2-Hydroxy-N-phenylbenzamides and Their Esters Inhibit Acetylcholinesterase and Butyrylcholinesterase

 ,
,  , and
, and
Abstract
1. Introduction
2. Materials and Methods
2.1. Chemistry
2.1.1. General Methods
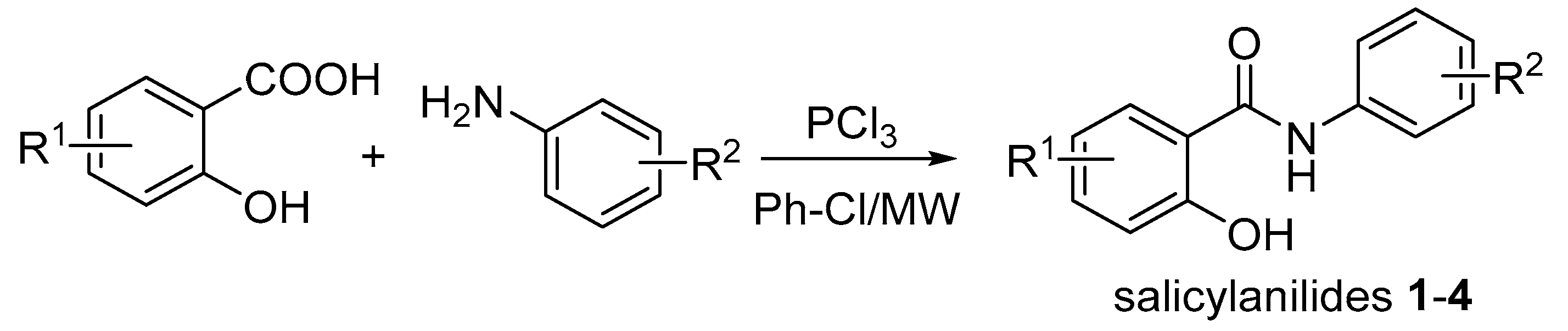
2.1.2. Synthesis of 2-Hydroxy-N-phenylbenzamides 1–4
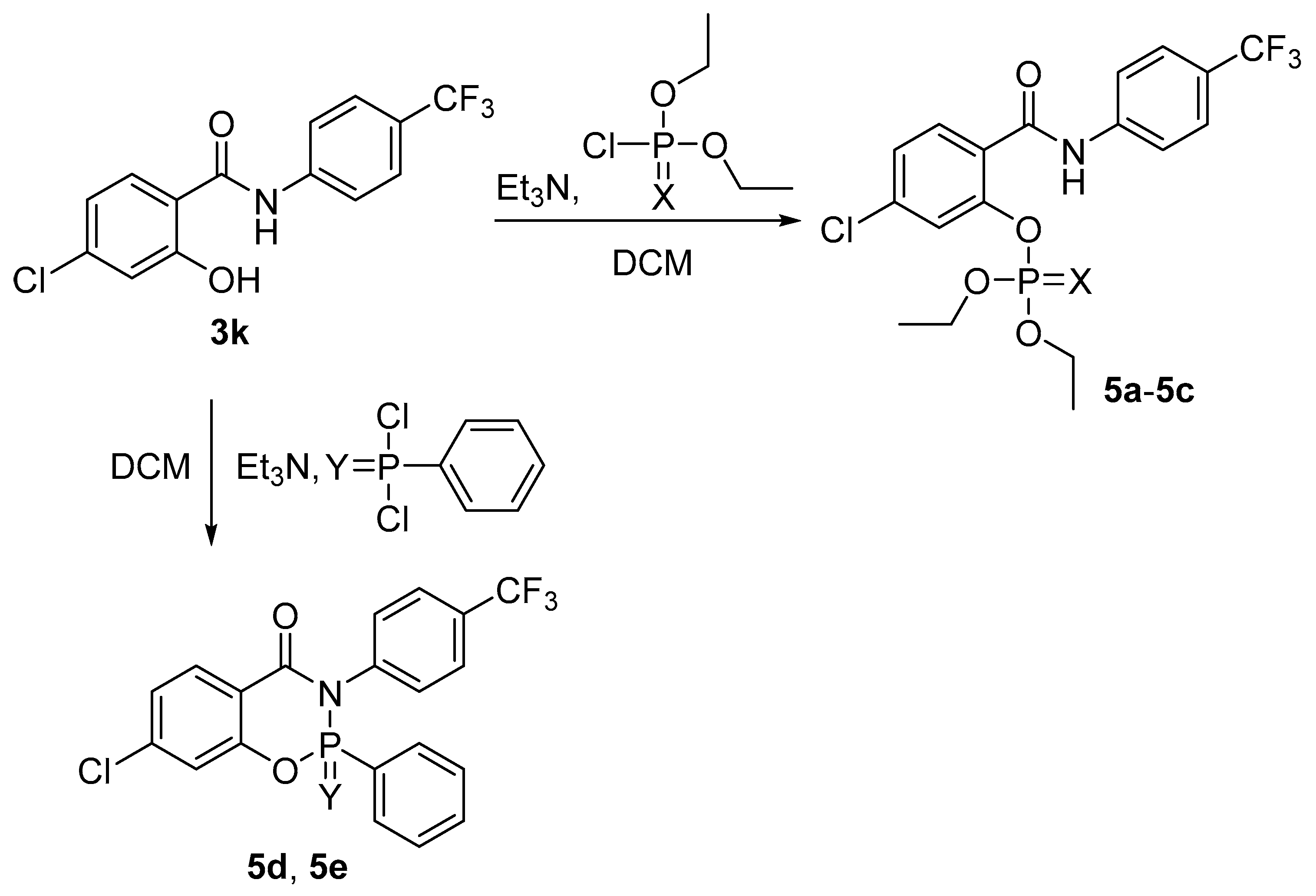
2.1.3. Synthesis of Phosphorus-Based Derivatives 5
2.1.4. Determination of Physicochemical Parameters
2.2. Determination of Cholinesterases Inhibition
3. Results and Discussion
3.1. Chemistry
3.2. In Vitro Inhibition of Acetylcholinesterase and Butyrylcholinesterase
- Slightly better inhibition is associated with derivatives of 5-substituted salicylic acid (series 2 and 4).
