Molecular Docking Studies of a Cyclic Octapeptide-Cyclosaplin from Sandalwood



Abstract
1. Introduction
2. Materials and Methods
2.1. Softwares and Tools
2.2. Ligand Preparation
2.3. Lipinski Rule for Ligands
- o
- Molecular mass less than 500 Da;
- o
- High lipophilicity (expressed as LogP less than 5);
- o
- Less than 5 hydrogen bond donors;
- o
- Less than 10 hydrogen bond acceptors;
- o
- Molar refractivity between 40 and 130.
2.4. Protein Preparation
2.5. Docking Studies Using AutoDock Vina
2.6. Protein–Ligand Interactions
3. Results
3.1. Ligand Preparation
3.2. Lipinski Rule
3.3. Protein Preparation
3.4. Docking Studies Using AutoDock Vina
3.5. Protein–Ligand Interactions
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Crowell, J.A. The chemopreventive agent development research program in the Division of Cancer Prevention of the US National Cancer Institute: An overview. Eur. J. Cancer 2005, 41, 1889–1910. [Google Scholar] [CrossRef] [PubMed]
- DiMasi, J.A.; Hansen, R.W.; Grabowski, H.G. The price of innovation: New estimates of drug development costs. J. Health Econ. 2002, 22, 151–185. [Google Scholar] [CrossRef]
- Guariento, S.; Tonelli, M.; Espinoza, S.; Gerasimov, A.S.; Gainetdinov, R.R.; Cichero, E. Rational design, chemical synthesis and biological evaluation of novel biguanides exploring species-specificity responsiveness of TAAR1 agonists. Eur. J. Med. Chem. 2018, 146, 171–184. [Google Scholar] [CrossRef] [PubMed]
- Franchini, S.; Manasieva, L.I.; Sorbi, C.; Battisti, U.M.; Fossa, P.; Cichero, E.; Denora, N.; Iacobazzi, R.M.; Cilia, A.; Pirona, L.; et al. Synthesis, biological evaluation and molecular modeling of 1-oxa-4-thiaspiro-and 1,4-dithiaspiro[4.5]decane derivatives as potent and selective 5-HT1A receptor agonists. Eur. J. Med. Chem. 2017, 125, 435–452. [Google Scholar] [CrossRef]
- Dileep, K.V.; Kelly, M.; Hardin, E. Approaches in the Chemoprevention of Breast Cancer. Cancer Sci. 2013, 5, 1948–5956. [Google Scholar]
- Agyei, D.; Danquah, M.K. Industrial-scale manufacturing of pharmaceuticalgrade bioactive peptides. Biotechnol. Adv. 2011, 29, 272–277. [Google Scholar] [CrossRef]
- Joo, S.H. Cyclic peptides as therapeutic agents and biochemical tools. Biomolecules 2012, 20, 19–26. [Google Scholar] [CrossRef]
- Gurrath, M.; Müller, G.; Kessler, H.; Aumailley, M.; Timpl, R. Conformation/activity studies of rationally designed potent anti-adhesive RGD peptides. Eur. J. Biochem. 1992, 210, 911–921. [Google Scholar] [CrossRef]
- Hotchkiss, R.D. The chemical nature of gramicidin and tyrocidine. J. Biol. Chem. 1941, 141, 171–185. [Google Scholar]
