Clerodane Diterpene Ameliorates Inflammatory Bowel Disease and Potentiates Cell Apoptosis of Colorectal Cancer

 ,
, 
Abstract
1. Introduction
2. Experimental Section
2.1. Study Design
2.2. Chemicals
2.3. In Vivo Test
2.3.1. Animal Source and Care
2.3.2. Inflammatory Bowel Disease Model Induction and HCD Treatment
2.4. In Vitro Test
2.4.1. Cell Culture
2.4.2. Cytotoxicity Assay
2.4.3. Cell Cycle Analysis
2.4.4. Western Blotting
2.5. Statistical Analysis
3. Results
3.1. Histological Change of Intestine Tissue after AOM/DSS Induction and HCD Treatment
3.2. Cytotoxicity Effects of HCD and 5-Fluorouracil on Colorectal Cancer Cells
3.3. Characteristics of HCD-Induced Cell Death
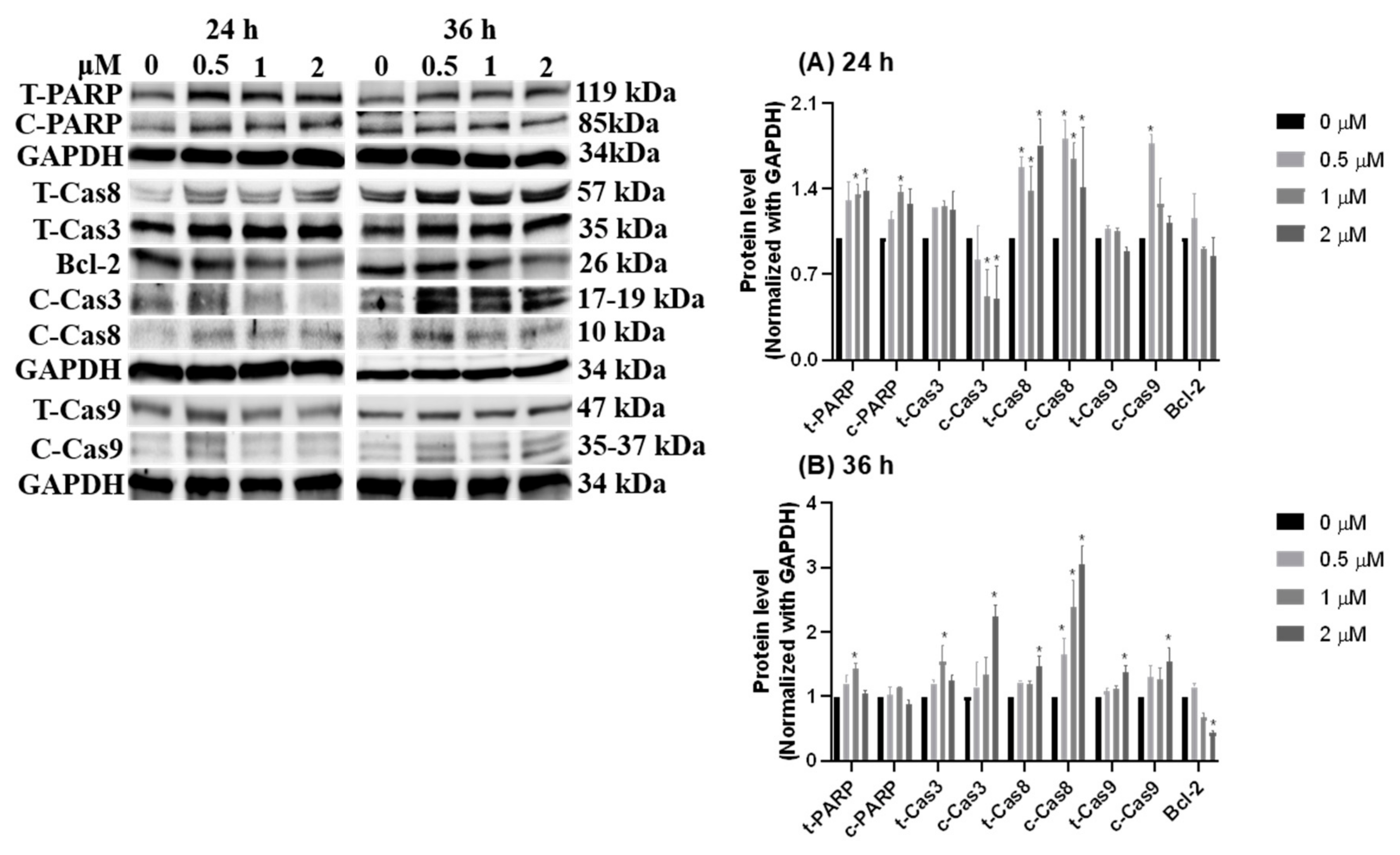
3.4. Growth Signal Reduced by HCD Triggered Caco-2 Cell Apoptosis
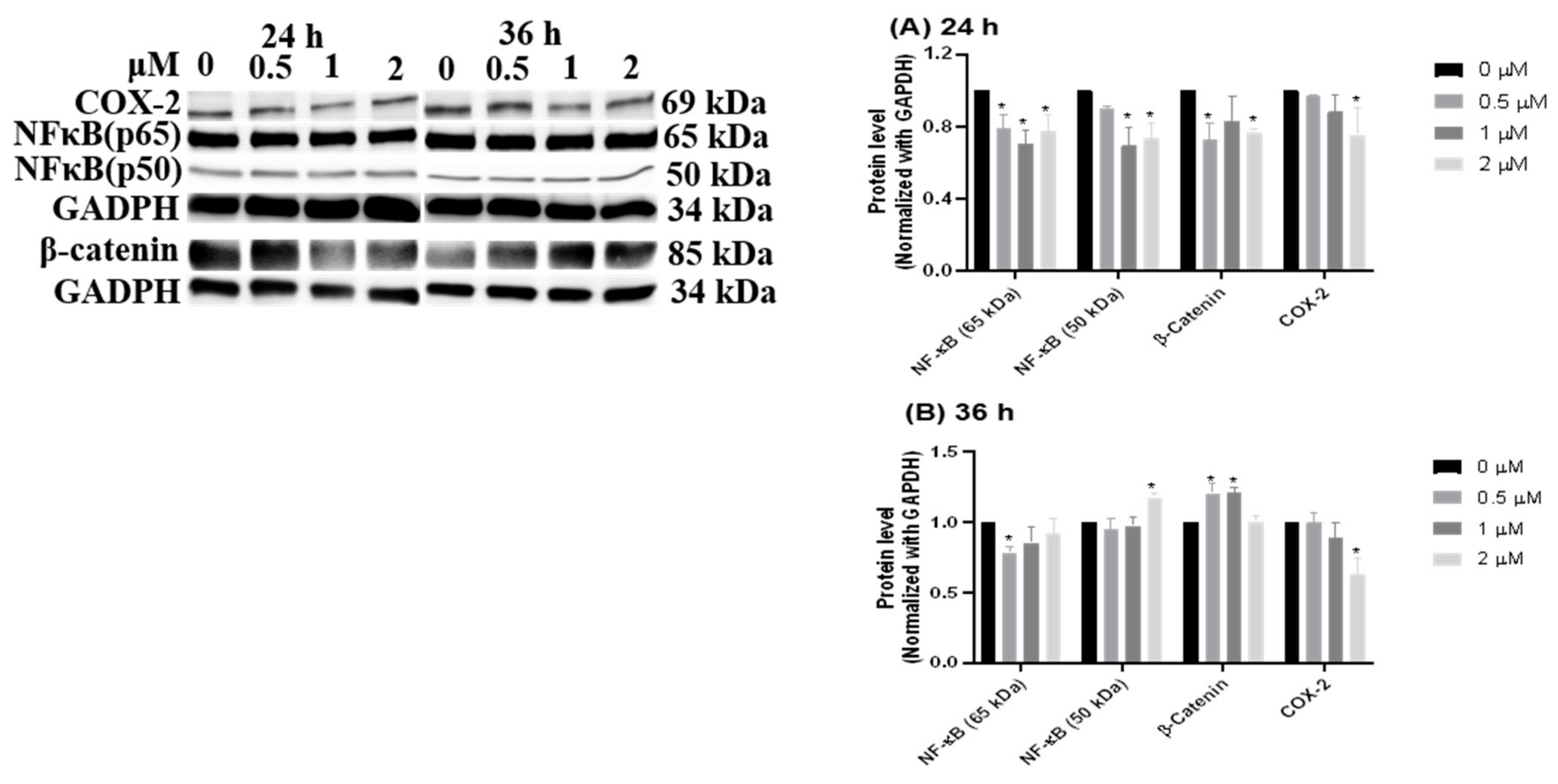
3.5. Inflammation-Suppressing Effect of HCD in Caco-2 Cells
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Chiang, T.Y.; Wang, C.H.; Lin, Y.F.; You, J.F.; Chen, J.S.; Chen, S.C. Colorectal cancer in Taiwan: A case-control retrospective analysis of the impact of a case management programme on refusal and discontinuation of treatment. J. Adv. Nurs. 2018, 74, 395–406. [Google Scholar] [CrossRef]
- Coppede, F. The role of epigenetics in colorectal cancer. Expert Rev. Gastroenterol. Hepatol. 2014, 8, 935–948. [Google Scholar] [CrossRef]
- Board, P. Colon Cancer Treatment (PDQ(R)): Health Professional Version. In PDQ Cancer Information Summaries; PDQ Adult Treatment Editorial Board—National Cancer Institute: Bethesda, MD, USA, 2002. [Google Scholar]
- Zippi, M.; Pica, R.; De Nitto, D.; Paoluzi, P. Biological therapy for dermatological manifestations of inflammatory bowel disease. World J. Clin. Cases 2013, 1, 74–78. [Google Scholar] [CrossRef]
- Ng, S.C.; Shi, H.Y.; Hamidi, N.; Underwood, F.E.; Tang, W.; Benchimol, E.I.; Panaccione, R.; Ghosh, S.; Wu, J.C.Y.; Chan, F.K.L.; et al. Worldwide incidence and prevalence of inflammatory bowel disease in the 21st century: a systematic review of population-based studies. Lancet 2017, 390, 2769–2778. [Google Scholar] [CrossRef]
- Neurath, M.F. Cytokines in inflammatory bowel disease. Nat. Rev. Immunol. 2014, 14, 329–342. [Google Scholar] [CrossRef] [PubMed]