- The substitution of the position 3 at the aniline ring led to improved activity when compared to the position 4 (majority of the series 3 and the pair 2h and 2i are partial exceptions).
- The order of preferred aniline substituents in the case of monosubstitution is as follows: CF3 > F > Br > Cl (series 4); Cl > Br > F > CF3 (series 3); F > Br > CF3 > Cl (series 2).
- The di- and tri-substitution of aniline (both by chlorine c–e and trifluoromethyl group l) resulted predominantly in significantly more potent inhibition of AChE when compared to monosubstituted analogues.
- Regarding polychlorinated anilides, the derivatives of 3,5-dichloroaniline d are superior to 3,4-dichloroanilides c and also to 3,4,5-trichloroanilides e; the additional substitution of the 3,5-dichloro compounds d by 4-chlorine (i.e., providing trichloro structures e) is detrimental but it is still superior to 3,4-dichloroanilides c. Series 3 is a partial exception.
- The most potent inhibition was exhibited by the derivatives of 5-bromosalicylic acid (series 4) followed by 5-chlorosalicylic acid (2).
- The order of preferred atoms on monosubstituted aniline ring is as follows: Br > CF3 > F > Cl (series 2); Cl > F > Br > CF3 (series 3); CF3 > F > Br > Cl (series 4; i.e., the identical results as obtained for AChE). There was no sharp difference in the activity of 3- and 4-substituted anilines.
- The presence of di- and tri-substituted anilines increases the BuChE inhibition predominantly, especially for trifluoromethyl derivatives.
- Regarding polychlorinated anilides within the series 3 and 4, the derivatives of 3,4-dichloroaniline c are superior to 3,5-dichloroanilides d and also to 3,4,5-trichloroanilides e. However, the reverse SAR was found in the series 2.
