Notch Signaling in Kidney Development, Maintenance, and Disease


Abstract
1. Introduction
2. The Myriad Functions of Notch Signaling during Kidney Development
2.1. Overview of Kidney Development
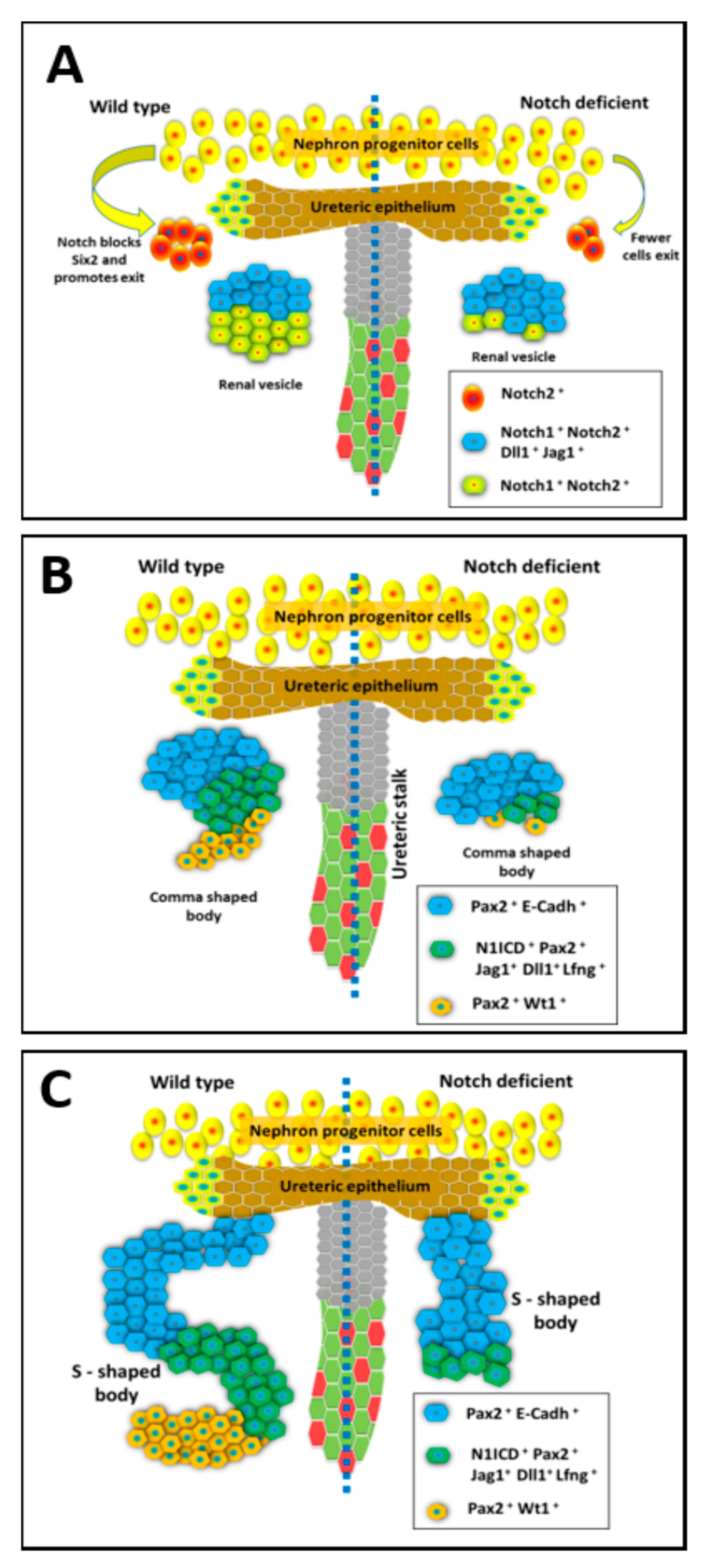
2.2. Expression Pattern of Notch Pathway Components during Kidney Development
2.3. Notch Signaling Represses Six2 to Promote Exit from Nephron Progenitor Niche
2.4. Notch Signaling Mediates Nephron Segmentation during Conversion of RV to SSB
2.5. Loss of Notch Signaling Allows for the Formation of Proximal Tubular Cysts and Microadenomas
2.6. Notch Signaling is Required for Patterning the Collecting Duct Cell Types
3. Notch Signaling in Adult Kidney Maintenance, Repair, and Fibrosis
3.1. Sustained Activation of Notch Signaling Promotes Chronic Kidney Disease
3.2. Opposing Roles for Notch Signaling Following Kidney Injury
3.3. Notch Signaling in the Maintenance of Mature Kidney Cell Types and Remodeling of Epithelial Segments
4. Human Kidney Diseases Associated with Altered Notch Signaling
4.1. Alagille Syndrome (ALGS)
4.2. Hajdu-Cheney Syndrome (HCS)
4.3. Congenital Anomalies of the Kidney and Urinary Tract (CAKUT)
4.4. Diabetic Nephropathy
4.5. Kidney Cancers
5. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Logeat, F.; Bessia, C.; Brou, C.; LeBail, O.; Jarriault, S.; Seidah, N.G.; Israel, A. The Notch1 receptor is cleaved constitutively by a furin-like convertase. Proc. Natl. Acad Sci. USA 1998, 95, 8108–8112. [Google Scholar] [CrossRef] [PubMed]
- Parks, A.L.; Klueg, K.M.; Stout, J.R.; Muskavitch, M.A. Ligand endocytosis drives receptor dissociation and activation in the Notch pathway. Development 2000, 127, 1373–1385. [Google Scholar]
- Brou, C.; Logeat, F.; Gupta, N.; Bessia, C.; LeBail, O.; Doedens, J.R.; Cumano, A.; Roux, P.; Black, R.A.; Israel, A. A novel proteolytic cleavage involved in Notch signaling: The role of the disintegrin-metalloprotease TACE. Mol. Cell 2000, 5, 207–216. [Google Scholar] [CrossRef]
- Ye, Y.; Lukinova, N.; Fortini, M.E. Neurogenic phenotypes and altered Notch processing in Drosophila Presenilin mutants. Nature 1999, 398, 525–529. [Google Scholar] [CrossRef] [PubMed]
- Struhl, G.; Greenwald, I. Presenilin is required for activity and nuclear access of Notch in Drosophila. Nature 1999, 398, 522–525. [Google Scholar] [CrossRef]
- De Strooper, B.; Annaert, W.; Cupers, P.; Saftig, P.; Craessaerts, K.; Mumm, J.S.; Schroeter, E.H.; Schrijvers, V.; Wolfe, M.S.; Ray, W.J.; et al. A presenilin-1-dependent gamma-secretase-like protease mediates release of Notch intracellular domain. Nature 1999, 398, 518–522. [Google Scholar] [CrossRef]
- Tamura, K.; Taniguchi, Y.; Minoguchi, S.; Sakai, T.; Tun, T.; Furukawa, T.; Honjo, T. Physical interaction between a novel domain of the receptor Notch and the transcription factor RBP-J kappa/Su(H). Curr. Biol. 1995, 5, 1416–1423. [Google Scholar] [CrossRef]
- Jarriault, S.; Brou, C.; Logeat, F.; Schroeter, E.H.; Kopan, R.; Israel, A. Signalling downstream of activated mammalian Notch. Nature 1995, 377, 355–358. [Google Scholar] [CrossRef]
- Kovall, R.A.; Gebelein, B.; Sprinzak, D.; Kopan, R. The Canonical Notch Signaling Pathway: Structural and Biochemical Insights into Shape, Sugar, and Force. Dev. Cell 2017, 41, 228–241. [Google Scholar] [CrossRef]
- Kopan, R.; Ilagan, M.X. The canonical Notch signaling pathway: Unfolding the activation mechanism. Cell 2009, 137, 216–233. [Google Scholar] [CrossRef]
- Fabris, L.; Fiorotto, R.; Spirli, C.; Cadamuro, M.; Mariotti, V.; Perugorria, M.J.; Banales, J.M.; Strazzabosco, M. Pathobiology of inherited biliary diseases: A roadmap to understand acquired liver diseases. Nat. Rev. Gastroenterol. HepatoL. 2019, 16, 497–511. [Google Scholar] [CrossRef] [PubMed]
- Adams, J.M.; Jafar-Nejad, H. The Roles of Notch Signaling in Liver Development and Disease. Biomolecules 2019, 9, 608. [Google Scholar] [CrossRef] [PubMed]
- Lasky, J.L.; Wu, H. Notch signaling, brain development, and human disease. Pediatr. Res. 2005, 57, 104R–109R. [Google Scholar] [CrossRef] [PubMed]
- Goel, P.N.; Moharrer, Y.; Hebb, J.H.; Egol, A.J.; Kaur, G.; Hankenson, K.D.; Ahn, J.; Ashley, J.W. Suppression of Notch Signaling in Osteoclasts Improves Bone Regeneration and Healing. J. Orthop. Res. 2019, 37, 2089–2103. [Google Scholar] [CrossRef] [PubMed]
- Meurette, O.; Mehlen, P. Notch Signaling in the Tumor Microenvironment. Cancer Cell 2018, 34, 536–548. [Google Scholar] [CrossRef] [PubMed]
- Vega, Q.C.; Worby, C.A.; Lechner, M.S.; Dixon, J.E.; Dressler, G.R. Glial cell line-derived neurotrophic factor activates the receptor tyrosine kinase RET and promotes kidney morphogenesis. Proc. Natl. Acad Sci. USA 1996, 93, 10657–10661. [Google Scholar] [CrossRef]
- Shakya, R.; Watanabe, T.; Costantini, F. The role of GDNF/Ret signaling in ureteric bud cell fate and branching morphogenesis. Dev. Cell 2005, 8, 65–74. [Google Scholar] [CrossRef]
