Hinge Region in DNA Packaging Terminase pUL15 of Herpes Simplex Virus: A Potential Allosteric Target for Antiviral Drugs

 , and
, and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Estimated Rate of Change
2.2. Phylogenetic Tree
2.3. Protein Structure Modeling
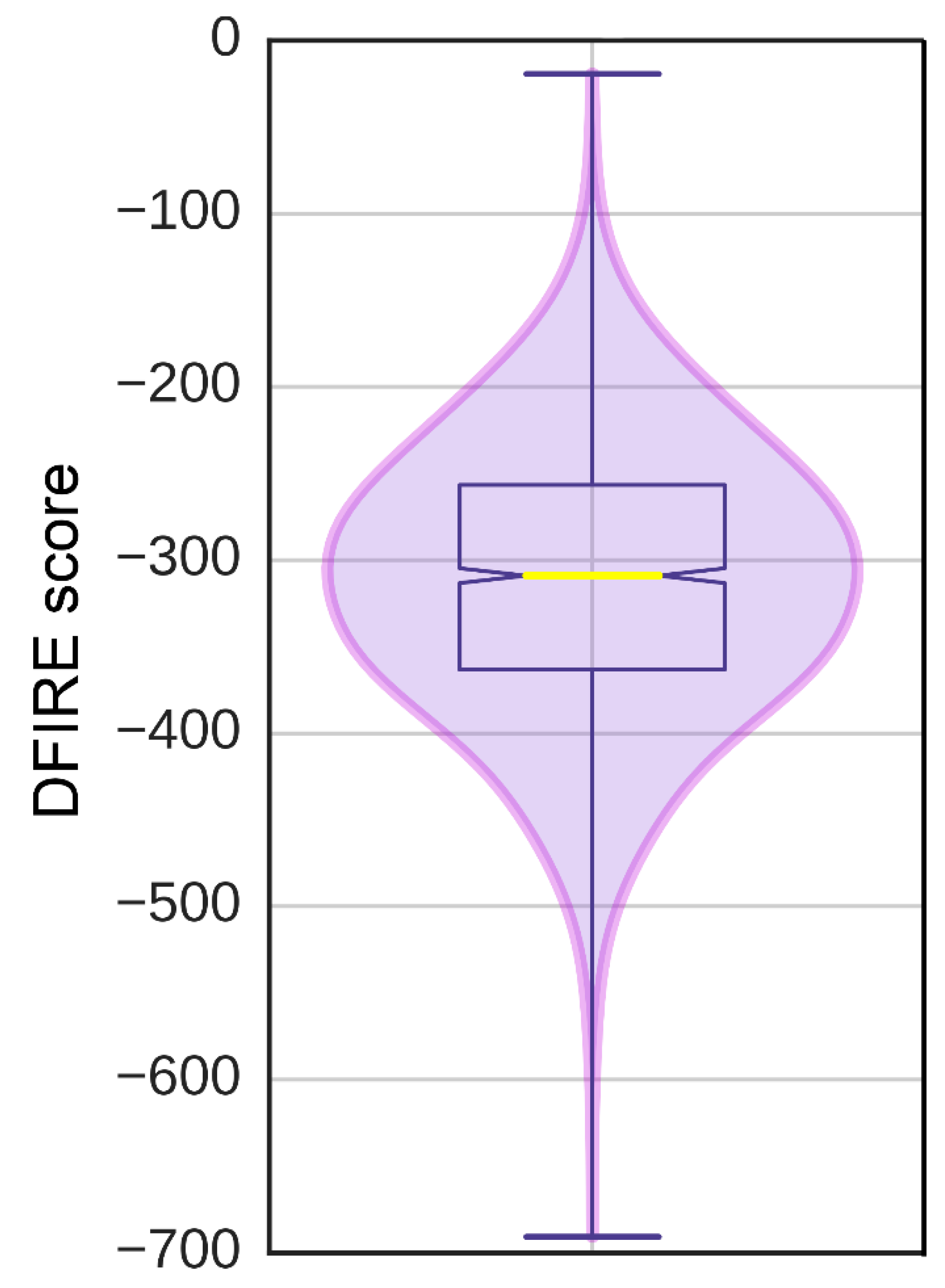
2.4. ATP-Binding Site Prediction
2.5. DNA-Binding Site Prediction
2.6. Cross-Correlation of Residue Fluctuations
3. Results
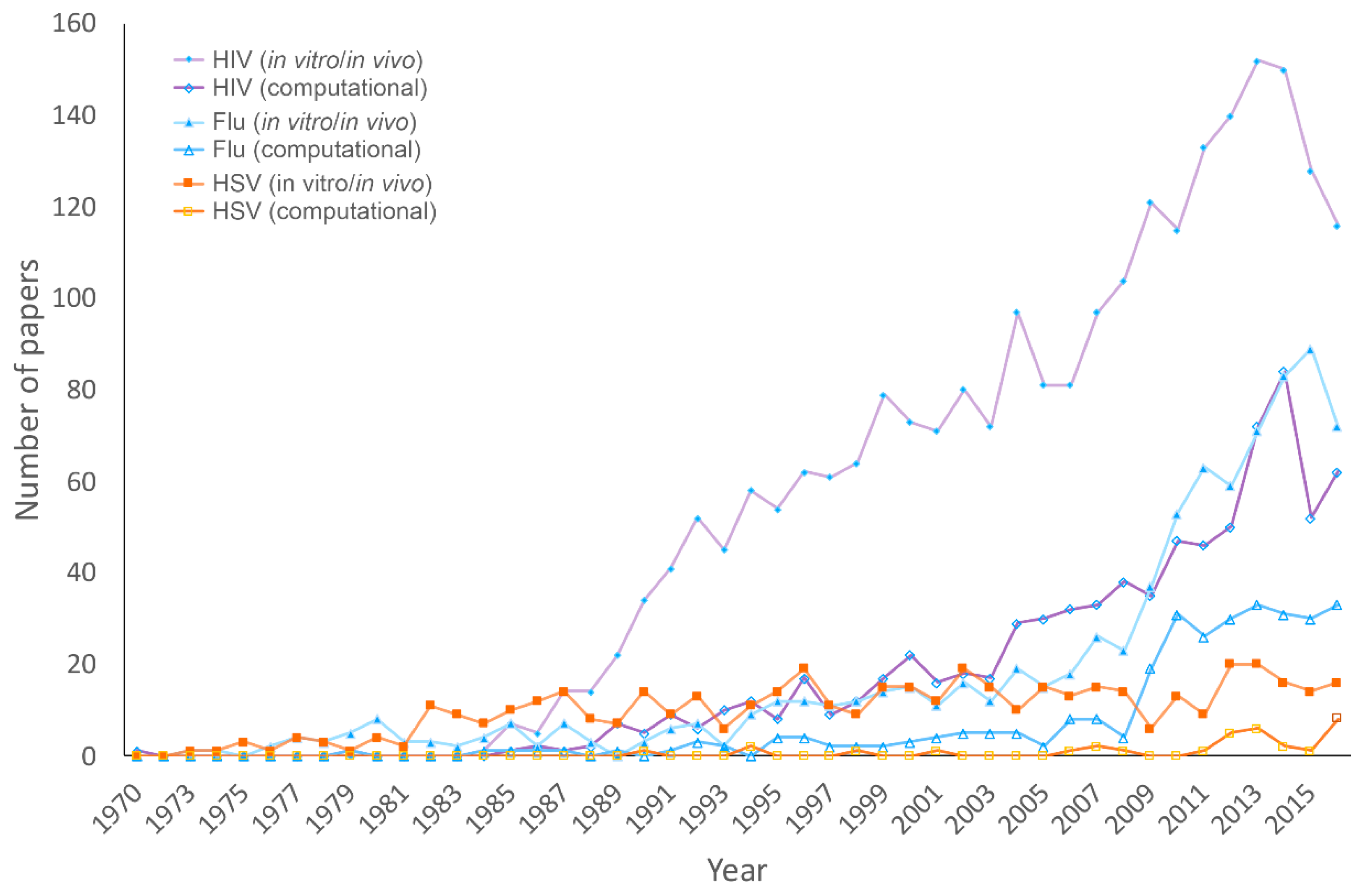
3.1. Publications on HHV, HIV, and Influenza
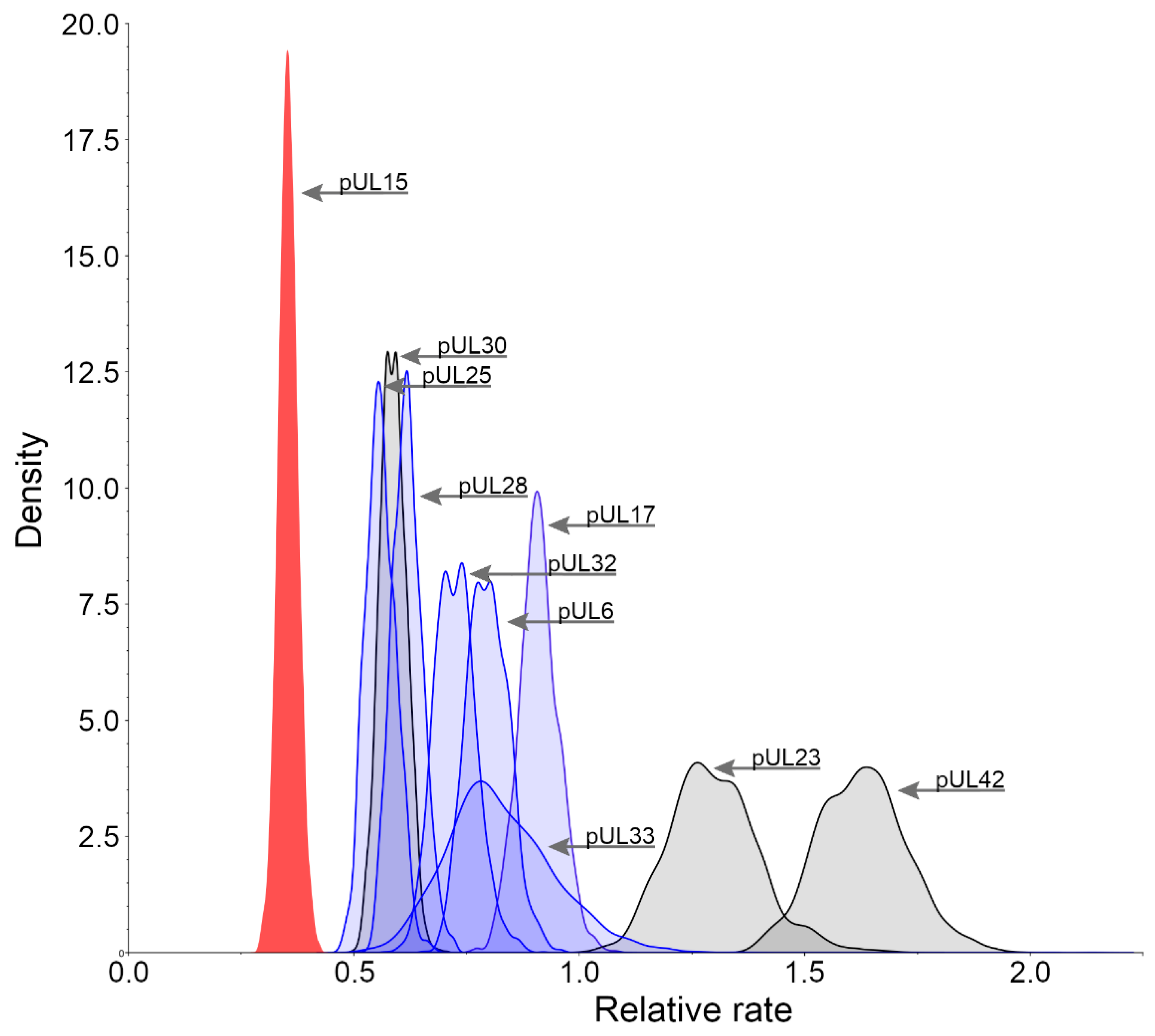
3.2. Relative Substitution Rates for HHV-1 Proteins
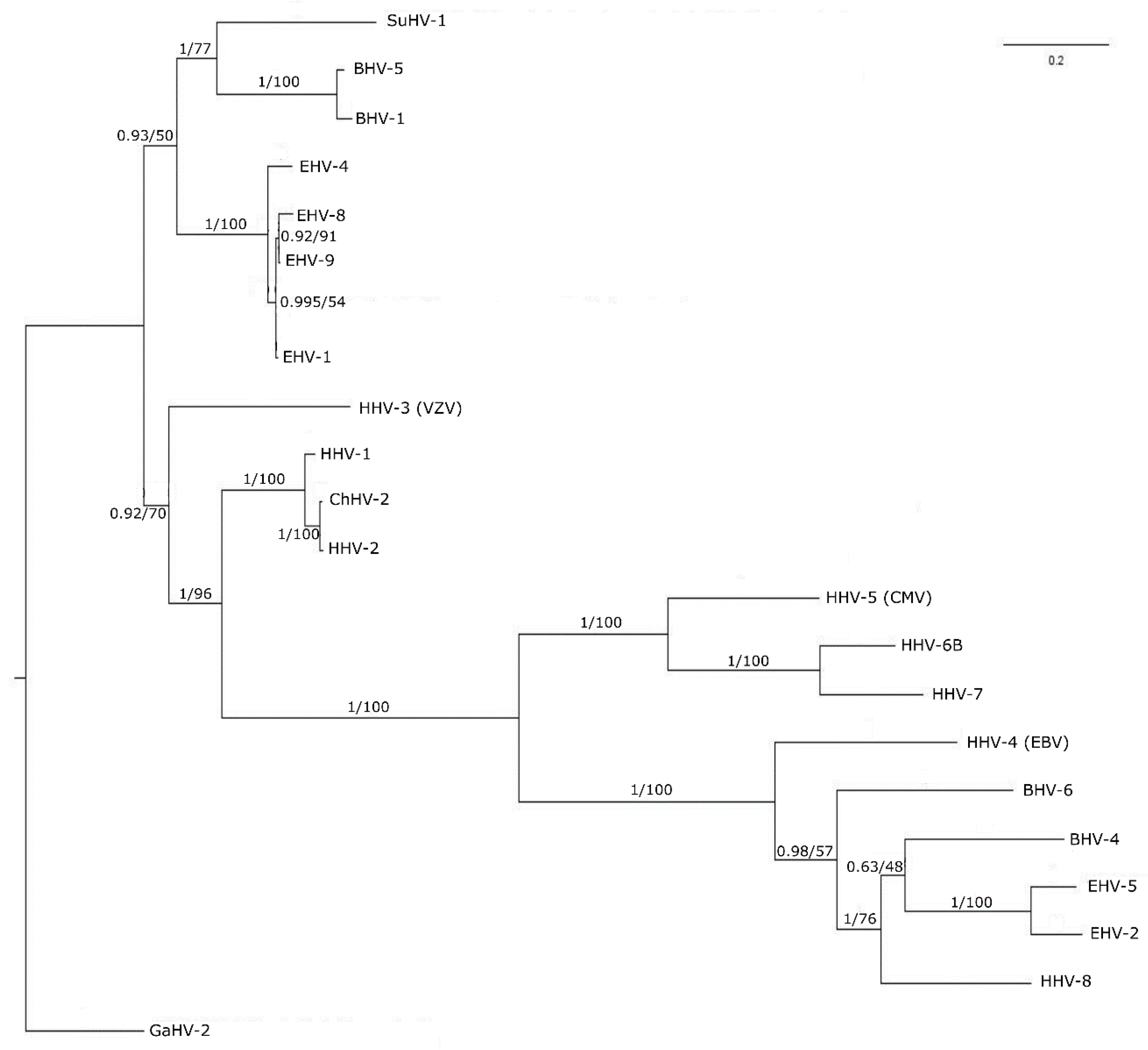
3.3. Phylogenetic Tree Analysis
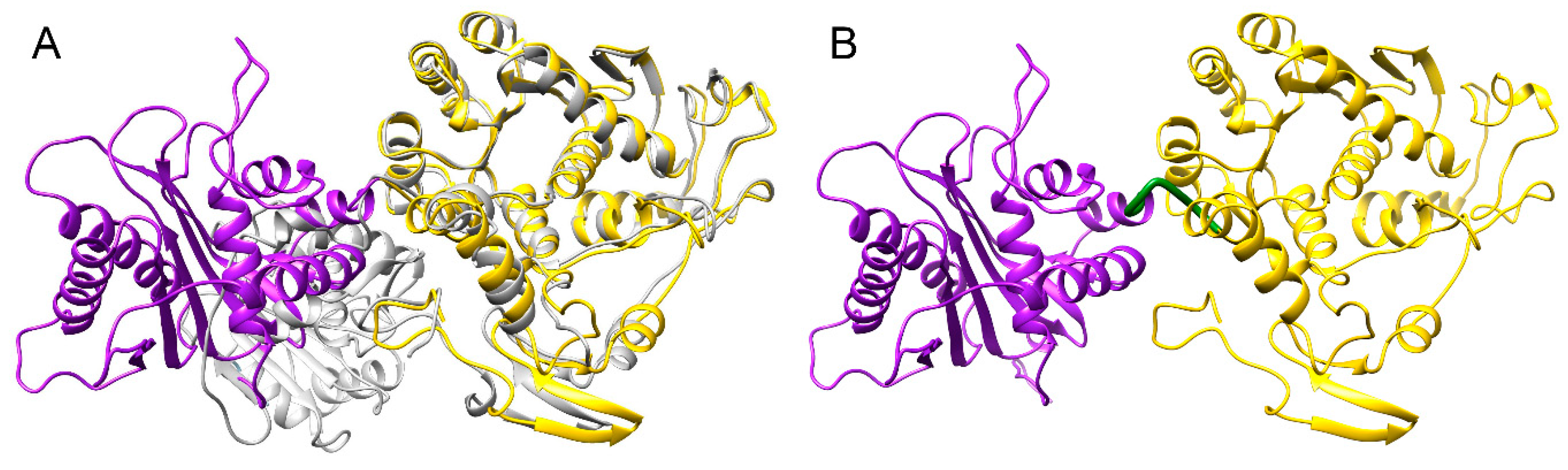
3.4. Structure Model of pUL15
3.5. Cross-Correlation between Residue Fluctuations
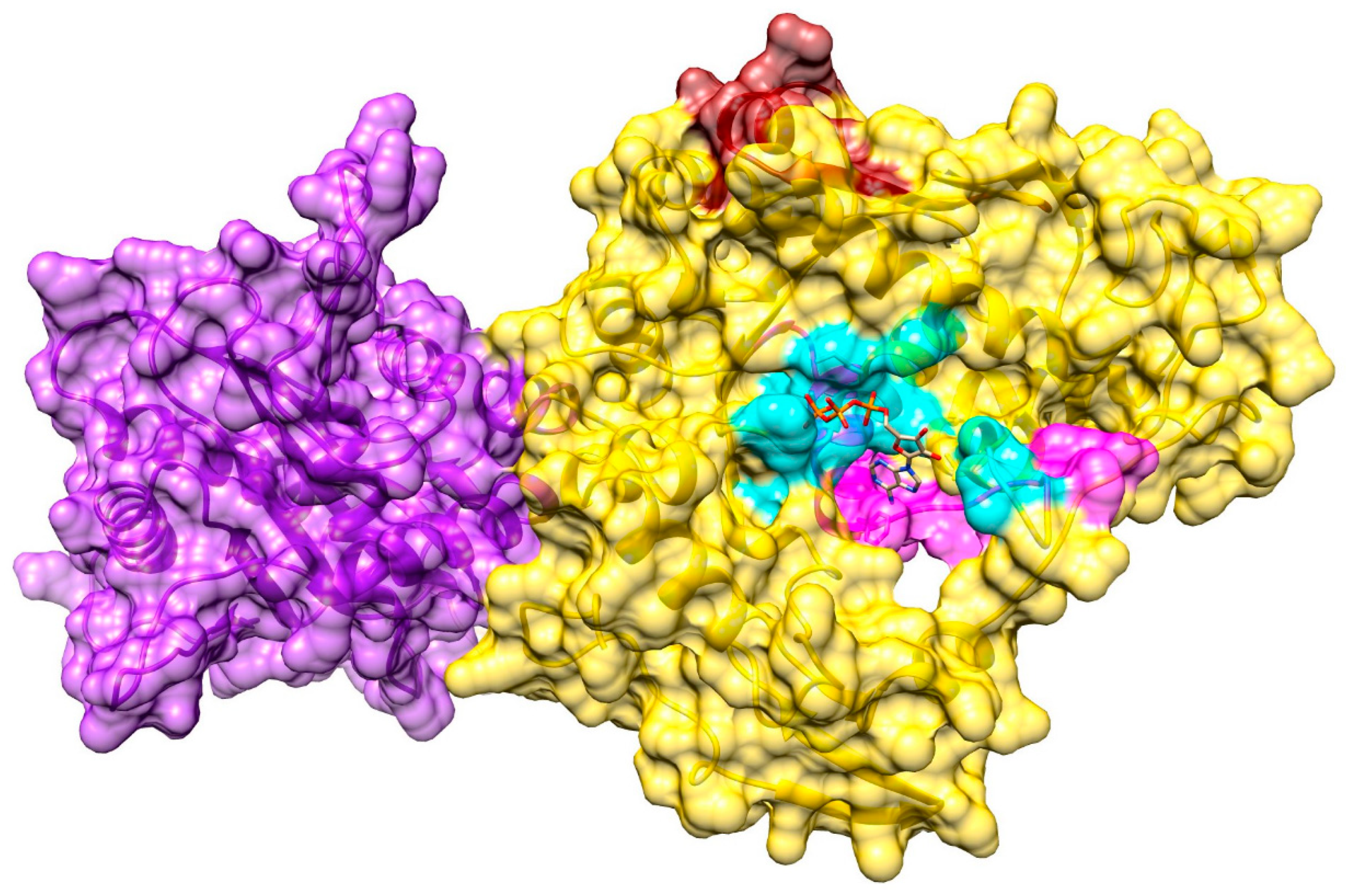
3.6. Ligand-Binding Site in pUL15
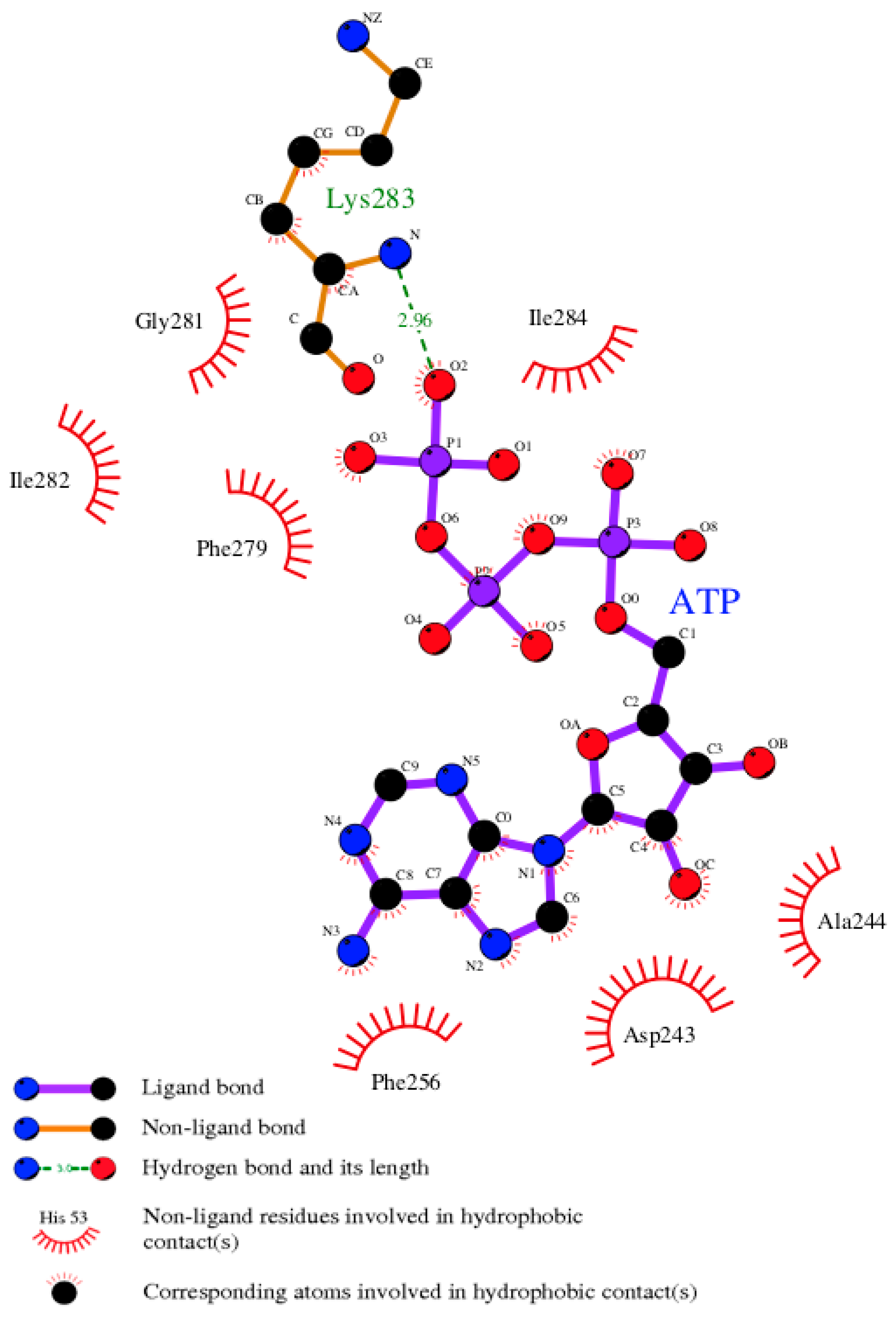
3.7. Model of pUL15 Complexed with ATP
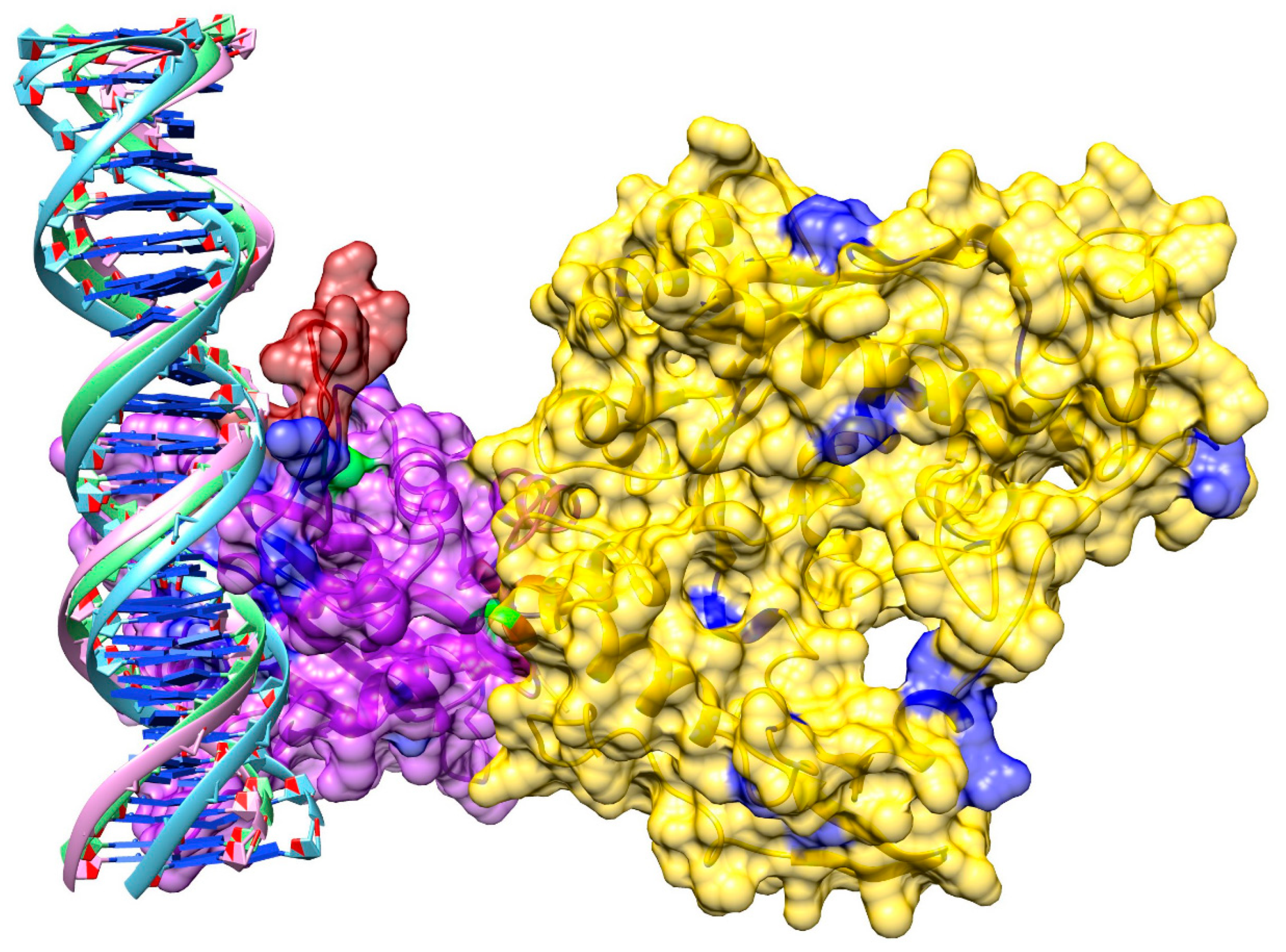
3.8. Model of pUL15 Bound to DNA
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wald, A.; Corey, L. Persistence in the population: Epidemiology, transmission. In Human Herpesviruses: Biology, Therapy, and Immunoprophylaxis; Arvin, A., Campadelli-Fiume, G., Mocarski, E., Eds.; Cambridge University Press: Cambridge, UK, 2007. [Google Scholar]
- Fishman, J.A.; Emery, V.; Freeman, R.; Pascual, M.; Rostaing, L.; Schlitt, H.J.; Sgarabotto, D.; Torre-Cisneros, J.; Uknis, M.E. Cytomegalovirus in transplantation - challenging the status quo. Clin. Transplant 2007, 21, 149–158. [Google Scholar] [CrossRef] [PubMed]
- Penkert, R.R.; Kalejta, R.F. Tegument protein control of latent herpesvirus establishment and animation. Herpesviridae 2011, 2, 3. [Google Scholar] [CrossRef] [PubMed]
- Chisholm, C.; Lopez, L. Cutaneous infections caused by herpesviridae: A review. Arch. Pathol. Lab. Med. 2011, 135, 1357–1362. [Google Scholar] [CrossRef] [PubMed]
- Grinde, B. Herpesviruses: Latency and reactivation - viral strategies and host response. J. Oral. Microbiol. 2013, 5, 22766. [Google Scholar] [CrossRef] [PubMed]
- Kotton, C.N. Management of cytomegalovirus infection in solid organ transplantation. Nat. Rev. Nephrol. 2010, 6, 711–721. [Google Scholar] [CrossRef]
- Whitley, R.J. Herpesviruses. In Medical Microbiology, 4th ed.; Baron, S., Ed.; University of Texas Medical Branch: Galveston, TX, USA, 1996. [Google Scholar]
- Boppana, S.B.; Fowler, K.B.; Britt, W.J.; Stagno, S.; Pass, R.F. Symptomatic congenital cytomegalovirus infection in infants born to mothers with preexisting immunity to cytomegalovirus. Pediatrics 1999, 104, 55–60. [Google Scholar] [CrossRef]