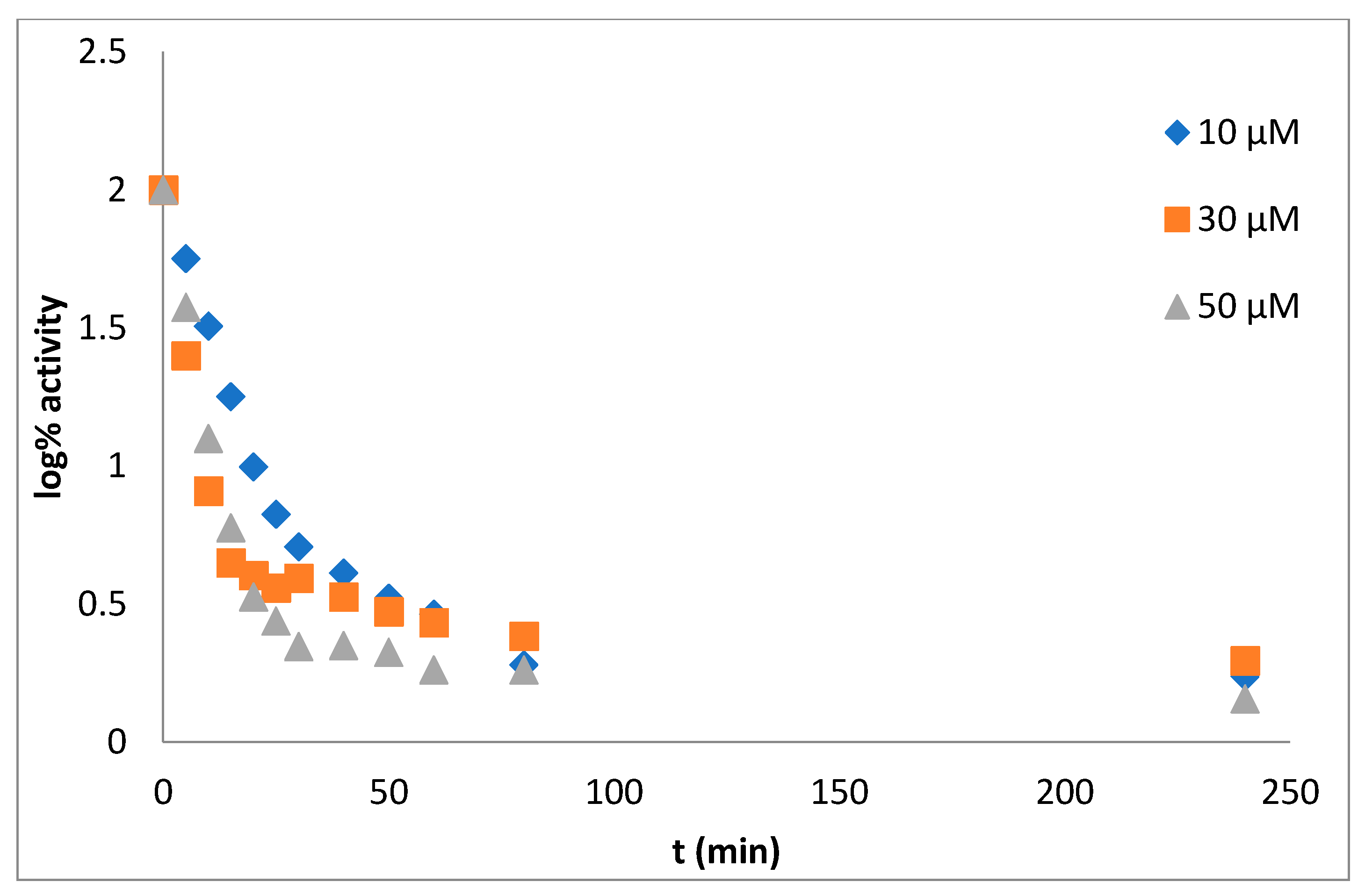
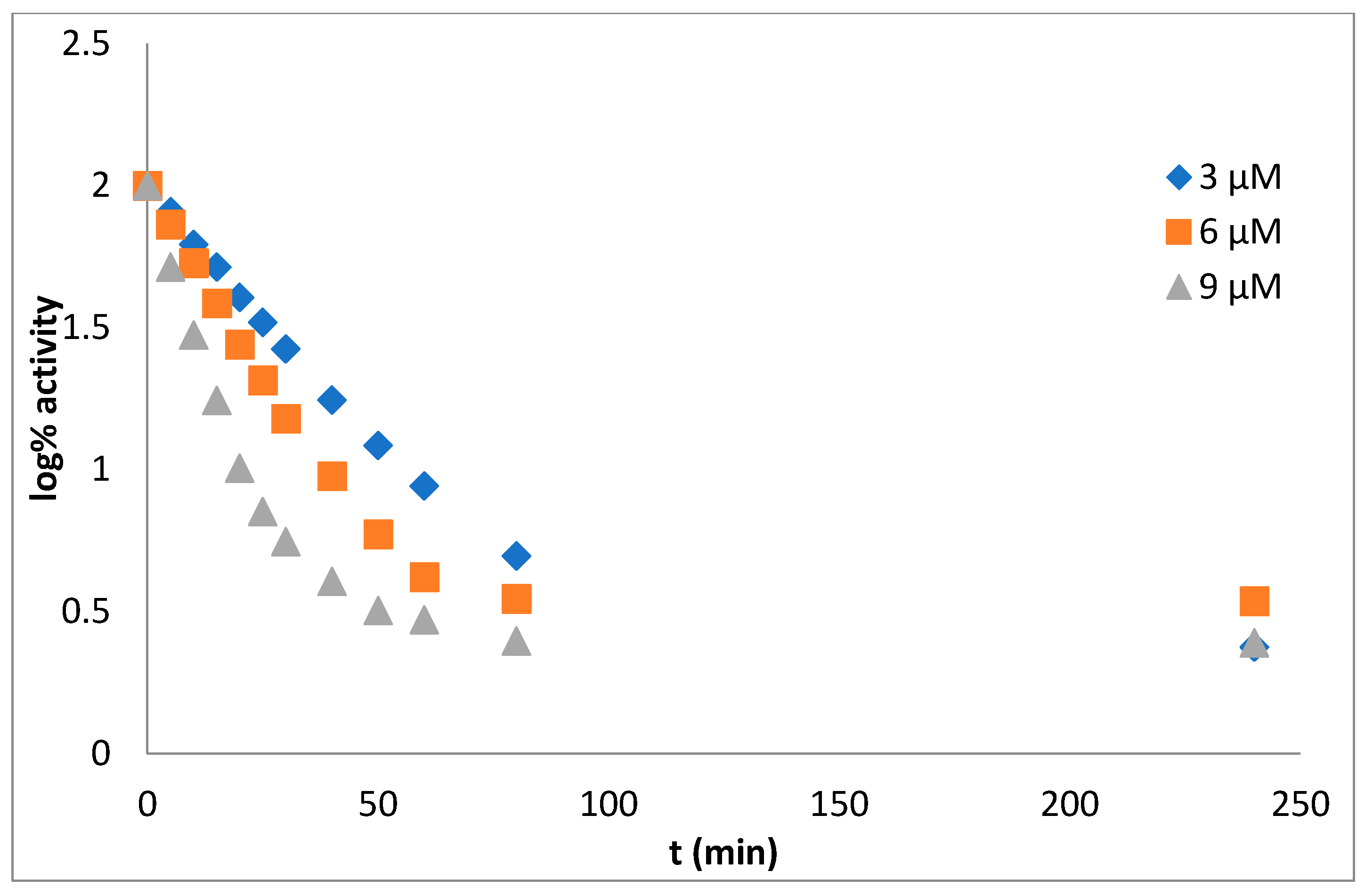
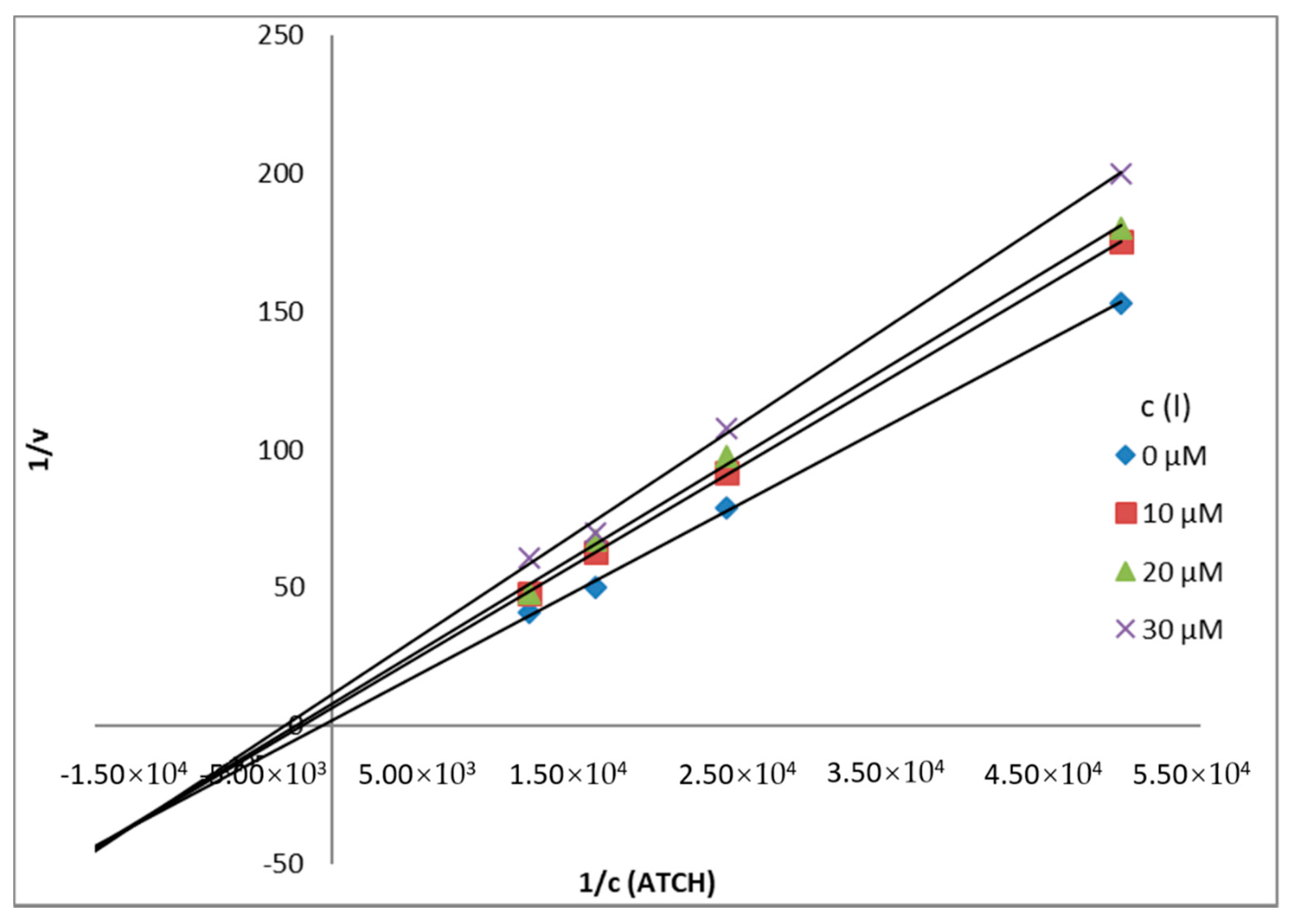
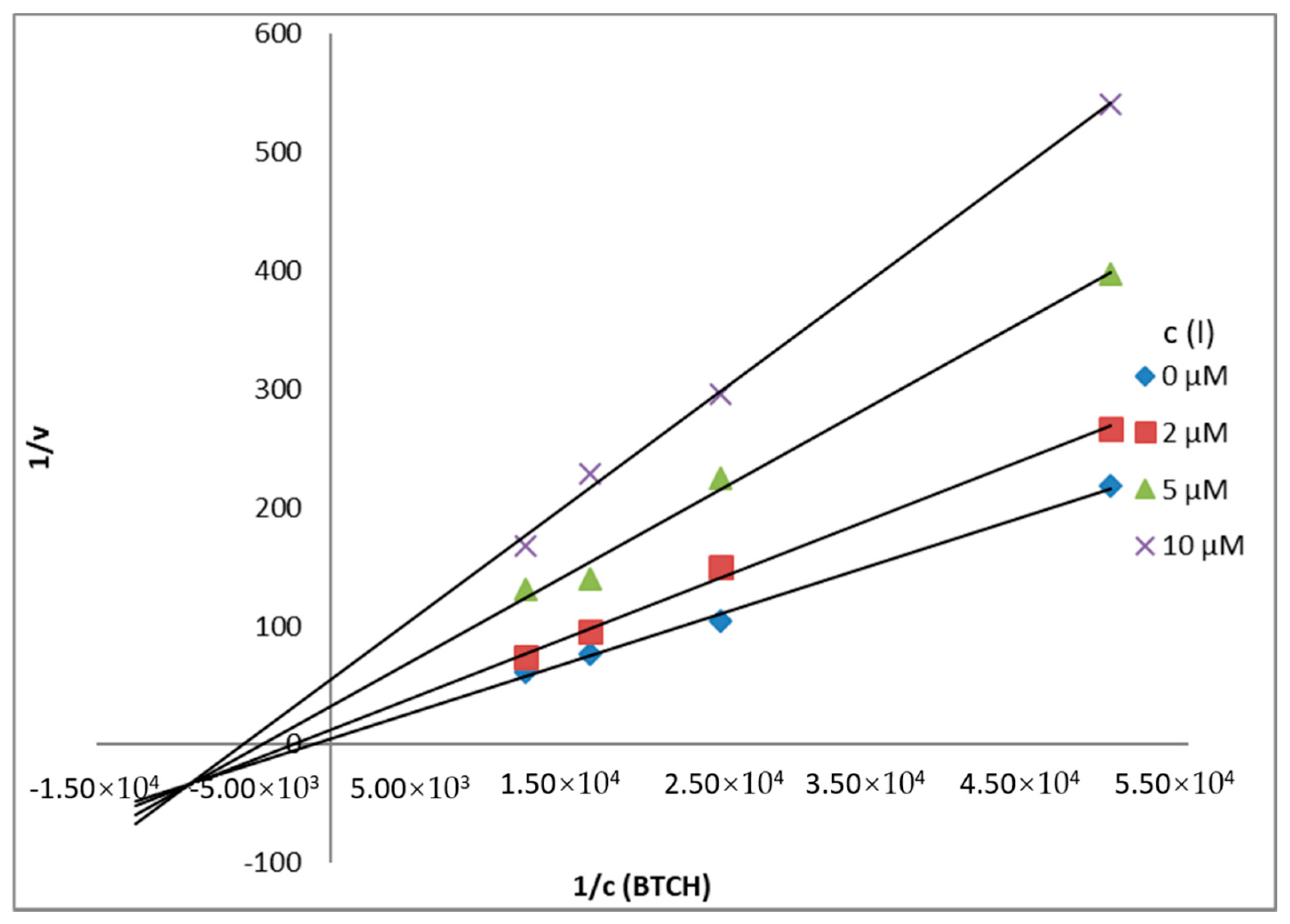
Mechanism and Type of Inhibition
3.3. Prediction of Physicochemical Parameters, Drug-Likeness and CNS Delivery
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Bondi, M.W.; Edmonds, E.C.; Salmon, D.P. Alzheimer’s Disease: Past, Present, and Future. J. Int. Neuropsychol. Soc. 2017, 23, 818–831. [Google Scholar] [CrossRef] [PubMed]
- Karlawish, J.; Jack, C.R.; Rocca, W.A.; Snyder, H.M.; Carrillo, M.C. Alzheimer’s disease: The next frontier-Special Report 2017. Alzheimers Dement. 2017, 13, 374–380. [Google Scholar] [CrossRef] [PubMed]
- World Alzheimer Report 2018. Available online: https://www.alz.co.uk/research/world-report-2018 (accessed on 20 June 2019).
