Honokiol Enhances TRAIL-Mediated Apoptosis through STAMBPL1-Induced Survivin and c-FLIP Degradation




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Transfection
2.2. Reagents, Antibodies, siRNAs, and Plasmids
2.3. FACS Analysis
2.4. Western Blotting
2.5. DNA Fragmentation and DEVDase Activity Assay for Detection of Apoptosis
2.6. Reverse Transcription Polymerase Chain Reaction (RT-PCR) and Quantitative PCR (qPCR)
2.7. Detection of DR5 Expression on Cell Surface
2.8. Deubiquitination Assay
2.9. Immunoprecipitation
2.10. Statistical Analysis
3. Results
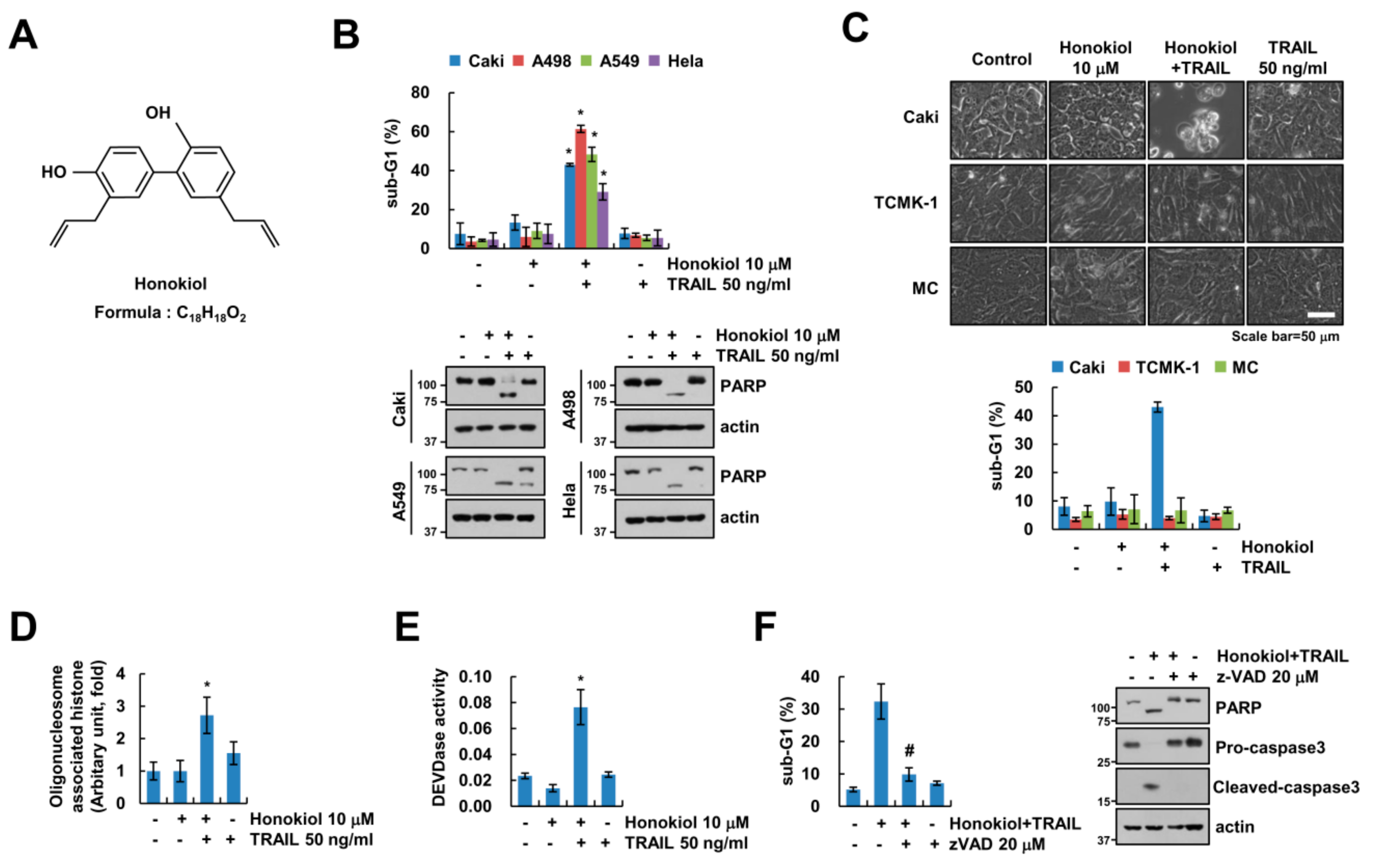
3.1. Honokiol Sensitizes Cancer Cells to TRAIL-Mediated Apoptosis, but Not Normal Cells
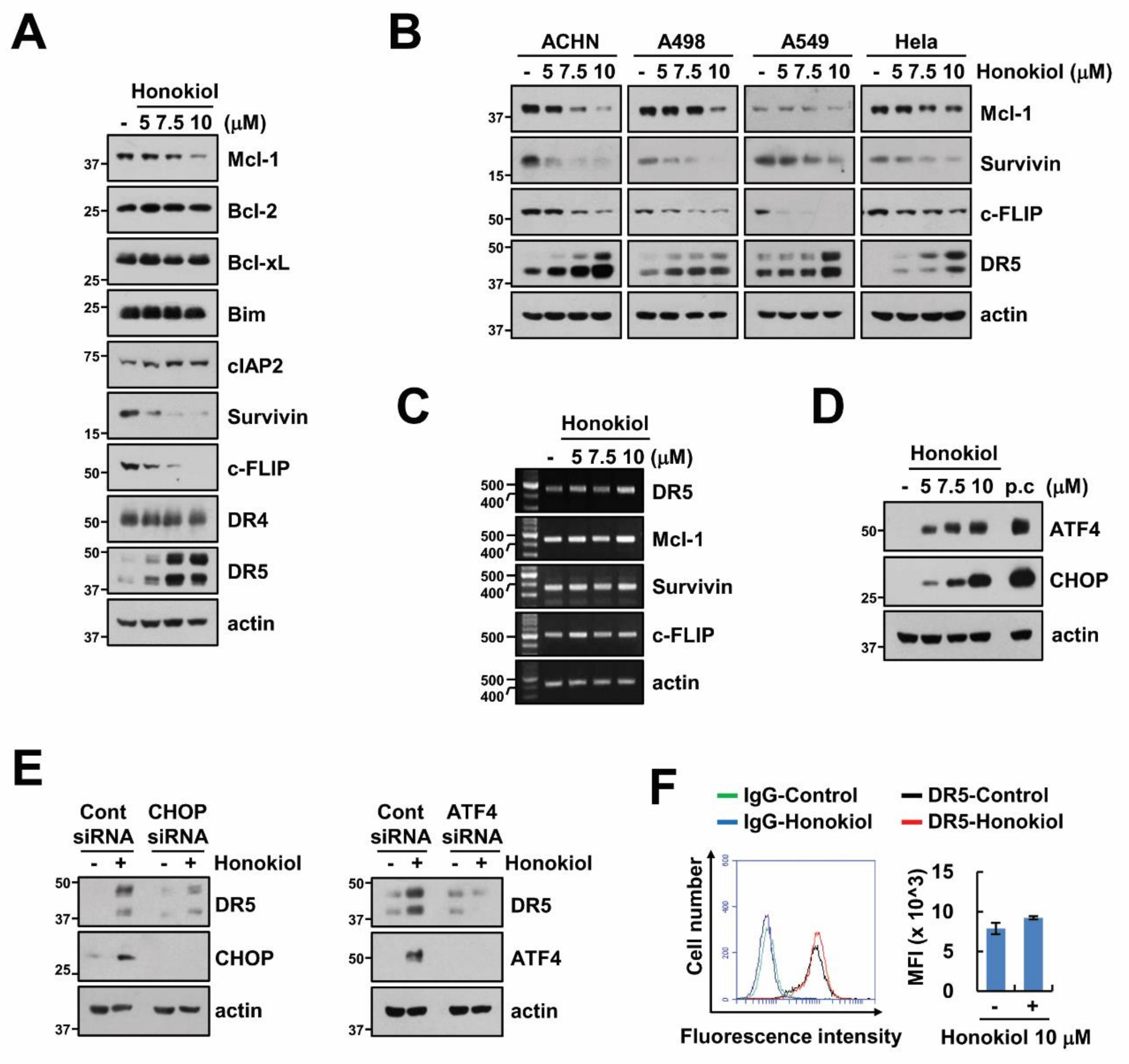
3.2. Upregulation of DR5 by Honokiol Is Not Involved in Enhancement of TRAIL Sensitivity
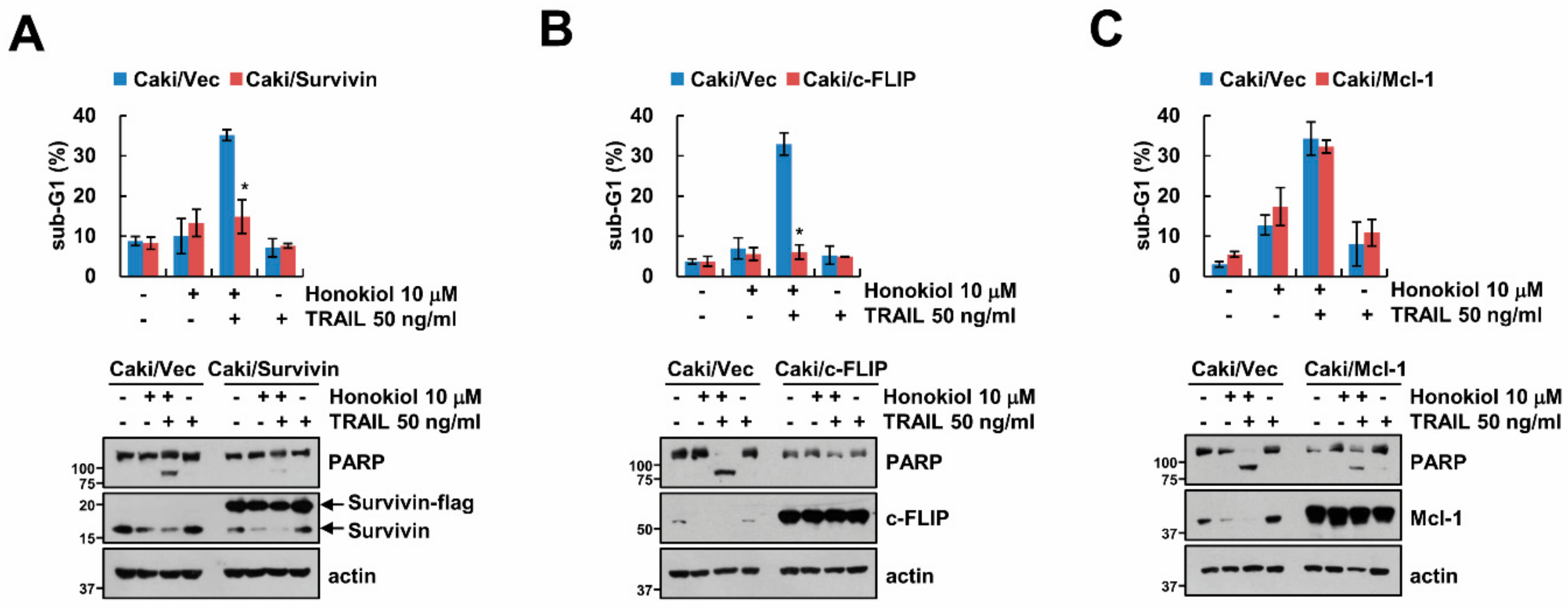
3.3. Downregulation of Survivin and c-FLIP Is Associated with Honokiol Plus TRAIL-Induced Apoptosis
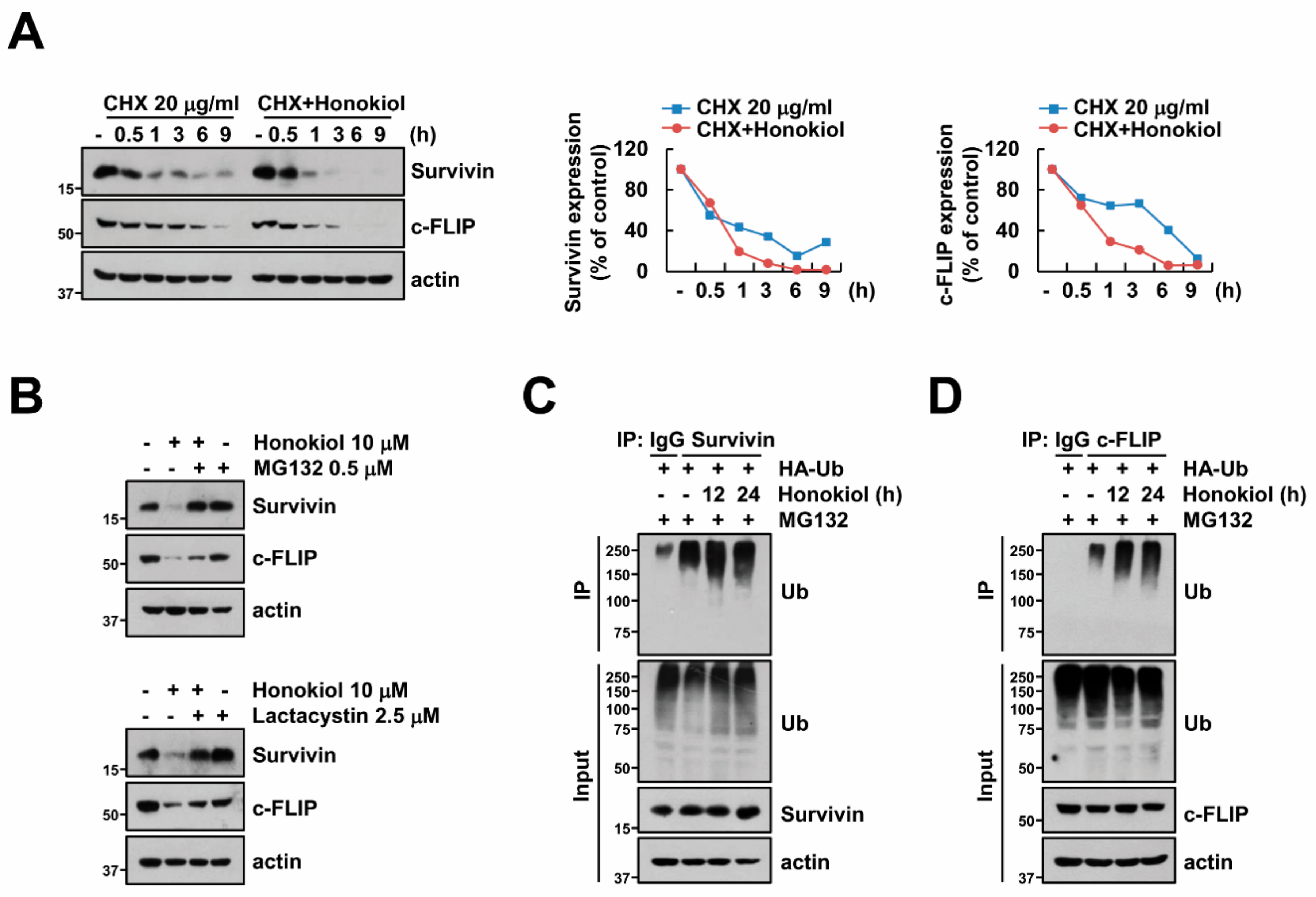
3.4. Honokiol Induces Survivin and c-FLIP Degradation through Activation of Ubiquitin-Proteasome System
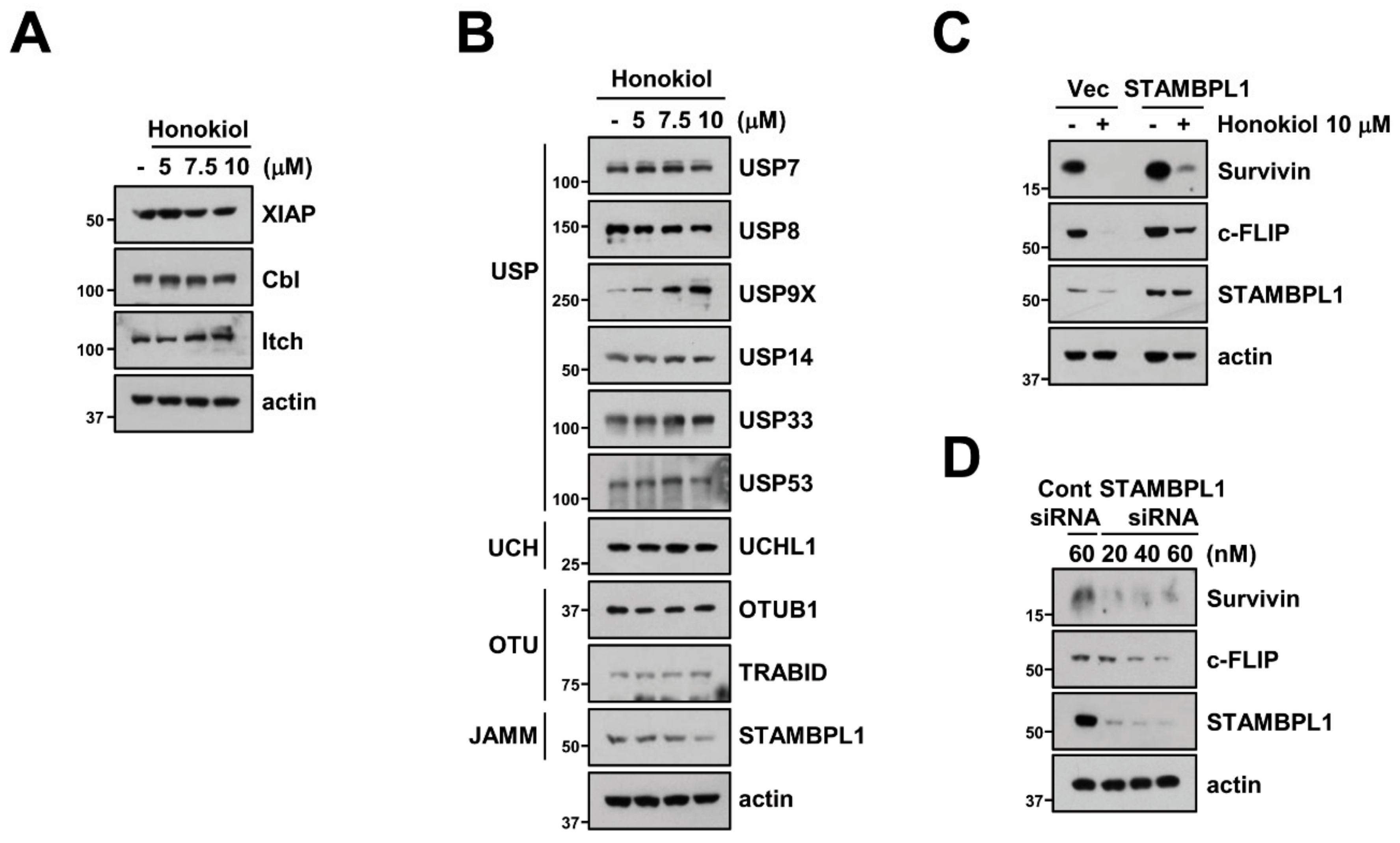
3.5. STAMBPL1 Can Regulate Survivin and c-FLIP Stability
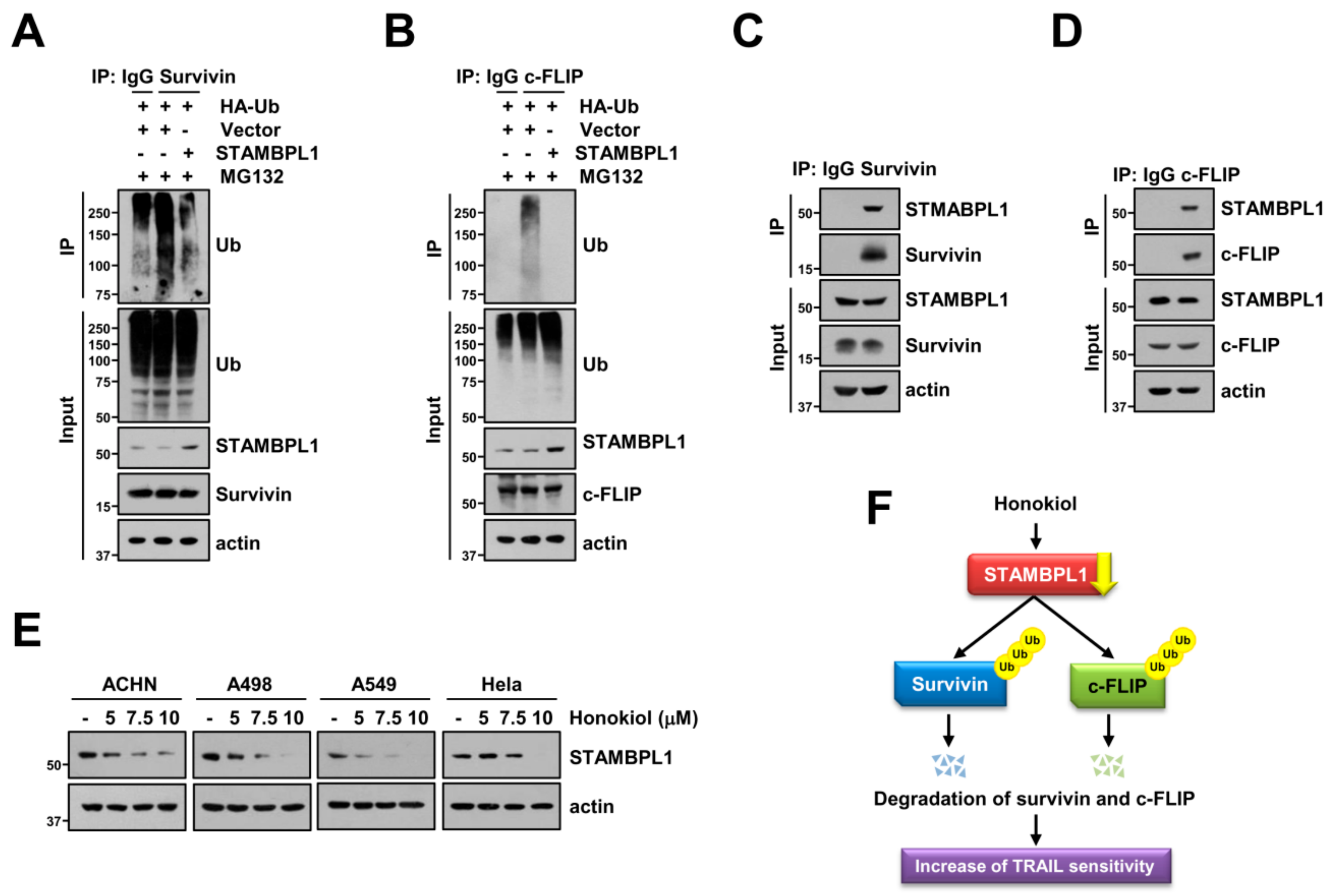
3.6. STAMBPL1 Interacts and Induces Deubiquitination of Survivin and c-FLIP
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Johnstone, R.W.; Frew, A.J.; Smyth, M.J. The TRAIL apoptotic pathway in cancer onset, progression and therapy. Nat. Rev. Cancer 2008, 8, 782–798. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Fang, B. Mechanisms of resistance to TRAIL-induced apoptosis in cancer. Cancer Gene Ther. 2005, 12, 228–237. [Google Scholar] [CrossRef] [PubMed]
- LeBlanc, H.N.; Ashkenazi, A. Apo2L/TRAIL and its death and decoy receptors. Cell Death Differ. 2003, 10, 66–75. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, B. TRAIL resistance of breast cancer cells is associated with constitutive endocytosis of death receptors 4 and 5. Mol. Cancer Res. 2008, 6, 1861–1871. [Google Scholar] [CrossRef]
