Dynamics of Solid Proteins by Means of Nuclear Magnetic Resonance Relaxometry

 , , ,
, , ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Theory: Decomposition of 1H Spin-Lattice Relaxation Profiles
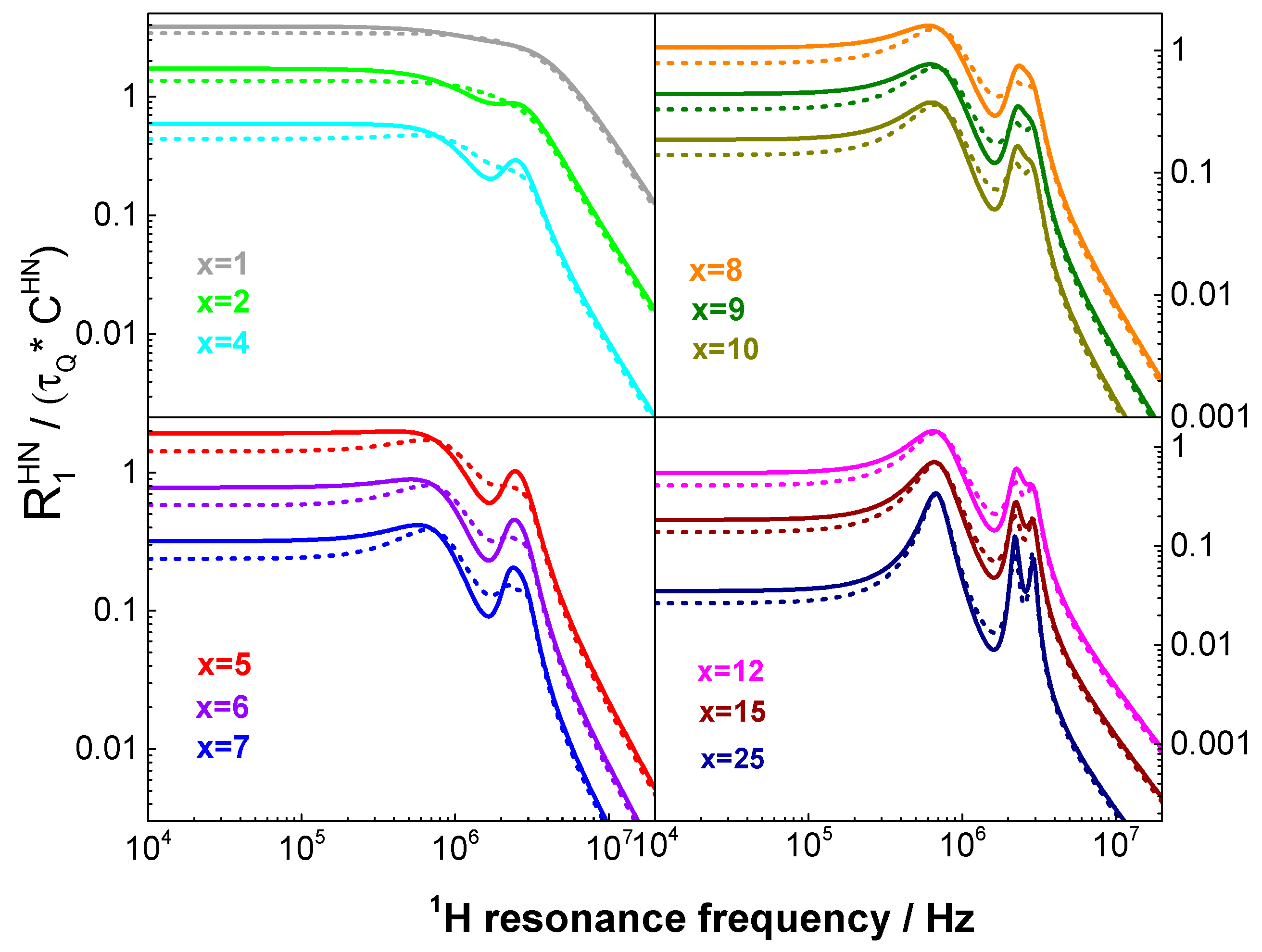
2.2. Theory: Validity Range of the Description of QRE effects
2.3. Experimental Details
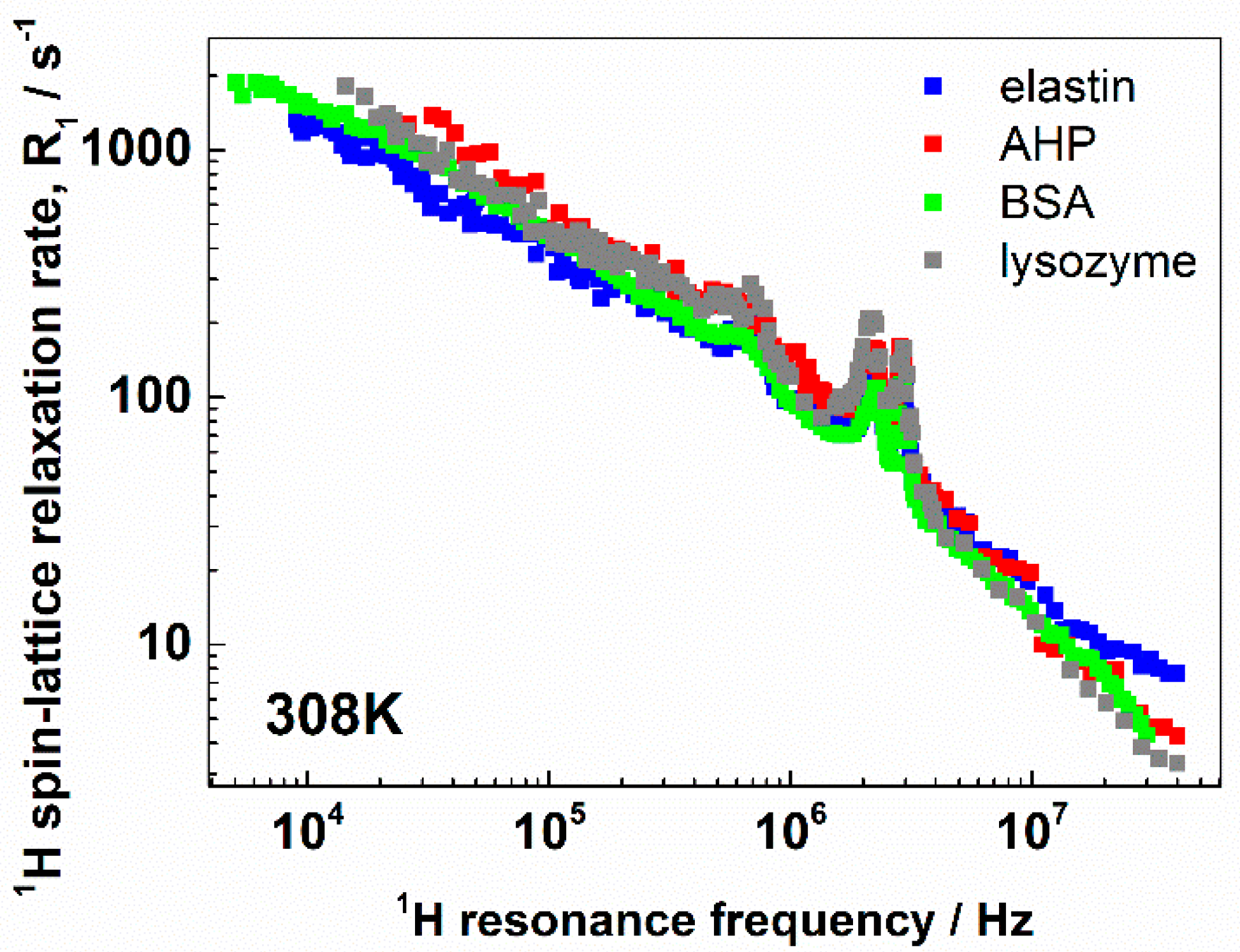
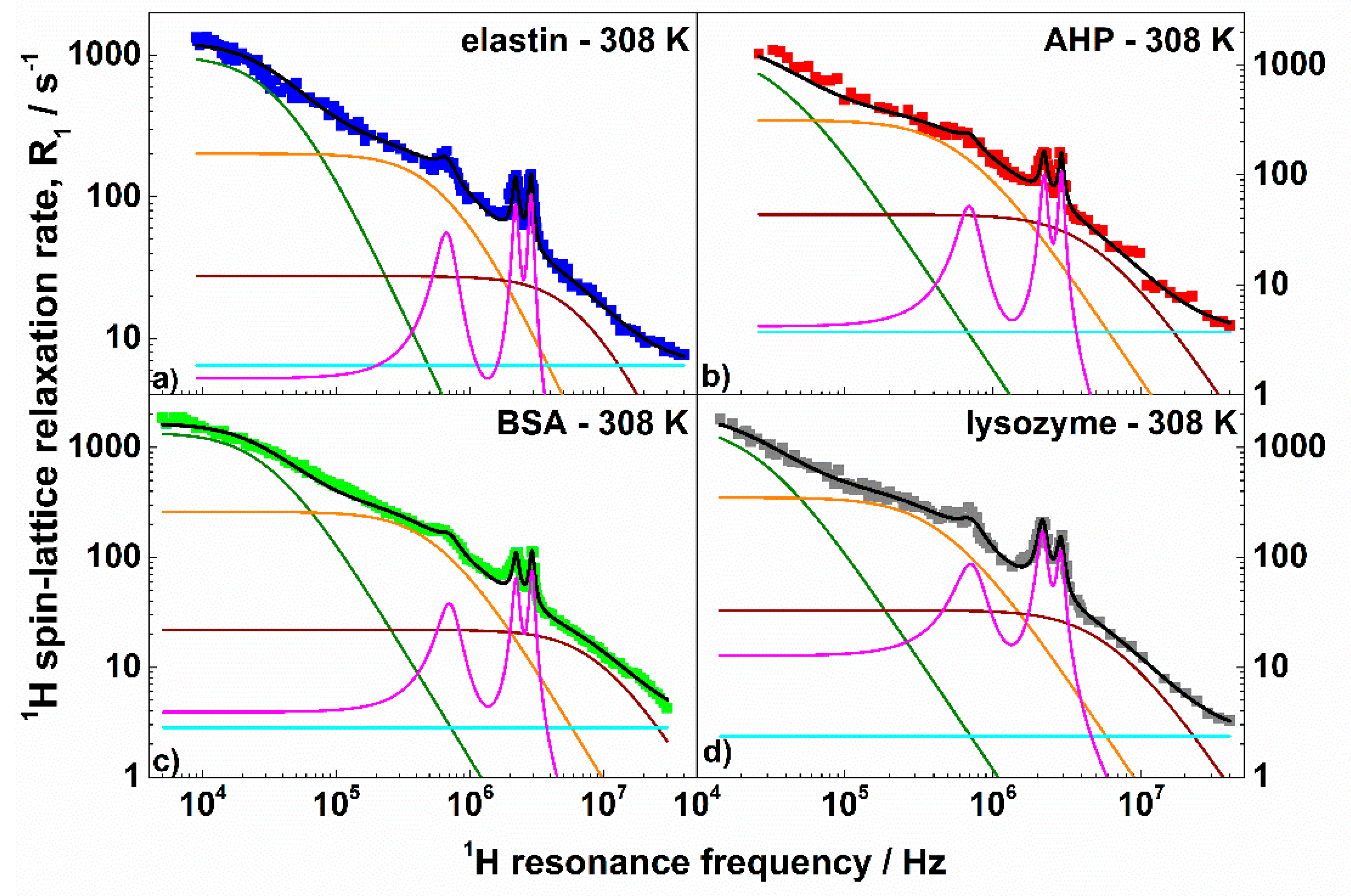
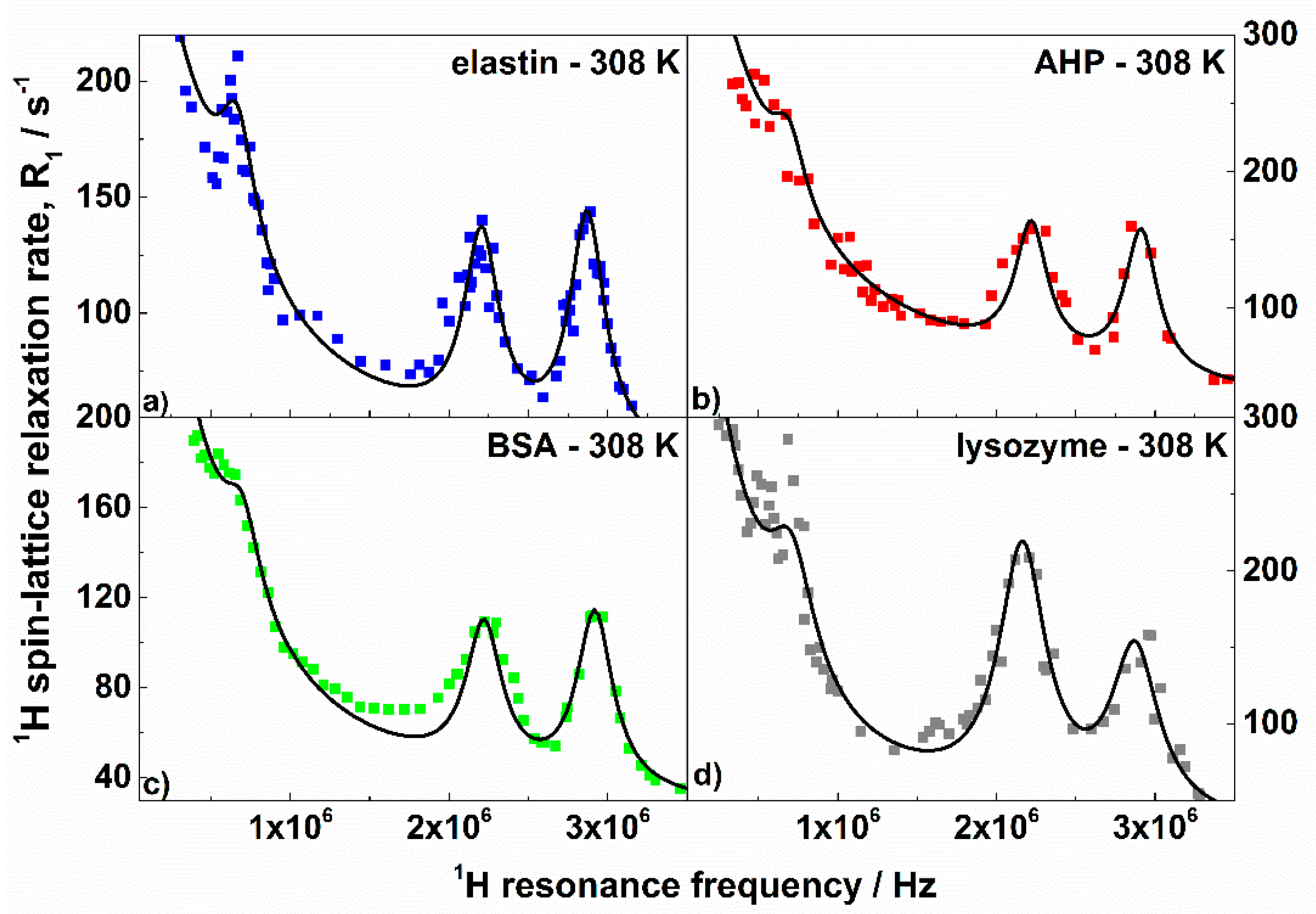
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Sugiki, T.; Kobayashi, N.; Fujiwara, T. Modern technologies of solution nuclear magnetic resonance spectroscopy for three-dimensional structure determination of proteins open avenues for life scientists. Comput. Struct. Biotechnol. J. 2017, 15, 328–339. [Google Scholar] [CrossRef]
- Sakakibara, D.; Sasaki, A.; Ikeya, T.; Hamatsu, J.; Hanashima, T.; Mishima, M.; Yoshimasu, M.; Hayashi, N.; Mikawa, T.; Wälchli, M.; et al. Protein structure determination in living cells by in-cell NMR spectroscopy. Nature 2009, 458, 102–105. [Google Scholar] [CrossRef] [PubMed]
- Wüthrich, K. The way to NMR structures of proteins. Nat. Struct. Biol. 2001, 8, 923–925. [Google Scholar] [CrossRef] [PubMed]
- Kruk, D.; Herrmann, A.; Rössler, E.A. Field-cycling NMR relaxometry of viscous liquids and polymers. Prog. Nucl. Magn. Reson. Spectrosc. 2012, 63, 33–64. [Google Scholar] [CrossRef]
- Kruk, D.; Florek-Wojciechowska, M.; Jakubas, R.; Chaurasia, S.K.; Brym, S. Dynamics of Molecular Crystals by Means of 1H NMR Relaxometry: Dynamical heterogeneity versus homogenous motion. ChemPhysChem 2016, 17, 2329–2339. [Google Scholar] [CrossRef]
- Field-cycling NMR Relaxometry: Instrumentation, Model Theories and Applications; Kimmich, R., Ed.; New Developments in NMR; The Royal Society of Chemistry: London, UK, 2019. [Google Scholar] [CrossRef]
- Kimmich, R.; Fatkullin, N. Polymer chain dynamics and NMR. Adv. Polym. Sci. 2004, 170, 1–113. [Google Scholar] [CrossRef]