- Sawatzky, E.; Wehle, S.; Kling, B.; Wendrich, J.; Bringmann, G.; Sotriffer, C.A.; Heilmann, J.; Decker, M. Discovery of Highly Selective and Nanomolar Carbamate-Based Butyrylcholinesterase Inhibitors by Rational Investigation into Their Inhibition Mode. J. Med. Chem. 2016, 59, 2067–2082. [Google Scholar] [CrossRef] [PubMed]
- Darvesh, S. Butyrylcholinesterase as a diagnostic and therapeutic target for Alzheimer’s disease. Curr. Alzheimer Res. 2016, 13, 1173–1177. [Google Scholar] [CrossRef]
- García-Ayllón, M.-S.; Small, D.H.; Avila, J.; Saez-Valero, J. Revisiting the Role of Acetylcholinesterase in Alzheimer’s Disease: Cross-Talk with P-tau and β-Amyloid. Front. Molec. Neurosci. 2011, 4, 22. [Google Scholar] [CrossRef]
- Li, Q.; Yang, H.Y.; Chen, Y.; Sun, H.P. Recent progress in the identification of selective butyrylcholinesterase inhibitors for Alzheimer’s disease. Eur. J. Med. Chem. 2017, 132, 294–309. [Google Scholar] [CrossRef]
- Mehta, M.; Adem, A.; Sabbagh, M. New Acetylcholinesterase Inhibitors for Alzheimer’s Disease. Int. J. Alzheimers Dis. 2012, 2012, 728983. [Google Scholar] [CrossRef]
- Colovic, M.B.; Krstic, D.Z.; Lazarevic-Pasti, T.D.; Bondzic, A.M.; Vasic, V.M. Acetylcholinesterase Inhibitors: Pharmacology and Toxicology. Curr. Neuropharmacol. 2013, 11, 315–335. [Google Scholar] [CrossRef]
- Horáková, E.; Drabina, P.; Brož, B.; Štěpánková, Š.; Vorčáková, K.; Královec, K.; Havelek, R.; Sedlák, M. Synthesis, characterization and in vitro evaluation of substituted N-(2-phenylcyclopropyl)carbamates as acetyl- and butyrylcholinesterase inhibitors. J. Enzyme Inhib. Med. Chem. 2016, 31, 173–179. [Google Scholar] [CrossRef][Green Version]
- Carrarini, C.; Russo, M.; Dono, F.; Di Pietro, M.; Rispoli, M.G.; Di Stefano, V.; Ferri, L.; Barbone, F.; Vitale, M.; Thomas, A.; et al. A Stage-Based Approach to Therapy in Parkinson’s Disease. Biomolecules 2019, 9, 388. [Google Scholar] [CrossRef]
- Bohnen, N.I.; Grothe, M.J.; Ray, N.J.; Müller, M.L.T.M.; Teipel, S.J. Recent Advances in Cholinergic Imaging and Cognitive Decline—Revisiting the Cholinergic Hypothesis of Dementia. Curr. Geri. Rep. 2018, 7, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Zhang, A.; Sun, H.; Han, Y.; Kong, L.; Wang, X. Two decades of new drug discovery and development for Alzheimer’s disease. RSC Adv. 2017, 7, 6046–6058. [Google Scholar] [CrossRef]
- Krátký, M.; Vinšová, J. Salicylanilide ester prodrugs as potential antimicrobial agents–a review. Curr. Pharm. Des. 2011, 17, 3494–3505. [Google Scholar] [CrossRef] [PubMed]
- Paraskevopoulos, G.; Monteiro, S.; Vosátka, R.; Krátký, M.; Navrátilová, L.; Trejtnar, F.; Stolaříková, J.; Vinšová, J. Novel salicylanilides from 4,5-dihalogenated salicylic acids: Synthesis, antimicrobial activity and cytotoxicity. Bioorg. Med. Chem. 2017, 25, 1524–1532. [Google Scholar] [CrossRef] [PubMed]
- Krátký, M.; Vinšová, J. Salicylanilide N-monosubstituted carbamates: Synthesis and in vitro antimicrobial activity. Bioorg. Med. Chem. 2016, 24, 1322–1330. [Google Scholar] [CrossRef] [PubMed]
- Vinšová, J.; Kozic, J.; Krátký, M.; Stolaříková, J.; Mandíková, J.; Trejtnar, F.; Buchta, V. Salicylanilide diethyl phosphates: Synthesis, antimicrobial activity and cytotoxicity. Bioorg. Med. Chem. 2014, 22, 728–737. [Google Scholar] [CrossRef] [PubMed]
- Krátký, M.; Vinšová, J.; Stolaříková, J. Antimycobacterial Assessment of Salicylanilide Benzoates including Multidrug-Resistant Tuberculosis Strains. Molecules 2012, 17, 12812–12820. [Google Scholar] [CrossRef]
- Swan, G.E. The pharmacology of halogenated salicylanilides and their anthelmintic use in animals. J. S. Afr. Vet. Assoc. 1999, 70, 61–70. [Google Scholar] [CrossRef]
- Rajamuthiah, R.; Fuchs, B.B.; Conery, A.L.; Kim, W.; Jayamani, E.; Kwon, B.; Ausubel, F.M.; Mylonakis, E. Repurposing salicylanilide anthelmintic drugs to combat drug resistant Staphylococcus aureus. PLoS ONE 2015, 10, e0124595. [Google Scholar] [CrossRef]
