustundag, B.; Yilmaz, E.; Gurgoze, M.K. Serum IL-1beta, IL-2, and IL-6 in insulin-dependent diabetic children. Mediat. Inflamm. 2006, 2006, 59206. [Google Scholar] [CrossRef]
- Kim, J.K.; Jin, H.S.; Suh, H.W.; Jo, E.K. Negative regulators and their mechanisms in NLRP3 inflammasome activation and signaling. Immunol. Cell Biol. 2017, 95, 584–592. [Google Scholar] [CrossRef]
- Devaraj, S.; Dasu, M.R.; Rockwood, J.; Winter, W.; Griffen, S.C.; Jialal, I. Increased toll-like receptor (TLR) 2 and TLR4 expression in monocytes from patients with type 1 diabetes: Further evidence of a proinflammatory state. J. Clin. Endocrinol. Metab. 2008, 93, 578–583. [Google Scholar] [CrossRef] [PubMed]
- Netea, M.G.; Simon, A.; van de Veerdonk, F.; Kullberg, B.J.; Van der Meer, J.W.; Joosten, L.A. IL-1beta processing in host defense: Beyond the inflammasomes. PLoS Pathog. 2010, 6, e1000661. [Google Scholar] [CrossRef] [PubMed]
- Wittmann, M.; Kingsbury, S.R.; McDermott, M.F. Is caspase 1 central to activation of interleukin-1? Jt. Bone Spine Rev. Rhum. 2011, 78, 327–330. [Google Scholar] [CrossRef] [PubMed]
- Shimada, K.; Crother, T.R.; Karlin, J.; Dagvadorj, J.; Chiba, N.; Chen, S.; Ramanujan, V.K.; Wolf, A.J.; Vergnes, L.; Ojcius, D.M.; et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 2012, 36, 401–414. [Google Scholar] [CrossRef] [PubMed]
- Carlos, D.; Costa, F.R.; Pereira, C.A.; Rocha, F.A.; Yaochite, J.N.; Oliveira, G.G.; Carneiro, F.S.; Tostes, R.C.; Ramos, S.G.; Zamboni, D.S.; et al. Mitochondrial DNA Activates the NLRP3 Inflammasome and Predisposes to Type 1 Diabetes in Murine Model. Front. Immunol. 2017, 8, 164. [Google Scholar] [CrossRef]
- Liu, H.; Xu, R.; Kong, Q.; Liu, J.; Yu, Z.; Zhao, C. Downregulated NLRP3 and NLRP1 inflammasomes signaling pathways in the development and progression of type 1 diabetes mellitus. Biomed. Pharmacother. 2017, 94, 619–626. [Google Scholar] [CrossRef]
- Spalinger, M.R.; Lang, S.; Gottier, C.; Dai, X.; Rawlings, D.J.; Chan, A.C.; Rogler, G.; Scharl, M. PTPN22 regulates NLRP3-mediated IL1B secretion in an autophagy-dependent manner. Autophagy 2017, 13, 1590–1601. [Google Scholar] [CrossRef]
- Birnbaum, Y.; Bajaj, M.; Qian, J.; Ye, Y. Dipeptidyl peptidase-4 inhibition by Saxagliptin prevents inflammation and renal injury by targeting the Nlrp3/ASC inflammasome. BMJ Open Diabetes Res. Care 2016, 4, e000227. [Google Scholar] [CrossRef]
- Zhou, R.; Tardivel, A.; Thorens, B.; Choi, I.; Tschopp, J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat. Immunol. 2010, 11, 136–140. [Google Scholar] [CrossRef]
- Dror, E.; Dalmas, E.; Meier, D.T.; Wueest, S.; Thevenet, J.; Thienel, C.; Timper, K.; Nordmann, T.M.; Traub, S.; Schulze, F.; et al. Postprandial macrophage-derived IL-1beta stimulates insulin, and both synergistically promote glucose disposal and inflammation. Nat. Immunol. 2017, 18, 283–292. [Google Scholar] [CrossRef]
- Sanna, S.; van Zuydam, N.R.; Mahajan, A.; Kurilshikov, A.; Vich Vila, A.; Vosa, U.; Mujagic, Z.; Masclee, A.A.M.; Jonkers, D.; Oosting, M.; et al. Causal relationships among the gut microbiome, short-chain fatty acids and metabolic diseases. Nat. Genet. 2019, 51, 600–605. [Google Scholar] [CrossRef] [PubMed]