- Sebastião, P.J. NMR Relaxometry in liquid crystals: Molecular organization and molecular dynamics interrelation. In Field-cycling NMR Relaxometry: Instrumentation, Model Theories and Applications; The Royal Society of Chemistry: London, UK, 2018; pp. 255–302. [Google Scholar] [CrossRef]
- Kimmich, R.; Anoardo, E. Field-cycling NMR relaxometry. Prog. Nucl. Magn. Reson. Spectrosc. 2004, 44, 257–320. [Google Scholar] [CrossRef]
- Fujara, F.; Kruk, D.; Privalov, A.F. Solid state Field-Cycling NMR relaxometry: Instrumental improvements and new applications. Prog. Nucl. Magn. Reson. Spectrosc. 2014, 82, 39–69. [Google Scholar] [CrossRef]
- Korb, J.P. Multi-scales nuclear spin relaxation of liquids in porous media. C. R. Phys. 2010, 11, 192–203. [Google Scholar] [CrossRef]
- Meier, R.; Kruk, D.; Gmeiner, J.; Rössler, E.A. Intermolecular relaxation in glycerol as revealed by field cycling 1H NMR relaxometry dilution experiments. J. Chem. Phys. 2012, 136, 03450. [Google Scholar] [CrossRef]
- Hwang, L.; Freed, J.H. Dynamic effects of pair correlation functions on spin relaxation by translational diffusion in liquids. J. Chem. Phys. 1975, 63, 4017–4025. [Google Scholar] [CrossRef]
- Ayant, Y.; Belorizky, E.; Aluzon, J.; Gallice, J. Calcul des densités spectrales résultant d’un mouvement aléatoire de translation en relaxation par interaction dipolaire magnétique dans les liquides. J. Phys. France 1975, 36, 991–1004. [Google Scholar] [CrossRef]
- Belorizky, E.; Fries, P.H.; Guillermo, A.; Poncelet, O. Almost ideal 1D water diffusion in imogolite nanotubes evidenced by NMR relaxometry. ChemPhysChem 2010, 11, 2021–2026. [Google Scholar] [CrossRef]
- Heitjans, P.; Indris, S.; Wilkening, M. Solid-state diffusion and NMR. Diffus. Fundam. 2005, 2, 45-1–45-20. [Google Scholar]
- Epp, V.; Wilkening, M. Fast Li diffusion in crystalline LiBH4 due to reduced dimensionality: Frequency-dependent NMR spectroscopy. Phys. Rev. B 2010, 82, 020301. [Google Scholar] [CrossRef]
- Kruk, D.; Wojciechowski, M.; Brym, S.; Singh, R.K. Dynamics of ionic liquids in bulk and in confinement by means of 1H NMR relaxometry-BMIM-OcSO4 in an SiO2 matrix as an example. Phys. Chem. Chem. Phys. 2016, 18, 23184–23194. [Google Scholar] [CrossRef]