- Garcia, C.; Burgain, A.; Chaillot, J.; Pic, E.; Khemiri, I.; Sellam, A. A phenotypic small-molecule screen identifies halogenated salicylanilides as inhibitors of fungal morphogenesis, biofilm formation and host cell invasion. Sci. Rep. 2018, 8, 11559. [Google Scholar] [CrossRef]
- Krátký, M.; Štěpánková, Š.; Vorčáková, K.; Vinšová, J. Salicylanilide Diethyl Phosphates as Cholinesterases Inhibitors. Bioorg. Med. Chem. 2015, 58, 48–52. [Google Scholar] [CrossRef] [PubMed]
- Krátký, M.; Vorčáková, K.; Vinšová, J.; Štěpánková, Š. Investigation of salicylanilide and 4-chlorophenol-based N-monosubstituted carbamates as potential inhibitors of acetyl- and butyrylcholinesterase. Bioorg. Chem. 2018, 80, 668–673. [Google Scholar] [CrossRef] [PubMed]
- Krátký, M.; Štěpánková, Š.; Vorčáková, K.; Švarcová, M.; Vinšová, J. Novel Cholinesterases Inhibitors Based on O-Aromatic N,N-Disubstituted Carbamates and Thiocarbamates. Molecules 2016, 21, 191. [Google Scholar] [CrossRef] [PubMed]
- Vinšová, J.; Krátký, M.; Komlóová, M.; Dadapeer, E.; Štěpánková, Š.; Vorčáková, K.; Stolaříková, J. Diethyl 2-(Phenylcarbamoyl)phenyl Phosphorothioates: Synthesis, Antimycobacterial Activity and Cholinesterase Inhibition. Molecules 2014, 19, 7152–7168. [Google Scholar] [CrossRef]
- Krátký, M.; Vinšová, J. Antifungal Activity of Salicylanilides and Their Esters with 4-(Trifluoromethyl)benzoic Acid. Molecules 2012, 17, 9426–9442. [Google Scholar] [CrossRef]
- Lee, I.Y.; Gruber, T.D.; Samuels, A.; Yun, M.; Nam, B.; Kang, M.; Crowley, K.; Winterroth, B.; Boshoff, H.I.; Barry, C.E. Structure-activity relationships of antitubercular salicylanilides consistent with disruption of the proton gradient via proton shuttling. Bioorg. Med. Chem. 2013, 21, 114–126. [Google Scholar] [CrossRef]
- Institute of Medicinal Molecular Design, Inc.; Muto, S.; Itai, A. Inhibitors against the Production and Release of Inflammatory Cytokines. Patent No. EP1512396, A1, 9 March 2005. [Google Scholar]
- Geigy, A.G.; Bindler, J.; Model, E. Poly Halo-Salicylanilides. Patent No. US2703332 (A), 1 March 1955. [Google Scholar]
- Charles University, Faculty of Pharmacy in Hradec Králové; Vinšová, J.; Krátký, M.; Paraskevopoulos, G. Substituted derivative of oxyphosphorus acids, its use and pharmaceutical preparation containing it. Patent Application No. WO 2016095878 A1, 23 June 2016. [Google Scholar]
- Zdrazilova, P.; Stepankova, S.; Komers, K.; Ventura, K.; Cegan, A. Half-inhibition concentrations of new cholinesterase inhibitors. Z. Naturforsch. C 2004, 59, 293–296. [Google Scholar] [CrossRef]
- Sinko, G.; Calic, M.; Bosak, A.; Kovarik, Z. Limitation of the Ellman method: Cholinesterase activity measurement in the presence of oximes. Anal. Biochem. 2007, 370, 223–227. [Google Scholar] [CrossRef]
- Lineweaver, H.; Burk, D. The Determination of Enzyme Dissociation Constants. J. Am. Chem. Soc. 1934, 56, 658–666. [Google Scholar] [CrossRef]
- Kandiah, N.; Pai, M.C.; Senanarong, V.; Looi, I.; Ampil, E.; Park, K.W.; Karanam, A.K.; Christopher, S. Rivastigmine: The advantages of dual inhibition of acetylcholinesterase and butyrylcholinesterase and its role in subcortical vascular dementia and Parkinson’s disease dementia. Clin. Interv. Aging 2017, 12, 697–707. [Google Scholar] [CrossRef]
- Szedlacsek, S.E.; Duggleby, R.G. [6] Kinetics of slow and tight-binding inhibitors. Methods Enzymol. 1995, 249, 144–180. [Google Scholar]
- Singh, J.; Petter, R.C.; Bailli, T.A.; Whitty, A. The resurgence of covalent drugs. Nat. Rev. Drug Discov. 2011, 10, 307–317. [Google Scholar] [CrossRef]
- Jann, M.W.; Shirley, K.L.; Small, G.W. Clinical Pharmacokinetics and Pharmacodynamics of Cholinesterase Inhibitors. Clin. Pharmacokinet. 2002, 41, 719–739. [Google Scholar] [CrossRef]
- Bartolini, M.; Cavrini, V.; Andrisano, V. Characterization of reversible and pseudo-irreversible acetylcholinesterase inhibitors by means of an immobilized enzyme reactor. J. Chromatogr. A 2007, 1144, 102–110. [Google Scholar] [CrossRef]
- Biochemistry. Fourth edition. Available online: http://gtu.ge/Agro-Lib/Reginald%20H.%20Garrett,%20Charles%20M.%20Grisham%20-%20Biochemistry%20(4th%20ed.)%20-%202010.pdf (accessed on 20 June 2019).