- Lu, B.C.; Cebrian, C.; Chi, X.; Kuure, S.; Kuo, R.; Bates, C.M.; Arber, S.; Hassell, J.; MacNeil, L.; Hoshi, M.; et al. Etv4 and Etv5 are required downstream of GDNF and Ret for kidney branching morphogenesis. Nat. Genet. 2009, 41, 1295–1302. [Google Scholar] [CrossRef]
- Riccio, P.; Cebrian, C.; Zong, H.; Hippenmeyer, S.; Costantini, F. Ret and Etv4 Promote Directed Movements of Progenitor Cells during Renal Branching Morphogenesis. PLoS Biol. 2016, 14, e1002382. [Google Scholar] [CrossRef]
- Kuure, S.; Chi, X.; Lu, B.; Costantini, F. The transcription factors Etv4 and Etv5 mediate formation of the ureteric bud tip domain during kidney development. Development 2010, 137, 1975–1979. [Google Scholar] [CrossRef]
- Watanabe, T.; Costantini, F. Real-time analysis of ureteric bud branching morphogenesis in vitro. Dev. Biol. 2004, 271, 98–108. [Google Scholar] [CrossRef] [PubMed]
- Self, M.; Lagutin, O.V.; Bowling, B.; Hendrix, J.; Cai, Y.; Dressler, G.R.; Oliver, G. Six2 is required for suppression of nephrogenesis and progenitor renewal in the developing kidney. EMBO J. 2006, 25, 5214–5228. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, A.; Valerius, M.T.; Mugford, J.W.; Carroll, T.J.; Self, M.; Oliver, G.; McMahon, A.P. Six2 defines and regulates a multipotent self-renewing nephron progenitor population throughout mammalian kidney development. Cell Stem Cell 2008, 3, 169–181. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, A.; Kwan, K.M.; Carroll, T.J.; McMahon, A.P.; Mendelsohn, C.L.; Behringer, R.R. Distinct and sequential tissue-specific activities of the LIM-class homeobox gene Lim1 for tubular morphogenesis during kidney development. Development 2005, 132, 2809–2823. [Google Scholar] [CrossRef] [PubMed]
- Chung, E.; Deacon, P.; Marable, S.; Shin, J.; Park, J.S. Notch signaling promotes nephrogenesis by downregulating Six2. Development 2016, 143, 3907–3913. [Google Scholar] [CrossRef] [PubMed]
- Kuure, S.; Sainio, K.; Vuolteenaho, R.; Ilves, M.; Wartiovaara, K.; Immonen, T.; Kvist, J.; Vainio, S.; Sariola, H. Crosstalk between Jagged1 and GDNF/Ret/GFRalpha1 signalling regulates ureteric budding and branching. Mech. Dev. 2005, 122, 765–780. [Google Scholar] [CrossRef] [PubMed]
- Kuure, S.; Vuolteenaho, R.; Vainio, S. Kidney morphogenesis: Cellular and molecular regulation. Mech. Dev. 2000, 92, 31–45. [Google Scholar] [CrossRef]
- Little, M.H.; McMahon, A.P. Mammalian kidney development: Principles, progress, and projections. Cold Spring Harb Perspect Biol. 2012, 4, a008300. [Google Scholar] [CrossRef]
- Hartman, H.A.; Lai, H.L.; Patterson, L.T. Cessation of renal morphogenesis in mice. Dev. Biol. 2007, 310, 379–387. [Google Scholar] [CrossRef]
- Cebrian, C.; Borodo, K.; Charles, N.; Herzlinger, D.A. Morphometric index of the developing murine kidney. Dev. Dyn. 2004, 231, 601–608. [Google Scholar] [CrossRef]
- Carroll, T.J.; Park, J.S.; Hayashi, S.; Majumdar, A.; McMahon, A.P. Wnt9b plays a central role in the regulation of mesenchymal to epithelial transitions underlying organogenesis of the mammalian urogenital system. Dev. Cell 2005, 9, 283–292. [Google Scholar] [CrossRef] [PubMed]
- McCright, B.; Gao, X.; Shen, L.; Lozier, J.; Lan, Y.; Maguire, M.; Herzlinger, D.; Weinmaster, G.; Jiang, R.; Gridley, T. Defects in development of the kidney, heart and eye vasculature in mice homozygous for a hypomorphic Notch2 mutation. Development 2001, 128, 491–502. [Google Scholar] [PubMed]
- Cheng, H.T.; Miner, J.H.; Lin, M.; Tansey, M.G.; Roth, K.; Kopan, R. Gamma-secretase activity is dispensable for mesenchyme-to-epithelium transition but required for podocyte and proximal tubule formation in developing mouse kidney. Development 2003, 130, 5031–5042. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Pereira, F.A.; Beasley, D.; Zheng, H. Presenilins are required for the formation of comma- and S-shaped bodies during nephrogenesis. Development 2003, 130, 5019–5029. [Google Scholar] [CrossRef]
- Chen, L.; Al-Awqati, Q. Segmental expression of Notch and Hairy genes in nephrogenesis. Am. J. Physiol. Ren. Physiol. 2005, 288, F939–F952. [Google Scholar] [CrossRef]
- Jeong, H.W.; Jeon, U.S.; Koo, B.K.; Kim, W.Y.; Im, S.K.; Shin, J.; Cho, Y.; Kim, J.; Kong, Y.Y. Inactivation of Notch signaling in the renal collecting duct causes nephrogenic diabetes insipidus in mice. J. Clin. Investig. 2009, 119, 3290–3300. [Google Scholar] [CrossRef]
- Guo, Q.; Wang, Y.; Tripathi, P.; Manda, K.R.; Mukherjee, M.; Chaklader, M.; Austin, P.F.; Surendran, K.; Chen, F. Adam10 Mediates the Choice between Principal Cells and Intercalated Cells in the Kidney. J. Am. Soc. Nephrol. 2015, 26, 149–159. [Google Scholar] [CrossRef]
- Leimeister, C.; Schumacher, N.; Gessler, M. Expression of Notch pathway genes in the embryonic mouse metanephros suggests a role in proximal tubule development. Gene Expr. Patterns 2003, 3, 595–598. [Google Scholar] [CrossRef]
- Lindstrom, N.O.; Tran, T.; Guo, J.; Rutledge, E.; Parvez, R.K.; Thornton, M.E.; Grubbs, B.; McMahon, J.A.; McMahon, A.P. Conserved and Divergent Molecular and Anatomic Features of Human and Mouse Nephron Patterning. J. Am. Soc. Nephrol. 2018, 29, 825–840. [Google Scholar] [CrossRef]