- Korb, J.-P.; Winterhalter, M.; McConnell, H.M. Theory of spin relaxation by translational diffusion in two-dimensional systems. J. Chem. Phys. 1984, 80, 1059–1068. [Google Scholar] [CrossRef]
- Westlund, P.O. Quadrupole-enhanced proton spin relaxation for a slow reorienting spin pair: (I)-(S). A stochastic Liouville approach. Mol. Phys. 2009, 107, 2141–2148. [Google Scholar] [CrossRef]
- Westlund, P.O. The quadrupole enhanced 1H spin–lattice relaxation of the amide proton in slow tumbling proteins. Phys. Chem. Chem. Phys. 2010, 12, 3136–3140. [Google Scholar] [CrossRef]
- Kruk, D.; Kubica, A.; Masierak, W.; Privalov, A.F.; Wojciechowski, M.; Medycki, W. Quadrupole relaxation enhancement—Application to molecular crystals. Solid State Nucl. Magn. Reson. 2011, 40, 114–120. [Google Scholar] [CrossRef]
- Kruk, D.; Umut, E.; Masiewicz, E.; Fischer, R.; Scharfetter, H. Multi-quantum quadrupole relaxation enhancement effects in 209Bi compounds. J. Chem. Phys. 2019, 150, 184309. [Google Scholar] [CrossRef]
- Kruk, D.; Privalov, A.; Medycki, W.; Uniszkiewicz, C.; Masierak, W.; Jakubas, R. NMR studies of Solid-State dynamics. In Annual Reports on NMR Spectroscopy; Elsevier Ltd.: Amsterdam, The Netherlands, 2012; Volume 76, pp. 67–138. [Google Scholar] [CrossRef]
- Florek-Wojciechowska, M.; Wojciechowski, M.; Jakubas, R.; Brym, S.; Kruk, D. 1H-NMR relaxometry and quadrupole relaxation enhancement as a sensitive probe of dynamical properties of solids—[C(NH2)3]3Bi2I9 as an example. J. Chem. Phys. 2016, 144, 054501. [Google Scholar] [CrossRef]
- Florek-Wojciechowska, M.; Jakubas, R.; Kruk, D. Structure and dynamics of [NH2 (CH3)2]3Sb2Cl9 by means of 1H NMR relaxometry—Quadrupolar relaxation enhancement effects. Phys. Chem. Chem. Phys. 2017, 19, 11197–11205. [Google Scholar] [CrossRef]
- Kruk, D.; Umut, E.; Masiewicz, E.; Sampl, C.; Fischer, R.; Spirk, S.; Gösweiner, C.; Scharfetter, H. 209Bi quadrupole relaxation enhancement in solids as a step towards new contrast mechanisms in magnetic resonance imaging. Phys. Chem. Chem. Phys. 2018, 20, 12710–12718. [Google Scholar] [CrossRef]