- Dinarello, C.A. Immunological and inflammatory functions of the interleukin-1 family. Annu. Rev. Immunol. 2009, 27, 519–550. [Google Scholar] [CrossRef] [PubMed]
- Okamura, H.; Tsutsi, H.; Komatsu, T.; Yutsudo, M.; Hakura, A.; Tanimoto, T.; Torigoe, K.; Okura, T.; Nukada, Y.; Hattori, K.; et al. Cloning of a new cytokine that induces IFN-gamma production by T cells. Nature 1995, 378, 88–91. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, K.; Yoshimoto, T.; Tsutsui, H.; Okamura, H. Interleukin-18 is a unique cytokine that stimulates both Th1 and Th2 responses depending on its cytokine milieu. Cytokine Growth Factor Rev. 2001, 12, 53–72. [Google Scholar] [CrossRef]
- Yamanishi, K.; Maeda, S.; Kuwahara-Otani, S.; Watanabe, Y.; Yoshida, M.; Ikubo, K.; Okuzaki, D.; El-Darawish, Y.; Li, W.; Nakasho, K.; et al. Interleukin-18-deficient mice develop dyslipidemia resulting in nonalcoholic fatty liver disease and steatohepatitis. Transl. Res. 2016, 173, 101–114. [Google Scholar] [CrossRef]
- Lindegaard, B.; Matthews, V.B.; Brandt, C.; Hojman, P.; Allen, T.L.; Estevez, E.; Watt, M.J.; Bruce, C.R.; Mortensen, O.H.; Syberg, S.; et al. Interleukin-18 activates skeletal muscle AMPK and reduces weight gain and insulin resistance in mice. Diabetes 2013, 62, 3064–3074. [Google Scholar] [CrossRef]
- Huang, Y.; Xu, M.; Hong, J.; Gu, W.; Bi, Y.; Li, X. -607 C/A polymorphism in the promoter of IL-18 gene is associated with 2 h post-loading plasma glucose level in Chinese. Endocrine 2010, 37, 507–512. [Google Scholar] [CrossRef]
- Van Greevenbroek, M.M.; Vermeulen, V.M.; Feskens, E.J.; Evelo, C.T.; Kruijshoop, M.; Hoebee, B.; van der Kallen, C.J.; de Bruin, T.W. Genetic variation in thioredoxin interacting protein (TXNIP) is associated with hypertriglyceridaemia and blood pressure in diabetes mellitus. Diabet. Med. 2007, 24, 498–504. [Google Scholar] [CrossRef]
- Maedler, K.; Storling, J.; Sturis, J.; Zuellig, R.A.; Spinas, G.A.; Arkhammar, P.O.; Mandrup-Poulsen, T.; Donath, M.Y. Glucose- and interleukin-1beta-induced beta-cell apoptosis requires Ca2+ influx and extracellular signal-regulated kinase (ERK) 1/2 activation and is prevented by a sulfonylurea receptor 1/inwardly rectifying K+ channel 6.2 (SUR/Kir6.2) selective potassium channel opener in human islets. Diabetes 2004, 53, 1706–1713. [Google Scholar] [CrossRef]
- Grant, R.W.; Dixit, V.D. Mechanisms of disease: Inflammasome activation and the development of type 2 diabetes. Front. Immunol. 2013, 4, 50. [Google Scholar] [CrossRef]
- Bell, P.M.; Cuthbertson, J.; Patterson, S.; O’Harte, F.P. Additive hypoglycaemic effect of nateglinide and exogenous glucagon-like peptide-1 in type 2 diabetes. Diabetes Res. Clin. Pract. 2011, 91, e68–e70. [Google Scholar] [CrossRef] [PubMed]
- Inzucchi, S.E.; Lipska, K.J.; Mayo, H.; Bailey, C.J.; McGuire, D.K. Metformin in patients with type 2 diabetes and kidney disease: A systematic review. JAMA 2014, 312, 2668–2675. [Google Scholar] [CrossRef] [PubMed]
- Duez, H.; Cariou, B.; Staels, B. DPP-4 inhibitors in the treatment of type 2 diabetes. Biochem. Pharmacol. 2012, 83, 823–832. [Google Scholar] [CrossRef] [PubMed]
