Curcumin Ameliorates Lead-Induced Hepatotoxicity by Suppressing Oxidative Stress and Inflammation, and Modulating Akt/GSK-3β Signaling Pathway

 ,
,
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Experimental Animals and Treatments
2.2. Determination of Liver Function Markers
2.3. Determination of LPO, NO, Antioxidants and TNF-α
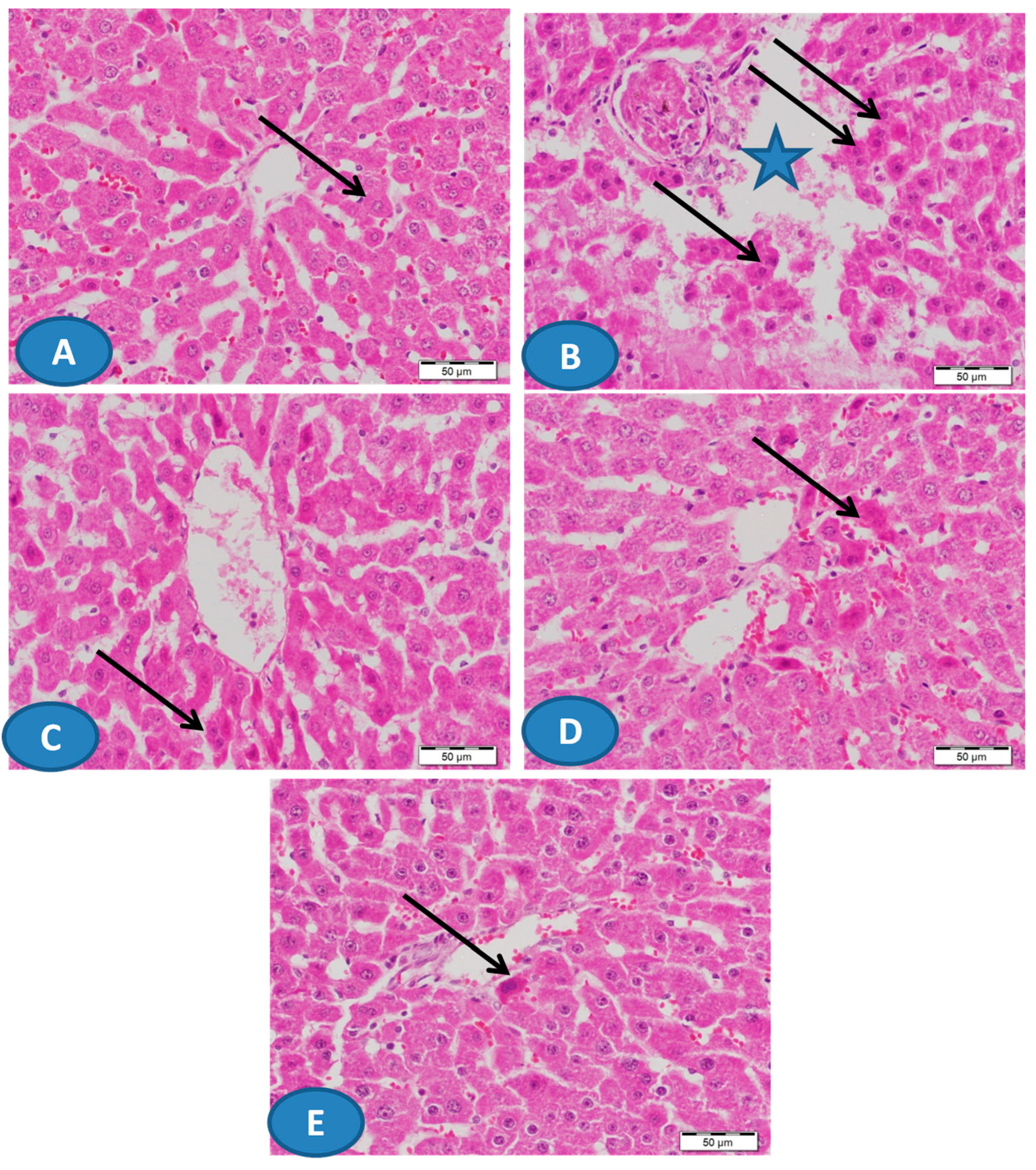
2.4. Histological Examination
2.5. Western Blot
2.6. Determination of DNA Fragmentation
2.7. Statistical Analysis
3. Results
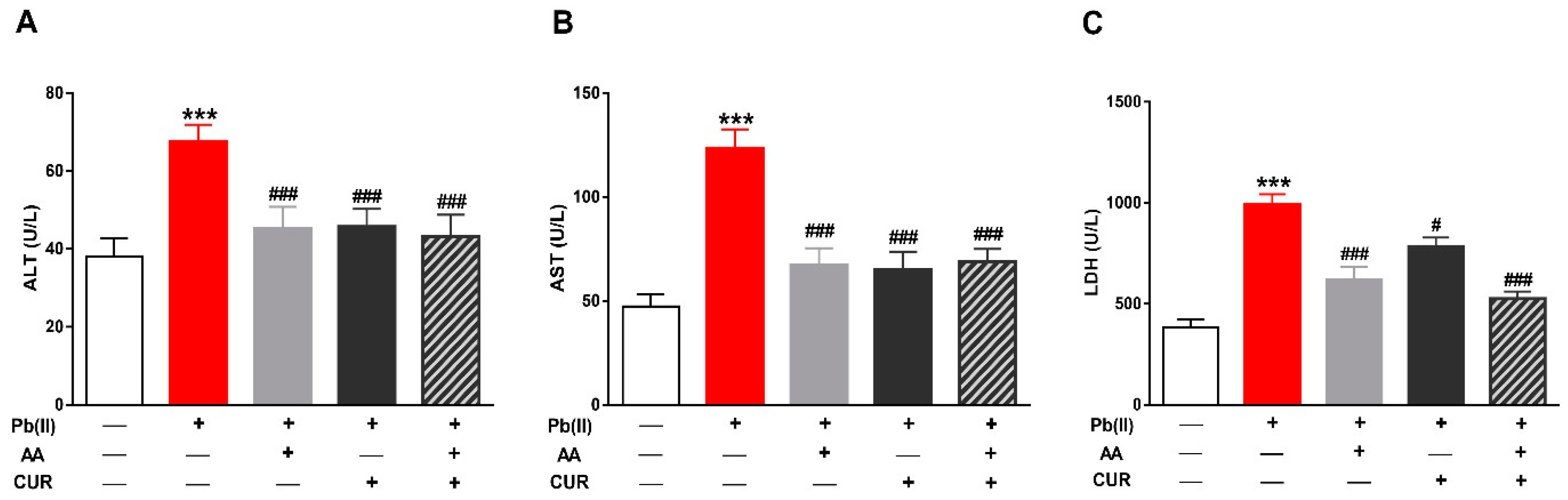
3.1. CUR Prevents Pb(II)-Induced Liver Injury
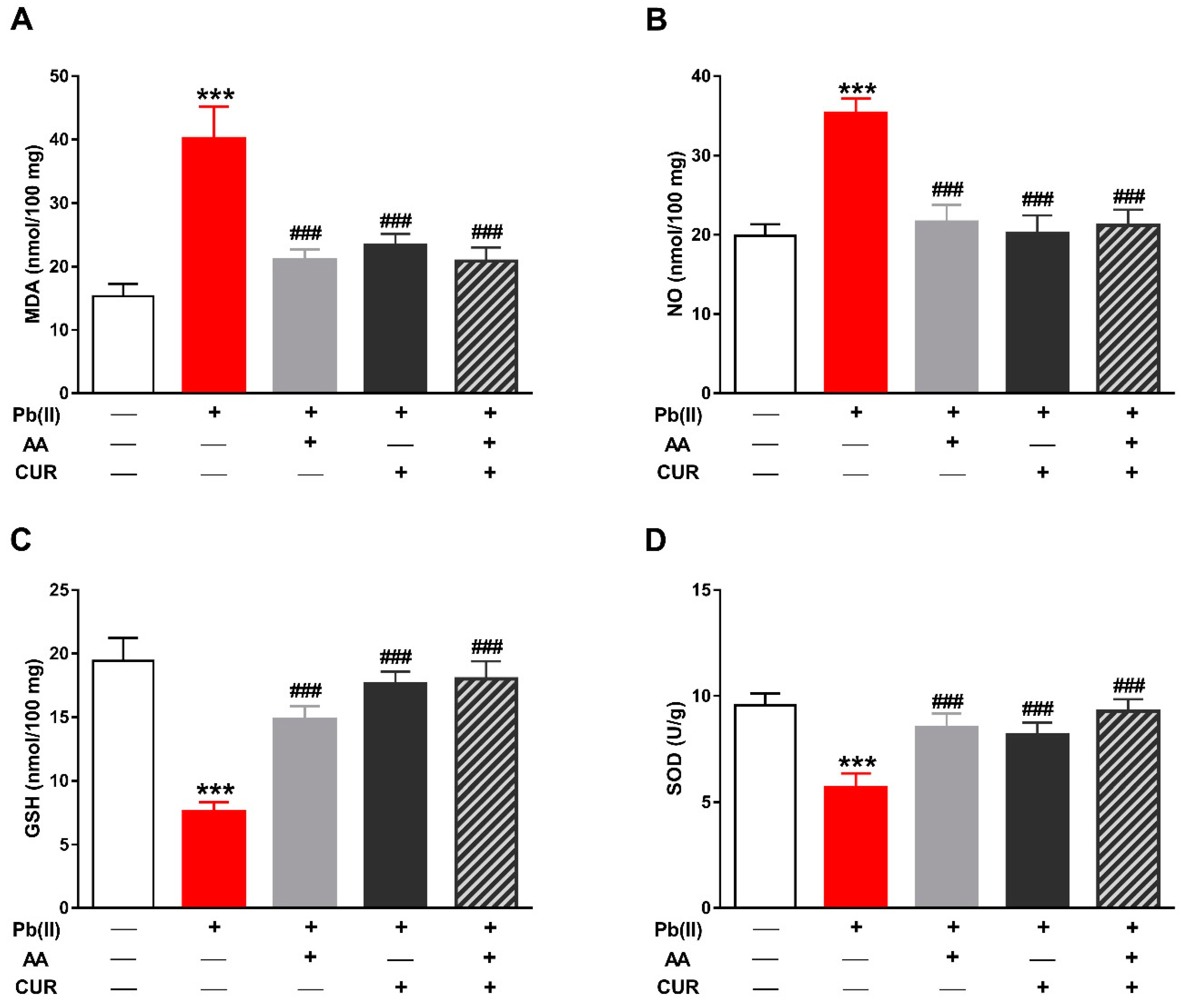
3.2. CUR Attenuates Oxidative Stress in Pb(II)-Induced Rats
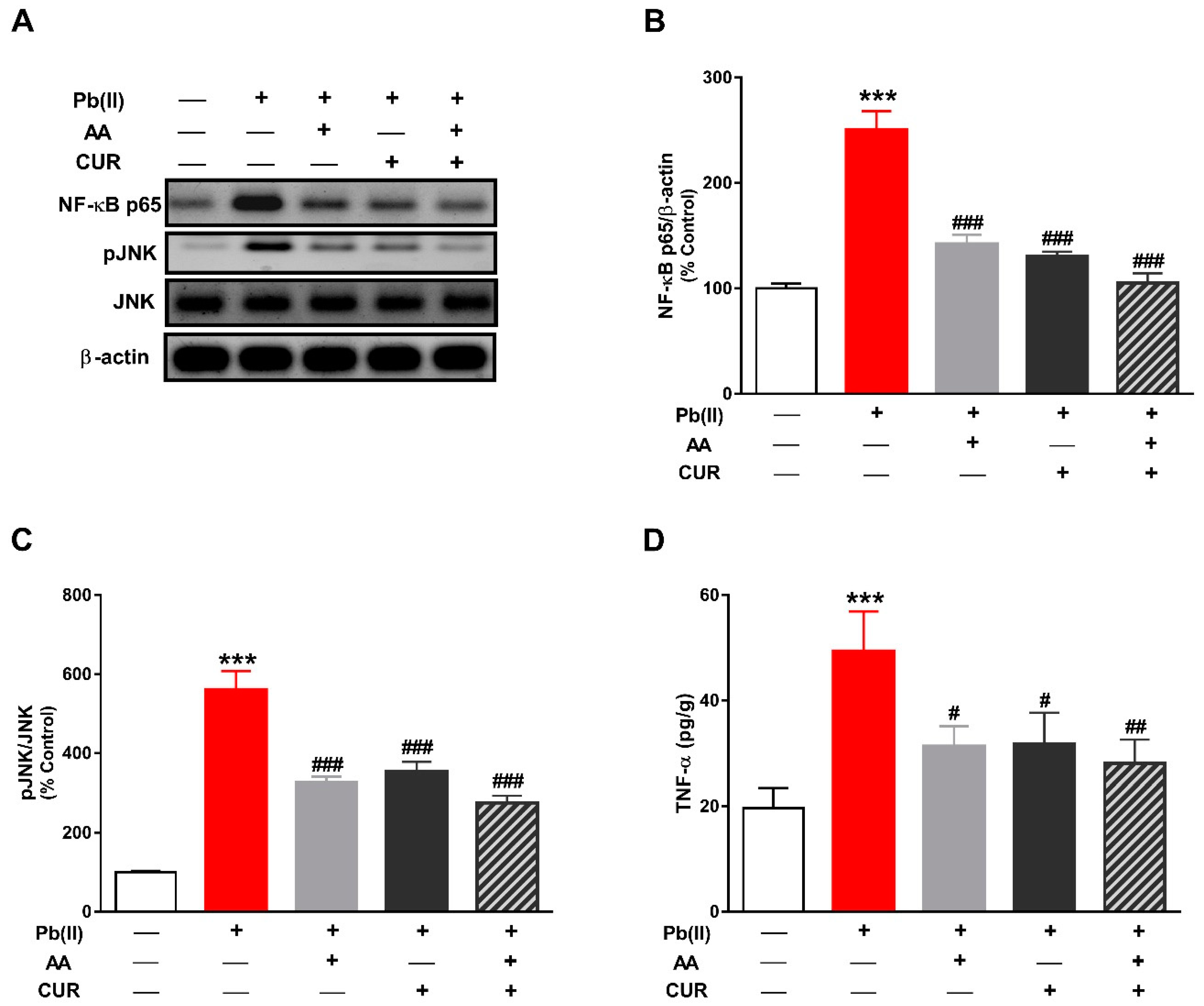
3.3. CUR Inhibits Inflammation in Pb(II)-Induced Rats
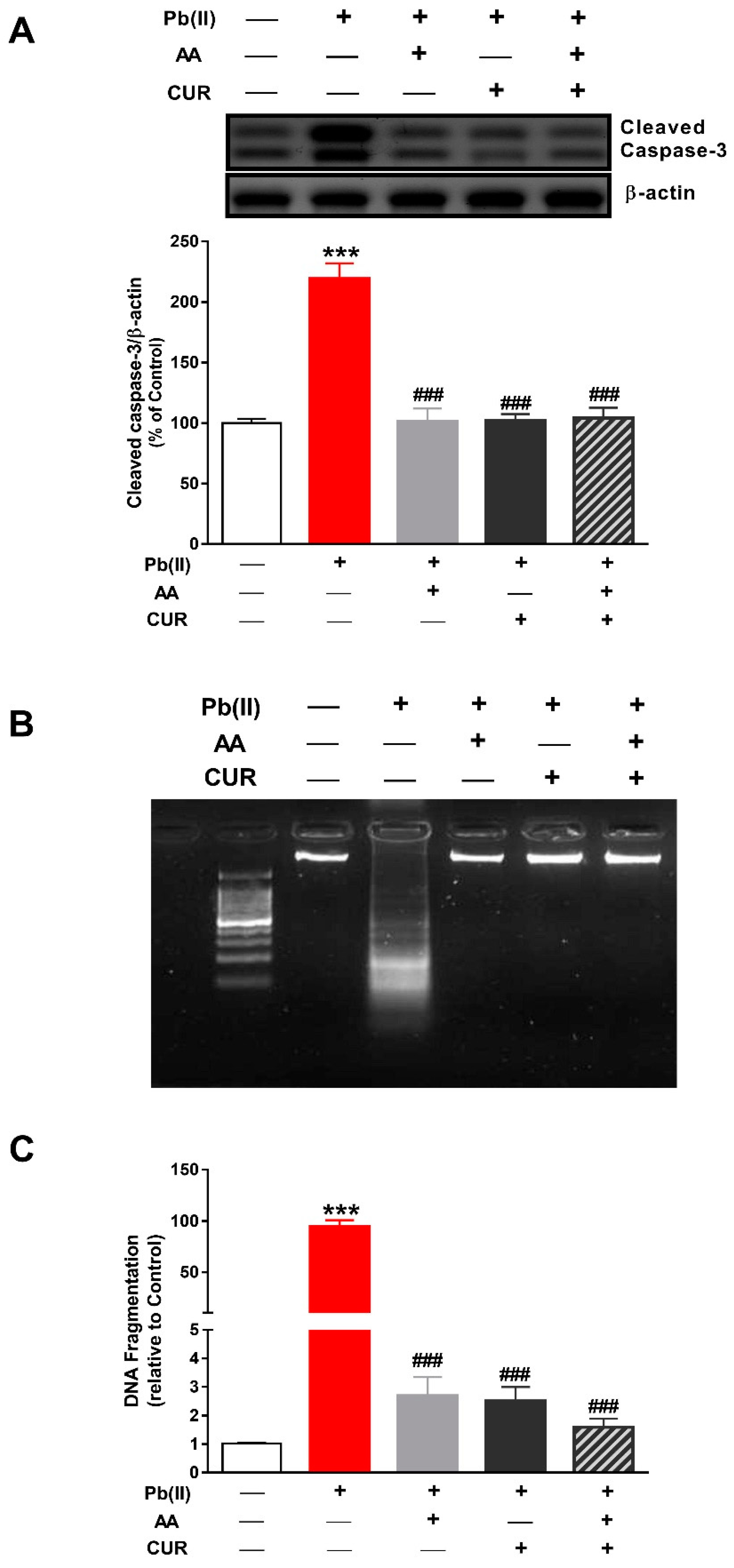
3.4. CUR Attenuates Apoptosis in Pb(II)-Induced Rats
3.5. CUR Up-Regulates Akt/GSK-3β Signaling in Pb(II)-Induced Rats
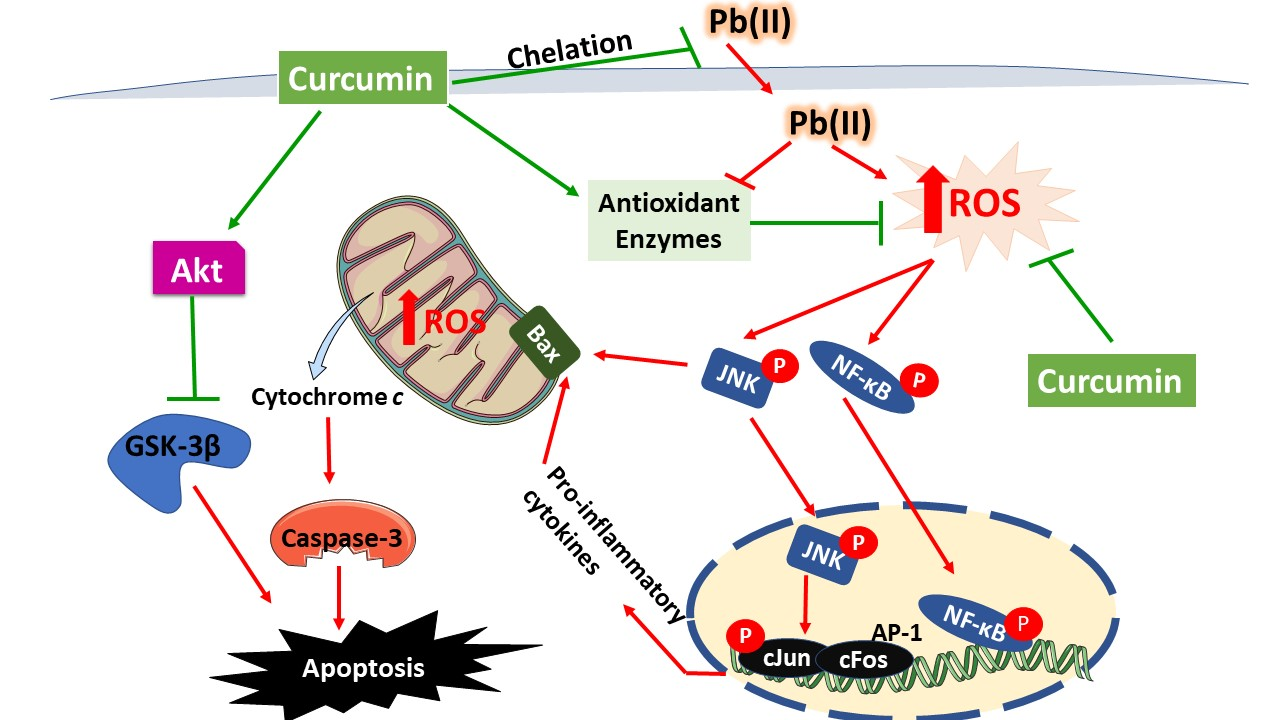
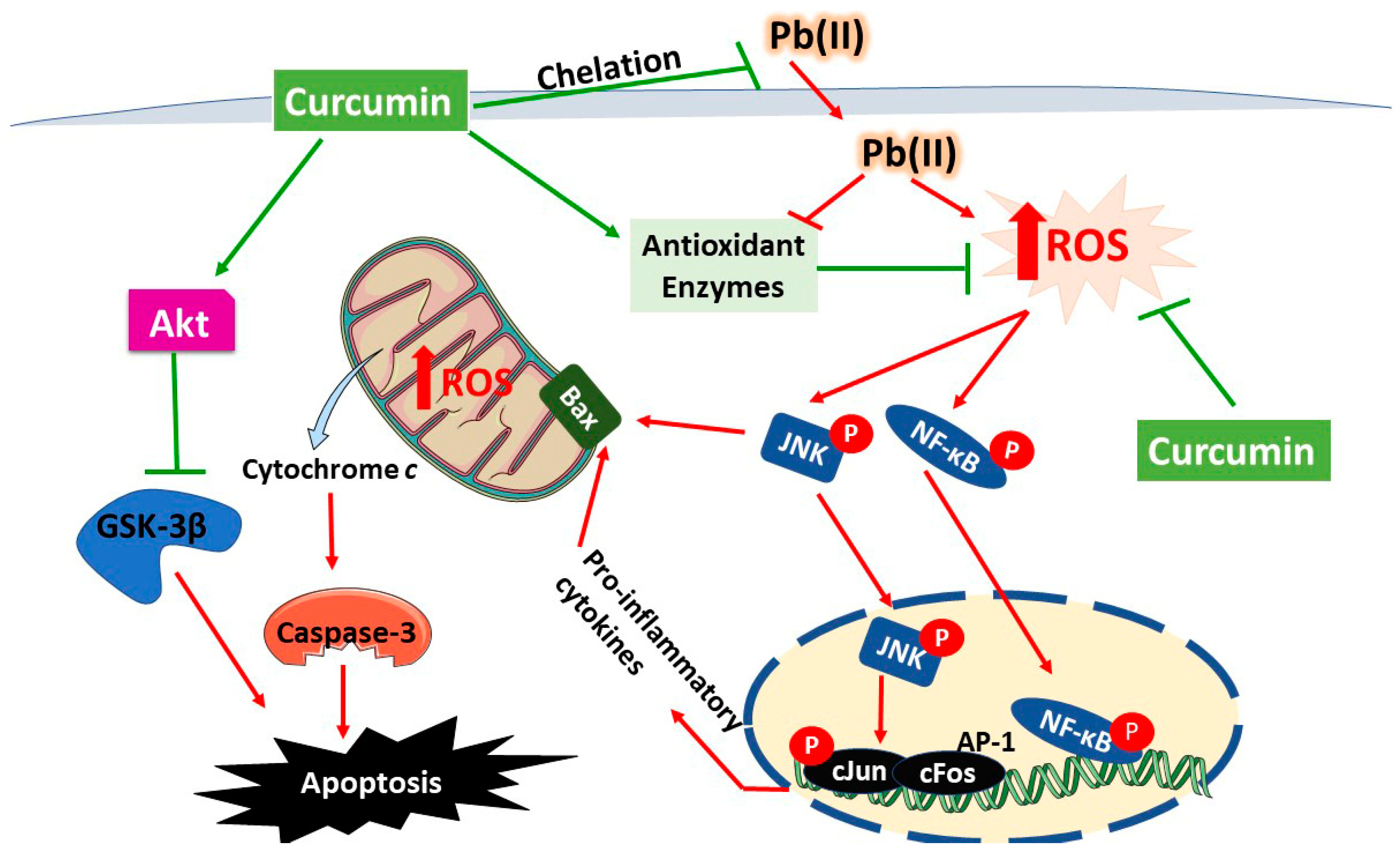
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Jarup, L. Hazards of heavy metal contamination. Br. Med. Bull. 2003, 68, 167–182. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.F.; Aarts, M.G. The molecular mechanism of zinc and cadmium stress response in plants. Cell. Mol. Life Sci. CMLS 2012, 69, 3187–3206. [Google Scholar] [CrossRef] [PubMed]
- Brzoska, M.M.; Moniuszko-Jakoniuk, J. Low-level exposure to cadmium during the lifetime increases the risk of osteoporosis and fractures of the lumbar spine in the elderly: Studies on a rat model of human environmental exposure. Toxicol. Sci. Off. J. Soc. Toxicol. 2004, 82, 468–477. [Google Scholar] [CrossRef] [PubMed]
- de Kok, T.M.; Hogervorst, J.G.; Briede, J.J.; van Herwijnen, M.H.; Maas, L.M.; Moonen, E.J.; Driece, H.A.; Kleinjans, J.C. Genotoxicity and physicochemical characteristics of traffic-related ambient particulate matter. Environ. Mol. Mutagenesis 2005, 46, 71–80. [Google Scholar] [CrossRef]
- Alissa, E.M.; Ferns, G.A. Heavy metal poisoning and cardiovascular disease. J. Toxicol. 2011, 2011, 870125. [Google Scholar] [CrossRef]
- World Health Organization. Lead Poisoning and Health; World Health Organization: Geneva, Switzerland, 2018. [Google Scholar]