- Kruk, D.; Medycki, W.; Mielczarek, A.; Jakubas, R.; Uniszkiewicz, C. Complex Nuclear Relaxation Processes in guanidinium compounds [C(NH2)3]3Sb2X9 (X = Br, Cl): Effects of Quadrupolar interactions. Appl. Magn. Reson. 2010, 39, 233–249. [Google Scholar] [CrossRef]
- Sunde, E.P.; Halle, B. Mechanism of 1H-14N cross-relaxation in immobilized proteins. J. Magn. Reson. 2010, 203, 257–273. [Google Scholar] [CrossRef]
- Winter, F.; Kimmich, R. 14N1H and 2H1H cross-relaxation in hydrated proteins. Biophys. J. 1985, 48, 331–335. [Google Scholar] [CrossRef]
- Lurie, D.J. Quadrupole-Dips measured by whole-body Field-Cycling Relaxometry and Imaging. Proc. Intl. Soc. Magn. Reson. Med. 1999, 7, 653. [Google Scholar]
- Lurie, D.J.; Davies, G.R.; Foster, M.A.; Hutchison, J.M.S. Field-cycled PEDRI imaging of free radicals with detection at 450 mT. Magn. Reson. Imaging 2005, 23, 175–181. [Google Scholar] [CrossRef]
- Lurie, D.J.; Aime, S.; Baroni, S.; Booth, N.A.; Broche, L.M.; Choi, C.H.; Davies, G.R.; Ismail, S.; Ó hÓgáin, D.; Pine, K.J. Fast field-cycling magnetic resonance imaging. C. R. Phys. 2010, 11, 136–148. [Google Scholar] [CrossRef]
- Korb, J.P.; Van-Quynh, A.; Bryant, R. Low-frequency localized spin-dynamical coupling in proteins. C. R. l’Academie des Sci. Ser. IIc Chem. 2001, 4, 833–837. [Google Scholar] [CrossRef]
- Goddard, Y.A.; Korb, J.P.; Bryant, R.G. Water molecule contributions to proton spin–lattice relaxation in rotationally immobilized proteins. J. Magn. Reson. 2009, 199, 68–74. [Google Scholar] [CrossRef]
- Korb, J.P.; Bryant, R.G. Magnetic field dependence of proton spin–lattice relaxation of confined proteins. C. R. Phys. 2004, 5, 349–357. [Google Scholar] [CrossRef]
- Goddard, Y.; Korb, J.-P.; Bryant, R.G. The magnetic field and temperature dependences of proton spin–lattice relaxation in proteins. J. Chem. Phys. 2007, 126, 175105. [Google Scholar] [CrossRef]