- Deacon, C.F.; Holst, J.J. Linagliptin, a xanthine-based dipeptidyl peptidase-4 inhibitor with an unusual profile for the treatment of type 2 diabetes. Expert Opin. Investig. Drugs 2010, 19, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Taslimi, P.; Aslan, H.E.; Demir, Y.; Oztaskin, N.; Maras, A.; Gulcin, I.; Beydemir, S.; Goksu, S. Diarylmethanon, bromophenol and diarylmethane compounds: Discovery of potent aldose reductase, alpha-amylase and alpha-glycosidase inhibitors as new therapeutic approach in diabetes and functional hyperglycemia. Int. J. Biol. Macromol. 2018, 119, 857–863. [Google Scholar] [CrossRef] [PubMed]
- Stienstra, R.; van Diepen, J.A.; Tack, C.J.; Zaki, M.H.; van de Veerdonk, F.L.; Perera, D.; Neale, G.A.; Hooiveld, G.J.; Hijmans, A.; Vroegrijk, I.; et al. Inflammasome is a central player in the induction of obesity and insulin resistance. Proc. Natl. Acad. Sci. USA 2011, 108, 15324–15329. [Google Scholar] [CrossRef] [PubMed]
- Vandanmagsar, B.; Youm, Y.H.; Ravussin, A.; Galgani, J.E.; Stadler, K.; Mynatt, R.L.; Ravussin, E.; Stephens, J.M.; Dixit, V.D. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat. Med. 2011, 17, 179–188. [Google Scholar] [CrossRef]
- Reynolds, C.M.; McGillicuddy, F.C.; Harford, K.A.; Finucane, O.M.; Mills, K.H.; Roche, H.M. Dietary saturated fatty acids prime the NLRP3 inflammasome via TLR4 in dendritic cells-implications for diet-induced insulin resistance. Mol. Nutr. Food Res. 2012, 56, 1212–1222. [Google Scholar] [CrossRef]
- Netea, M.G.; Joosten, L.A.; Lewis, E.; Jensen, D.R.; Voshol, P.J.; Kullberg, B.J.; Tack, C.J.; van Krieken, H.; Kim, S.H.; Stalenhoef, A.F.; et al. Deficiency of interleukin-18 in mice leads to hyperphagia, obesity and insulin resistance. Nat. Med. 2006, 12, 650–656. [Google Scholar] [CrossRef]
- Zorrilla, E.P.; Sanchez-Alavez, M.; Sugama, S.; Brennan, M.; Fernandez, R.; Bartfai, T.; Conti, B. Interleukin-18 controls energy homeostasis by suppressing appetite and feed efficiency. Proc. Natl. Acad. Sci. USA 2007, 104, 11097–11102. [Google Scholar] [CrossRef]
- Watt, M.J.; Dzamko, N.; Thomas, W.G.; Rose-John, S.; Ernst, M.; Carling, D.; Kemp, B.E.; Febbraio, M.A.; Steinberg, G.R. CNTF reverses obesity-induced insulin resistance by activating skeletal muscle AMPK. Nat. Med. 2006, 12, 541–548. [Google Scholar] [CrossRef] [PubMed]
- Hotamisligil, G.S. Inflammation and metabolic disorders. Nature 2006, 444, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Jung, C.; Gerdes, N.; Fritzenwanger, M.; Figulla, H.R. Circulating levels of interleukin-1 family cytokines in overweight adolescents. Mediat. Inflamm. 2010, 2010, 958403. [Google Scholar] [CrossRef] [PubMed]
- Opstad, T.B.; Pettersen, A.A.; Arnesen, H.; Seljeflot, I. Circulating levels of IL-18 are significantly influenced by the IL-18 +183 A/G polymorphism in coronary artery disease patients with diabetes type 2 and the metabolic syndrome: An observational study. Cardiovasc. Diabetol. 2011, 10, 110. [Google Scholar] [CrossRef]
- Kim, D.H.; Kim, S.M.; Lee, B.; Lee, E.K.; Chung, K.W.; Moon, K.M.; An, H.J.; Kim, K.M.; Yu, B.P.; Chung, H.Y. Effect of betaine on hepatic insulin resistance through FOXO1-induced NLRP3 inflammasome. J. Nutr. Biochem. 2017, 45, 104–114. [Google Scholar] [CrossRef]
- Xu, X.; Chen, Y.; Song, J.; Hou, F.; Ma, X.; Liu, B.; Huang, F. Mangiferin suppresses endoplasmic reticulum stress in perivascular adipose tissue and prevents insulin resistance in the endothelium. Eur. J. Nutr. 2018, 57, 1563–1575. [Google Scholar] [CrossRef]