- The New Top Six Toxic Threats: A Priority List for Remediation, World’s Worst Pollution Problems Report; Pure Earth: New York, NY, USA, 2015.
- El-Nekeety, A.A.; El-Kady, A.A.; Soliman, M.S.; Hassan, N.S.; Abdel-Wahhab, M.A. Protective effect of Aquilegia vulgaris (L.) against lead acetate-induced oxidative stress in rats. Food Chem. Toxicol. Int. J. Publ. Br. Ind. Biol. Res. Assoc. 2009, 47, 2209–2215. [Google Scholar] [CrossRef]
- Jia, Q.; Ha, X.; Yang, Z.; Hui, L.; Yang, X. Oxidative stress: A possible mechanism for lead-induced apoptosis and nephrotoxicity. Toxicol. Mech. Methods 2012, 22, 705–710. [Google Scholar] [CrossRef]
- El-Tantawy, W.H. Antioxidant effects of spirulina supplement against lead acetate-induced hepatic injury in rats. J. Tradit. Complement. Med. 2016, 6, 327–331. [Google Scholar] [CrossRef]
- Jaishankar, M.; Tseten, T.; Anbalagan, N.; Mathew, B.B.; Beeregowda, K.N. Toxicity, mechanism and health effects of some heavy metals. Interdiscip. Toxicol. 2014, 7, 60–72. [Google Scholar] [CrossRef]
- Flora, S.J.S.; Flora, G.; Saxena, G. Environmental occurrence, health effects and management of lead poisoning. In Lead Chemistry, Analytical Aspects, Environmental Impacts and Health Effects; Cascas, S.B., Sordo, J., Eds.; Elsevier Publication: Amsterdam, The Netherlands, 2006; pp. 158–228. [Google Scholar]
- Adegbesan, B.O.; Adenuga, G.A. Effect of lead exposure on liver lipid peroxidative and antioxidant defense systems of protein-undernourished rats. Biol. Trace Elem. Res. 2007, 116, 219–225. [Google Scholar] [CrossRef]
- Sirivarasai, J.; Wananukul, W.; Kaojarern, S.; Chanprasertyothin, S.; Thongmung, N.; Ratanachaiwong, W.; Sura, T.; Sritara, P. Association between inflammatory marker, environmental lead exposure, and glutathione s-transferase gene. BioMed Res. Int. 2013, 2013, 474963. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Lee, K.H.; Yoo, D.H.; Kang, D.; Cho, S.H.; Hong, Y.C. GSTM1 and TNF-alpha gene polymorphisms and relations between blood lead and inflammatory markers in a non-occupational population. Mutat. Res. 2007, 629, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Khan, D.A.; Qayyum, S.; Saleem, S.; Khan, F.A. Lead-induced oxidative stress adversely affects health of the occupational workers. Toxicol. Ind. Health 2008, 24, 611–618. [Google Scholar] [CrossRef] [PubMed]