- Robles, A.I.; Traverso, G.; Zhang, M.; Roberts, N.J.; Khan, M.A.; Joseph, C.; Lauwers, G.Y.; Selaru, F.M.; Popoli, M.; Pittman, M.E.; et al. Whole-Exome Sequencing Analyses of Inflammatory Bowel Disease-Associated Colorectal Cancers. Gastroenterology 2016, 150, 931–943. [Google Scholar] [CrossRef] [PubMed]
- Gracie, D.J.; Irvine, A.J.; Sood, R.; Mikocka-Walus, A.; Hamlin, P.J.; Ford, A.C. Effect of psychological therapy on disease activity, psychological comorbidity, and quality of life in inflammatory bowel disease: a systematic review and meta-analysis. Lancet Gastroenterol. Hepatol. 2017, 2, 189–199. [Google Scholar] [CrossRef]
- Van der Sloot, K.W.J.; Amini, M.; Peters, V.; Dijkstra, G.; Alizadeh, B.Z. Inflammatory Bowel Diseases: Review of Known Environmental Protective and Risk Factors Involved. Inflamm. Bowel Dis. 2017, 23, 1499–1509. [Google Scholar] [CrossRef]
- Gecse, K.B.; Vermeire, S. Differential diagnosis of inflammatory bowel disease: imitations and complications. Lancet Gastroenterol. Hepatol. 2018, 3, 644–653. [Google Scholar] [CrossRef]
- Newman, D.J.; Cragg, G.M. Natural Products as Sources of New Drugs from 1981 to 2014. J. Nat. Prod. 2016, 79, 629–661. [Google Scholar] [CrossRef]
- Katkar, K.V.; Suthar, A.C.; Chauhan, V.S. The chemistry, pharmacologic, and therapeutic applications of Polyalthia longifolia. Pharm. Rev. 2010, 4, 62–68. [Google Scholar] [CrossRef]
- Shih, Y.T.; Hsu, Y.Y.; Chang, F.R.; Wu, Y.C.; Lo, Y.C. 6-Hydroxycleroda-3,13-dien-15,16-olide protects neuronal cells from lipopolysaccharide-induced neurotoxicity through the inhibition of microglia-mediated inflammation. Planta Med. 2010, 76, 120–127. [Google Scholar] [CrossRef] [PubMed]
- Yao, L.J.; Jalil, J.; Attiq, A.; Hui, C.C.; Zakaria, N.A. The medicinal uses, toxicities and anti-inflammatory activity of Polyalthia species (Annonaceae). J. Ethnopharmacol. 2019, 229, 303–325. [Google Scholar] [CrossRef] [PubMed]
- Thiyagarajan, V.; Sivalingam, K.S.; Viswanadha, V.P.; Weng, C.F. 16-hydroxy-cleroda-3,13-dien-16,15-olide induced glioma cell autophagy via ROS generation and activation of p38 MAPK and ERK-1/2. Env. Toxicol. Pharm. 2016, 45, 202–211. [Google Scholar] [CrossRef]