- Cheng, H.T.; Kim, M.; Valerius, M.T.; Surendran, K.; Schuster-Gossler, K.; Gossler, A.; McMahon, A.P.; Kopan, R. Notch2, but not Notch1, is required for proximal fate acquisition in the mammalian nephron. Development 2007, 134, 801–811. [Google Scholar] [CrossRef]
- Liu, Z.; Chen, S.; Boyle, S.; Zhu, Y.; Zhang, A.; Piwnica-Worms, D.R.; Ilagan, M.X.; Kopan, R. The extracellular domain of Notch2 increases its cell-surface abundance and ligand responsiveness during kidney development. Dev. Cell 2013, 25, 585–598. [Google Scholar] [CrossRef] [PubMed]
- Surendran, K.; Boyle, S.; Barak, H.; Kim, M.; Stomberski, C.; McCright, B.; Kopan, R. The contribution of Notch1 to nephron segmentation in the developing kidney is revealed in a sensitized Notch2 background and can be augmented by reducing Mint dosage. Dev. Biol. 2010, 337, 386–395. [Google Scholar] [CrossRef] [PubMed]
- Grimm, P.R.; Lazo-Fernandez, Y.; Delpire, E.; Wall, S.M.; Dorsey, S.G.; Weinman, E.J.; Coleman, R.; Wade, J.B.; Welling, P.A. Integrated compensatory network is activated in the absence of NCC phosphorylation. J. Clin. Investig. 2015, 125, 2136–2150. [Google Scholar] [CrossRef]
- Dressler, G.R. Another niche for Notch. Kidney Int. 2008, 73, 1207–1209. [Google Scholar] [CrossRef] [PubMed]
- Fujimura, S.; Jiang, Q.; Kobayashi, C.; Nishinakamura, R. Notch2 activation in the embryonic kidney depletes nephron progenitors. J. Am. Soc. Nephrol. 2010, 21, 803–810. [Google Scholar] [CrossRef]
- Oxburgh, L.; Muthukrishnan, S.D.; Brown, A. Growth Factor Regulation in the Nephrogenic Zone of the Developing Kidney. Results Probl. Cell Differ. 2017, 60, 137–164. [Google Scholar]
- Waters, A.M.; Wu, M.Y.; Onay, T.; Scutaru, J.; Liu, J.; Lobe, C.G.; Quaggin, S.E.; Piscione, T.D. Ectopic notch activation in developing podocytes causes glomerulosclerosis. J. Am. Soc. Nephrol. 2008, 19, 1139–1157. [Google Scholar] [CrossRef]
- Surendran, K.; Selassie, M.; Liapis, H.; Krigman, H.; Kopan, R. Reduced Notch signaling leads to renal cysts and papillary microadenomas. J. Am. Soc. Nephrol. 2010, 21, 819–832. [Google Scholar] [CrossRef]
- Bonegio, R.G.; Beck, L.H.; Kahlon, R.K.; Lu, W.; Salant, D.J. The fate of Notch-deficient nephrogenic progenitor cells during metanephric kidney development. Kidney Int. 2011, 79, 1099–1112. [Google Scholar] [CrossRef]
- Grassmeyer, J.; Mukherjee, M.; deRiso, J.; Hettinger, C.; Bailey, M.; Sinha, S.; Visvader, J.E.; Zhao, H.; Fogarty, E.; Surendran, K. Elf5 is a principal cell lineage specific transcription factor in the kidney that contributes to Aqp2 and Avpr2 gene expression. Dev. Biol. 2017, 424, 77–89. [Google Scholar] [CrossRef]
- Chung, E.; Deacon, P.; Park, J.S. Notch is required for the formation of all nephron segments and primes nephron progenitors for differentiation. Development 2017, 144, 4530–4539. [Google Scholar] [CrossRef] [PubMed]
- Werth, M.; Schmidt-Ott, K.M.; Leete, T.; Qiu, A.; Hinze, C.; Viltard, M.; Paragas, N.; Shawber, C.J.; Yu, W.; Lee, P.; et al. Transcription factor TFCP2L1 patterns cells in the mouse kidney collecting ducts. Elife 2017, 6, e24265. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Willet, S.G.; Bankaitis, E.D.; Xu, Y.; Wright, C.V.; Gu, G. Non-parallel recombination limits Cre-LoxP-based reporters as precise indicators of conditional genetic manipulation. Genesis 2013, 51, 436–442. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, M.; Ratnayake, I.; Janga, M.; Fogarty, E.; Scheidt, S.; Grassmeyer, J.; deRiso, J.; Chandrasekar, I.; Ahrenkiel, P.; Kopan, R.; et al. Notch signaling regulates Akap12 expression and primary cilia length during renal tubule morphogenesis. bioRxiv 2019, 760181. [Google Scholar] [CrossRef]
- Artavanis-Tsakonas, S.; Simpson, P. Choosing a cell fate: A view from the Notch locus. Trends Genet. 1991, 7, 403–408. [Google Scholar] [CrossRef]
- Bielesz, B.; Sirin, Y.; Si, H.; Niranjan, T.; Gruenwald, A.; Ahn, S.; Kato, H.; Pullman, J.; Gessler, M.; Haase, V.H.; et al. Epithelial Notch signaling regulates interstitial fibrosis development in the kidneys of mice and humans. J. Clin. Investig. 2010, 120, 4040–4054. [Google Scholar] [CrossRef]
- Niranjan, T.; Bielesz, B.; Gruenwald, A.; Ponda, M.P.; Kopp, J.B.; Thomas, D.B.; Susztak, K. The Notch pathway in podocytes plays a role in the development of glomerular disease. Nat. Med. 2008, 14, 290–298. [Google Scholar] [CrossRef]
- Vooijs, M.; Ong, C.T.; Hadland, B.; Huppert, S.; Liu, Z.; Korving, J.; van den Born, M.; Stappenbeck, T.; Wu, Y.; Clevers, H.; et al. Mapping the consequence of Notch1 proteolysis in vivo with NIP-CRE. Development 2007, 134, 535–544. [Google Scholar] [CrossRef]
- Niranjan, T.; Murea, M.; Susztak, K. The pathogenic role of Notch activation in podocytes. Nephron. Exp. Nephrol. 2009, 111, e73–e79. [Google Scholar] [CrossRef]
