Caffeine May Abrogate LPS-Induced Oxidative Stress and Neuroinflammation by Regulating Nrf2/TLR4 in Adult Mouse Brains

 , ,
, ,
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Material and Methods
2.1. Reagents and Antibodies
2.2. Animal Grouping and Drug Treatment
2.3. Protein Extraction from Mouse Brains
2.4. Immunofluorescence Staining
2.5. Determination of Reactive Oxygen Species
2.6. Determination of Lipid Peroxidation
2.7. Western Blot Analysis
2.8. Data Analysis
3. Results
3.1. Caffeine Prevents LPS-Induced Oxidative Stress in Mouse Brain
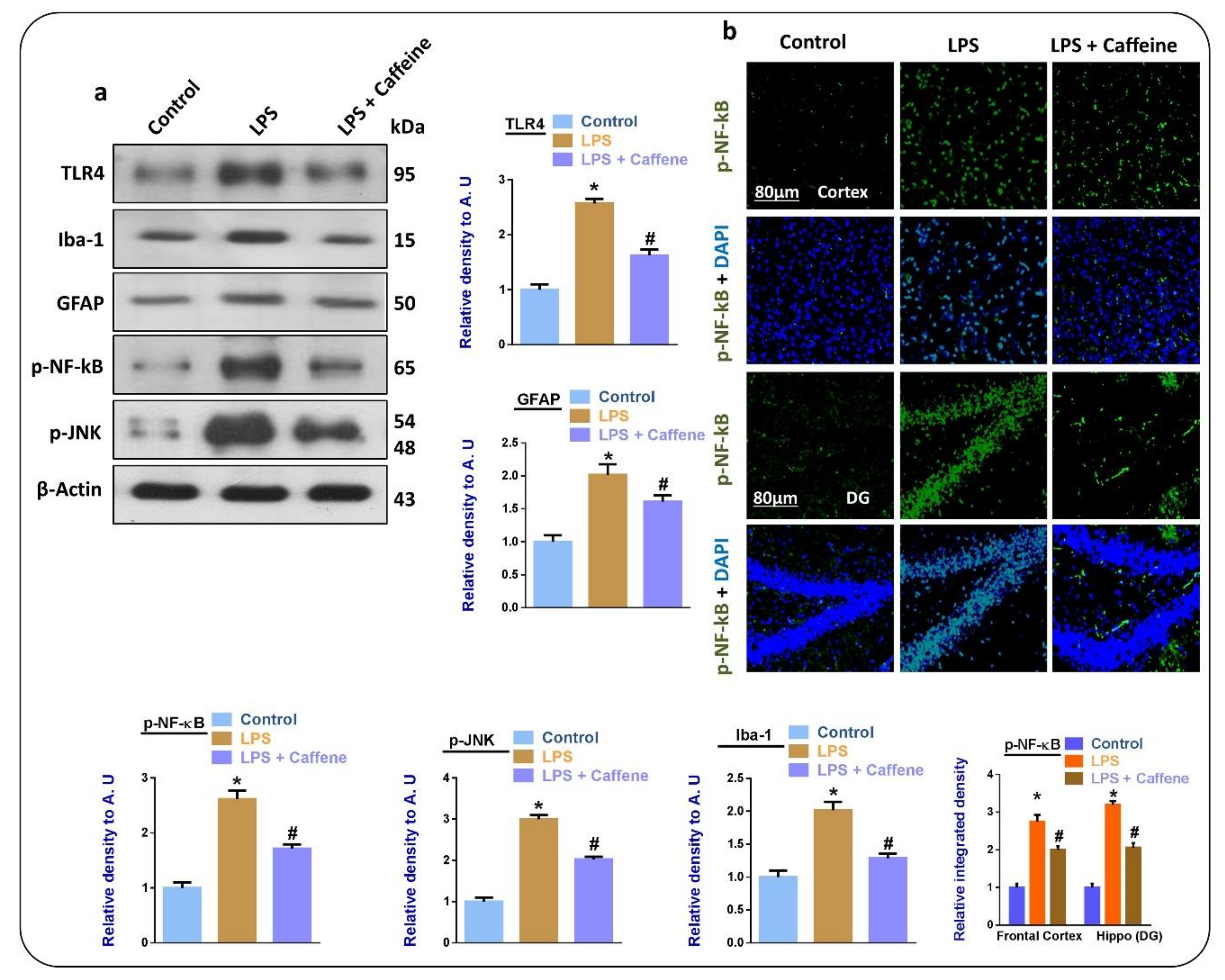
3.2. Caffeine Suppresses LPS-Induced Activated Glial Cell Markers and Inflammatory Mediators in Mouse Brain
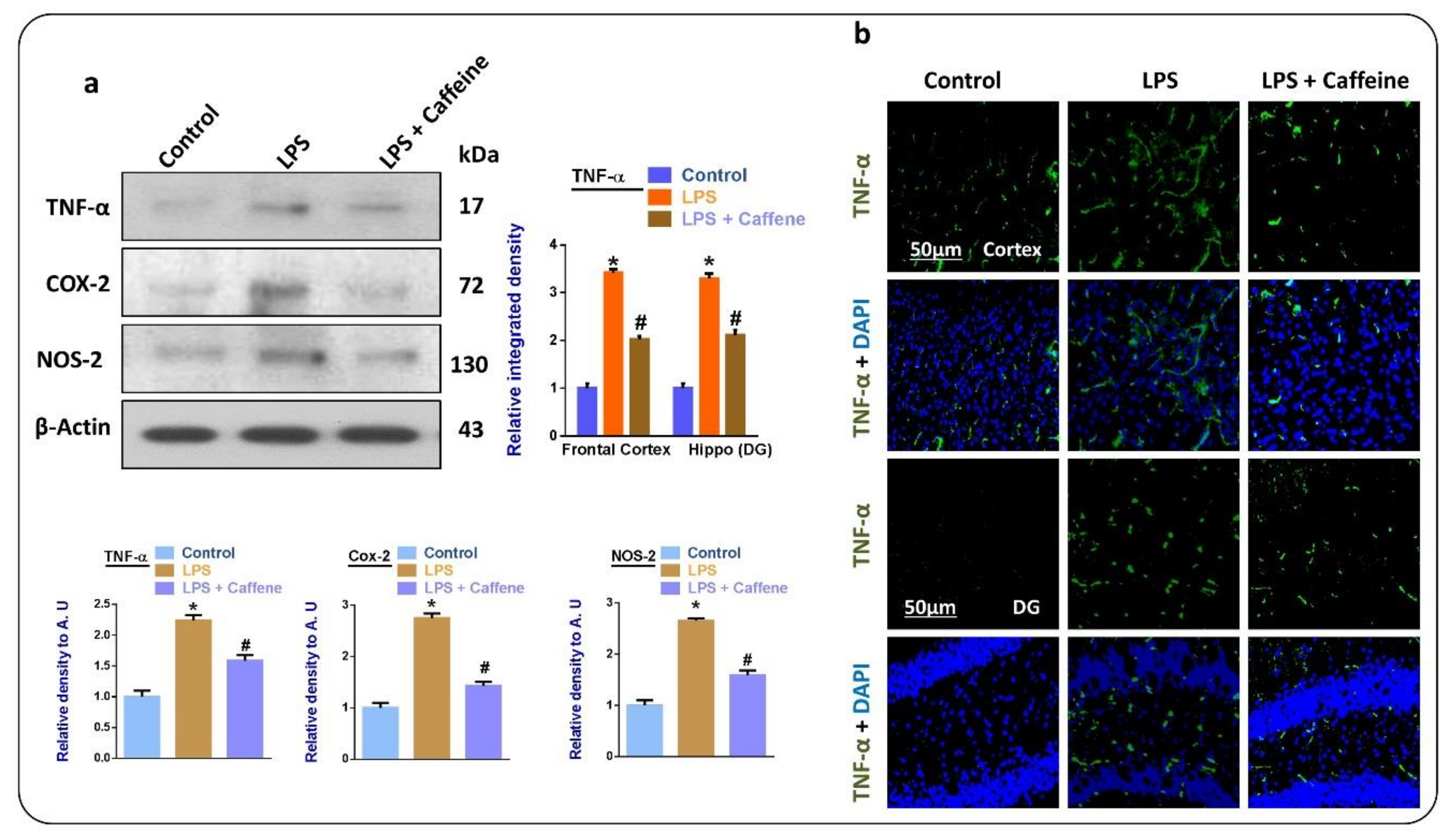
3.3. Caffeine Attenuates the LPS-Induced Release of Inflammatory Cytokines in Mouse Brain
3.4. Caffeine Attenuates LPS-Induced Mitochondrial Apoptosis in Mouse Brains
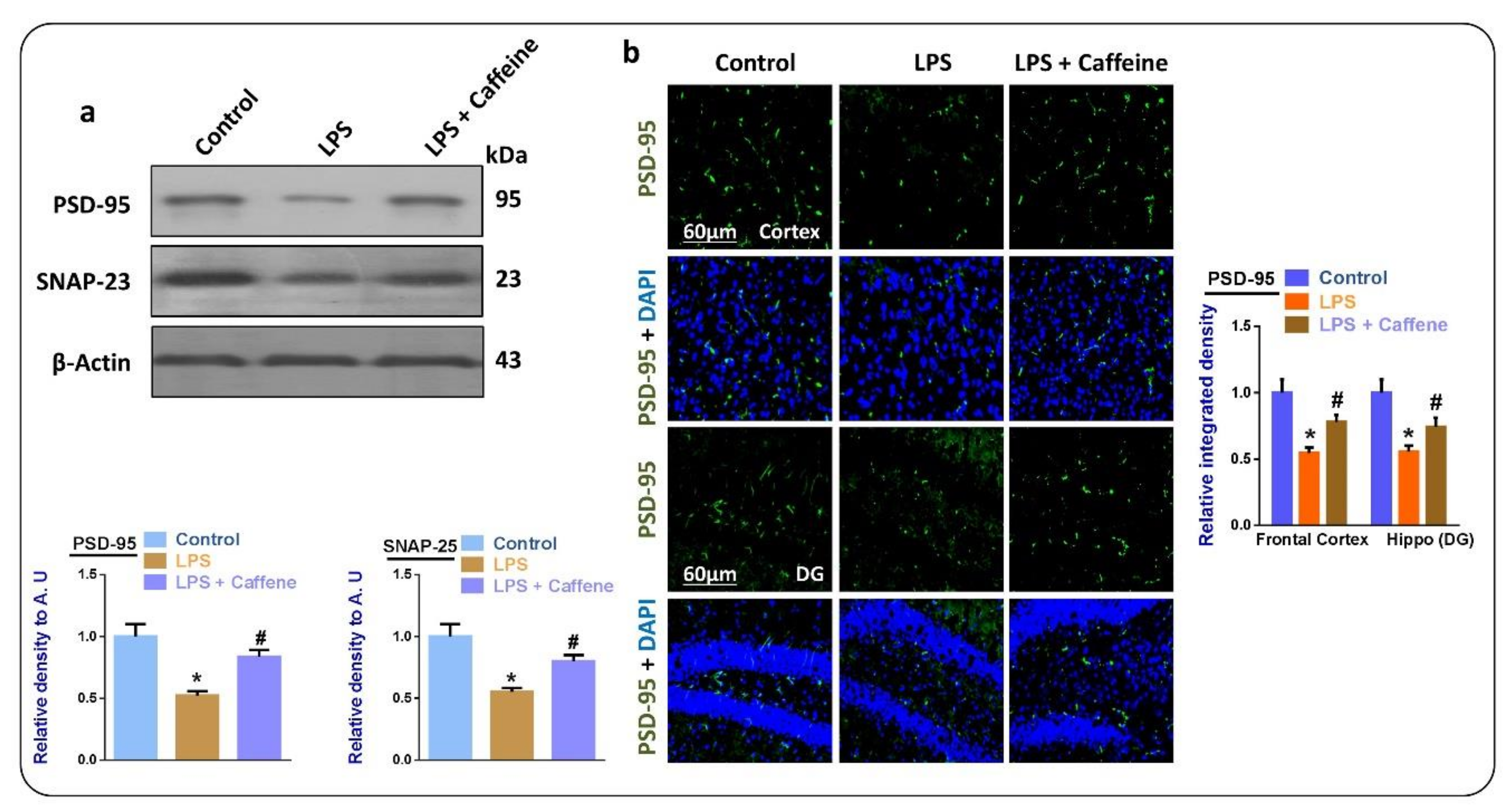
3.5. Caffeine May Attenuate LPS-Induced Synaptic Dysfunction
4. Discussion
5. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Jo, M.G.; Ikram, M.; Jo, M.H.; Yoo, L.; Chung, K.C.; Nah, S.Y.; Hwang, H.; Rhim, H.; Kim, M.O. Gintonin Mitigates MPTP-Induced Loss of Nigrostriatal Dopaminergic Neurons and Accumulation of alpha-Synuclein via the Nrf2/HO-1 Pathway. Mol. Neurobiol. 2019, 56, 39–55. [Google Scholar] [CrossRef]
- Ikram, M.; Saeed, K.; Khan, A.; Muhammad, T.; Khan, M.S.; Jo, M.G.; Rehman, S.U.; Kim, M.O. Natural Dietary Supplementation of Curcumin Protects Mice Brains against Ethanol-Induced Oxidative Stress-Mediated Neurodegeneration and Memory Impairment via Nrf2/TLR4/RAGE Signaling. Nutrients 2019, 11, 1082. [Google Scholar] [CrossRef]