- Kaidanovich-Beilin, O.; Woodgett, J.R. GSK-3: Functional insights from cell biology and animal models. Front. Mol. Neurosci. 2011, 4, 40. [Google Scholar] [CrossRef]
- Jope, R.S.; Johnson, G.V. The glamour and gloom of glycogen synthase kinase-3. Trends Biochem. Sci. 2004, 29, 95–102. [Google Scholar] [CrossRef]
- Beurel, E.; Jope, R.S. The paradoxical pro- and anti-apoptotic actions of GSK3 in the intrinsic and extrinsic apoptosis signaling pathways. Prog. Neurobiol. 2006, 79, 173–189. [Google Scholar] [CrossRef]
- McManus, E.J.; Sakamoto, K.; Armit, L.J.; Ronaldson, L.; Shpiro, N.; Marquez, R.; Alessi, D.R. Role that phosphorylation of GSK3 plays in insulin and wnt signalling defined by knockin analysis. Embo J. 2005, 24, 1571–1583. [Google Scholar] [CrossRef]
- Wang, H.; Brown, J.; Martin, M. Glycogen synthase kinase 3: A point of convergence for the host inflammatory response. Cytokine 2011, 53, 130–140. [Google Scholar] [CrossRef]
- Bhushan, B.; Poudel, S.; Manley, M.W., Jr.; Roy, N.; Apte, U. Inhibition of glycogen synthase kinase 3 accelerated liver regeneration after acetaminophen-induced hepatotoxicity in mice. Am. J. Pathol. 2017, 187, 543–552. [Google Scholar] [CrossRef]
- Fu, H.; Xu, H.; Chen, H.; Li, Y.; Li, W.; Zhu, Q.; Zhang, Q.; Yuan, H.; Liu, F.; Wang, Q.; et al. Inhibition of glycogen synthase kinase 3 ameliorates liver ischemia/reperfusion injury via an energy-dependent mitochondrial mechanism. J. Hepatol. 2014, 61, 816–824. [Google Scholar] [CrossRef]
- Ren, F.; Zhang, L.; Zhang, X.; Shi, H.; Wen, T.; Bai, L.; Zheng, S.; Chen, Y.; Chen, D.; Li, L.; et al. Inhibition of glycogen synthase kinase 3β promotes autophagy to protect mice from acute liver failure mediated by peroxisome proliferator-activated receptor α. Cell Death Dis. 2016, 7, e2151. [Google Scholar] [CrossRef] [PubMed]
- Mahmoud, A.M.; Ahmed, O.M.; Galaly, S.R. Thymoquinone and curcumin attenuate gentamicin-induced renal oxidative stress, inflammation and apoptosis in rats. EXCLI J. 2014, 13, 98–110. [Google Scholar] [PubMed]
- Galaly, S.R.; Ahmed, O.M.; Mahmoud, A.M. Thymoquinone and curcumin prevent gentamicin-induced liver injury by attenuating oxidative stress, inflammation and apoptosis. J. Physiol. Pharm. 2014, 65, 823–832. [Google Scholar]