- Boppana, S.B.; Pass, R.F.; Britt, W.J.; Stagno, S.; Alford, C.A. Symptomatic congenital cytomegalovirus infection: Neonatal morbidity and mortality. Pediatr. Infect. Dis. J. 1992, 11, 93–99. [Google Scholar] [CrossRef]
- Coll, O.; Benoist, G.; Ville, Y.; Weisman, L.E.; Botet, F.; Anceschi, M.M.; Greenough, A.; Gibbs, R.S.; Carbonell-Estrany, X.; Group, W.P.I.W. Guidelines on CMV congenital infection. J. Perinat. Med. 2009, 37, 433–445. [Google Scholar] [CrossRef]
- Peritz, D.C.; Duncan, C.; Kurek, K.; Perez-Atayde, A.R.; Lehmann, L.E. Visceral varicella zoster virus (VZV) after allogeneic hematopoietic stem cell transplant (HSCT) in pediatric patients with chronic graft-versus-host disease (cGVHD). J. Pediatr. Hematol. Oncol. 2008, 30, 931–934. [Google Scholar] [CrossRef]
- Bonnet, M.; Guinebretiere, J.M.; Kremmer, E.; Grunewald, V.; Benhamou, E.; Contesso, G.; Joab, I. Detection of Epstein-Barr virus in invasive breast cancers. J. Natl. Cancer Inst. 1999, 91, 1376–1381. [Google Scholar] [CrossRef]
- Wong, M.; Pagano, J.S.; Schiller, J.T.; Tevethia, S.S.; Raab-Traub, N.; Gruber, J. New associations of human papillomavirus, Simian virus 40, and Epstein-Barr virus with human cancer. J. Natl. Cancer Inst. 2002, 94, 1832–1836. [Google Scholar] [CrossRef]
- Schulz, T.F. The pleiotropic effects of Kaposi’s sarcoma herpesvirus. J. Pathol. 2006, 208, 187–198. [Google Scholar] [CrossRef]
- Sunil, M.; Reid, E.; Lechowicz, M.J. Update on HHV-8-Associated Malignancies. Curr. Infect. Dis. Rep. 2010, 12, 147–154. [Google Scholar] [CrossRef]
- McGeoch, D.J.; Dalrymple, M.A.; Davison, A.J.; Dolan, A.; Frame, M.C.; McNab, D.; Perry, L.J.; Scott, J.E.; Taylor, P. The complete DNA sequence of the long unique region in the genome of herpes simplex virus type 1. J. Gen. Virol. 1988, 69, 1531–1574. [Google Scholar] [CrossRef] [PubMed]
- Macdonald, S.J.; Mostafa, H.H.; Morrison, L.A.; Davido, D.J. Genome sequence of herpes simplex virus 1 strain KOS. J. Virol. 2012, 86, 6371–6372. [Google Scholar] [CrossRef] [PubMed]
- Macdonald, S.J.; Mostafa, H.H.; Morrison, L.A.; Davido, D.J. Genome sequence of herpes simplex virus 1 strain McKrae. J. Virol. 2012, 86, 9540–9541. [Google Scholar] [CrossRef]
- Davison, A.J. Comparative analysis of the genomes. In Human Herpesviruses: Biology, Therapy, and Immunoprophylaxis; Campadelli-Fiume, A.A., Mocarski, G.E., Eds.; Cambridge University Press: Cambridge, UK, 2007. [Google Scholar]
- Karamitros, T.; Harrison, I.; Piorkowska, R.; Katzourakis, A.; Magiorkinis, G.; Mbisa, J.L. De novo assembly of human herpes virus type 1 (HHV-1) genome, mining of non-canonical structures and detection of novel drug-resistance mutations using short- and long-read next generation sequencing technologies. PLoS ONE 2016, 11, e0157600. [Google Scholar] [CrossRef]