- Mukherjee, M.; deRiso, J.; Otterpohl, K.; Ratnayake, I.; Kota, D.; Ahrenkiel, P.; Chandrasekar, I.; Surendran, K. Endogenous Notch Signaling in Adult Kidneys Maintains Segment-Specific Epithelial Cell Types of the Distal Tubules and Collecting Ducts to Ensure Water Homeostasis. J. Am. Soc. Nephrol. 2019, 30, 110–126. [Google Scholar] [CrossRef]
- Piscione, T.D.; Wu, M.Y.; Quaggin, S.E. Expression of Hairy/Enhancer of Split genes, Hes1 and Hes5, during murine nephron morphogenesis. Gene Expr. Patterns 2004, 4, 707–711. [Google Scholar] [CrossRef] [PubMed]
- Murea, M.; Park, J.K.; Sharma, S.; Kato, H.; Gruenwald, A.; Niranjan, T.; Si, H.; Thomas, D.B.; Pullman, J.M.; Melamed, M.L.; et al. Expression of Notch pathway proteins correlates with albuminuria, glomerulosclerosis, and renal function. Kidney Int. 2010, 78, 514–522. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Shrestha, R.; Qiu, C.; Kondo, A.; Huang, S.; Werth, M.; Li, M.; Barasch, J.; Susztak, K. Single-cell transcriptomics of the mouse kidney reveals potential cellular targets of kidney disease. Science 2018, 360, 758–763. [Google Scholar] [CrossRef] [PubMed]
- Han, S.H.; Wu, M.Y.; Nam, B.Y.; Park, J.T.; Yoo, T.H.; Kang, S.W.; Park, J.; Chinga, F.; Li, S.Y.; Susztak, K. PGC-1alpha Protects from Notch-Induced Kidney Fibrosis Development. J. Am. Soc. Nephrol. 2017, 28, 3312–3322. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Park, J.; Qiu, C.; Chung, K.W.; Li, S.Y.; Sirin, Y.; Han, S.H.; Taylor, V.; Zimber-Strobl, U.; Susztak, K. Jagged1/Notch2 controls kidney fibrosis via Tfam-mediated metabolic reprogramming. PLoS Biol. 2018, 16, e2005233. [Google Scholar] [CrossRef] [PubMed]
- Bellomo, R.; Ronco, C.; Kellum, J.A.; Mehta, R.L.; Palevsky, P.; Acute Dialysis Quality Initiative Workgroup. Acute renal failure-definition, outcome measures, animal models, fluid therapy and information technology needs: The Second International Consensus Conference of the Acute Dialysis Quality Initiative, (ADQI) Group. Crit. Care 2004, 8, R204–R212. [Google Scholar] [CrossRef] [PubMed]
- Coca, S.G.; Singanamala, S.; Parikh, C.R. Chronic kidney disease after acute kidney injury: A systematic review and meta-analysis. Kidney Int. 2012, 81, 442–448. [Google Scholar] [CrossRef]
- Ishani, A.; Xue, J.L.; Himmelfarb, J.; Eggers, P.W.; Kimmel, P.L.; Molitoris, B.A.; Collins, A.J. Acute kidney injury increases risk of ESRD among elderly. J. Am. Soc. Nephrol. 2009, 20, 223–228. [Google Scholar] [CrossRef]
- Singh, A.P.; Junemann, A.; Muthuraman, A.; Jaggi, A.S.; Singh, N.; Grover, K.; Dhawan, R. Animal models of acute renal failure. Pharm. Rep. 2012, 64, 31–44. [Google Scholar] [CrossRef]
- Ortiz, A.; Sanchez-Nino, M.D.; Izquierdo, M.C.; Martin-Cleary, C.; Garcia-Bermejo, L.; Moreno, J.A.; Ruiz-Ortega, M.; Draibe, J.; Cruzado, J.M.; Garcia-Gonzalez, M.A.; et al. Translational value of animal models of kidney failure. Eur. J. Pharm. 2015, 759, 205–220. [Google Scholar] [CrossRef]
- Sharp, C.N.; Siskind, L.J. Developing better mouse models to study cisplatin-induced kidney injury. Am. J. Physiol. Ren. Physiol. 2017, 313, F835–F841. [Google Scholar] [CrossRef] [PubMed]
- Morrissey, J.; Guo, G.; Moridaira, K.; Fitzgerald, M.; McCracken, R.; Tolley, T.; Klahr, S. Transforming growth factor-beta induces renal epithelial jagged-1 expression in fibrotic disease. J. Am. Soc. Nephrol. 2002, 13, 1499–1508. [Google Scholar] [CrossRef] [PubMed]
- Kaneto, H.; Morrissey, J.; Klahr, S. Increased expression of TGF-beta 1 mRNA in the obstructed kidney of rats with unilateral ureteral ligation. Kidney Int. 1993, 44, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, T.; Terada, Y.; Kuwana, H.; Tanaka, H.; Okado, T.; Kuwahara, M.; Tohda, S.; Sakano, S.; Sasaki, S. Expression and function of the Delta-1/Notch-2/Hes-1 pathway during experimental acute kidney injury. Kidney Int. 2008, 73, 1240–1250. [Google Scholar] [CrossRef] [PubMed]
- Bonventre, J.V. Mechanisms of ischemic acute renal failure. Kidney Int. 1993, 43, 1160–1178. [Google Scholar] [CrossRef]
- Gupta, S.; Li, S.; Abedin, M.J.; Wang, L.; Schneider, E.; Najafian, B.; Rosenberg, M. Effect of Notch activation on the regenerative response to acute renal failure. Am. J. Physiol. Ren. Physiol. 2010, 298, F209–F215. [Google Scholar] [CrossRef][Green Version]
- Huang, R.; Zhou, Q.; Veeraragoo, P.; Yu, H.; Xiao, Z. Notch2/Hes-1 pathway plays an important role in renal ischemia and reperfusion injury-associated inflammation and apoptosis and the gamma-secretase inhibitor DAPT has a nephroprotective effect. Ren. Fail 2011, 33, 207–216. [Google Scholar] [CrossRef]
- Chen, J.; Chen, J.K.; Conway, E.M.; Harris, R.C. Survivin mediates renal proximal tubule recovery from AKI. J. Am. Soc. Nephrol. 2013, 24, 2023–2033. [Google Scholar] [CrossRef]
- Humphreys, B.D.; Valerius, M.T.; Kobayashi, A.; Mugford, J.W.; Soeung, S.; Duffield, J.S.; McMahon, A.P.; Bonventre, J.V. Intrinsic epithelial cells repair the kidney after injury. Cell Stem Cell 2008, 2, 284–291. [Google Scholar] [CrossRef]