- Tsuda, T. Curcumin as a functional food-derived factor: Degradation products, metabolites, bioactivity, and future perspectives. Food Funct. 2018, 9, 705–714. [Google Scholar] [CrossRef] [PubMed]
- Abubakar, K.; Muhammad Mailafiya, M.; Danmaigoro, A.; Musa Chiroma, S.; Abdul Rahim, E.B.; Abu Bakar Zakaria, M.Z. Curcumin attenuates lead-induced cerebellar toxicity in rats via chelating activity and inhibition of oxidative stress. Biomolecules 2019, 9. [Google Scholar] [CrossRef] [PubMed]
- McCubrey, J.A.; Lertpiriyapong, K.; Steelman, L.S.; Abrams, S.L.; Cocco, L.; Ratti, S.; Martelli, A.M.; Candido, S.; Libra, M.; Montalto, G.; et al. Regulation of GSK-3 activity by curcumin, berberine and resveratrol: Potential effects on multiple diseases. Adv. Biol. Regul. 2017, 65, 77–88. [Google Scholar] [CrossRef] [PubMed]
- Bustanji, Y.; Taha, M.O.; Almasri, I.M.; Al-Ghussein, M.A.; Mohammad, M.K.; Alkhatib, H.S. Inhibition of glycogen synthase kinase by curcumin: Investigation by simulated molecular docking and subsequent in vitro/in vivo evaluation. J. Enzym. Inhib. Med. Chem. 2009, 24, 771–778. [Google Scholar] [CrossRef]
- Mishra, H.; Kesharwani, R.K.; Singh, D.B.; Tripathi, S.; Dubey, S.K.; Misra, K. Computational simulation of inhibitory effects of curcumin, retinoic acid and their conjugates on GSK-3 beta. Netw. Model. Anal. Health Inform. Bioinform. 2019, 8, 3. [Google Scholar] [CrossRef]
- Soliman, M.M.; Baiomy, A.A.; Yassin, M.H. Molecular and histopathological study on the ameliorative effects of curcumin against lead acetate-induced hepatotoxicity and nephrototoxicity in wistar rats. Biol. Trace Elem. Res. 2015, 167, 91–102. [Google Scholar] [CrossRef]
- Farshid, A.A.; Tamaddonfard, E.; Ranjbar, S. Oral administration of vitamin c and histidine attenuate cyclophosphamide-induced hemorrhagic cystitis in rats. Indian J. Pharmacol. 2013, 45, 126–129. [Google Scholar] [CrossRef]
- Preuss, H.G.; Jarrell, S.T.; Scheckenbach, R.; Lieberman, S.; Anderson, R.A. Comparative effects of chromium, vanadium and gymnema sylvestre on sugar-induced blood pressure elevations in SHR. J. Am. Coll. Nutr. 1998, 17, 116–123. [Google Scholar] [CrossRef] [PubMed]
- Grisham, M.B.; Johnson, G.G.; Lancaster, J.R., Jr. Quantitation of nitrate and nitrite in extracellular fluids. Methods Enzymol. 1996, 268, 237–246. [Google Scholar] [PubMed]