- Andrei, G.; Snoeck, R. Herpes simplex virus drug-resistance: New mutations and insights. Curr. Opin. Infect. Dis. 2013, 26, 551–560. [Google Scholar] [CrossRef] [PubMed]
- Razonable, R.R. Antiviral drugs for viruses other than human immunodeficiency virus. Mayo. Clin. Proc. 2011, 86, 1009–1026. [Google Scholar] [CrossRef]
- Griffiths, P.D.; Boeck, M. Antiviral therapy for human cytomegalovirus. In Human Herpesviruses: Biology, Therapy, and Immunoprophylaxis; Arvin, A., Campadelli-Fiume, G., Mocarski, E., Moore, P.S., Roizman, B., Whitley, R., Yamanishi, K., Eds.; Cambridge University Press: Cambridge, UK, 2007. [Google Scholar]
- Kimberlin, D.W.; Whitley, R.J. Antiviral therapy of HSV-1 and -2. In Human Herpesviruses: Biology, Therapy, and Immunoprophylaxis; Arvin, A., Campadelli-Fiume, G., Mocarski, E., Moore, P.S., Roizman, B., Whitley, R., Yamanishi, K., Eds.; Cambridge University Press: Cambridge, UK, 2007. [Google Scholar]
- Turner, N.; Strand, A.; Grewal, D.S.; Cox, G.; Arif, S.; Baker, A.W.; Maziarz, E.K.; Saullo, J.H.; Wolfe, C.R. Use of letermovir as salvage therapy for drug-resistant cytomegalovirus retinitis. Antimicrob. Agents Chemother. 2019, 63, e02337-18. [Google Scholar] [CrossRef] [PubMed]
- Morfin, F.; Thouvenot, D. Herpes simplex virus resistance to antiviral drugs. J. Clin. Virol. 2003, 26, 29–37. [Google Scholar] [CrossRef]
- Lurain, N.S.; Chou, S. Antiviral drug resistance of human cytomegalovirus. Clin. Microbiol. Rev. 2010, 23, 689–712. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, A.; Harter, G.; Schubert, A.; Bunjes, D.; Mertens, T.; Michel, D. Antiviral treatment of cytomegalovirus infection and resistant strains. Expert. Opin. Pharmacother. 2009, 10, 191–209. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Yang, Q.; Wang, M.; Jia, R.; Chen, S.; Zhu, D.; Liu, M.; Wu, Y.; Zhao, X.; Zhang, S.; et al. Terminase large subunit provides a new drug target for herpesvirus treatment. Viruses 2019, 11, 219. [Google Scholar] [CrossRef]
- Dittmer, A.; Woskobojnik, I.; Adfeldt, R.; Drach, J.C.; Townsend, L.B.; Voigt, S.; Bogner, E. Tetrahalogenated benzimidazole D-ribonucleosides are active against rat cytomegalovirus. Antiviral. Res. 2017, 137, 102–107. [Google Scholar] [CrossRef]
- Goldner, T.; Hewlett, G.; Ettischer, N.; Ruebsamen-Schaeff, H.; Zimmermann, H.; Lischka, P. The novel anticytomegalovirus compound AIC246 (Letermovir) inhibits human cytomegalovirus replication through a specific antiviral mechanism that involves the viral terminase. J. Virol. 2011, 85, 10884–10893. [Google Scholar] [CrossRef]
- Melendez, D.P.; Razonable, R.R. Letermovir and inhibitors of the terminase complex: A promising new class of investigational antiviral drugs against human cytomegalovirus. Infect. Drug Resist. 2015, 8, 269–277. [Google Scholar]