- Liu, J.; Krautzberger, A.M.; Sui, S.H.; Hofmann, O.M.; Chen, Y.; Baetscher, M.; Grgic, I.; Kumar, S.; Humphreys, B.D.; Hide, W.A.; et al. Cell-specific translational profiling in acute kidney injury. J. Clin. Investig. 2014, 124, 1242–1254. [Google Scholar] [CrossRef]
- Kang, H.M.; Huang, S.; Reidy, K.; Han, S.H.; Chinga, F.; Susztak, K. Sox9-Positive Progenitor Cells Play a Key Role in Renal Tubule Epithelial Regeneration in Mice. Cell Rep. 2016, 14, 861–871. [Google Scholar] [CrossRef] [PubMed]
- Challen, G.A.; Bertoncello, I.; Deane, J.A.; Ricardo, S.D.; Little, M.H. Kidney side population reveals multilineage potential and renal functional capacity but also cellular heterogeneity. J. Am. Soc. Nephrol. 2006, 17, 1896–1912. [Google Scholar] [CrossRef] [PubMed]
- Chiba, S. Notch signaling in stem cell systems. Stem Cells 2006, 24, 2437–2447. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.; Wang, Y.; Zhang, T.; Zuo, W. Notch-mediated Sox9(+) cell activation contributes to kidney repair after partial nephrectomy. Life Sci. 2018, 193, 104–109. [Google Scholar] [CrossRef]
- Blomqvist, S.R.; Vidarsson, H.; Fitzgerald, S.; Johansson, B.R.; Ollerstam, A.; Brown, R.; Persson, A.E.; Bergstrom, G.G.; Enerback, S. Distal renal tubular acidosis in mice that lack the forkhead transcription factor Foxi1. J. Clin. Investig. 2004, 113, 1560–1570. [Google Scholar] [CrossRef]
- Stone, K.A. Lithium-induced nephrogenic diabetes insipidus. J. Am. Board Fam Pr. 1999, 12, 43–47. [Google Scholar] [CrossRef]
- Edeling, M.; Ragi, G.; Huang, S.; Pavenstadt, H.; Susztak, K. Developmental signalling pathways in renal fibrosis: The roles of Notch, Wnt and Hedgehog. Nat. Rev. Nephrol. 2016, 12, 426–439. [Google Scholar] [CrossRef]
- Asfahani, R.I.; Tahoun, M.M.; Miller-Hodges, E.V.; Bellerby, J.; Virasami, A.K.; Sampson, R.D.; Moulding, D.; Sebire, N.J.; Hohenstein, P.; Scambler, P.J.; et al. Activation of podocyte Notch mediates early Wt1 glomerulopathy. Kidney Int. 2018, 93, 903–920. [Google Scholar] [CrossRef]
- Sorensen-Zender, I.; Rong, S.; Susnik, N.; Zender, S.; Pennekamp, P.; Melk, A.; Haller, H.; Schmitt, R. Renal tubular Notch signaling triggers a prosenescent state after acute kidney injury. Am. J. Physiol. Ren. Physiol. 2014, 306, F907–F915. [Google Scholar] [CrossRef]
- McDaniell, R.; Warthen, D.M.; Sanchez-Lara, P.A.; Pai, A.; Krantz, I.D.; Piccoli, D.A.; Spinner, N.B. NOTCH2 mutations cause Alagille syndrome, a heterogeneous disorder of the notch signaling pathway. Am. J. Hum. Genet. 2006, 79, 169–173. [Google Scholar] [CrossRef]
- Kamath, B.M.; Bauer, R.C.; Loomes, K.M.; Chao, G.; Gerfen, J.; Hutchinson, A.; Hardikar, W.; Hirschfield, G.; Jara, P.; Krantz, I.D.; et al. NOTCH2 mutations in Alagille syndrome. J. Med Genet. 2012, 49, 138–144. [Google Scholar] [CrossRef] [PubMed]
- van der Ven, A.T.; Connaughton, D.M.; Ityel, H.; Mann, N.; Nakayama, M.; Chen, J.; Vivante, A.; Hwang, D.-y.; Schulz, J.; Braun, D.A.; et al. Whole-Exome Sequencing Identifies Causative Mutations in Families with Congenital Anomalies of the Kidney and Urinary Tract. J. Am. Soc. Nephrol. 2018, 29, 2348. [Google Scholar] [CrossRef] [PubMed]
- Gray, M.J.; Kim, C.A.; Bertola, D.R.; Arantes, P.R.; Stewart, H.; Simpson, M.A.; Irving, M.D.; Robertson, S.P. Serpentine fibula polycystic kidney syndrome is part of the phenotypic spectrum of Hajdu-Cheney syndrome. Eur. J. Hum. Genet. EJHG 2012, 20, 122–124. [Google Scholar] [CrossRef] [PubMed]
- Piccoli, D.A.; Spinner, N.B. Alagille Syndrome and the Jagged1 Gene; Seminars in Liver Disease, Copyright© 2001 by Thieme Medical Publishers: New York, NY, USA, 2001; pp. 525–534. [Google Scholar]
- Kamath, B.M.; Podkameni, G.; Hutchinson, A.L.; Leonard, L.D.; Gerfen, J.; Krantz, I.D.; Piccoli, D.A.; Spinner, N.B.; Loomes, K.M.; Meyers, K. Renal anomalies in Alagille syndrome: A disease-defining feature. Am. J. Med. Genet. Part A 2012, 158A, 85–89. [Google Scholar] [CrossRef] [PubMed]
- Barakat, A.J.; Saba, C.; Rennert, O.M. Kidney abnormalities in Hajdu-Cheney syndrome. Pediatric Nephrol. 1996, 10, 712. [Google Scholar] [CrossRef] [PubMed]
- Battelino, N.; Writzl, K.; Bratanič, N.; Irving, M.D.; Novljan, G. End-Stage Renal Disease in an Infant with Hajdu-Cheney Syndrome. Ther. Apher. Dial. 2016, 20, 318–321. [Google Scholar] [CrossRef]
- Kusaba, T.; Hatta, T.; Kimura, T.; Sonomura, K.; Tanda, S.; Kishimoto, N.; Kameyama, H.; Okigaki, M.; Mori, Y.; Ishigami, N.; et al. Renal involvement in cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy, (CADASIL). Clin. Nephrol. 2007, 67, 182–187. [Google Scholar] [CrossRef]
- Guerrot, D.; Francois, A.; Boffa, J.J.; Boulos, N.; Hanoy, M.; Legallicier, B.; Triquenot-Bagan, A.; Guyant-Marechal, L.; Laquerriere, A.; Freguin-Bouilland, C.; et al. Nephroangiosclerosis in cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy: Is NOTCH3 mutation the common culprit? Am. J. Kidney Dis. 2008, 52, 340–345. [Google Scholar] [CrossRef]