- Cheng, M.F.; Lin, S.R.; Tseng, F.J.; Huang, Y.C.; Tsai, M.J.; Fu, Y.S.; Weng, C.F. The autophagic inhibition oral squamous cell carcinoma cancer growth of 16-hydroxy-cleroda-3,14-dine-15,16-olide. Oncotarget 2017, 8, 78379–78396. [Google Scholar] [CrossRef]
- Chen, Y.C.; Huang, B.M.; Lee, W.C.; Chen, Y.C. 16-Hydroxycleroda-3,13-dien-15,16-olide induces anoikis in human renal cell carcinoma cells: involvement of focal adhesion disassembly and signaling. Onco Targets 2018, 11, 7679–7690. [Google Scholar] [CrossRef]
- Liu, C.; Lee, W.C.; Huang, B.M.; Chia, Y.C.; Chen, Y.C.; Chen, Y.C. 16-Hydroxycleroda-3, 13-dien-15, 16-olide inhibits the proliferation and induces mitochondrial-dependent apoptosis through Akt, mTOR, and MEK-ERK pathways in human renal carcinoma cells. Phytomedicine 2017, 36, 95–107. [Google Scholar] [CrossRef]
- Velmurugan, B.K.; Wang, P.C.; Weng, C.F. 16-Hydroxycleroda-3,13-dien-15,16-olide and N-Methyl-Actinodaphine Potentiate Tamoxifen-Induced Cell Death in Breast Cancer. Molecules 2018, 23, 1966. [Google Scholar] [CrossRef]
- Chen, C.Y.; Chang, F.R.; Shih, Y.C.; Hsieh, T.J.; Chia, Y.C.; Tseng, H.Y.; Chen, H.C.; Chen, S.J.; Hsu, M.C.; Wu, Y.C. Cytotoxic constituents of Polyalthia longifolia var. pendula. J. Nat. Prod. 2000, 63, 1475–1478. [Google Scholar] [CrossRef]
- Parang, B.; Barrett, C.W.; Williams, C.S. AOM/DSS Model of Colitis-Associated Cancer. In Gastrointestinal Physiology and Diseases; Ivanov, A., Ed.; Humana Press: New York, NY, USA, 2016; Volume 1422, pp. 297–307. [Google Scholar]
- Janakiram, N.B.; Rao, C.V. The Role of Inflammation in Colon Cancer. In Inflammation and Cancer; Aggarwal, B., Sung, B., Gupta, S., Eds.; Springer: Basel, Switzerland, 2014; Volume 816, pp. 25–52. [Google Scholar]
- Ma, B.; Hottiger, M.O. Crosstalk between Wnt/beta-Catenin and NF-kappaB Signaling Pathway during Inflammation. Front. Immunol. 2016, 7, 378. [Google Scholar] [CrossRef]
- Wheat, C.L.; Ko, C.W.; Clark-Snustad, K.; Grembowski, D.; Thornton, T.A.; Devine, B. Inflammatory Bowel Disease (IBD) pharmacotherapy and the risk of serious infection: a systematic review and network meta-analysis. Bmc Gastroenterol. 2017, 17, 52. [Google Scholar] [CrossRef] [PubMed]