- Lipinski, C.A. Lead- and drug-like compounds: The rule-of-five revolution. Drug. Discov. Today Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, Y.; Zhang, T.; Zhang, J.; Wu, B. Significantly enhanced bioavailability of niclosamide through submicron lipid emulsions with or without PEG-lipid: A comparative study. J. Microencapsul. 2015, 32, 496–502. [Google Scholar] [CrossRef]
- Hitchcock, S.A.; Pennington, L.D. Structure-Brain Exposure Relationships. J. Med. Chem. 2006, 49, 7559–7583. [Google Scholar] [CrossRef]
- Ghose, A.K.; Herbertz, T.; Hudkins, R.L.; Dorsey, B.D.; Mallamo, J.P. Knowledge-Based, Central Nervous System (CNS) Lead Selection and Lead Optimization for CNS Drug Discovery. ACS Chem. Neurosci. 2012, 3, 50–68. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||
|---|---|---|---|---|---|
| Code | R1 | R2 | IC50 (EeAChE) [µM] | IC50 (EqBuChE) [µM] | Selectivity AChE/BuChE |
| 1 | H | H | 49.71 ± 3.73 | 205.73 ± 12.15 | 0.24 |
| 2a | 5-Cl | 3-Cl | 59.50 ± 3.80 | 228.42 ± 11.29 | 0.26 |
| 2b | 5-Cl | 4-Cl | 76.53 ± 2.17 | 169.60 ± 3.09 | 0.45 |
| 2c | 5-Cl | 3,4-diCl | 60.79 ± 6.88 | 186.47 ± 15.69 | 0.33 |
| 2d | 5-Cl | 3,5-diCl | 46.21 ± 0.63 | 111.59 ± 7.98 | 0.41 |
| 2e | 5-Cl | 3,4,5-triCl | 51.06 ± 0.49 | 102.72 ± 0.97 | 0.50 |
| 2f | 5-Cl | 3-Br | 50.03 ± 0.15 | 141.04 ± 1.13 | 0.35 |
| 2g | 5-Cl | 4-Br | 58.25 ± 4.87 | 118.74 ± 6.65 | 0.49 |
| 2h | 5-Cl | 3-F | 58.65 ± 3.98 | 154.40 ± 8.38 | 0.38 |
| 2i | 5-Cl | 4-F | 48.27 ± 8.90 | 132.99 ± 15.81 | 0.36 |
| 2j | 5-Cl | 3-CF3 | 54.41 ± 0.25 | 120.30 ± 3.02 | 0.45 |
| 2k | 5-Cl | 4-CF3 | 58.47 ± 2.80 | 154.65 ± 3.58 | 0.38 |
| 2l | 5-Cl | 3,5-bis-CF3 | 50.18 ± 0.88 | 64.44 ± 1.34 | 0.78 |
| 3a | 4-Cl | 3-Cl | 58.08 ± 1.20 | 134.29 ± 0.24 | 0.43 |
| 3b | 4-Cl | 4-Cl | 56.19 ± 3.23 | 152.00 ± 3.13 | 0.37 |
| 3c | 4-Cl | 3,4-diCl | 46.26 ± 0.56 | 100.27 ± 1.15 | 0.46 |
| 3d | 4-Cl | 3,5-diCl | 50.15 ± 0.26 | 141.10 ± 2.80 | 0.36 |
| 3e | 4-Cl | 3,4,5-triCl | 57.78 ± 4.05 | 132.36 ± 4.31 | 0.44 |
| 3f | 4-Cl | 3-Br | 60.03 ± 0.42 | 167.80 ± 2.06 | 0.36 |
| 3g | 4-Cl | 4-Br | 58.32 ± 1.57 | 173.99 ± 4.02 | 0.34 |
| 3h | 4-Cl | 3-F | 60.79 ± 1.74 | 145.66 ± 4.07 | 0.42 |
| 3i | 4-Cl | 4-F | 62.85 ± 1.53 | 151.76 ± 7.58 | 0.41 |
| 3j | 4-Cl | 3-CF3 | 68.28 ± 3.78 | 163.18 ± 11.68 | 0.42 |
| 3k | 4-Cl | 4-CF3 | 60.29 ± 2.39 | 199.53 ± 2.02 | 0.30 |
| 3l | 4-Cl | 3,5-bis-CF3 | 54.72 ± 2.05 | 150.50 ± 0.87 | 0.36 |
| 4a | 5-Br | 3-Cl | 62.04 ± 6.48 | 134.00 ± 1.63 | 0.46 |
| 4b | 5-Br | 4-Cl | 85.75 ± 6.10 | 130.61 ± 3.99 | 0.66 |
| 4c | 5-Br | 3,4-diCl | 60.83 ± 3.65 | 122.60 ± 7.20 | 0.50 |
| 4d | 5-Br | 3,5-diCl | 33.13 ± 0.47 | 135.92 ± 0.14 | 0.24 |
| 4e | 5-Br | 3,4,5-triCl | 42.08 ± 2.41 | 140.07 ± 6.20 | 0.30 |
| 4f | 5-Br | 3-Br | 69.72 ± 9.97 | 145.78 ± 3.53 | 0.48 |
| 4g | 5-Br | 4-Br | 70.30 ± 2.85 | 112.16 ± 1.79 | 0.63 |
| 4h | 5-Br | 3-F | 54.44 ± 0.73 | 119.24 ± 2.53 | 0.46 |
| 4i | 5-Br | 4-F | 61.22 ± 1.08 | 136.57 ± 1.47 | 0.45 |
| 4j | 5-Br | 3-CF3 | 42.67 ± 0.18 | 116.91 ± 2.23 | 0.36 |
| 4k | 5-Br | 4-CF3 | 44.79 ± 3.44 | 134.81 ± 0.86 | 0.33 |
| 4l | 5-Br | 3,5-bis-CF3 | 48.46 ± 1.07 | 53.46 ± 3.20 | 0.91 |
| Rivastigmine | 56.10 ± 1.41 | 38.40 ± 1.97 | 1.46 | ||
 | ||||
|---|---|---|---|---|
| Code | X | IC50 (EeAChE) [µM] | IC50 (EqBuChE) [µM] | Selectivity AChE/BuChE |
| 5a [22] | O | 86.3 ± 4.9 | 9.84 ± 0.06 | 8.77 |
| 5b [25] | S | 9.16 ± 0.17 | 18.3 ± 1.0 | 0.50 |
| 5c | − | 66.37 ± 1.14 | 2.37 ± 0.01 | 28.00 |
| 5d | O | 48.13 ± 1.70 | 25.00 ± 0.30 | 1.93 |
| 5e | S | 63.48 ± 1.41 | 170.10 ± 13.58 | 0.37 |
| 3k | 60.29 ± 2.39 | 199.53 ± 2.02 | 0.30 | |
| Rivastigmine | 56.10 ± 1.41 | 38.40 ± 1.97 | 1.46 | |