- Halle, B.; Jóhannesson, H.; Venu, K. Model-Free Analysis of Stretched Relaxation Dispersions. J. Magn. Reson. 1998, 135, 1–13. [Google Scholar] [CrossRef]
- Bertini, I.; Fragai, M.; Luchinat, C.; Parigi, G. 1H NMRD profiles of diamagnetic proteins: A model-freeanalysis. Magn. Reson. Chem. 2000, 38, 543–550. [Google Scholar] [CrossRef]
- Ravera, E.; Parigi, G.; Mainz, A.; Religa, T.L.; Reif, B.; Luchinat, C. Experimental determination of microsecond reorientation correlation times in protein solutions. J. Phys. Chem. B 2013, 117, 3548–3553. [Google Scholar] [CrossRef]
- Luchinat, C.; Parigi, G.; Ravera, E. Water and protein dynamics in sedimented systems: A relaxometric investigation. ChemPhysChem 2013, 14, 3156–3161. [Google Scholar] [CrossRef]
- Ravera, E.; Fragai, M.; Parigi, G.; Luchinat, C. Differences in dynamics between crosslinked and non-crosslinked hyaluronates measured by using Fast Field-Cycling Relaxometry. ChemPhysChem 2015, 16, 2803–2809. [Google Scholar] [CrossRef]
- Slichter, C.P. Principles of Magnetic Resonance, 3rd ed.; Springer: Berlin, Germany, 1990. [Google Scholar]
- Kowalewski, J.; Maler, L. Nuclear Spin Relaxation in Liquids: Theory, Experiments, and Applications, 2nd ed.; CRC Press, Taylor & Francis Group: Boca Raton, FL, USA, 2017. [Google Scholar]
- Kruk, D. Theory of Evolution and Relaxation of Multi-spin Systems. In Application to Nuclear Magnetic Resonance (NMR) and Electron Spin Resonance (ESR); Abramis Academic, Arima Publishing: Suffolk, UK, 2007. [Google Scholar]
- Fries, P.H.; Belorizky, E. Simple expressions of the nuclear relaxation rate enhancement due to quadrupole nuclei in slowly tumbling molecules. J. Chem. Phys. 2015, 143, 044202-1–044202-17. [Google Scholar] [CrossRef]
- Westlund, P.O. Nuclear Paramagnetic Spin Relaxation theory: Paramagnetic spinprobes in homogeneous and microheterogeneous solutions. In Dynamics of Solutions and Fluid Mixtures by NMR; Delpuech, J.-J., Ed.; WILEY: Singapore, 1995. [Google Scholar]