- Yang, K.; Dang, X.; Baines, J.D. A domain of herpes simplex virus pUL33 required to release monomeric viral genomes from cleaved concatemeric DNA. J. Virol 2017, 91, e00854-17. [Google Scholar] [CrossRef]
- Krosky, P.M.; Underwood, M.R.; Turk, S.R.; Feng, K.W.; Jain, R.K.; Ptak, R.G.; Westerman, A.C.; Biron, K.K.; Townsend, L.B.; Drach, J.C. Resistance of human cytomegalovirus to benzimidazole ribonucleosides maps to two open reading frames: UL89 and UL56. J. Virol. 1998, 72, 4721–4728. [Google Scholar]
- Underwood, M.R.; Harvey, R.J.; Stanat, S.C.; Hemphill, M.L.; Miller, T.; Drach, J.C.; Townsend, L.B.; Biron, K.K. Inhibition of human cytomegalovirus DNA maturation by a benzimidazole ribonucleoside is mediated through the UL89 gene product. J. Virol. 1998, 72, 717–725. [Google Scholar]
- Biron, K.K. Antiviral drugs for cytomegalovirus diseases. Antiviral. Res. 2006, 71, 154–163. [Google Scholar] [CrossRef] [PubMed]
- Good, S.S.; Owens, B.S.; Townsend, L.B.; Drach, J.C. The disposition in rats and monkey of 2-bromo-5,6-dichloro-1-(ß-D-ribofuranosyl)-benzimidazole (BDCRB) and its 2,5,6-trichloro conger (TCRB). Antiviral. Res. 1994, 23, 103. [Google Scholar]
- Hwang, J.S.; Kregler, O.; Schilf, R.; Bannert, N.; Drach, J.C.; Townsend, L.B.; Bogner, E. Identification of acetylated, tetrahalogenated benzimidazole D-ribonucleosides with enhanced activity against human cytomegalovirus. J. Virol. 2007, 81, 11604–11611. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.S.; Schilf, R.; Drach, J.C.; Townsend, L.B.; Bogner, E. Susceptibilities of human cytomegalovirus clinical isolates and other herpesviruses to new acetylated, tetrahalogenated benzimidazole D-ribonucleosides. Antimicrob. Agents Chemother. 2009, 53, 5095–5101. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Dittmer, A.; Drach, J.C.; Townsend, L.B.; Fischer, A.; Bogner, E. Interaction of the putative human cytomegalovirus portal protein pUL104 with the large terminase subunit pUL56 and its inhibition by benzimidazole-D-ribonucleosides. J. Virol. 2005, 79, 14660–14667. [Google Scholar] [CrossRef]
- Zhou, B.; Yang, K.; Wills, E.; Tang, L.; Baines, J.D. A mutation in the DNA polymerase accessory factor of herpes simplex virus 1 restores viral DNA replication in the presence of raltegravir. J. Virol. 2014, 88, 11121–11129. [Google Scholar] [CrossRef]
- Goldner, T.; Zimmermann, H.; Lischka, P. Phenotypic characterization of two naturally occurring human Cytomegalovirus sequence polymorphisms located in a distinct region of ORF UL56 known to be involved in in vitro resistance to letermovir. Antiviral. Res. 2015, 116, 48–50. [Google Scholar] [CrossRef]
- Cherrier, L.; Nasar, A.; Goodlet, K.J.; Nailor, M.D.; Tokman, S.; Chou, S. Emergence of letermovir resistance in a lung transplant recipient with ganciclovir-resistant cytomegalovirus infection. Am. J. Transplant. 2018, 18, 3060–3064. [Google Scholar] [CrossRef]