- Beutler, E.; Duron, O.; Kelly, B.M. Improved method for the determination of blood glutathione. J. Lab. Clin. Med. 1963, 61, 882–888. [Google Scholar]
- Marklund, S.; Marklund, G. Involvement of the superoxide anion radical in the autoxidation of pyrogallol and a convenient assay for superoxide dismutase. Eur. J. Biochem. 1974, 47, 469–474. [Google Scholar] [CrossRef] [PubMed]
- Hickey, E.J.; Raje, R.R.; Reid, V.E.; Gross, S.M.; Ray, S.D. Diclofenac induced in vivo nephrotoxicity may involve oxidative stress-mediated massive genomic DNA fragmentation and apoptotic cell death. Free Radic. Biol. Med. 2001, 31, 139–152. [Google Scholar] [CrossRef]
- Mudipalli, A. Lead hepatotoxicity & potential health effects. Indian J. Med. Res. 2007, 126, 518–527. [Google Scholar]
- Mehana, E.E.; Meki, A.R.M.A.; Fazili, K.M. Ameliorated effects of green tea extract on lead induced liver toxicity in rats. Exp. Toxicol. Pathol. 2012, 64, 291–295. [Google Scholar] [CrossRef]
- Martindale, J.L.; Holbrook, N.J. Cellular response to oxidative stress: Signaling for suicide and survival. J. Cell. Physiol. 2002, 192, 1–15. [Google Scholar] [CrossRef]
- Peng, X.; Dai, C.; Liu, Q.; Li, J.; Qiu, J. Curcumin attenuates on carbon tetrachloride-induced acute liver injury in mice via modulation of the Nrf2/HO-1 and TGF-β1/Smad3 pathway. Molecules (Baselswitzerland) 2018, 23, 215. [Google Scholar] [CrossRef]
- Wang, H.; Muhammad, I.; Li, W.; Sun, X.; Cheng, P.; Zhang, X. Sensitivity of arbor acres broilers and chemoprevention of aflatoxin b1-induced liver injury by curcumin, a natural potent inducer of phase-ii enzymes and Nrf2. Environ. Toxicol. Pharmacol. 2018, 59, 94–104. [Google Scholar] [CrossRef]
- Xie, Y.L.; Chu, J.G.; Jian, X.M.; Dong, J.Z.; Wang, L.P.; Li, G.X.; Yang, N.B. Curcumin attenuates lipopolysaccharide/d-galactosamine-induced acute liver injury by activating Nrf2 nuclear translocation and inhibiting NF-kB activation. Biomed. Pharmacother. 2017, 91, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Satta, S.; Mahmoud, A.M.; Wilkinson, F.L.; Yvonne Alexander, M.; White, S.J. The role of Nrf2 in cardiovascular function and disease. Oxid. Med. Cell. Longev. 2017, 2017, 9237263. [Google Scholar] [CrossRef] [PubMed]