- Chawla-Sarkar, M.; Bae, S.I.; Reu, F.J.; Jacobs, B.S.; Lindner, D.J.; Borden, E.C. Downregulation of Bcl-2, FLIP or IAPs (XIAP and survivin) by siRNAs sensitizes resistant melanoma cells to Apo2L/TRAIL-induced apoptosis. Cell Death Differ. 2004, 11, 915–923. [Google Scholar] [CrossRef]
- Kruyt, F.A. TRAIL and cancer therapy. Cancer Lett. 2008, 263, 14–25. [Google Scholar] [CrossRef]
- De Miguel, D.; Lemke, J.; Anel, A.; Walczak, H.; Martinez-Lostao, L. Onto better TRAILs for cancer treatment. Cell Death Differ. 2016, 23, 733–747. [Google Scholar] [CrossRef]
- Cho, J.H.; Jeon, Y.J.; Park, S.M.; Shin, J.C.; Lee, T.H.; Jung, S.; Park, H.; Ryu, J.; Chen, H.; Dong, Z.; et al. Multifunctional effects of honokiol as an anti-inflammatory and anti-cancer drug in human oral squamous cancer cells and xenograft. Biomaterials 2015, 53, 274–284. [Google Scholar] [CrossRef]
- Bai, X.; Cerimele, F.; Ushio-Fukai, M.; Waqas, M.; Campbell, P.M.; Govindarajan, B.; Der, C.J.; Battle, T.; Frank, D.A.; Ye, K.; et al. Honokiol, a small molecular weight natural product, inhibits angiogenesis in vitro and tumor growth in vivo. J. Biol. Chem. 2003, 278, 35501–35507. [Google Scholar] [CrossRef]
- Zhao, C.; Liu, Z.Q. Comparison of antioxidant abilities of magnolol and honokiol to scavenge radicals and to protect DNA. Biochimie 2011, 93, 1755–1760. [Google Scholar] [CrossRef]
- Sun, L.; Liao, K.; Hang, C.; Wang, D. Honokiol induces reactive oxygen species-mediated apoptosis in Candida albicans through mitochondrial dysfunction. PLoS ONE 2017, 12, e0172228. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; Zhang, Q.; Liu, Q.; Komas, S.M.; Kalyanaraman, B.; Lubet, R.A.; Wang, Y.; You, M. Honokiol inhibits lung tumorigenesis through inhibition of mitochondrial function. Cancer Prev. Res. (Phila.) 2014, 7, 1149–1159. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Jiang, F.; Zhang, X.; Alitongbieke, G.; Shi, X.; Meng, M.; Xu, Y.; Ren, A.; Wang, J.; Cai, L.; et al. Honokiol sensitizes breast cancer cells to TNF-alpha induction of apoptosis by inhibiting Nur77 expression. Br. J. Pharmacol. 2016, 173, 344–356. [Google Scholar] [CrossRef] [PubMed]