- Marschall, M.; Stamminger, T.; Urban, A.; Wildum, S.; Ruebsamen-Schaeff, H.; Zimmermann, H.; Lischka, P. In vitro evaluation of the activities of the novel anticytomegalovirus compound AIC246 (letermovir) against herpesviruses and other human pathogenic viruses. Antimicrob. Agents Chemother. 2012, 56, 1135–1137. [Google Scholar] [CrossRef]
- Caruso Brown, A.E.; Cohen, M.N.; Tong, S.; Braverman, R.S.; Rooney, J.F.; Giller, R.; Levin, M.J. Pharmacokinetics and safety of intravenous cidofovir for life-threatening viral infections in pediatric hematopoietic stem cell transplant recipients. Antimicrob. Agents Chemother. 2015, 59, 3718–3725. [Google Scholar] [CrossRef]
- Chen, K.; Cheng, M.P.; Hammond, S.P.; Einsele, H.; Marty, F.M. Antiviral prophylaxis for cytomegalovirus infection in allogeneic hematopoietic cell transplantation. Blood Adv. 2018, 2, 2159–2175. [Google Scholar] [CrossRef] [PubMed]
- Clark, K.; Karsch-Mizrachi, I.; Lipman, D.J.; Ostell, J.; Sayers, E.W. GenBank. Nucleic Acids Res. 2016, 44, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, G. Estimating the dimension of a model. Ann. Stat. 1978, 6, 461–464. [Google Scholar] [CrossRef]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. ProtTest 3: Fast selection of best-fit models of protein evolution. Bioinformatics 2011, 27, 1164–1165. [Google Scholar] [CrossRef]
- Jones, D.T.; Taylor, W.R.; Thornton, J.M. The rapid generation of mutation data matrices from protein sequences. Comput. Appl. Biosci. 1992, 8, 275–282. [Google Scholar] [CrossRef]
- Ronquist, F.; Huelsenbeck, J.P. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 2003, 19, 1572–1574. [Google Scholar] [CrossRef]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior summarization in bayesian phylogenetics using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- O’Leary, N.A.; Wright, M.W.; Brister, J.R.; Ciufo, S.; Haddad, D.; McVeigh, R.; Rajput, B.; Robbertse, B.; Smith-White, B.; Ako-Adjei, D.; et al. Reference sequence (RefSeq) database at NCBI: Current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016, 44, 733–745. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Abascal, F.; Zardoya, R.; Posada, D. ProtTest: Selection of best-fit models of protein evolution. Bioinformatics 2005, 21, 2104–2105. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Huelsenbeck, J.P.; Ronquist, F.; Nielsen, R.; Bollback, J.P. Bayesian inference of phylogeny and its impact on evolutionary biology. Science 2001, 294, 2310–2314. [Google Scholar] [CrossRef]
- Fig Tree, Molecular Evolution, Phylogenetics and Epidemiology. Available online: http://tree.bio.ed.ac.uk/software/figtree (accessed on 9 October 2019).