- Tada, M.; Itoh, S.; Ishii-Watabe, A.; Suzuki, T.; Kawasaki, N. Functional analysis of the Notch ligand Jagged1 missense mutant proteins underlying Alagille syndrome. FEBS J. 2012, 279, 2096–2107. [Google Scholar] [CrossRef]
- Andersson, E.R.; Chivukula, I.V.; Hankeova, S.; Sjoqvist, M.; Tsoi, Y.L.; Ramskold, D.; Masek, J.; Elmansuri, A.; Hoogendoorn, A.; Vazquez, E.; et al. Mouse Model of Alagille Syndrome and Mechanisms of Jagged1 Missense Mutations. Gastroenterology 2018, 154, 1080–1095. [Google Scholar] [CrossRef]
- Saleh, M.; Kamath, B.M.; Chitayat, D. Alagille syndrome: Clinical perspectives. Appl. Clin. Genet. 2016, 9, 75–82. [Google Scholar] [PubMed]
- Mitchell, E.; Gilbert, M.; Loomes, K.M. Alagille syndrome. Clin. Liver Dis. 2018, 22, 625–641. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, M.A.; Bauer, R.C.; Rajagopalan, R.; Grochowski, C.M.; Chao, G.; McEldrew, D.; Nassur, J.A.; Rand, E.B.; Krock, B.L.; Kamath, B.M.; et al. Alagille syndrome mutation update: Comprehensive overview of JAG1 and NOTCH2 mutation frequencies and insight into missense variant classification. Hum. Mutat. 2019, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Majewski, J.; Schwartzentruber, J.A.; Caqueret, A.; Patry, L.; Marcadier, J.; Fryns, J.P.; Boycott, K.M.; Ste-Marie, L.G.; McKiernan, F.E.; Marik, I. Mutations in NOTCH2 in families with Hajdu-Cheney syndrome. Hum. Mutat. 2011, 32, 1114–1117. [Google Scholar] [CrossRef] [PubMed]
- Canalis, E.; Zanotti, S. Hajdu-Cheney syndrome: A review. Orphanet J. Rare Dis. 2014, 9, 200. [Google Scholar] [CrossRef]
- Brennan, A.M.; Pauli, R.M. Hajdu-Cheney syndrome: Evolution of phenotype and clinical problems. Am. J. Med. Genet. 2001, 100, 292–310. [Google Scholar] [CrossRef]
- Van den Houten, B.; Ten Kate, L.; Gerding, J. The Hajdu-Cheney syndrome: A review of the literature and report of 3 cases. Int. J. Oral Surg. 1985, 14, 113–125. [Google Scholar] [CrossRef]
- Bugeon, L.; Taylor, H.B.; Progatzky, F.; Lin, M.I.; Ellis, C.D.; Welsh, N.; Smith, E.; Vargesson, N.; Gray, C.; Renshaw, S.A.; et al. The NOTCH pathway contributes to cell fate decision in myelopoiesis. Haematologica 2011, 96, 1753–1760. [Google Scholar] [CrossRef]
- Ramos, F.J.; Kaplan, B.S.; Bellah, R.D.; Zackai, E.H.; Kaplan, P. Further evidence that the Hajdu-Cheney syndrome and the “serpentine fibula-polycystic kidney syndrome” are a single entity. Am. J. Med Genet. 1998, 78, 474–481. [Google Scholar] [CrossRef]
- Toka, H.R.; Toka, O.; Hariri, A.; Nguyen, H.T. Congenital anomalies of kidney and urinary tract. Semin. Nephrol. 2010, 30, 374–386. [Google Scholar] [CrossRef]
- Farber, G.; Hurtado, R.; Loh, S.; Monette, S.; Mtui, J.; Kopan, R.; Quaggin, S.; Meyer-Schwesinger, C.; Herzlinger, D.; Scott, R.P.; et al. Glomerular endothelial cell maturation depends on ADAM10, a key regulator of Notch signaling. Angiogenesis 2018, 21, 335–347. [Google Scholar] [CrossRef] [PubMed]
- Alicic, R.Z.; Rooney, M.T.; Tuttle, K.R. Diabetic kidney disease: Challenges, progress, and possibilities. Clin. J. Am. Soc. Nephrol. 2017, 12, 2032–2045. [Google Scholar] [CrossRef] [PubMed]
- Akchurin, O.; Du, Z.; Ramkellawan, N.; Dalal, V.; Han, S.H.; Pullman, J.; Musch, A.; Susztak, K.; Reidy, K.J. Partitioning-Defective 1a/b Depletion Impairs Glomerular and Proximal Tubule Development. J. Am. Soc. Nephrol. 2016, 27, 3725–3737. [Google Scholar] [CrossRef] [PubMed]
- Ahn, S.H.; Susztak, K. Getting a notch closer to understanding diabetic kidney disease. Diabetes 2010, 59, 1865–1867. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Rojas, J.D.; Lin, F.; Chiang, Y.C.; Chytil, A.; Chong, D.C.; Bautch, V.L.; Rathmell, W.K.; Dayton, P.A. Ultrasound Molecular Imaging of VEGFR-2 in Clear-Cell Renal Cell Carcinoma Tracks Disease Response to Antiangiogenic and Notch-Inhibition Therapy. Theranostics 2018, 8, 141–155. [Google Scholar] [CrossRef] [PubMed]
- Ljungberg, B.; Campbell, S.C.; Choi, H.Y.; Jacqmin, D.; Lee, J.E.; Weikert, S.; Kiemeney, L.A. The epidemiology of renal cell carcinoma. Eur. Urol. 2011, 60, 615–621. [Google Scholar] [CrossRef]
- Ai, Q.; Ma, X.; Huang, Q.; Liu, S.; Shi, T.; Zhang, C.; Zhu, M.; Zhang, Y.; Wang, B.; Ni, D.; et al. High-Level Expression of Notch1 Increased the Risk of Metastasis in T1 Stage Clear Cell Renal Cell Carcinoma. PLoS ONE 2012, 7, e35022. [Google Scholar] [CrossRef]
- Jędroszka, D.; Orzechowska, M.; Bednarek, A.K. Predictive values of Notch signalling in renal carcinoma. Arch. Med Sci. AMS 2017, 13, 1249–1254. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Regulatory Region Driving Cre/Time Point and Place in Renal System Where Inactivation Occurs | Mouse Model: Genetically Modified Gene and Observed Phenotype | Reference |
|---|---|---|
| Hypomorphic Notch2 alleles (Notch2 del1/del1) | Notch2 del1/del1: Perinatal lethality with hypoplastic kidneys, vascular lesions near cortical region, defective glomerulogenesis, and lack of proper glomeruli | [32] |
| Notch 2 +/del1, Jag1 +/−: Half sized kidneys, decrease in glomeruli number, defective glomeruli | ||