- Schneider, D.J.; Freed, J.H. Spin relaxation and motional dynamics. In Advances in Chemical Physics: Lasers, Molecules, and Methods; Hirschfelder, J.O., Wyatt, R.E., Coalson, R.D., Eds.; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 1989; Volume 73, p. 387. [Google Scholar] [CrossRef]
- Benetis, N.; Kowalewski, J.; Nordenskiöld, L.; Wennerström, H.; Westlund, P.O. Nuclear spin relaxation in paramagnetic systems. The slow motion problem for electron spin relaxation. Mol. Phys. 1983, 48, 329–346. [Google Scholar] [CrossRef]
- Nilsson, T.; Kowalewski, J. Slow-Motion Theory of nuclear spin relaxation in paramagnetic low-symmetry complexes: A generalization to High Electron Spin. J. Magn. Reson. 2000, 146, 345–358. [Google Scholar] [CrossRef]
- Kruk, D. Understanding Spin Dynamics; CRC Press, Pan Stanford Publishing: Boca Raton, FL, USA, 2015. [Google Scholar]
- Kruk, D.; Masiewicz, E.; Umut, E.; Petrovic, A.; Kargl, R.; Scharfetter, H. Estimation of the magnitude of quadrupole relaxation enhancement in the context of magnetic resonance imaging contrast. J. Chem. Phys. 2019, 150, 184306. [Google Scholar] [CrossRef]
- Gromiha, M.M. Proteins. In Protein Bioinformatics from Sequence to Function; Elsevier: New Delhi, Amsterdam, The Netherlands, 2010; pp. 1–27. [Google Scholar]
- Alberts, B.; Johnson, A.; Lewis, J. The shape and structure of proteins. In Molecular Biology of the Cell; Garland Science: New York, NY, USA, 2002. [Google Scholar]
- Feng, R.; Konishi, Y.; Bell, A.W. High accuracy molecular weight determination and variation characterization of proteins up to 80 ku by ionspray mass spectrometry. J. Am. Soc. Mass Spectrom. 1991, 2, 387–401. [Google Scholar] [CrossRef]
- Meloun, B.; Morávek, L.; Kostka, V. Complete amino acid sequence of human serum albumin. FEBS Lett. 1975, 58, 134–137. [Google Scholar] [CrossRef]
- Majorek, K.A.; Porebski, P.J.; Dayal, A.; Zimmerman, M.D.; Jablonska, K.; Stewart, A.J.; Chruszcz, M.; Minor, W. Structural and immunologic characterization of bovine, horse, and rabbit serum albumins. Mol. Immunol. 2012, 52, 174–182. [Google Scholar] [CrossRef]
- Abrosimova, K.V.; Shulenina, O.V.; Paston, S.V. FTIR study of secondary structure of bovine serum albumin and ovalbumin. J. Phys. Conf. Ser. 2016, 769, 012016. [Google Scholar] [CrossRef]