| Code | R1 | R2 | MW | LogP | H-bond Donors | H-bond Acceptors | Number of Violations | tPSA [Å2] |
|---|---|---|---|---|---|---|---|---|
| 1 | H | H | 213.24 | 2.45 | 2 | 3 | 0 | 49.33 |
| 2a | 5-Cl | 3-Cl | 282.12 | 3.57 | 2 | 3 | 0 | 49.33 |
| 2b | 5-Cl | 4-Cl | 282.12 | 3.57 | 2 | 3 | 0 | 49.33 |
| 2c | 5-Cl | 3,4-diCl | 316.56 | 4.12 | 2 | 3 | 0 | 49.33 |
| 2d | 5-Cl | 3,5-diCl | 316.56 | 4.12 | 2 | 3 | 0 | 49.33 |
| 2e | 5-Cl | 3,4,5-triCl | 351.00 | 4.68 | 2 | 3 | 0 | 49.33 |
| 2f | 5-Cl | 3-Br | 326.57 | 3.84 | 2 | 3 | 0 | 49.33 |
| 2g | 5-Cl | 4-Br | 326.57 | 3.84 | 2 | 3 | 0 | 49.33 |
| 2h | 5-Cl | 3-F | 265.67 | 3.17 | 2 | 3 | 0 | 49.33 |
| 2i | 5-Cl | 4-F | 265.67 | 3.17 | 2 | 3 | 0 | 49.33 |
| 2j | 5-Cl | 3-CF3 | 315.68 | 3.93 | 2 | 3 | 0 | 49.33 |
| 2k | 5-Cl | 4-CF3 | 315.68 | 3.93 | 2 | 3 | 0 | 49.33 |
| 2l | 5-Cl | 3,5-bis-CF3 | 383.67 | 4.85 | 2 | 3 | 0 | 49.33 |
| 3a | 4-Cl | 3-Cl | 282.12 | 3.57 | 2 | 3 | 0 | 49.33 |
| 3b | 4-Cl | 4-Cl | 282.12 | 3.57 | 2 | 3 | 0 | 49.33 |
| 3c | 4-Cl | 3,4-diCl | 316.56 | 4.12 | 2 | 3 | 0 | 49.33 |
| 3d | 4-Cl | 3,5-diCl | 316.56 | 4.12 | 2 | 3 | 0 | 49.33 |
| 3e | 4-Cl | 3,4,5-triCl | 351.00 | 4.68 | 2 | 3 | 0 | 49.33 |
| 3f | 4-Cl | 3-Br | 326.57 | 3.84 | 2 | 3 | 0 | 49.33 |
| 3g | 4-Cl | 4-Br | 326.57 | 3.84 | 2 | 3 | 0 | 49.33 |
| 3h | 4-Cl | 3-F | 265.67 | 3.17 | 2 | 3 | 0 | 49.33 |
| 3i | 4-Cl | 4-F | 265.67 | 3.17 | 2 | 3 | 0 | 49.33 |
| 3j | 4-Cl | 3-CF3 | 315.68 | 3.93 | 2 | 3 | 0 | 49.33 |
| 3k | 4-Cl | 4-CF3 | 315.68 | 3.93 | 2 | 3 | 0 | 49.33 |
| 3l | 4-Cl | 3,5-bis-CF3 | 383.67 | 4.85 | 2 | 3 | 0 | 49.33 |
| 4a | 5-Br | 3-Cl | 326.57 | 3.84 | 2 | 3 | 0 | 49.33 |
| 4b | 5-Br | 4-Cl | 326.57 | 3.84 | 2 | 3 | 0 | 49.33 |
| 4c | 5-Br | 3,4-diCl | 361.02 | 4.4 | 2 | 3 | 0 | 49.33 |
| 4d | 5-Br | 3,5-diCl | 361.02 | 4.4 | 2 | 3 | 0 | 49.33 |
| 4e | 5-Br | 3,4,5-triCl | 395.46 | 4.95 | 2 | 3 | 0 | 49.33 |
| 4f | 5-Br | 3-Br | 371.03 | 4.11 | 2 | 3 | 0 | 49.33 |
| 4g | 5-Br | 4-Br | 371.03 | 4.11 | 2 | 3 | 0 | 49.33 |
| 4h | 5-Br | 3-F | 310.12 | 3.44 | 2 | 3 | 0 | 49.33 |
| 4i | 5-Br | 4-F | 310.12 | 3.44 | 2 | 3 | 0 | 49.33 |
| 4j | 5-Br | 3-CF3 | 360.13 | 4.2 | 2 | 3 | 0 | 49.33 |
| 4k | 5-Br | 4-CF3 | 360.13 | 4.2 | 2 | 3 | 0 | 49.33 |
| 4l | 5-Br | 3,5-bis-CF3 | 428.13 | 5.12 | 2 | 3 | 1 | 49.33 |
| 5a | - | - | 451.76 | 5.08 | 1 | 6 | 1 | 73.86 |
| 5b | - | - | 467.82 | 5.82 | 1 | 5 | 1 | 56.79 |
| 5c | - | - | 435.76 | 4.70 | 1 | 5 | 0 | 56.79 |
| 5d | - | - | 437.74 | 5.64 | 0 | 4 | 1 | 46.61 |
| 5e | - | - | 453.80 | 6.37 | 0 | 3 | 1 | 29.54 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krátký, M.; Štěpánková, Š.; Houngbedji, N.-H.; Vosátka, R.; Vorčáková, K.; Vinšová, J. 2-Hydroxy-N-phenylbenzamides and Their Esters Inhibit Acetylcholinesterase and Butyrylcholinesterase. Biomolecules 2019, 9, 698. https://doi.org/10.3390/biom9110698
Krátký M, Štěpánková Š, Houngbedji N-H, Vosátka R, Vorčáková K, Vinšová J. 2-Hydroxy-N-phenylbenzamides and Their Esters Inhibit Acetylcholinesterase and Butyrylcholinesterase. Biomolecules. 2019; 9(11):698. https://doi.org/10.3390/biom9110698
Chicago/Turabian StyleKrátký, Martin, Šárka Štěpánková, Neto-Honorius Houngbedji, Rudolf Vosátka, Katarína Vorčáková, and Jarmila Vinšová. 2019. "2-Hydroxy-N-phenylbenzamides and Their Esters Inhibit Acetylcholinesterase and Butyrylcholinesterase" Biomolecules 9, no. 11: 698. https://doi.org/10.3390/biom9110698
APA StyleKrátký, M., Štěpánková, Š., Houngbedji, N.-H., Vosátka, R., Vorčáková, K., & Vinšová, J. (2019). 2-Hydroxy-N-phenylbenzamides and Their Esters Inhibit Acetylcholinesterase and Butyrylcholinesterase. Biomolecules, 9(11), 698. https://doi.org/10.3390/biom9110698