- Muhammad, T.; Ikram, M.; Ullah, R.; Rehman, S.U.; Kim, M.O. Hesperetin, a Citrus Flavonoid, Attenuates LPS-Induced Neuroinflammation, Apoptosis and Memory Impairments by Modulating TLR4/NF-kappaB Signaling. Nutrients 2019, 11, 648. [Google Scholar] [CrossRef]
- Mayer, H.; Tharanathan, R.; Weckesser, J. 6 Analysis of Lipopolysaccharides of Gram-Negative Bacteria. In Methods in Microbiology; Elsevier: Amsterdam, The Netherlands, 1985; Volume 18, pp. 157–207. [Google Scholar]
- Noworyta-Sokolowska, K.; Gorska, A.; Golembiowska, K. LPS-induced oxidative stress and inflammatory reaction in the rat striatum. Pharmacol. Rep. 2013, 65, 863–869. [Google Scholar] [CrossRef]
- Chang, C.W.; Chen, Y.S.; Tsay, Y.G.; Han, C.L.; Chen, Y.J.; Yang, C.C.; Hung, K.F.; Lin, C.H.; Huang, T.Y.; Kao, S.Y.; et al. ROS-independent ER stress-mediated NRF2 activation promotes warburg effect to maintain stemness-associated properties of cancer-initiating cells. Cell Death Dis. 2018, 9, 194. [Google Scholar] [CrossRef]
- Rehman, S.U.; Ikram, M.; Ullah, N.; Alam, S.I.; Park, H.Y.; Badshah, H.; Choe, K.; Kim, M.O. Neurological Enhancement Effects of Melatonin against Brain Injury-Induced Oxidative Stress, Neuroinflammation, and Neurodegeneration via AMPK/CREB Signaling. Cells 2019, 8, 760. [Google Scholar] [CrossRef]
- Li, S.; Dong, P.; Wang, J.; Zhang, J.; Gu, J.; Wu, X.; Wu, W.; Fei, X.; Zhang, Z.; Wang, Y.; et al. Icariin, a natural flavonol glycoside, induces apoptosis in human hepatoma SMMC-7721 cells via a ROS/JNK-dependent mitochondrial pathway. Cancer Lett. 2010, 298, 222–230. [Google Scholar] [CrossRef]