| Notch 2 +/del1, Dll1 +/−: No kidney defects | ||
| Global deletions and global overexpression | Psen1 −/−, Psen2 −/− with rescue of severe pre-natal lethality by PSEN1 human expression: Lethality at 2 h post birth, smaller kidneys that lack comma and S-shaped bodies, glomeruli, proximal tubules, and distal nephron tubule begins formation but does not fully mature | [34] |
| Hoxb7->Cre/collecting duct | Human Jag1 gene overexpression: Variable phenotypes including cysts, decreased nephrons, hypoplastic kidneys, hydropelvises, hydroureters along with drastically lowered GDNF expression levels | [26] |
| Pax3->Cre/Metanephrogenic Mesenchyme Pre-11.5 | Notch2 f/f: Lethality between P1 and P2 due to renal failure with no filtration apparatus present, smaller kidneys with collapsed renal pelvis, no proximal tubules with intact distal tubules, and lack of proximal podocytes | [40] |
| Pax2-Cre/Pre-10.5 renal development | Notch2 f/f: No podocytes or proximal tubules present in kidneys of mutants | |
| RBPJ f/f: Death by E13.5, explant culture revealed lack of proximal tubule and podocytes Notch1f/f: Explant kidney shows no phenotype | ||
| Six2->GFP-Cre/E12.5 onwards in cap mesenchyme | NICD overexpression: Hypoplastic kidneys with only one ureteric branch | |
| Neph->Cre/Podocytes only | NICD overexpression: Proteinuria caused by impaired glomerular filtration selective permeability with progressive glomerulosclerosis and a decrease in mature marker expression (Wt1, Nphs1, and Nphs2) with increased cell cycle activity and increased Pax2 expression | [47] |
| Podocin->Cre/Podocytes only | RBPJ f/−: No observable phenotype | |
| RBPJ f/−, NICD overexpression: Rescue of severe selective filtration defect from increased Notch and rescue of Glomerulosclerosis | ||
| Hoxb7->Cre/collecting duct | Mib1 f/f: Unilateral or bilateral hydronephrosis of distended kidneys at P17, reduced number of principal cells, and increased number of intercalated cells | [36] |
| Six2->GFP-Cre/E12.5 onwards in cap mesenchyme surrounding ureteric bud tips | N2-ICD overexpression: Lethality after birth and kidneys have glomerular cysts, dilated renal tubules, and thin cortexes | [45] |
| Six2->GFP-Cre/E12.5 onwards in cap mesenchyme surrounding ureteric bud tips | Notch2 f/f: Low percentage of renal cysts at P0 with a formation of micro adenomas (proliferating cells) by 52 weeks of age | [48] |
| Notch1 f/f: 30% of mutant mice have renal cysts at P0 | ||
| RBPJ f/f: Lethality by P2 in mutants, kidneys have few glomeruli and proximal tubules | ||
| Rarb2->Cre/Condensing mesenchyme | RBPJ f/f: Large proximal tubule cysts present in mutant kidneys | |
| Six2->GFP-Cre/E12.5 onwards in cap mesenchyme surrounding ureteric bud tips | Notch2 f/f: 31% of mutants have smaller kidneys with fewer glomeruli | [42] |
| Notch1 +/f; Notch2f/f: 67% of mutants have smaller kidneys with fewer glomeruli, increased blood urea nitrogen levels at birth, reduced life span | ||
| Notch1 f/f, Notch2 f/f: Lethality at P1 with compromised renal function (blood urea nitrogen), few proximal tubules with very few glomeruli | ||
| RBPJ f/f: Mutants mice die at P2 due to insufficient filtration in small kidneys Kidneys have few mature nephrons and no S-Shaped body during development with few proximal tubules | ||
| Pax3->Cre/Metanephric Mesenchyme Pre-11.5 | Notch2 f/f, Mint f/f: Mint inactivation partially rescues Notch2-deficient phenotype by increasing the number of proximal nephron segments forms | |
| Rarb2->Cre/Condensing mesenchyme | RBPJ f/deletion: Hypoplastic kidneys that develop cysts with death due to increased blood urea levels causing renal failure Fewer proximal tubules formed, were dilated and cyst-like, with few glomeruli that were functioning Note: Some progenitors escape RBPJ inactivation leading to the development of the present proximal tubules | [49] |
| Hoxb7->Cre/Collecting duct | Adam10 f/f: Hydronephrosis in 30% of mutants with increased water intake, increase urine output, and decreased osmolality; decrease in principal cells and an increase in intercalated cells in collecting duct | [37] |
| Six2->GFP-Cre/ E12.5 onwards in cap mesenchyme surrounding ureteric bud tips | Six2-3XFlag overexpression: Decreased differentiation from mesenchymal progenitors to epithelial tubules | [25] |
| RBPJ f/f: Increased Six2+ cells found deeper in medullary, decreased nephron endowment | ||
| N1 f/f, N2 f/f: Increased Six2+ cells found deeper in medullary, decrease in the number nephrons, lack of development of proper nephrons, and renal vesicles failed to form S-shaped bodies Important note: Mosaicism in Cre positive cells formed some nephrons | ||
| Hoxb7-Cre/Collecting duct | Rbpj f/−: Increased intercalated cells in collecting duct, decreased expression of Elf5 | [50] |
| PS1 −/f and Ps2 −/−: Increased intercalated cell gene expression with decreased principal cell gene expression | ||
| Rosa +/NICD: Increased principal cell gene expression including Elf5 | ||