- Vlieghe, P.; Lisowski, V.; Martinez, J.; Khrestchatisky, M. Synthetic therapeutic peptides: Science and market. Drug Discov. Today 2010, 15, 40–56. [Google Scholar] [CrossRef]
- Mishra, A.; Gauri, S.S.; Mukhopadhyay, S.K.; Chatterjee, S.; Das, S.S.; Mandal, S.M.; Dey, S. Identification and structural characterization of a new pro-apoptotic cyclic octapeptide cyclosaplin from somatic seedling of Santalum album L. Peptides 2014, 54, 148–158. [Google Scholar] [CrossRef] [PubMed]
- Gilanski, K.; Rychewlski, L. Protein structure prediction of CAPS5 comparative modelling and fold recognition targets using consensus alignment and approach and 3D assessment. Proteins 2003, 53, 410–417. [Google Scholar]
- Bikadi, Z.; Hazai, E. Application of the PM6 semi-empirical method to modeling proteins enhances docking accuracy of AutoDock. J. Cheminform. 2009, 1, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Vila, M.C.; Giodarno, R.J.; Sidman, R.L.; Bronk, L.F. From combinatorial peptide selection to drug prototype (II): Targeting the epidermal growth factor receptor pathway. Proc. Nat. Acad. Sci. USA 2010, 107, 5118–5123. [Google Scholar] [CrossRef]
- Alghisi, G.; Ponsonnet, L.; Rüegg, C. The integrin antagonist cilengitide activates αVβ3, disrupts VE-cadherin localization at cell junctions and enhances permeability in endothelial cells. Public Libr. Sci. One 2009, 4, e4449. [Google Scholar] [CrossRef] [PubMed]
- Tal-Gan, Y.; Hurevich, M.; Klein, S.; Ben-Shimon, A.; Rosenthal, D.; Hazan, C.; Shalev, D.E.; Niv, M.Y.; Levitzki, A.; Gilon, C. Backbone cyclic peptide inhibitors of protein kinase B (PKB/Akt). J. Med. Chem. 2011, 54, 5154–5164. [Google Scholar] [CrossRef] [PubMed]
- Gill, K.; Nigam, L.; Singh, R.; Kumar, S.; Dey, S. The Rational Design of Specific Peptide Inhibitor against p38α MAPK at Allosteric-Site: A Therapeutic Modality for HNSCC. PLoS ONE 2014, 9, e101525. [Google Scholar] [CrossRef]
- Zhu, Z.; Jia, J.; Lu, R.; Lu, Y.; Fu, Z.; Zhao, L.; Wang, L.; Jin, M.; Zhao, L.; Gao, W.; et al. Expression of PTEN, p27, p21 and AKT mRNA and protein in human BEL-7402 hepatocarcinoma cells in transplanted tumors of nude mice treated with the tripeptide tyroservatide (YSV). Int. J. Cancer 2006, 118, 1539–1544. [Google Scholar] [CrossRef]
- Koivunen, E.; Arap, W.; Valtanen, H.; Rainisalo, A.; Medina, O.P.; Heikkilä, P.; Sorsa, T. Tumor targeting with a selective gelatinase inhibitor. Nat. Biotechnol. 1999, 17, 768–774. [Google Scholar] [CrossRef]
- Aguzzi, M.S.; Giampeitri, C.; Marchis, F.D. RGDS peptide induces caspase 8 and caspase 9 activation in human endothelial cells. Blood 2004, 103, 4180–4187. [Google Scholar] [CrossRef]