- Zhou, H.; Feng, L.; Xu, F.; Sun, Y.; Ma, Y.; Zhang, X.; Liu, H.; Xu, G.; Wu, X.; Shen, Y.; et al. Berberine inhibits palmitate-induced NLRP3 inflammasome activation by triggering autophagy in macrophages: A new mechanism linking berberine to insulin resistance improvement. Biomed. Pharmacother. 2017, 89, 864–874. [Google Scholar] [CrossRef]
- Finucane, O.M.; Lyons, C.L.; Murphy, A.M.; Reynolds, C.M.; Klinger, R.; Healy, N.P.; Cooke, A.A.; Coll, R.C.; McAllan, L.; Nilaweera, K.N.; et al. Monounsaturated fatty acid-enriched high-fat diets impede adipose NLRP3 inflammasome-mediated IL-1beta secretion and insulin resistance despite obesity. Diabetes 2015, 64, 2116–2128. [Google Scholar] [CrossRef]
- Yang, X.; Li, L.; Fang, K.; Dong, R.; Li, J.; Zhao, Y.; Dong, H.; Yi, P.; Huang, Z.; Chen, G.; et al. Wu-Mei-Wan Reduces Insulin Resistance via Inhibition of NLRP3 Inflammasome Activation in HepG2 Cells. Evid. Based Complement. Altern. Med. 2017, 2017, 7283241. [Google Scholar] [CrossRef]
- Chen, Y.; Qian, Q.; Yu, J. Carbenoxolone ameliorates insulin sensitivity in obese mice induced by high fat diet via regulating the IkappaB-alpha/NF-kappaB pathway and NLRP3 inflammasome. Biomed. Pharmacother. 2019, 115, 108868. [Google Scholar] [CrossRef]
- Lu, C.P.; Huang, C.Y.; Wang, S.H.; Chiu, C.H.; Li, L.H.; Hua, K.F.; Wu, T.H. Improvement of hyperglycemia in a murine model of insulin resistance and high glucose- and inflammasome-mediated IL-1beta expressions in macrophages by silymarin. Chem. Biol. Interact. 2018, 290, 12–18. [Google Scholar] [CrossRef] [PubMed]
- Nakae, J.; Cao, Y.; Oki, M.; Orba, Y.; Sawa, H.; Kiyonari, H.; Iskandar, K.; Suga, K.; Lombes, M.; Hayashi, Y. Forkhead transcription factor FoxO1 in adipose tissue regulates energy storage and expenditure. Diabetes 2008, 57, 563–576. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, N.; Asada, R.; Saito, A.; Kanemoto, S.; Imaizumi, K. Obesity-induced endoplasmic reticulum stress causes chronic inflammation in adipose tissue. Sci. Rep. 2012, 2, 799. [Google Scholar] [CrossRef] [PubMed]
- Suganami, T.; Ogawa, Y. Adipose tissue macrophages: Their role in adipose tissue remodeling. J. Leukoc. Biol. 2010, 88, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Hotamisligil, G.S. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell 2010, 140, 900–917. [Google Scholar] [CrossRef]
- Scheuner, D.; Kaufman, R.J. The unfolded protein response: A pathway that links insulin demand with beta-cell failure and diabetes. Endocr. Rev. 2008, 29, 317–333. [Google Scholar] [CrossRef]
- Lerner, A.G.; Upton, J.P.; Praveen, P.V.; Ghosh, R.; Nakagawa, Y.; Igbaria, A.; Shen, S.; Nguyen, V.; Backes, B.J.; Heiman, M.; et al. IRE1alpha induces thioredoxin-interacting protein to activate the NLRP3 inflammasome and promote programmed cell death under irremediable ER stress. Cell Metab. 2012, 16, 250–264. [Google Scholar] [CrossRef]
- Ron, D.; Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529. [Google Scholar] [CrossRef]
- Chaudhari, N.; Talwar, P.; Parimisetty, A.; Lefebvre d’Hellencourt, C.; Ravanan, P. A molecular web: Endoplasmic reticulum stress, inflammation, and oxidative stress. Front. Cell. Neurosci. 2014, 8, 213. [Google Scholar] [CrossRef]