- Bramanti, E.; Benedetti, E. Determination of the secondary structure of isomeric forms of human serum albumin by a particular frequency deconvolution procedure applied to Fourier transform IR analysis. Biopolymers 1996, 38, 639–653. [Google Scholar] [CrossRef]
- Palmer, K.J.; Ballantyne, M.; Galvin, J.A. The molecular weight of lysozyme determined by the x-ray diffraction method. J. Am. Chem. Soc. 1948, 70, 906–908. [Google Scholar] [CrossRef]
- White, F.H., Jr. Studies on secondary structure in chicken egg-white lysozyme after reductive cleavage of disulfide bondst. Biochemistry 1976, 15, 2906–2912. [Google Scholar] [CrossRef]
- Debelle, L.; Alix, A.J.P.; Wei, S.M.; Jacob, M.P.; Huvenne, J.P.; Berjot, M.; Legrand, P. The secondary structure and architecture of human elastin. Eur. J. Biochem. 1998, 258, 533–539. [Google Scholar] [CrossRef] [PubMed]
- Keeley, F.W.; Bellingham, C.M.; Woodhouse, K.A. Elastin as a self-organizing biomaterial: Use of recombinantly expressed human elastin polypeptides as a model for investigations of structure and self-assembly of elastin. Philos. Trans. R. Soc. B Biol. Sci. 2002, 357, 185–189. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Zhou, H.X. Protein allostery and conformational dynamics. Chem. Rev. 2016, 116, 6503–6515. [Google Scholar] [CrossRef] [PubMed]
- Khodadadi, S.; Pawlus, S.; Roh, J.H.; Garcia Sakai, V.; Mamontov, E.; Sokolov, A.P. The origin of the dynamic transition in proteins. J. Chem. Phys. 2008, 128, 195106. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Havenith, M. Perspective: Watching low-frequency vibrations of water in biomolecular recognition by THz spectroscopy. J. Chem. Phys. 2015, 143, 170901. [Google Scholar] [CrossRef] [PubMed]
- Yao, L.; Vögeli, B.; Ying, J.; Bax, A. NMR determination of amide N-H equilibrium bond length from concerted dipolar coupling measurements. J. Am. Chem. Soc. 2008, 130, 16518–16520. [Google Scholar] [CrossRef]