- Luo, J.F.; Shen, X.Y.; Lio, C.K.; Dai, Y.; Cheng, C.S.; Liu, J.X.; Yao, Y.D.; Yu, Y.; Xie, Y.; Luo, P.; et al. Activation of Nrf2/HO-1 Pathway by Nardochinoid C Inhibits Inflammation and Oxidative Stress in Lipopolysaccharide-Stimulated Macrophages. Front. Pharmacol. 2018, 9, 911. [Google Scholar] [CrossRef] [PubMed]
- Ali, T.; Kim, T.; Rehman, S.U.; Khan, M.S.; Amin, F.U.; Khan, M.; Ikram, M.; Kim, M.O. Natural Dietary Supplementation of Anthocyanins via PI3K/Akt/Nrf2/HO-1 Pathways Mitigate Oxidative Stress, Neurodegeneration, and Memory Impairment in a Mouse Model of Alzheimer’s Disease. Mol. Neurobiol. 2018, 55, 6076–6093. [Google Scholar] [CrossRef]
- Montes Diaz, G.; Hupperts, R.; Fraussen, J.; Somers, V. Dimethyl fumarate treatment in multiple sclerosis: Recent advances in clinical and immunological studies. Autoimmun. Rev. 2018, 17, 1240–1250. [Google Scholar] [CrossRef] [PubMed]
- Vringer, E.; Tait, S.W.G. Mitochondria and Inflammation: Cell Death Heats Up. Front. Cell Dev. Biol. 2019, 7, 100. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Ikram, M.; Muhammad, T.; Park, J.; Kim, M.O. Caffeine Modulates Cadmium-Induced Oxidative Stress, neuroinflammation, and Cognitive Impairments by Regulating Nrf-2/HO-1 In Vivo and In Vitro. J. Clin. Med. 2019, 8, 680. [Google Scholar] [CrossRef] [PubMed]
- Tonelli, C.; Chio, I.I.C.; Tuveson, D.A. Transcriptional Regulation by Nrf2. Antioxid Redox Signal 2018, 29, 1727–1745. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Ali, T.; Rehman, S.U.; Khan, M.S.; Alam, S.I.; Ikram, M.; Muhammad, T.; Saeed, K.; Badshah, H.; Kim, M.O. Neuroprotective Effect of Quercetin Against the Detrimental Effects of LPS in the Adult Mouse Brain. Front. Pharmacol. 2018, 9, 1383. [Google Scholar] [CrossRef]