- Aladaileh, S.H.; Abukhalil, M.H.; Saghir, S.A.M.; Hanieh, H.; Alfwuaires, M.A.; Almaiman, A.A.; Bin-Jumah, M.; Mahmoud, A.M. Galangin activates Nrf2 signaling and attenuates oxidative damage, inflammation, and apoptosis in a rat model of cyclophosphamide-induced hepatotoxicity. Biomolecules 2019, 9, 346. [Google Scholar] [CrossRef] [PubMed]
- Mahmoud, A.M.; Mohammed, H.M.; Khadrawy, S.M.; Galaly, S.R. Hesperidin protects against chemically induced hepatocarcinogenesis via modulation of Nrf2/are/HO-1, ppargamma and TGF-β1/Smad3 signaling, and amelioration of oxidative stress and inflammation. Chem. Biol. Interact. 2017, 277, 146–158. [Google Scholar] [CrossRef] [PubMed]
- Mahmoud, A.M.; Hozayen, W.G.; Ramadan, S.M. Berberine ameliorates methotrexate-induced liver injury by activating Nrf2/HO-1 pathway and ppargamma, and suppressing oxidative stress and apoptosis in rats. Biomed. Pharm. 2017, 94, 280–291. [Google Scholar] [CrossRef] [PubMed]
- Mahmoud, A.M.; Hussein, O.E.; Hozayen, W.G.; Abd El-Twab, S.M. Methotrexate hepatotoxicity is associated with oxidative stress, and down-regulation of ppargamma and Nrf2: Protective effect of 18β-glycyrrhetinic acid. Chem. Biol. Interact. 2017, 270, 59–72. [Google Scholar] [CrossRef] [PubMed]
- Yan, C.; Zhang, Y.; Zhang, X.; Aa, J.; Wang, G.; Xie, Y. Curcumin regulates endogenous and exogenous metabolism via nrf2-fxr-lxr pathway in nafld mice. Biomed. Pharmacother. 2018, 105, 274–281. [Google Scholar] [CrossRef]
- Mishra, P.; Paital, B.; Jena, S.; Swain, S.S.; Kumar, S.; Yadav, M.K.; Chainy, G.B.N.; Samanta, L. Possible activation of NRF2 by Vitamin E/Curcumin against altered thyroid hormone induced oxidative stress via NFkB/AKT/mTOR/KEAP1 signalling in rat heart. Sci. Rep. 2019, 9, 7408. [Google Scholar] [CrossRef]
- Turner, M.D.; Nedjai, B.; Hurst, T.; Pennington, D.J. Cytokines and chemokines: At the crossroads of cell signalling and inflammatory disease. Biochim. Et Biophys. Acta 2014, 1843, 2563–2582. [Google Scholar] [CrossRef]
- Guo, T.L.; Mudzinski, S.P.; Lawrence, D.A. The heavy metal lead modulates the expression of both TNF-α and TNF-α receptors in lipopolysaccharide-activated human peripheral blood mononuclear cells. J. Leukoc. Biol. 1996, 59, 932–939. [Google Scholar] [CrossRef]
- Dooley, S.; ten Dijke, P. TGF-β in progression of liver disease. Cell Tissue Res. 2012, 347, 245–256. [Google Scholar] [CrossRef] [PubMed]