- Kennedy, S.D.; Bryant, R.G. Structural effects of hydration: Studies of lysozyme by 13C solids NMR. Biopolymers 1990, 29, 1801–1806. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Elastin | AHP | BSA | Lysozyme |
|---|---|---|---|---|
| /Hz2 | 7.85 × 107 | 9.91 × 107 | 8.95 × 107 | 8.89 × 107 |
| /s | 2.55 × 10−6 (2%) | 2.93 × 10−6 (8%) | 3.06 × 10−6 (2%) | 3.79 × 10−6 (7%) |
| /Hz2 | 2.84 × 108 | 4.16 × 108 | 3.01 × 108 | 3.36 × 108 |
| /s | 1.43 × 10−7 (5%) | 1.52 × 10−7 (9%) | 1.72 × 10−7 (3%) | 2.09 × 10−7 (5%) |
| /Hz2 | 4.21 × 108 | 4.52 × 108 | 4.37 × 108 | 4.24 × 108 |
| /s | 1.31 × 10−8 (12%) | 1.94 × 10−8 (11%) | 1.10 × 10−8 (5%) | 1.56 × 10−8 (7%) |
| /s−1 | 6.47 | 3.73 | 3.04 | 2.36 |
| * /MHz | 3.38 | 3.42 | 3.43 | 3.36 |
| * | 0.39 | 0.40 | 0.41 | 0.42 |
| /s | 1.19 × 10−6 (8%) | 1.27 × 10−6 (12%) | 1.16 × 10−6 (7%) | 8.91 × 10−7 (8%) |
| /Hz2 | 1.01 × 108 | 1.01 × 108 | 7.81 × 107 | 1.93 × 108 |
| /o | 69 | 75 | 72 | 68 |
| /o | 50 | 47 | 49 | 33 |
| relative error (%) | 6.9 | 10.1 | 6.1 | 7.4 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kruk, D.; Masiewicz, E.; Borkowska, A.M.; Rochowski, P.; Fries, P.H.; Broche, L.M.; Lurie, D.J. Dynamics of Solid Proteins by Means of Nuclear Magnetic Resonance Relaxometry. Biomolecules 2019, 9, 652. https://doi.org/10.3390/biom9110652
Kruk D, Masiewicz E, Borkowska AM, Rochowski P, Fries PH, Broche LM, Lurie DJ. Dynamics of Solid Proteins by Means of Nuclear Magnetic Resonance Relaxometry. Biomolecules. 2019; 9(11):652. https://doi.org/10.3390/biom9110652
Chicago/Turabian StyleKruk, Danuta, Elzbieta Masiewicz, Anna M. Borkowska, Pawel Rochowski, Pascal H. Fries, Lionel M. Broche, and David J. Lurie. 2019. "Dynamics of Solid Proteins by Means of Nuclear Magnetic Resonance Relaxometry" Biomolecules 9, no. 11: 652. https://doi.org/10.3390/biom9110652
APA StyleKruk, D., Masiewicz, E., Borkowska, A. M., Rochowski, P., Fries, P. H., Broche, L. M., & Lurie, D. J. (2019). Dynamics of Solid Proteins by Means of Nuclear Magnetic Resonance Relaxometry. Biomolecules, 9(11), 652. https://doi.org/10.3390/biom9110652