- Inkscape, Draw Freely. Available online: https://inkscape.org/. (accessed on 9 October 2019).
- Selvarajan Sigamani, S.; Zhao, H.; Kamau, Y.N.; Baines, J.D.; Tang, L. The structure of the herpes simplex virus DNA-packaging terminase pUL15 nuclease domain suggests an evolutionary lineage among eukaryotic and prokaryotic viruses. J. Virol. 2013, 87, 7140–7148. [Google Scholar] [CrossRef]
- Berman, H.M.; Battistuz, T.; Bhat, T.N.; Bluhm, W.F.; Bourne, P.E.; Burkhardt, K.; Feng, Z.; Gilliland, G.L.; Iype, L.; Jain, S.; et al. The Protein Data Bank. Acta Crystallogr. D Biol. Crystallogr. 2002, 58, 899–907. [Google Scholar] [CrossRef]
- Sun, S.; Kondabagil, K.; Gentz, P.M.; Rossmann, M.G.; Rao, V.B. The structure of the ATPase that powers DNA packaging into bacteriophage T4 procapsids. Mol. Cell. 2007, 25, 943–949. [Google Scholar] [CrossRef]
- Zimmermann, L.; Stephens, A.; Nam, S.Z.; Rau, D.; Kubler, J.; Lozajic, M.; Gabler, F.; Soding, J.; Lupas, A.N.; Alva, V. A completely reimplemented MPI Bioinformatics Toolkit with a new HHpred server at its core. J. Mol. Biol. 2018, 430, 2237–2243. [Google Scholar] [CrossRef]
- Sun, S.; Kondabagil, K.; Draper, B.; Alam, T.I.; Bowman, V.D.; Zhang, Z.; Hegde, S.; Fokine, A.; Rossmann, M.G.; Rao, V.B. The structure of the phage T4 DNA packaging motor suggests a mechanism dependent on electrostatic forces. Cell 2008, 135, 1251–1262. [Google Scholar] [CrossRef]
- Pandit, S.B.; Skolnick, J. Fr-TM-align: A new protein structural alignment method based on fragment alignments and the TM-score. BMC Bioinformatics 2008, 9, 531. [Google Scholar] [CrossRef]
- Braberg, H.; Webb, B.M.; Tjioe, E.; Pieper, U.; Sali, A.; Madhusudhan, M.S. SALIGN: A web server for alignment of multiple protein sequences and structures. Bioinformatics 2012, 28, 2072–2073. [Google Scholar] [CrossRef] [PubMed]
- Webb, B.; Sali, A. Comparative protein structure modeling using MODELLER. Curr. Protoc. Protein Sci. 2014, 47, 5–6. [Google Scholar] [CrossRef] [PubMed]
- Brylinski, M.; Feinstein, W.P. eFindSite: Improved prediction of ligand binding sites in protein models using meta-threading, machine learning and auxiliary ligands. J. Comput. Aided. Mol. Des. 2013, 27, 551–567. [Google Scholar] [CrossRef] [PubMed]
- Feinstein, W.P.; Brylinski, M. eFindSite: Enhanced fingerprint-based virtual screening against predicted ligand binding sites in protein models. Mol. Inform. 2014, 33, 135–150. [Google Scholar] [CrossRef]
- Brylinski, M. Nonlinear scoring functions for similarity-based ligand docking and binding affinity prediction. J. Chem. Inf. Model. 2013, 53, 3097–3112. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple ligand-protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 2011, 51, 2778–2786. [Google Scholar] [CrossRef]