- Mahmoud, A.M.; Hozayen, W.G.; Hasan, I.H.; Shaban, E.; Bin-Jumah, M. Umbelliferone ameliorates CCl4-induced liver fibrosis in rats by upregulating ppargamma and attenuating oxidative stress, inflammation, and TGF-β1/Smad3 signaling. Inflammation 2019, 42, 1103–1116. [Google Scholar] [CrossRef] [PubMed]
- Plotnikov, A.; Zehorai, E.; Procaccia, S.; Seger, R. The mapk cascades: Signaling components, nuclear roles and mechanisms of nuclear translocation. Biochim. Biophys. Acta 2011, 1813, 1619–1633. [Google Scholar] [CrossRef] [PubMed]
- Seki, E.; Brenner, D.A.; Karin, M. A liver full of JNK: Signaling in regulation of cell function and disease pathogenesis, and clinical approaches. Gastroenterology 2012, 143, 307–320. [Google Scholar] [CrossRef] [PubMed]
- Micheau, O.; Tschopp, J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell 2003, 114, 181–190. [Google Scholar] [CrossRef]
- Win, S.; Than, T.A.; Han, D.; Petrovic, L.M.; Kaplowitz, N. c-Jun N-terminal kinase (JNK)-dependent acute liver injury from acetaminophen or tumor necrosis factor (TNF) requires mitochondrial sab protein expression in mice. J. Biol. Chem. 2011, 286, 35071–35078. [Google Scholar] [CrossRef]
- Wang, Y.; Singh, R.; Lefkowitch, J.H.; Rigoli, R.M.; Czaja, M.J. Tumor necrosis factor-induced toxic liver injury results from JNK2-dependent activation of caspase-8 and the mitochondrial death pathway. J. Biol. Chem. 2006, 281, 15258–15267. [Google Scholar] [CrossRef]
- Jobin, C.; Bradham, C.A.; Russo, M.P.; Juma, B.; Narula, A.S.; Brenner, D.A.; Sartor, R.B. Curcumin blocks cytokine-mediated NF-kappa B activation and proinflammatory gene expression by inhibiting inhibitory factor I-Kappa B kinase activity. J. Immunol. 1999, 163, 3474–3483. [Google Scholar]
- Wang, L.; Li, N.; Lin, D.; Zang, Y. Curcumin protects against hepatic ischemia/reperfusion induced injury through inhibiting TLR4/NF-kappaB pathway. Oncotarget 2017, 8, 65414–65420. [Google Scholar]
- Geng, S.; Wang, S.; Zhu, W.; Xie, C.; Li, X.; Wu, J.; Zhu, J.; Jiang, Y.; Yang, X.; Li, Y.; et al. Curcumin suppresses JNK pathway to attenuate BPA-induced insulin resistance in LO2 cells. Biomed. Pharmacother. 2018, 97, 1538–1543. [Google Scholar] [CrossRef]
- Maurer, U.; Preiss, F.; Brauns-Schubert, P.; Schlicher, L.; Charvet, C. GSK-3—at the crossroads of cell death and survival. J. Cell Sci. 2014, 127, 1369. [Google Scholar] [CrossRef] [PubMed]