- Yenisetti, S.C. Beneficial Role of Coffee and Caffeine in Neurodegenerative Diseases: A Minireview. AIMS Public Health 2016, 3, 407–422. [Google Scholar] [CrossRef]
- Xu, K.; Xu, Y.H.; Chen, J.F.; Schwarzschild, M.A. Caffeine’s neuroprotection against 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine toxicity shows no tolerance to chronic caffeine administration in mice. Neurosci. Lett. 2002, 322, 13–16. [Google Scholar] [CrossRef]
- Idrees, M.; Xu, L.; Song, S.H.; Joo, M.D.; Lee, K.L.; Muhammad, T.; El Sheikh, M.; Sidrat, T.; Kong, I.K. PTPN11 (SHP2) Is Indispensable for Growth Factors and Cytokine Signal Transduction During Bovine Oocyte Maturation and Blastocyst Development. Cells 2019, 8, 1272. [Google Scholar] [CrossRef]
- Ikram, M.; Muhammad, T.; Rehman, S.U.; Khan, A.; Jo, M.G.; Ali, T.; Kim, M.O. Hesperetin Confers Neuroprotection by Regulating Nrf2/TLR4/NF-kappaB Signaling in an Abeta Mouse Model. Mol. Neurobiol. 2019, 56, 6293–6309. [Google Scholar] [CrossRef]
- Walter, S.; Letiembre, M.; Liu, Y.; Heine, H.; Penke, B.; Hao, W.; Bode, B.; Manietta, N.; Walter, J.; Schulz-Schuffer, W.; et al. Role of the toll-like receptor 4 in neuroinflammation in Alzheimer’s disease. Cell. Physiol. Biochem. 2007, 20, 947–956. [Google Scholar] [CrossRef]
- Metro, D.; Cernaro, V.; Santoro, D.; Papa, M.; Buemi, M.; Benvenga, S.; Manasseri, L. Beneficial effects of oral pure caffeine on oxidative stress. J. Clin. Transl. Endocrinol. 2017, 10, 22–27. [Google Scholar] [CrossRef]
- Jung, S.; Kim, M.H.; Park, J.H.; Jeong, Y.; Ko, K.S. Cellular antioxidant and anti-inflammatory effects of coffee extracts with different roasting levels. J. Med. Food 2017, 20, 626–635. [Google Scholar] [CrossRef] [PubMed]