- Malhotra, J.D.; Kaufman, R.J. Endoplasmic reticulum stress and oxidative stress: A vicious cycle or a double-edged sword? Antioxid. Redox Signal. 2007, 9, 2277–2293. [Google Scholar] [CrossRef]
- Sharp, W.W.; Fang, Y.H.; Han, M.; Zhang, H.J.; Hong, Z.; Banathy, A.; Morrow, E.; Ryan, J.J.; Archer, S.L. Dynamin-related protein 1 (Drp1)-mediated diastolic dysfunction in myocardial ischemia-reperfusion injury: Therapeutic benefits of Drp1 inhibition to reduce mitochondrial fission. FASEB J. 2014, 28, 316–326. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Zhang, S.; Li, J.; Liu, K.; Huang, F.; Liu, B. Metformin and resveratrol inhibit Drp1-mediated mitochondrial fission and prevent ER stress-associated NLRP3 inflammasome activation in the adipose tissue of diabetic mice. Mol. Cell. Endocrinol. 2016, 434, 36–47. [Google Scholar] [CrossRef] [PubMed]
- Cotillard, A.; Kennedy, S.P.; Kong, L.C.; Prifti, E.; Pons, N.; Le Chatelier, E.; Almeida, M.; Quinquis, B.; Levenez, F.; Galleron, N.; et al. Dietary intervention impact on gut microbial gene richness. Nature 2013, 500, 585–588. [Google Scholar] [CrossRef] [PubMed]
- Leiva-Gea, I.; Sanchez-Alcoholado, L.; Martin-Tejedor, B.; Castellano-Castillo, D.; Moreno-Indias, I.; Urda-Cardona, A.; Tinahones, F.J.; Fernandez-Garcia, J.C.; Queipo-Ortuno, M.I. Gut Microbiota Differs in Composition and Functionality Between Children With Type 1 Diabetes and MODY2 and Healthy Control Subjects: A Case-Control Study. Diabetes Care 2018, 41, 2385–2395. [Google Scholar] [CrossRef]
- Henao-Mejia, J.; Elinav, E.; Jin, C.; Hao, L.; Mehal, W.Z.; Strowig, T.; Thaiss, C.A.; Kau, A.L.; Eisenbarth, S.C.; Jurczak, M.J.; et al. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature 2012, 482, 179–185. [Google Scholar] [CrossRef]
- Lee, H.M.; Kim, J.J.; Kim, H.J.; Shong, M.; Ku, B.J.; Jo, E.K. Upregulated NLRP3 inflammasome activation in patients with type 2 diabetes. Diabetes 2013, 62, 194–204. [Google Scholar] [CrossRef]
- Yao, X.; Zhang, C.; Xing, Y.; Xue, G.; Zhang, Q.; Pan, F.; Wu, G.; Hu, Y.; Guo, Q.; Lu, A.; et al. Remodelling of the gut microbiota by hyperactive NLRP3 induces regulatory T cells to maintain homeostasis. Nat. Commun. 2017, 8, 1896. [Google Scholar] [CrossRef]
- Elinav, E.; Strowig, T.; Kau, A.L.; Henao-Mejia, J.; Thaiss, C.A.; Booth, C.J.; Peaper, D.R.; Bertin, J.; Eisenbarth, S.C.; Gordon, J.I.; et al. NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell 2011, 145, 745–757. [Google Scholar] [CrossRef]
- Pahwa, R.; Balderas, M.; Jialal, I.; Chen, X.; Luna, R.A.; Devaraj, S. Gut Microbiome and Inflammation: A Study of Diabetic Inflammasome-Knockout Mice. J. Diabetes Res. 2017, 2017, 6519785. [Google Scholar] [CrossRef]
- Mantovani, A.; Sica, A.; Sozzani, S.; Allavena, P.; Vecchi, A.; Locati, M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004, 25, 677–686. [Google Scholar] [CrossRef]
- Koh, A.; Molinaro, A.; Stahlman, M.; Khan, M.T.; Schmidt, C.; Manneras-Holm, L.; Wu, H.; Carreras, A.; Jeong, H.; Olofsson, L.E.; et al. Microbially Produced Imidazole Propionate Impairs Insulin Signaling through mTORC1. Cell 2018, 175, 947–961. [Google Scholar] [CrossRef]


{kind=link}
| Type of Diabetes | Model | NLRP | Functional Consequences | Reference |