- Zhang, C.; Liu, S.; Zhu, Q.; Zhou, Y. A knowledge-based energy function for protein-ligand, protein-protein, and protein-DNA complexes. J. Med. Chem. 2005, 48, 2325–2335. [Google Scholar] [CrossRef]
- Krissinel, E.; Henrick, K. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 2007, 372, 774–797. [Google Scholar] [CrossRef]
- Tjong, H.; Zhou, H.X. DISPLAR: An accurate method for predicting DNA-binding sites on protein surfaces. Nucleic Acids Res. 2007, 35, 1465–1477. [Google Scholar] [CrossRef]
- Hwang, S.; Gou, Z.; Kuznetsov, I.B. DP-Bind: A web server for sequence-based prediction of DNA-binding residues in DNA-binding proteins. Bioinformatics 2007, 23, 634–636. [Google Scholar] [CrossRef]
- Van Dijk, M.; Bonvin, A.M. 3D-DART: A DNA structure modelling server. Nucleic Acids Res. 2009, 37, 235–239. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Chang, Y.Y.; Lee, J.Y.; Bahar, I.; Yang, L.W. DynOmics: Dynamics of structural proteome and beyond. Nucleic Acids Res. 2017, 45, 374–380. [Google Scholar]
- Sato, H.; Yokoyama, M.; Toh, H. Genomics and computational science for virus research. Front Microbiol. 2013, 4, 42. [Google Scholar] [CrossRef] [PubMed]
- Mann, J.M. The World Health Organization’s global strategy for the prevention and control of AIDS. West J. Med. 1987, 147, 732–734. [Google Scholar]
- World Health Organization. Global Health Sector Strategy on HIV, 2016–2021; WHO: Geneva, Switzerland, 2016. [Google Scholar]
- Johnston, C.; Gottlieb, S.L.; Wald, A. Status of vaccine research and development of vaccines for herpes simplex virus. Vaccine 2016, 34, 2948–2952. [Google Scholar] [CrossRef]
- Deshpande, S.P.; Kumaraguru, U.; Rouse, B.T. Why do we lack an effective vaccine against herpes simplex virus infections? Microbes. Infect. 2000, 2, 973–978. [Google Scholar] [CrossRef]
- Looker, K.J.; Magaret, A.S.; Turner, K.M.; Vickerman, P.; Gottlieb, S.L.; Newman, L.M. Global estimates of prevalent and incident herpes simplex virus type 2 infections in 2012. PLoS ONE 2015, 10, e114989. [Google Scholar] [CrossRef]
- World Health Organization. Available online: https://www.who.int/news-room/fact-sheets/detail/herpes-simplex-virus (accessed on 9 October 2019).
- McQuillan, G.; Kruszon-Moran, D.; Flagg, E.W.; Paulose-Ram, R. In Prevalence of Herpes Simplex Virus Type 1 and Type 2 in Persons Aged 14-49: USA, 2015-2016. NCHS Data Brief. 2018, 304, 1–8. [Google Scholar]
- Przech, A.J.; Yu, D.; Weller, S.K. Point mutations in exon I of the herpes simplex virus putative terminase subunit, UL15, indicate that the most conserved residues are essential for cleavage and packaging. J. Virol. 2003, 77, 9613–9621. [Google Scholar] [CrossRef]
- Rao, V.B.; Mitchell, M.S. The N-terminal ATPase site in the large terminase protein gp17 is critically required for DNA packaging in bacteriophage T4. J. Mol. Biol. 2001, 314, 401–411. [Google Scholar] [CrossRef]