- Heins, M.S.; Quax, W.J. Implications of a Newly Discovered DR5 Specific Antagonistic Peptide for Neurodegenerative Disorders. Mol. Cell. Pharmacol. 2010, 277, 1653–1665. [Google Scholar]
- Hirohashi, Y.; Torigoe, T.; Maeda, A.; Nabeta, Y. An HLA-A24-restricted Cytotoxic T Lymphocyte Epitope of a Tumor-associated Protein, Survivin. Clin. Cancer Res. 2002, 8, 1731. [Google Scholar] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Gfeller, D.; Grosdidier, A.; Wirth, M. Swiss Target Prediction: A web server for target prediction of bioactive small molecules. Nucleic Acid Res. 2013, 42, 32–38. [Google Scholar] [CrossRef]
- Berman, H.M.; Henrick, K.; Nakamura, H. Announcing the world wide Protein Data Bank. Nat. Struct. Biol. 2000, 10, 980. [Google Scholar]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Wallace, A.C.; Laskowski, R.A.; Thornton, J.M. LigPlot: A program to generate schematic diagrams of protein-ligand interactions. Protein Eng. 1995, 8, 127–134. [Google Scholar] [CrossRef]
- Roxin, A.; Zheng, G. Flexible or fixed: A comparative review of linear and cyclic cancer-targeting peptides. Future Med. Chem. 2012, 4, 1601–1618. [Google Scholar] [CrossRef]
- KCathcart, J.; Pulkoski-Gross, A.; Cao, J. Targeting matrix metalloproteinases in cancer: Bringing new life to old ideas. Genes Dis. 2015, 2, 26–34. [Google Scholar] [CrossRef]
- Koskimaki, J.E.; Pandey, N.B.; Tamiz, A.P.; Popel, A.S. Structure-activity relationship study of collagen-derived anti-angiogenic biomimetic peptides. Chem. Biol. Drug Des. 2012, 1, 27–37. [Google Scholar]
- Pero, S.C.; Shukla, G.S.; Armstrong, A.L.; Peterson, D.; Fuller, S.P.; Godin, K.; Kingsley-Richards, S.L.; Weaver, D.L.; Bond, J.; Krag, D.N. Identification of a small peptide that inhibits the phosphorylation of ErbB2 and proliferation of ErbB2 overexpressing breast cancer cells. Int. J. Cancer 2004, 111, 951–960. [Google Scholar] [CrossRef] [PubMed]
- Ubol, S.; Kramyu, J.; Masrinoul, P.; Kachangchaeng, C.; Pittayanurak, P.; Sophasan, S.; Reutrakul, V. A novel cycloheptapeptide exerts strong anticancer activity via stimulation of multiple apoptotic pathways in Caspase-3 deficient cancer cells. Anticancer Res. 2007, 27, 2473–2480. [Google Scholar] [PubMed]
- Janin, J.; Henrick, K.; Moult, J. A critical assessment of predicted interaction. Proteins Struct. Funct. Genet. 2003, 52, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, N.; Morpurgo, N.; Linial, M. Novel families of toxin-like peptides in insects and mammals: A computational approach. J. Mol. Biol. 2007, 369, 553–566. [Google Scholar] [CrossRef] [PubMed]