- Kolahdouzan, M.; Hamadeh, M.J. The neuroprotective effects of caffeine in neurodegenerative diseases. CNS Neurosci. Ther. 2017, 23, 272–290. [Google Scholar] [CrossRef] [PubMed]
- Keshavarz, M.; Farrokhi, M.R.; Amiri, A. Caffeine neuroprotective mechanism against β-amyloid neurotoxicity in SHSY5Y cell line: Involvement of adenosine, ryanodine, and N-methyl-D-aspartate receptors. Adv Pharm. Bull. 2017, 7, 579. [Google Scholar] [CrossRef] [PubMed]
- Ullah, F.; Ali, T.; Ullah, N.; Kim, M.O. Caffeine prevents d-galactose-induced cognitive deficits, oxidative stress, neuroinflammation and neurodegeneration in the adult rat brain. Neurochem. Int. 2015, 90, 114–124. [Google Scholar] [CrossRef] [PubMed]
- Brothers, H.M.; Marchalant, Y.; Wenk, G.L. Caffeine attenuates lipopolysaccharide-induced neuroinflammation. Neurosci. Lett. 2010, 480, 97–100. [Google Scholar] [CrossRef] [PubMed]
- Azam, S.; Hadi, N.; Khan, N.U.; Hadi, S.M. Antioxidant and prooxidant properties of caffeine, theobromine and xanthine. Med. Sci. Monit. 2003, 9, BR325–BR330. [Google Scholar] [PubMed]
- Aparicio-Soto, M.; Alarcon-De-La-Lastra, C.; Cárdeno, A.; Sánchez-Fidalgo, S.; Sanchez-Hidalgo, M. Melatonin modulates microsomal PGE synthase 1 and NF-E2-related factor-2-regulated antioxidant enzyme expression in LPS-induced murine peritoneal macrophages. Br. J. Pharmacol. 2014, 171, 134–144. [Google Scholar] [CrossRef]
- Shah, S.A.; Khan, M.; Jo, M.H.; Jo, M.G.; Amin, F.U.; Kim, M.O. Melatonin Stimulates the SIRT1/Nrf2 Signaling Pathway Counteracting Lipopolysaccharide (LPS)-Induced Oxidative Stress to Rescue Postnatal Rat Brain. CNS Neurosci. Ther. 2017, 23, 33–44. [Google Scholar] [CrossRef]