- Triantafillidis, J.K.; Merikas, E.; Georgopoulos, F. Current and emerging drugs for the treatment of inflammatory bowel disease. Drug Des. Devel. 2011, 5, 185–210. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Li, C.J.; Yang, J.Z.; Ma, J.; Wu, L.Q.; Wang, W.J.; Zhang, D.M. Phenylpropanoid and lignan glycosides from the aerial parts of Lespedeza cuneata. Phytochemistry 2016, 121, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Vecchi Brumatti, L.; Marcuzzi, A.; Tricarico, P.M.; Zanin, V.; Girardelli, M.; Bianco, A.M. Curcumin and inflammatory bowel disease: potential and limits of innovative treatments. Molecules 2014, 19, 21127–21153. [Google Scholar] [CrossRef]
- Lewis, S.N.; Brannan, L.; Guri, A.J.; Lu, P.; Hontecillas, R.; Bassaganya-Riera, J.; Bevan, D.R. Dietary alpha-eleostearic acid ameliorates experimental inflammatory bowel disease in mice by activating peroxisome proliferator-activated receptor-gamma. PLoS ONE 2011, 6, e24031. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef]
- Tummers, B.; Green, D.R. Caspase-8: regulating life and death. Immunol. Rev. 2017, 277, 76–89. [Google Scholar] [CrossRef]
- Li, P.; Zhou, L.; Zhao, T.; Liu, X.; Zhang, P.; Liu, Y.; Zheng, X.; Li, Q. Caspase-9: structure, mechanisms and clinical application. Oncotarget 2017, 8, 23996–24008. [Google Scholar] [CrossRef]
- Czabotar, P.E.; Lessene, G.; Strasser, A.; Adams, J.M. Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy. Nat. Rev. Mol. Cell Biol. 2014, 15, 49–63. [Google Scholar] [CrossRef]
- Lin, Y.H.; Lee, C.C.; Chan, W.L.; Chang, W.H.; Wu, Y.C.; Chang, J.G. 16-Hydroxycleroda-3,13-dien-15,16-olide deregulates PI3K and Aurora B activities that involve in cancer cell apoptosis. Toxicology 2011, 285, 72–80. [Google Scholar] [CrossRef]
- Lin, Y.H.; Lee, C.C.; Chang, F.R.; Chang, W.H.; Wu, Y.C.; Chang, J.G. 16-hydroxycleroda-3,13-dien-15,16-olide regulates the expression of histone-modifying enzymes PRC2 complex and induces apoptosis in CML K562 cells. Life Sci. 2011, 89, 886–895. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, C.; Feng, Z. Tumor suppressor p53 and its gain-of-function mutants in cancer. Acta Biochim. Biophys. Sin. (Shanghai) 2014, 46, 170–179. [Google Scholar] [CrossRef] [PubMed]