- Raja, S.M.; Chen, S.; Yue, P.; Acker, T.M.; Lefkove, B.; Arbiser, J.L.; Khuri, F.R.; Sun, S.Y. The natural product honokiol preferentially inhibits cellular FLICE-inhibitory protein and augments death receptor-induced apoptosis. Mol. Cancer Ther. 2008, 7, 2212–2223. [Google Scholar] [CrossRef] [PubMed]
- Ishitsuka, K.; Hideshima, T.; Hamasaki, M.; Raje, N.; Kumar, S.; Hideshima, H.; Shiraishi, N.; Yasui, H.; Roccaro, A.M.; Richardson, P.; et al. Honokiol overcomes conventional drug resistance in human multiple myeloma by induction of caspase-dependent and -independent apoptosis. Blood 2005, 106, 1794–1800. [Google Scholar] [CrossRef] [PubMed]
- Ji, N.; Jiang, L.; Deng, P.; Xu, H.; Chen, F.; Liu, J.; Li, J.; Liao, G.; Zeng, X.; Lin, Y.; et al. Synergistic effect of honokiol and 5-fluorouracil on apoptosis of oral squamous cell carcinoma cells. J. Oral Pathol. Med. 2017, 46, 201–207. [Google Scholar] [CrossRef]
- Chio, C.C.; Tai, Y.T.; Mohanraj, M.; Liu, S.H.; Yang, S.T.; Chen, R.M. Honokiol enhances temozolomide-induced apoptotic insults to malignant glioma cells via an intrinsic mitochondrion-dependent pathway. Phytomedicine 2018, 49, 41–51. [Google Scholar] [CrossRef]
- Wang, X.; Beitler, J.J.; Wang, H.; Lee, M.J.; Huang, W.; Koenig, L.; Nannapaneni, S.; Amin, A.R.; Bonner, M.; Shin, H.J.; et al. Honokiol enhances paclitaxel efficacy in multi-drug resistant human cancer model through the induction of apoptosis. PLoS ONE 2014, 9, e86369. [Google Scholar] [CrossRef]
- He, Z.; Subramaniam, D.; Ramalingam, S.; Dhar, A.; Postier, R.G.; Umar, S.; Zhang, Y.; Anant, S. Honokiol radiosensitizes colorectal cancer cells: Enhanced activity in cells with mismatch repair defects. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 301, G929–G937. [Google Scholar] [CrossRef]
- Tian, W.; Deng, Y.; Li, L.; He, H.; Sun, J.; Xu, D. Honokiol synergizes chemotherapy drugs in multidrug resistant breast cancer cells via enhanced apoptosis and additional programmed necrotic death. Int. J. Oncol. 2013, 42, 721–732. [Google Scholar] [CrossRef]