- Mader, J.S.; Salsman, J.; Conrad, D.M.; Hoskin, D.W. Bovine lactoferricin inhibits basic fibroblast growth factor and vascular endothelial growth factor 165-induced angiogenesis by competing for heparin-like binding sites on endothelial sites. Am. J. Pathol. 2006, 169, 1753–1766. [Google Scholar] [CrossRef]
- Ruegg, C. Vascular integrins in tumor angiogenesis: Mediators and therapeutic targets. Endothelium 2006, 13, 113–135. [Google Scholar]
- DeLorbe, J.E.; Clements, J.H.; Teresk, M.G.; Befield, A.P.; Plake, H.R.; Millspaugh, L.E.; Martin, S.F. Thermodynamic and structural effects of conformational constraints in protein-ligand interactions: Entropic paradoxy associated with ligand pre-organization. J. Am. Chem. Soc. 2009, 131, 16758–16770. [Google Scholar] [CrossRef]
- Yang, K.S.; Macdonald-Obermann, J.L.; Linda, J.P. Asp-960/Glu-961 controls the movement of the c-terminal tail of the epidermal growth factor receptor to regulate asymmetric dimer formation. J. Biol. Chem. 2010, 285, 24014–24022. [Google Scholar] [CrossRef]
- Ward, C.W.; Gough, K.H.; Rashke, M.; Wan, S.S.; Tribbick, G.; Wang, J. Systematic mapping of potential binding sites for Shc and Grb2 SH2 domains on insulin receptor substrate-1 and the receptors for insulin, epidermal growth factor, platelet-derived growth factor, and fibroblast growth factor. J. Biol. Chem. 1996, 271, 5603–5609. [Google Scholar] [CrossRef]
- Stamos, J.; Swilkowski, M.X.; Eigenbrot, C. Structure of the epidermal growth factor receptor kinase domain alone and in complex with a 4 anilinoquinazoline inhibitor. J. Biol. Chem. 2002, 277, 46265–46272. [Google Scholar] [CrossRef]
- Kyte, J.; Doolittle, R.F. A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 1982, 157, 105–132. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, N.; Han, S.; Wang, D.; Mo, S.; Yu, L.; Huang, H.; Tsui, K.; Shen, J.; Chen, J. Dietary compound Isoliquiritigenin inhibits breast cancer neoangiogenesis via VEGF/VEGFR-2 Signaling Pathway. PLoS ONE 2013, 8, e68566. [Google Scholar] [CrossRef] [PubMed]
- Olsson, A.K.; Dimberg, A.; Kreuger, J.; Claesson-Welsh, L. VEGF receptor signalling-in control of vascular function. Nat. Rev. Mol. Cell Biol. 2006, 7, 359–371. [Google Scholar] [CrossRef] [PubMed]
- Chan, T.O.; Zhang, J.; Rodeck, U.; Pascal, J.M.; Armen, R.S.; Spring, M.; Dumitru, C.D.; Myers, V.; Li, X.; Cheung, J.Y.; et al. Resistance of Akt-kinases to dephosphorylation through ATP-dependent conformational plasticity. Proc. Natl. Acad. Sci. USA 2011, 108, e1120–e1127. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Cron, P.; Good, V.M.; Thompson, V.; Hemmings, B.A.; Barford, D. Crystal structure of an activated Akt/protein kinase B ternary complex with GSK3-peptide, A.M.P.P.N.P. Nat. J. Struct. Biol. 2002, 12, 940–944. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Harkins, P.C.; Ulevitch, R.J.; Han, J.; Cobb, M.H.; Goldsmith, E.J. The structure of mitogen-activated protein kinase p38 at 2.1-Å resolution. Proc. Natl. Acad. Sci. USA 1997, 94, 2327–2332. [Google Scholar]