- Pap, M.; Cooper, G.M. Role of glycogen synthase kinase-3 in the phosphatidylinositol 3-Kinase/Akt cell survival pathway. J. Biol. Chem. 1998, 273, 19929–19932. [Google Scholar] [CrossRef] [PubMed]
- Linseman, D.A.; Butts, B.D.; Precht, T.A.; Phelps, R.A.; Le, S.S.; Laessig, T.A.; Bouchard, R.J.; Florez-McClure, M.L.; Heidenreich, K.A. Glycogen synthase kinase-3β phosphorylates bax and promotes its mitochondrial localization during neuronal apoptosis. J. Neurosci. 2004, 24, 9993–10002. [Google Scholar] [CrossRef] [PubMed]
- Hongisto, V.; Smeds, N.; Brecht, S.; Herdegen, T.; Courtney, M.J.; Coffey, E.T. Lithium blocks the c-jun stress response and protects neurons via its action on glycogen synthase kinase 3. Mol. Cell. Biol. 2003, 23, 6027–6036. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Wu, J.; Cai, F.; Xiang, J.; Zha, W.; Fan, D.; Guo, S.; Ming, Z.; Liu, C. Curcumin alleviates diabetic cardiomyopathy in experimental diabetic rats. PLoS ONE 2012, 7, e52013. [Google Scholar] [CrossRef] [PubMed]








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alhusaini, A.; Fadda, L.; Hasan, I.H.; Zakaria, E.; Alenazi, A.M.; Mahmoud, A.M. Curcumin Ameliorates Lead-Induced Hepatotoxicity by Suppressing Oxidative Stress and Inflammation, and Modulating Akt/GSK-3β Signaling Pathway. Biomolecules 2019, 9, 703. https://doi.org/10.3390/biom9110703
Alhusaini A, Fadda L, Hasan IH, Zakaria E, Alenazi AM, Mahmoud AM. Curcumin Ameliorates Lead-Induced Hepatotoxicity by Suppressing Oxidative Stress and Inflammation, and Modulating Akt/GSK-3β Signaling Pathway. Biomolecules. 2019; 9(11):703. https://doi.org/10.3390/biom9110703
Chicago/Turabian StyleAlhusaini, Ahlam, Laila Fadda, Iman H. Hasan, Enas Zakaria, Abeer M. Alenazi, and Ayman M. Mahmoud. 2019. "Curcumin Ameliorates Lead-Induced Hepatotoxicity by Suppressing Oxidative Stress and Inflammation, and Modulating Akt/GSK-3β Signaling Pathway" Biomolecules 9, no. 11: 703. https://doi.org/10.3390/biom9110703
APA StyleAlhusaini, A., Fadda, L., Hasan, I. H., Zakaria, E., Alenazi, A. M., & Mahmoud, A. M. (2019). Curcumin Ameliorates Lead-Induced Hepatotoxicity by Suppressing Oxidative Stress and Inflammation, and Modulating Akt/GSK-3β Signaling Pathway. Biomolecules, 9(11), 703. https://doi.org/10.3390/biom9110703