- Wang, Y.; Yang, Z.; Zhao, X. Honokiol induces paraptosis and apoptosis and exhibits schedule-dependent synergy in combination with imatinib in human leukemia cells. Toxicol. Mech. Methods 2010, 20, 234–241. [Google Scholar] [CrossRef] [PubMed]
- Finley, D.; Ciechanover, A.; Varshavsky, A. Ubiquitin as a central cellular regulator. Cell 2004, 116, S29–S32. [Google Scholar] [CrossRef]
- Burger, A.M.; Seth, A.K. The ubiquitin-mediated protein degradation pathway in cancer: Therapeutic implications. Eur. J. Cancer 2004, 40, 2217–2229. [Google Scholar] [CrossRef] [PubMed]
- Hoeller, D.; Dikic, I. Targeting the ubiquitin system in cancer therapy. Nature 2009, 458, 438–444. [Google Scholar] [CrossRef] [PubMed]
- Deshaies, R.J.; Joazeiro, C.A. RING domain E3 ubiquitin ligases. Annu. Rev. Biochem 2009, 78, 399–434. [Google Scholar] [CrossRef] [PubMed]
- Leznicki, P.; Kulathu, Y. Mechanisms of regulation and diversification of deubiquitylating enzyme function. J. Cell Sci. 2017, 130, 1997–2006. [Google Scholar] [CrossRef]
- Huang, X.; Dixit, V.M. Drugging the undruggables: Exploring the ubiquitin system for drug development. Cell Res. 2016, 26, 484–498. [Google Scholar] [CrossRef]
- Komander, D.; Clague, M.J.; Urbe, S. Breaking the chains: Structure and function of the deubiquitinases. Nat. Rev. Mol. Cell Biol. 2009, 10, 550–563. [Google Scholar] [CrossRef]
- Clague, M.J.; Barsukov, I.; Coulson, J.M.; Liu, H.; Rigden, D.J.; Urbe, S. Deubiquitylases from genes to organism. Physiol. Rev. 2013, 93, 1289–1315. [Google Scholar] [CrossRef]
- McCullough, J.; Clague, M.J.; Urbe, S. AMSH is an endosome-associated ubiquitin isopeptidase. J. Cell Biol. 2004, 166, 487–492. [Google Scholar] [CrossRef]
- Kikuchi, K.; Ishii, N.; Asao, H.; Sugamura, K. Identification of AMSH-LP containing a Jab1/MPN domain metalloenzyme motif. Biochem. Biophys. Res. Commun. 2003, 306, 637–643. [Google Scholar] [CrossRef]