- De Azevedo, W.F.J.; Dieckmann, H.J.M.; Gahmen, U.S.; Worland, P.J.; Sausville, E.; Kim, S.H. Structural basis for specificity and potency of a flavonoid inhibitor of human CDK2, a cell cycle kinase. Proc. Natl. Acad. Sci. USA 1996, 83, 2735–2740. [Google Scholar] [CrossRef]
- Wilson, K.P.; Fitzgibbon, M.J.; Caron, P.N.; Griffith, J.P.; Chen, W.Y.; McCaffrey, P.G.; Chambers, S.P.; Su, M.S.S. Crystal structure of p38 mitogen-activated protein kinase. J. Biol. Chem. 1996, 271, 27696–27700. [Google Scholar] [CrossRef]
- Han, X.; Xu, Y.; Yang, Y.; Xi, J.; Tian, W. Discovery and characterization of a novel cyclic peptide that effectively inhibits ephrin binding to the EphA4 receptor and displays anti-angiogenesis activity. PLoS ONE 2013, 8, e80183. [Google Scholar] [CrossRef]
- Mishra, A.; Mukhopadhyay, S.K.; Dey, S. Evaluation of Cyclosaplin Efficacy Using a Silk Based 3D Tumor Model. Biomolecules 2019, 9, 123. [Google Scholar] [CrossRef]







{kind=link}
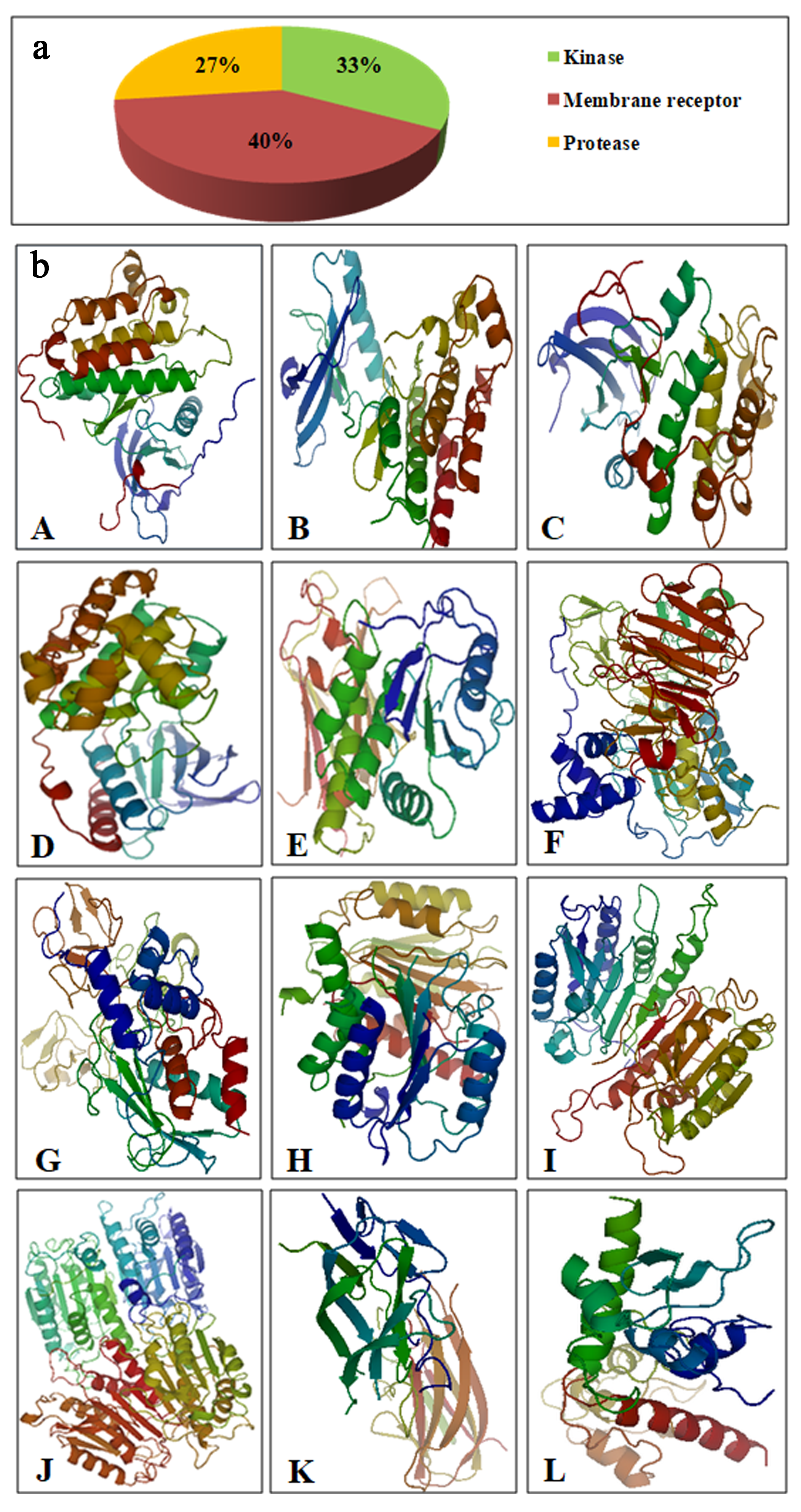{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S.No. | Ligand | References |
|---|---|---|
| 1 | CVRACGAD (Cyclic) | [14] |
| 2 | Cilengitide (Cyclic) | [15] |
| 3 | RPRTSSF (Cyclic) | [16] |
| 4 | FWCS (Linear) | [17] |
| 5 | YSV (Linear) | [18] |
| 6 | CTTHWGFTLC (Cyclic) | [19] |
| 7 | CRRHWGFEFC (Cyclic) | [19] |
| 8 | RGDS (Linear) | [20] |
| 9 | CKVILTHRC (Cyclic) | [21] |
| 10 | AYACNTSTL (Linear) | [22] |
| 11 | Cyclosaplin (Cyclic) | [11] |
| S.No. | Ligand | Molecular Weight (Da) | Molecular Formula |
|---|---|---|---|
| 1 | CVRACGAD (Cyclic) | 791.9 | C29H49N11O11S2 |
| 2 | Cilengitide (Cyclic) | 588.6 | C27H40N8O7 |
| 3 | RPRTSSF (Cyclic) | 875.0 | C39H66N14O9 |
| 4 | FWCS (Linear) | 541.6 | C26H31N5O6S1 |
| 5 | YSV (Linear) | 367.4 | C17H25N3O6 |
| 6 | CTTHWGFTLC (Cyclic) | 1166.3 | C52H71N13O14S2 |
| 7 | CRRHWGFEFC (Cyclic) | 1338.5 | C60H79N19O13S2 |
| 8 | RGDS (Linear) | 433.4 | C15H27N7O8 |
| 9 | CKVILTHRC (Cyclic) | 1070.3 | C45H79N15O11S2 |
| 10 | AYACNTSTL (Linear) | 943.0 | C39H62N10O15S1 |
| 11 | Cyclosaplin (Cyclic) | 858.9 | C33H60N14O12S1 |
| Ligand | Molecular Weight (Da) | Hydrogen Bond Donor | Hydrogen Bond Acceptor | LogP | Molar Refractivity | Rules Satisfied |
|---|---|---|---|---|---|---|
| CVRACGAD | 791.9 | 13 | 13 | −4.7 | 206.6 | 1/5 |