- Goretsky, T.; Dirisina, R.; Sinh, P.; Mittal, N.; Managlia, E.; Williams, D.B.; Posca, D.; Ryu, H.; Katzman, R.B.; Barrett, T.A. p53 mediates TNF-induced epithelial cell apoptosis in IBD. Am. J. Pathol. 2012, 181, 1306–1315. [Google Scholar] [CrossRef] [PubMed]
- Pastor, D.M.; Irby, R.B.; Poritz, L.S. Tumor necrosis factor alpha induces p53 up-regulated modulator of apoptosis expression in colorectal cancer cell lines. Dis. Colon Rectum 2010, 53, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Yashiro, M. Molecular Alterations of Colorectal Cancer with Inflammatory Bowel Disease. Dig. Dis. Sci. 2015, 60, 2251–2263. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, D.; Eide, P.W.; Eilertsen, I.A.; Danielsen, S.A.; Eknaes, M.; Hektoen, M.; Lind, G.E.; Lothe, R.A. Epigenetic and genetic features of 24 colon cancer cell lines. Oncogenesis 2013, 2, e71. [Google Scholar] [CrossRef] [PubMed]
- Triki, M.; Lapierre, M.; Cavailles, V.; Mokdad-Gargouri, R. Expression and role of nuclear receptor coregulators in colorectal cancer. World J. Gastroenterol. 2017, 23, 4480–4490. [Google Scholar] [CrossRef] [PubMed]
- Koch, S. Extrinsic control of Wnt signaling in the intestine. Differentiation 2017, 97, 1–8. [Google Scholar] [CrossRef]
- Asif, M.; Shafaei, A.; Abdul Majid, A.S.; Ezzat, M.O.; Dahham, S.S.; Ahamed, M.B.K.; Oon, C.E.; Abdul Majid, A.M.S. Mesua ferrea stem bark extract induces apoptosis and inhibits metastasis in human colorectal carcinoma HCT 116 cells, through modulation of multiple cell signalling pathways. Chin. J. Nat. Med. 2017, 15, 505–514. [Google Scholar] [CrossRef]
- Lee, M.A.; Kim, W.K.; Park, H.J.; Kang, S.S.; Lee, S.K. Anti-proliferative activity of hydnocarpin, a natural lignan, is associated with the suppression of Wnt/beta-catenin signaling pathway in colon cancer cells. Bioorg. Med. Chem. Lett. 2013, 23, 5511–5514. [Google Scholar] [CrossRef]
- Hseu, Y.C.; Chao, Y.H.; Lin, K.Y.; Way, T.D.; Lin, H.Y.; Thiyagarajan, V.; Yang, H.L. Antrodia camphorata inhibits metastasis and epithelial-to-mesenchymal transition via the modulation of claudin-1 and Wnt/beta-catenin signaling pathways in human colon cancer cells. J. Ethnopharmacol. 2017, 208, 72–83. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein Name | Molecular Weight (KDa) | Host | Manufacture | Dilution Factor |