- Ibarrola, N.; Kratchmarova, I.; Nakajima, D.; Schiemann, W.P.; Moustakas, A.; Pandey, A.; Mann, M. Cloning of a novel signaling molecule, AMSH-2, that potentiates transforming growth factor beta signaling. BMC Cell Biol. 2004, 5, 2. [Google Scholar] [CrossRef] [PubMed]
- Lavorgna, A.; Harhaj, E.W. An RNA interference screen identifies the Deubiquitinase STAMBPL1 as a critical regulator of human T-cell leukemia virus type 1 tax nuclear export and NF-kappaB activation. J. Virol. 2012, 86, 3357–3369. [Google Scholar] [CrossRef]
- Chen, X.; Shi, H.; Bi, X.; Li, Y.; Huang, Z. Targeting the deubiquitinase STAMBPL1 triggers apoptosis in prostate cancer cells by promoting XIAP degradation. Cancer Lett. 2019, 456, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Shahriyar, S.A.; Woo, S.M.; Seo, S.U.; Min, K.J.; Kwon, T.K. Cepharanthine Enhances TRAIL-Mediated Apoptosis through STAMBPL1-Mediated Downregulation of Survivin Expression in Renal Carcinoma Cells. Int. J. Mol. Sci. 2018, 19, 3280. [Google Scholar] [CrossRef]
- Ittiudomrak, T.; Puthong, S.; Roytrakul, S.; Chanchao, C. alpha-Mangostin and Apigenin Induced Cell Cycle Arrest and Programmed Cell Death in SKOV-3 Ovarian Cancer Cells. Toxicol. Res. 2019, 35, 167–179. [Google Scholar] [CrossRef]
- Seo, S.U.; Kim, T.H.; Kim, D.E.; Min, K.J.; Kwon, T.K. NOX4-mediated ROS production induces apoptotic cell death via down-regulation of c-FLIP and Mcl-1 expression in combined treatment with thioridazine and curcumin. Redox Biol. 2017, 13, 608–622. [Google Scholar] [CrossRef]
- Kim, S.; Woo, S.M.; Min, K.J.; Seo, S.U.; Lee, T.J.; Kubatka, P.; Kim, D.E.; Kwon, T.K. WP1130 Enhances TRAIL-Induced Apoptosis through USP9X-Dependent miR-708-Mediated Downregulation of c-FLIP. Cancers 2019, 11, 344. [Google Scholar] [CrossRef]
- Woo, S.M.; Seo, S.U.; Min, K.J.; Kwon, T.K. BIX-01294 sensitizes renal cancer Caki cells to TRAIL-induced apoptosis through downregulation of survivin expression and upregulation of DR5 expression. Cell Death Discov. 2018, 4, 29–39. [Google Scholar] [CrossRef]