| Cilengitide | 588.6 | 7 | 8 | −1.4 | 170.9 | 2/5 |
| RPRTSSF | 875.0 | 15 | 12 | −5.9 | 236.7 | 0/5 |
| FWCS | 541.6 | 8 | 7 | −0.7 | 143.2 | 2/5 |
| YSV | 367.4 | 6 | 6 | −1.0 | 93.14 | 4/5 |
| CTTHWGFTLC | 1166.3 | 16 | 17 | −4.0 | 331.1 | 1/5 |
| CRRHWGFEFC | 1338.5 | 20 | 17 | −3.6 | 381.6 | 1/5 |
| RGDS | 433.4 | 10 | 8 | −4.7 | 99.7 | 4/5 |
| CKVILTHRC | 1070.3 | 16 | 16 | −3.3 | 306.4 | 1/5 |
| AYACNTSTL | 943.0 | 16 | 16 | −6.1 | 230.0 | 0/5 |
| Cyclosaplin | 858.9 | 17 | 13 | −6.5 | 243.0 | 0/5 |
| S.No. | Receptor | Ligand | Binding Affinity (kcal/mol) |
|---|---|---|---|
| 1 | Epidermal Growth Factor Receptor Kinase | CVRACGAD Cyclosaplin | −7.7 −6.8 |
| 2 | Vascular Endothelial Growth Factor r 2 Receptor Kinase | Cilengitide Cyclosaplin | −8.1 −7.8 |
| 3 | Protein Kinase B | RPRTSSF Cyclosaplin | −7.5 −8.1 |
| 4 | p38 (Mitogen Activated Protein Kinase) | FWCS Cyclosaplin | −8.9 −8.3 |
| 5 | PTEN | YSV Cyclosaplin | −7.8 6.3 |
| 6 | Matrix metalloproteinase-2 (MMP-2) | CTTHWGFTLC Cyclosaplin | −7.8 −8.2 |
| 7 | Matrix metalloproteinase-9 (MMP-9) | CRRHWGFEFC Cyclosaplin | −8.4 −7.3 |
| 8 | Procaspase 3 | Cilengitide Cyclosaplin | −8.1 −7.8 |
| 9 | Procaspase 7 | RGDS Cyclosaplin | −6.8 −8.7 |
| 10 | Caspase 9 | RGDS Cyclosaplin | −6.7 −8.9 |
| 11 | TRAIL | CKVILTHRC Cyclosaplin | −6.4 −8.2 |
| 12 | SURVIVIN | AYACNTSTL Cyclosaplin | −7.2 −7.4 |
| S.No. | Protein | Ligand | Hydrophilic Interactions | Hydrophobic Contacts | No. of H-Bonds |
|---|---|---|---|---|---|
| 1 | EGFR Kinase | CVRACGAD | Lys721, Thr766 | Ala719, Asp831, Gly695, Gly772, Leu694, Leu768 | 2 |
| Cyclosaplin | Glu961, Asp960 (4) | Arg962, Asp950, Gln788, Gln952, Gly786, Met963, Pro951, Ser787, Tyr789 | 5 | ||
| 2 | VEGFR 2 Kinase | Cilengitide | Asn923, Asp 1046, Thr926 | Ala866, Arg1032, Arg1066, Asp1064, Cys919, Cys1045, Gly841, Leu840, Phe918, Ser925, Val848 | 3 |
| Cyclosaplin | Arg842, Arg842, Glu885 | Ala1065, Asn923, Asp1046, Asp1064, Gly843, Gly846, Gln847, Leu840, Leu1035, Lys868, Thr926, Val848 | 3 | ||
| 3 | Protein Kinase B | RPRTSSF | Arg274, Glu315, Lys181, Lys277, Tyr327 | Asp275, Glu200, Gly295, Leu317, Lys160, Phe163, Thr162, Thr199, Val198 | 5 |
| Cyclosaplin | Arg274, Arg274, Asp275, Asp293, Val272, Val272 | Ile188, Leu183, Lys181, Phe163, Thr199, Tyr273 | 6 | ||
| 4 | p38 (Mitogen Activated Protein Kinase) | FWCS | Arg173, Arg 67 | Asp168, Glu71, Glu178, His64, Leu74, Leu75, Leu171, Ly53, Phe169, Thr68, Tyr35 | 2 |
| Cyclosaplin | Asp112, Asp112, Asp150, Asn115, Lys53, Phe169, Phe169, Ser154 | Arg173, Asn114, Asn155, Asp168, Gly170, Leu167, Met109, Tyr35, Val38 | 8 | ||