|---|---|---|---|---|
| iNOS | 131 | Rabbit | Genetex | 1:1000 |
| PARP | 119/85 | + | Cell Signaling | + |
| PI3K | 85 | + | Genetex | + |
| β-catenin | 85 | + | + | + |
| COX-2 | 69 | + | + | + |
| NF-κB (p65) | 65 | + | + | + |
| Akt | 60 | + | Cell Signaling | + |
| caspase-8 | 57/10 | + | + | + |
| p53 | 53-55 | + | + | + |
| NF-kB (p50) | 50 | + | Genetex | + |
| caspase-9 | 47/35-37 | Mouse | Cell Signaling | + |
| WNT11 | 39 | Rabbit | Genetex | + |
| cyclin D- | 36 | + | Cell Signaling | + |
| PCNA | 36 | Mouse | + | + |
| caspase-3 | 35/17-19 | Rabbit | + | + |
| GADPH | 34 | + | Genetex | + |
| p27 | 27 | + | Cell Signaling | + |
| Bcl-2 | 26 | + | + | + |
| Bad | 23 | + | + | + |
| p21 | 21 | + | Genetex | + |
| Bax | 20 | + | Cell Signaling | + |
| Anti-mouse HRP-conjugated 2nd Ab | Goat | Merck Millipore | 1:5000 | |
| Anti-mouse HRP-conjugated 2nd Ab | + | + | + |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zheng, J.-H.; Lin, S.-R.; Tseng, F.-J.; Tsai, M.-J.; Lue, S.-I.; Chia, Y.-C.; Woon, M.; Fu, Y.-S.; Weng, C.-F. Clerodane Diterpene Ameliorates Inflammatory Bowel Disease and Potentiates Cell Apoptosis of Colorectal Cancer. Biomolecules 2019, 9, 762. https://doi.org/10.3390/biom9120762
Zheng J-H, Lin S-R, Tseng F-J, Tsai M-J, Lue S-I, Chia Y-C, Woon M, Fu Y-S, Weng C-F. Clerodane Diterpene Ameliorates Inflammatory Bowel Disease and Potentiates Cell Apoptosis of Colorectal Cancer. Biomolecules. 2019; 9(12):762. https://doi.org/10.3390/biom9120762
Chicago/Turabian StyleZheng, Jia-Huei, Shian-Ren Lin, Feng-Jen Tseng, May-Jywan Tsai, Sheng-I Lue, Yi-Chen Chia, Mindar Woon, Yaw-Syan Fu, and Ching-Feng Weng. 2019. "Clerodane Diterpene Ameliorates Inflammatory Bowel Disease and Potentiates Cell Apoptosis of Colorectal Cancer" Biomolecules 9, no. 12: 762. https://doi.org/10.3390/biom9120762
APA StyleZheng, J.-H., Lin, S.-R., Tseng, F.-J., Tsai, M.-J., Lue, S.-I., Chia, Y.-C., Woon, M., Fu, Y.-S., & Weng, C.-F. (2019). Clerodane Diterpene Ameliorates Inflammatory Bowel Disease and Potentiates Cell Apoptosis of Colorectal Cancer. Biomolecules, 9(12), 762. https://doi.org/10.3390/biom9120762