- Woo, S.M.; Min, K.J.; Seo, B.R.; Seo, Y.H.; Jeong, Y.J.; Kwon, T.K. YM155 enhances ABT-737-mediated apoptosis through Mcl-1 downregulation in Mcl-1-overexpressed cancer cells. Mol. Cell. Biochem. 2017, 429, 91–102. [Google Scholar] [CrossRef]
- Yamaguchi, H.; Wang, H.G. CHOP is involved in endoplasmic reticulum stress-induced apoptosis by enhancing DR5 expression in human carcinoma cells. J. Biol. Chem. 2004, 279, 45495–45502. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Hu, T.; Liang, Y.; Li, P.; Chen, X.; Zhang, J.; Ma, Y.; Hao, Q.; Wang, J.; Zhang, P.; et al. Neddylation Inhibition Activates the Extrinsic Apoptosis Pathway through ATF4-CHOP-DR5 Axis in Human Esophageal Cancer Cells. Clin. Cancer Res. 2016, 22, 4145–4157. [Google Scholar] [CrossRef] [PubMed]
- Chiu, C.S.; Tsai, C.H.; Hsieh, M.S.; Tsai, S.C.; Jan, Y.J.; Lin, W.Y.; Lai, D.W.; Wu, S.M.; Hsing, H.Y.; Arbiser, J.L.; et al. Exploiting Honokiol-induced ER stress CHOP activation inhibits the growth and metastasis of melanoma by suppressing the MITF and beta-catenin pathways. Cancer Lett. 2019, 442, 113–125. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Xu, S.; Gao, W.; Feng, J.; Zhao, G. Honokiol induces endoplasmic reticulum stress-mediated apoptosis in human lung cancer cells. Life Sci. 2019, 221, 204–211. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.G.; Wang, J.; Yang, X.; Hsu, H.C.; Mountz, J.D. Regulation of apoptosis proteins in cancer cells by ubiquitin. Oncogene 2004, 23, 2009–2015. [Google Scholar] [CrossRef]
- Liu, Y.; Lear, T.; Iannone, O.; Shiva, S.; Corey, C.; Rajbhandari, S.; Jerome, J.; Chen, B.B.; Mallampalli, R.K. The Proapoptotic F-box Protein Fbxl7 Regulates Mitochondrial Function by Mediating the Ubiquitylation and Proteasomal Degradation of Survivin. J. Biol. Chem. 2015, 290, 11843–11852. [Google Scholar] [CrossRef]
- Song, X.; Kim, S.Y.; Zhou, Z.; Lagasse, E.; Kwon, Y.T.; Lee, Y.J. Hyperthermia enhances mapatumumab-induced apoptotic death through ubiquitin-mediated degradation of cellular FLIP(long) in human colon cancer cells. Cell Death Dis. 2013, 4, e577. [Google Scholar] [CrossRef]