- Camhi, S.L.; Alam, J.; Otterbein, L.; Sylvester, S.L.; Choi, A.M. Induction of heme oxygenase-1 gene expression by lipopolysaccharide is mediated by AP-1 activation. Am. J. Respir. Cell Mol. Biol. 1995, 13, 387–398. [Google Scholar] [CrossRef]
- Otterbein, L.; Sylvester, S.L.; Choi, A.M. Hemoglobin provides protection against lethal endotoxemia in rats: The role of heme oxygenase-1. Am. J. Respir. Cell Mol. Biol. 1995, 13, 595–601. [Google Scholar] [CrossRef]
- Kielian, T. Toll-like receptors in central nervous system glial inflammation and homeostasis. J. Neurosci. Res. 2006, 83, 711–730. [Google Scholar] [CrossRef] [PubMed]
- Fang, W.; Bi, D.; Zheng, R.; Cai, N.; Xu, H.; Zhou, R.; Lu, J.; Wan, M.; Xu, X. Identification and activation of TLR4-mediated signalling pathways by alginate-derived guluronate oligosaccharide in RAW264. 7 macrophages. Sci. Rep. 2017, 7, 1663. [Google Scholar] [CrossRef] [PubMed]
- Varma, S.; Hegde, K.; Kovtun, S. Oxidative stress in lens in vivo: Inhibitory effect of caffeine. A preliminary report. Mol. Vis. 2010, 16, 501. [Google Scholar] [PubMed]
- Kang, C.H.; Jayasooriya, R.G.; Dilshara, M.G.; Choi, Y.H.; Jeong, Y.K.; Kim, N.D.; Kim, G.Y. Caffeine suppresses lipopolysaccharide-stimulated BV2 microglial cells by suppressing Akt-mediated NF-kappaB activation and ERK phosphorylation. Food Chem. Toxicol. 2012, 50, 4270–4276. [Google Scholar] [CrossRef] [PubMed]
- Belyagoubi-Benhammou, N.; Belyagoubi, L.; Gismondi, A.; Di Marco, G.; Canini, A.; Bekkara, F.A. GC/MS analysis, and antioxidant and antimicrobial activities of alkaloids extracted by polar and apolar solvents from the stems of Anabasis articulata. Med. Chem. Res. 2019, 28, 754–767. [Google Scholar] [CrossRef]