| Cdh16->Cre/Collecting duct and connecting tubule | Elf5 F/del: Slight decrease in principal cell gene expression | |
| Wnt4->GFP-Cre/Pre-tubular aggregates | Notch1 f/f, Notch2 f/f: Lack of developed nephrons and not just proximal tubules as previously noted, no premature depletion of mesenchymal nephron progenitors | [51] |
| NICD overexpression: No effect on nephron differentiation, glomerulocystic kidney phenotype | ||
| Six2->GFP-Cre/E12.5 onwards in cap mesenchyme surrounding ureteric bud tips | Rosa LacZ/NICD: Glomerulocystic kidneys within mutants; heterogeneous nephron cell population segmenting in the nephron, and not just proximal tubule population as previously found | |
| Cdh16->Cre/Collecting duct and connecting tubule | Jag1 f/f: Increase in collecting duct cell types expressing both principal and intercalated proteins with tubules enlarged as well as enclosing fragments of the tubule; hydronephrosis present in adult mice | [52] |
| Tfcp2l1 f/f: Absence of intercalated cell development in collecting ducts | ||
| Atp6v1b1->Cre/Intercalated cells of the collecting duct (possibly leaky expression in principal cells) | Jag1 f/f: Increase in cells expressing both principal and intercalated proteins within the collecting duct with decreased principal cells | |
| Tfcp2l1 f/f: Standard ratio of principal to intercalated cells in collecting duct at two weeks of age; by two months the collecting duct contains half of the intercalated cells with decreased intercalated protein expression when compared to wild-type | ||
| EllA-Cre/ Early embryogenesis; one cell zygote stage | Tfcp2l1 f/f: Lethality post birth; elimination of intercalated cells in collecting duct |
| Human Disease | Genetic Mutation | Kidney Defect | References |
|---|---|---|---|
| Alagille Syndrome | NOTCH2 p.Cys444Tyr (C444Y)/ECD | Small congenital cystic kidney disease | [90] |
| NOTCH2 c.5930−1G→A/ICD | Tubular acidosis and dysplastic kidneys | [90] | |
| NOTCH2 p.Cys373Arg/ECD | Vesico-ureteric reflux | [91] | |
| NOTCH2 p.Arg2003X/ICD | Echogenicity of kidneys | [91] | |
| CAKUT | NOTCH2 p.Tyr1186Asn/ECD | Vesicoureteral reflux | [92] |
| NOTCH2 p.Arg2256His (R2256H)/ICD | Small dysplastic kidney, ureterovesical junction obstruction | [92] | |
| NOTCH2 p.Arg2298Trp/ICD | Hydronephrosis | [92] | |
| Hajdu–Cheney syndrome | NOTCH2 (Gln2389X)/ICD | Polycystic kidneys | [93] |
| Gene | Disease | Kidney Disease | References |
|---|---|---|---|
| JAG1 | ALGS | Dysplasia (generalized, focal, with vesicoureteral reflux, with renal insufficiency), renal tubular acidosis, vesicoureteral reflux, hydronephrosis, obstruction (retero-pelvic junction, with hydronephrosis), chronic renal failure, endstage renal disease, acute kidney injury, renal lipidosis, renal artery stenosis (bilateral), focal segmental glomerulosclerosis, duplex collecting system | [94,95] |
| NOTCH2 | ALGS | Severe infantile renal disease (small kidneys with cysts bilaterally, renal tubular acidosis, and renal insufficiency), proteinuria that resulted in renal failure, tubular acidosis and dysplastic kidneys, vesicoureteral reflux, echogenicity of kidneys, Neonatal renal failure | [90,91] |
| NOTCH2 | HCS/SFPKS | Cystic disease, hypoplasia, vesicoureteral reflux, glomerulonephritis, hypertension, chronic renal failure, bilateral dysplastic kidneys with numerous, small parenchymal cysts, associated with bilateral, high-grade vesicoureteral reflux | [96,97] |
| NOTCH3 | CADASIL | Focal segmental mesangial proliferation, the loss and degeneration of arterial medial smooth muscle cells and arterial intimal thickening. Immunofluorescence analysis of glomeruli revealed IgA deposition in the mesangial area. Electron microscope analysis revealed electron-dense deposition also in the mesangial area. In addition, granular osmophilic material (GOM) was observed in the extra-glomerular mesangial area and around the vascular smooth muscle cells | [98] |
| NOTCH3 | CADASIL | Chronic kidney disease, renal histological analysis showed severe arteriosclerosis and mild interstitial fibrosis | [99] |
| NOTCH1 | DKD | Higher Notch1 expression observed in glomerulosclerosis | [62] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mukherjee, M.; Fogarty, E.; Janga, M.; Surendran, K. Notch Signaling in Kidney Development, Maintenance, and Disease. Biomolecules 2019, 9, 692. https://doi.org/10.3390/biom9110692
Mukherjee M, Fogarty E, Janga M, Surendran K. Notch Signaling in Kidney Development, Maintenance, and Disease. Biomolecules. 2019; 9(11):692. https://doi.org/10.3390/biom9110692
Chicago/Turabian StyleMukherjee, Malini, Eric Fogarty, Madhusudhana Janga, and Kameswaran Surendran. 2019. "Notch Signaling in Kidney Development, Maintenance, and Disease" Biomolecules 9, no. 11: 692. https://doi.org/10.3390/biom9110692
APA StyleMukherjee, M., Fogarty, E., Janga, M., & Surendran, K. (2019). Notch Signaling in Kidney Development, Maintenance, and Disease. Biomolecules, 9(11), 692. https://doi.org/10.3390/biom9110692