| 5 | PTEN | YSV | Arg172, Arg173 | Asp324, Leu318, Leu320, Phe279, Tyr176, Tyr177, Tyr180, Val275 | 2 |
| Cyclosaplin | Ala72, Ala72, Gln87, Gln97 | Glu91, Glu99, Leu100, Pro89, Tyr88 | 4 | ||
| 6 | MMP-2 | CTTHWGFTLC | Arg98, Gly371, Gly394, Thr 511, Thr547 | Gln393, Gly216, Lys99, Lys372, Met373, Ser365, Ser546, Tyr395, Tyr425, Tyr427 | 6 |
| Cyclosaplin | Gly394, Tyr425 | Asp392, Glu515, Gln393, Gly216, Gly371, Phe512, Pro100, Pro514, Leu 548, Ser546, Thr426, Tyr427, Thr511, Tyr277, Tyr395 | 2 | ||
| 7 | MMP-9 | CRRHWGFEFC | Leu371, Arg2, Cys1 | Arg370, Arg424, Glu427, Gln391, Gly392, Lys92, Phe425, Pro97, Pro233, Ser240, Ser242, Thr426 Tyr393, Tyr423 | 3 |
| Cyclosaplin | Arg221, Thr331 | Arg279, Asp226, Asp284, Gly227, Gly285, Pro219, Pro272, Thr220, | 4 | ||
| 8 | Procaspase 3 | Cilengitide | Ala33, Arg238, Asn32, Ser36 (2), Tyr37, Tyr274, Tyr276 | Asn35, Glu272, Leu230, Lys38, His234 | 8 |
| Cyclosaplin | Gln261, Glu124, Lys186 | Arg164, Gly125, Ile126, Ile187, Leu136, Lys186, Pro188, Tyr197, Val189 | 3 | ||
| 9 | Procaspase 7 | RGDS | Arg87, Asn88, Gly228, Gln184 Thr189, Tyr229 | Arg187, Gly188, Gln287, His144, Lys285, Pro227, Ser239, Thr90 | 6 |
| Cyclosaplin | Arg170, Glu176, Glu284, Gly168, Phe282, Phe282, Ser277 | Ala169, Ala217, Arg167, Asp204, Gln276, Lys286, Leu175, Gln287, Ile288, Val215, Glu216, His283 | 7 | ||
| 10 | Caspase 9 | RGDS | Gly269, Ser339 | Ala149, Arg408, Asp150, Asp340, Gly276, Gly277, Gln399, Ile154, Ile396, Lys398, Met400, Thr337 | 2 |
| Cyclosaplin | Pro273, Ser144, Ser144 | Arg146, Asp228, Glu143, Gly147, Gly225, Gly276, Ile154, Leu155, Lys278, Lys414, Ser274 | 3 | ||
| 11 | SURVIVIN | AYACNTSTL | Arg18, Arg37, Cys31, Gly30 | Glu29, Glu36, Glu40, Gln92, Ile74, Leu14, Leu96, Leu104, Lys15, Lys90, Met1, Phe13, Phe93, Thr34 | 4 |
| Cyclosaplin | Gln92, Glu94, Lys91, Phe13 | Asp16, Gly2, Leu14, Leu96, Leu102, Lys15, Phe 93, Pro4 | 4 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mishra, A.; Dey, S. Molecular Docking Studies of a Cyclic Octapeptide-Cyclosaplin from Sandalwood. Biomolecules 2019, 9, 740. https://doi.org/10.3390/biom9110740
Mishra A, Dey S. Molecular Docking Studies of a Cyclic Octapeptide-Cyclosaplin from Sandalwood. Biomolecules. 2019; 9(11):740. https://doi.org/10.3390/biom9110740
Chicago/Turabian StyleMishra, Abheepsa, and Satyahari Dey. 2019. "Molecular Docking Studies of a Cyclic Octapeptide-Cyclosaplin from Sandalwood" Biomolecules 9, no. 11: 740. https://doi.org/10.3390/biom9110740
APA StyleMishra, A., & Dey, S. (2019). Molecular Docking Studies of a Cyclic Octapeptide-Cyclosaplin from Sandalwood. Biomolecules, 9(11), 740. https://doi.org/10.3390/biom9110740