|---|---|---|---|---|
| T1DM | PBMCs; GCs | NLRP3; NLRP1 | Progression of the disease | [67] |
| T1DM | NLRP3−/− mice | NLRP3 | Suppressed T-cell activation and modulated pathogenic T-cell migration to the pancreatic islets via regulating the expression of chemokine receptors CCR5 and CXCR3 by NLRP3 | [16] |
| T1DM | NLRP3−/− mice | NLRP3 | Increased the myeloid-derived suppressor cell and mast cell numbers of pancreatic lymph nodes in conjunction with an ascent of the IL-6, IL-10, and IL-4 in pancreatic tissue of NLRP3-deficient mice | [66] |
| T1DM | THP-1 cells | NLRP3 | Promoted sequestration into phagophores | [68] |
| T1DM/T2DM | Wild-type mice | NLRP3/ASC | Progression of the disease | [69] |
| T1DM | NLRP3−/− mice | NLRP3 | Reduced glucose tolerance and insulin sensitivity | [70] |
| Subjects | Model | Regulatory Mechanism | Functional Consequences | Reference |
|---|---|---|---|---|
| Betaine | Hep 2 cells, db/db mice | Decreased the production of IL-1β via interactions with FOXO1 and TXNIP and inhibited the activation of the NLRP3 inflammasome | Inhibition of RS-induced activation of the NLRP3 inflammasome in diabetic livers | [96] |
| Mangiferin | Perivascular adipose tissue | Increased LKB1-dependent AMPK activity; inhibited NLRP3 inflammasome activation; reduced the secretion of IL-1β | Prevention of endothelial insulin resistance | [97] |
| Berberine | ob/ob mice | Activation of AMPK-dependent autophagy in adipose tissue-resident macrophages | Alleviation of insulin resistance | [98] |
| MUFA | Mice; Bone marrow-derived cells | Decreased the formation of pro-IL-1β, reduced the secretion of IL-1β and maintained the activation of adipose AMPK | Improved insulin resistance mediated by IL-1β and adipose dysfunction | [99] |
| SFA | Mice; BMDC; | Improved the expression of IL-1R1, TLR4, and caspase-1 and increased the secretion of IL-1β | Decreased insulin levels in adipose | [89] |
| WMW | HepG2 cells | Regulated the downstream insulin signaling pathway via reduced IR and IRS-1; decreased IL-1β and NFκB | Alleviation of insulin resistance | [100] |
| CBX | Mice | Suppressed the IκB-α/NF-κB pathway and inhibited the activation of the NLRP3 inflammasome | Alleviation of insulin resistance in the liver and skeletal muscle | [101] |
| Silymarin | Hnf-1α- knockout mice | Reduced expression of IL-1β mediated by HG and inflammasome | Alleviation of insulin resistance | [102] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ding, S.; Xu, S.; Ma, Y.; Liu, G.; Jang, H.; Fang, J. Modulatory Mechanisms of the NLRP3 Inflammasomes in Diabetes. Biomolecules 2019, 9, 850. https://doi.org/10.3390/biom9120850
Ding S, Xu S, Ma Y, Liu G, Jang H, Fang J. Modulatory Mechanisms of the NLRP3 Inflammasomes in Diabetes. Biomolecules. 2019; 9(12):850. https://doi.org/10.3390/biom9120850
Chicago/Turabian StyleDing, Sujuan, Sheng Xu, Yong Ma, Gang Liu, Hongmei Jang, and Jun Fang. 2019. "Modulatory Mechanisms of the NLRP3 Inflammasomes in Diabetes" Biomolecules 9, no. 12: 850. https://doi.org/10.3390/biom9120850
APA StyleDing, S., Xu, S., Ma, Y., Liu, G., Jang, H., & Fang, J. (2019). Modulatory Mechanisms of the NLRP3 Inflammasomes in Diabetes. Biomolecules, 9(12), 850. https://doi.org/10.3390/biom9120850