- Berndsen, C.E.; Wolberger, C. New insights into ubiquitin E3 ligase mechanism. Nat. Struct. Mol. Biol. 2014, 21, 301–307. [Google Scholar] [CrossRef]
- Mevissen, T.E.T.; Komander, D. Mechanisms of Deubiquitinase Specificity and Regulation. Annu. Rev. Biochem. 2017, 86, 159–192. [Google Scholar] [CrossRef]
- Bhatt, J.R.; Finelli, A. Landmarks in the diagnosis and treatment of renal cell carcinoma. Nat. Rev. Urol. 2014, 11, 517–525. [Google Scholar] [CrossRef]
- Brodaczewska, K.K.; Szczylik, C.; Fiedorowicz, M.; Porta, C.; Czarnecka, A.M. Choosing the right cell line for renal cell cancer research. Mol. Cancer 2016, 15, 83. [Google Scholar] [CrossRef] [PubMed]
- Sinha, R.; Winer, A.G.; Chevinsky, M.; Jakubowski, C.; Chen, Y.B.; Dong, Y.; Tickoo, S.K.; Reuter, V.E.; Russo, P.; Coleman, J.A.; et al. Analysis of renal cancer cell lines from two major resources enables genomics-guided cell line selection. Nat. Commun. 2017, 8, 15165. [Google Scholar] [CrossRef] [PubMed]
- D’Avella, C.; Abbosh, P.; Pal, S.K.; Geynisman, D.M. Mutations in renal cell carcinoma. Urol. Oncol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, J.J.; Purdue, M.P.; Signoretti, S.; Swanton, C.; Albiges, L.; Schmidinger, M.; Heng, D.Y.; Larkin, J.; Ficarra, V. Renal cell carcinoma. Nat. Rev. Dis. Primers 2017, 3, 17009. [Google Scholar] [CrossRef]
- Sano, R.; Reed, J.C. ER stress-induced cell death mechanisms. Biochim. Biophys. Acta 2013, 1833, 3460–3470. [Google Scholar] [CrossRef]
- Lai, Y.J.; Lin, C.I.; Wang, C.L.; Chao, J.I. Expression of survivin and p53 modulates honokiol-induced apoptosis in colorectal cancer cells. J. Cell. Biochem. 2014, 115, 1888–1899. [Google Scholar] [CrossRef]
- Arora, V.; Cheung, H.H.; Plenchette, S.; Micali, O.C.; Liston, P.; Korneluk, R.G. Degradation of survivin by the X-linked inhibitor of apoptosis (XIAP)-XAF1 complex. J. Biol. Chem. 2007, 282, 26202–26209. [Google Scholar] [CrossRef]