- Noschang, C.G.; Krolow, R.; Pettenuzzo, L.F.; Ávila, M.C.; Fachin, A.; Arcego, D.; von Pozzer Toigo, E.; Crema, L.M.; Diehl, L.A.; Vendite, D.; et al. Interactions between chronic stress and chronic consumption of caffeine on the enzymatic antioxidant system. Neurochem. Res. 2009, 34, 1568–1574. [Google Scholar] [CrossRef]
- Devasagayam, T.; Kamat, J.; Mohan, H.; Kesavan, P.C. Caffeine as an antioxidant: Inhibition of lipid peroxidation induced by reactive oxygen species. Biochim. Biophys. Acta 1996, 1282, 63–70. [Google Scholar] [CrossRef]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Badshah, H.; Ikram, M.; Ali, W.; Ahmad, S.; Hahm, J.R.; Kim, M.O. Caffeine May Abrogate LPS-Induced Oxidative Stress and Neuroinflammation by Regulating Nrf2/TLR4 in Adult Mouse Brains. Biomolecules 2019, 9, 719. https://doi.org/10.3390/biom9110719
Badshah H, Ikram M, Ali W, Ahmad S, Hahm JR, Kim MO. Caffeine May Abrogate LPS-Induced Oxidative Stress and Neuroinflammation by Regulating Nrf2/TLR4 in Adult Mouse Brains. Biomolecules. 2019; 9(11):719. https://doi.org/10.3390/biom9110719
Chicago/Turabian StyleBadshah, Haroon, Muhammad Ikram, Waqar Ali, Sareer Ahmad, Jong Ryeal Hahm, and Myeong Ok Kim. 2019. "Caffeine May Abrogate LPS-Induced Oxidative Stress and Neuroinflammation by Regulating Nrf2/TLR4 in Adult Mouse Brains" Biomolecules 9, no. 11: 719. https://doi.org/10.3390/biom9110719
APA StyleBadshah, H., Ikram, M., Ali, W., Ahmad, S., Hahm, J. R., & Kim, M. O. (2019). Caffeine May Abrogate LPS-Induced Oxidative Stress and Neuroinflammation by Regulating Nrf2/TLR4 in Adult Mouse Brains. Biomolecules, 9(11), 719. https://doi.org/10.3390/biom9110719