- Seo, B.R.; Min, K.J.; Woo, S.M.; Choe, M.; Choi, K.S.; Lee, Y.K.; Yoon, G.; Kwon, T.K. Inhibition of Cathepsin S Induces Mitochondrial ROS That Sensitizes TRAIL-Mediated Apoptosis through p53-Mediated Downregulation of Bcl-2 and c-FLIP. Antioxid. Redox Signal. 2017, 27, 215–233. [Google Scholar] [CrossRef]
- Chang, L.; Kamata, H.; Solinas, G.; Luo, J.L.; Maeda, S.; Venuprasad, K.; Liu, Y.C.; Karin, M. The E3 ubiquitin ligase itch couples JNK activation to TNFalpha-induced cell death by inducing c-FLIP(L) turnover. Cell 2006, 124, 601–613. [Google Scholar] [CrossRef]
- Jeong, M.; Lee, E.W.; Seong, D.; Seo, J.; Kim, J.H.; Grootjans, S.; Kim, S.Y.; Vandenabeele, P.; Song, J. USP8 suppresses death receptor-mediated apoptosis by enhancing FLIPL stability. Oncogene 2017, 36, 458–470. [Google Scholar] [CrossRef]
- Chen, H.; Yang, F.; Li, X.; Gong, Z.J.; Wang, L.W. Long noncoding RNA LNC473 inhibits the ubiquitination of survivin via association with USP9X and enhances cell proliferation and invasion in hepatocellular carcinoma cells. Biochem. Biophys. Res. Commun. 2018, 499, 702–710. [Google Scholar] [CrossRef] [PubMed]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Woo, S.M.; Seo, S.U.; Kubatka, P.; Min, K.-j.; Kwon, T.K. Honokiol Enhances TRAIL-Mediated Apoptosis through STAMBPL1-Induced Survivin and c-FLIP Degradation. Biomolecules 2019, 9, 838. https://doi.org/10.3390/biom9120838
Woo SM, Seo SU, Kubatka P, Min K-j, Kwon TK. Honokiol Enhances TRAIL-Mediated Apoptosis through STAMBPL1-Induced Survivin and c-FLIP Degradation. Biomolecules. 2019; 9(12):838. https://doi.org/10.3390/biom9120838
Chicago/Turabian StyleWoo, Seon Min, Seung Un Seo, Peter Kubatka, Kyoung-jin Min, and Taeg Kyu Kwon. 2019. "Honokiol Enhances TRAIL-Mediated Apoptosis through STAMBPL1-Induced Survivin and c-FLIP Degradation" Biomolecules 9, no. 12: 838. https://doi.org/10.3390/biom9120838
APA StyleWoo, S. M., Seo, S. U., Kubatka, P., Min, K.-j., & Kwon, T. K. (2019). Honokiol Enhances TRAIL-Mediated Apoptosis through STAMBPL1-Induced Survivin and c-FLIP Degradation. Biomolecules, 9(12), 838. https://doi.org/10.3390/biom9120838