- Govindaraj, R.G.; Brylinski, M. Comparative assessment of strategies to identify similar ligand-binding pockets in proteins. BMC Bioinformatics 2018, 19, 91. [Google Scholar] [CrossRef] [PubMed]
- Pi, F.; Zhao, Z.; Chelikani, V.; Yoder, K.; Kvaratskhelia, M.; Guo, P. Development of Potent Antiviral Drugs Inspired by Viral Hexameric DNA-Packaging Motors with Revolving Mechanism. J. Virol. 2016, 90, 8036–8046. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Rao, V.B.; Rossmann, M.G. Genome packaging in viruses. Curr. Opin. Struct. Biol. 2010, 20, 114–120. [Google Scholar] [CrossRef]
- Waters, J.T.; Kim, H.D.; Gumbart, J.C.; Lu, X.J.; Harvey, S.C. DNA Scrunching in the Packaging of Viral Genomes. J. Phys. Chem. B. 2016, 120, 6200–6207. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Herpesvirus | Abbreviation | Accession Number | % Identity with HHV-1 |
|---|---|---|---|
| Human Alphaherpesvirus 1 | HHV-1 | YP 009137089.1 | 100.0% |
| Human Alphaherpesvirus 2 | HHV-2 | YP_009137166.1 | 94.8% |
| Human Alphaherpesvirus 3 | HHV-3 | NP_040165.1 | 59.0% |
| Human Gammaherpesvirus 4 | HHV-4 | YP 401690.1 | 32.2% |
| Human Betaherpesvirus Type 5 | HHV-5 | YP_081537.1 | 36.0% |
| Human Betaherpesvirus 6B | HHV-6B | NP_050241.1 | 34.5% |
| Human Betaherpesvirus 7 | HHV-7 | YP_073802.1 | 33.0% |
| Human Gammaherpesvirus 8 | HHV-8 | YP_001129382.1 | 31.6% |
| Chimpanzee Herpesvirus Strain 105640 | ChHV-2 | YP_009011001.1 | 95.0% |
| Cercopithecine Herpesvirus 2 | CeHV-2 | YP_164457.1 | 87.3% |
| Equid Alphaherpesvirus 1 | EHV-1 | YP_053090.1 | 63.9% |
| Equid Gammaherpesvirus 2 | EHV-2 | NP_042630.2 | 30.6% |
| Equid Alphaherpesvirus 4 | EHV-4 | NP_045262.1 | 63.7% |
| Equid Gammaherpesvirus 5 | EHV-5 | YP_009118419.1 | 31.6% |
| Equid Alphaherpesvirus 8 | EHV-8 | YP_006273027.1 | 29.6% |
| Equid Alphaherpesvirus 9 | EHV-9 | YP_002333526.2 | 64.1% |
| Felid Herpesvirus 1 | FHV-1 | YP_003331564.1 | 64.3% |
| Fruit bat Alphaherpesvirus 1 | FBAHV-1 | YP_009042077.1 | 82.6% |
| Bovine Alphaherpesvirus 1 | BHV-1 | NP 045342.1 | 60.4% |
| Bovine Gammaherpesvirus 4 | BHV-4 | NP 076521.1 | 31.3% |
| Bovine Alphaherpesvirus 5 | BHV-5 | YP 003662508.1 | 60.6% |
| Bovine Gammaherpesvirus 6 | BHV-6 | YP 009042007.1 | 31.0% |
| Gallid Alphaherpesvirus 2 | GaHV-2 | AAF66813.1 | 53.9% |
| Suid Alphaherpesvirus 1 | SuHV-1 | YP 068358.1 | 55.2% |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thaljeh, L.F.; Rothschild, J.A.; Naderi, M.; Coghill, L.M.; Brown, J.M.; Brylinski, M. Hinge Region in DNA Packaging Terminase pUL15 of Herpes Simplex Virus: A Potential Allosteric Target for Antiviral Drugs. Biomolecules 2019, 9, 603. https://doi.org/10.3390/biom9100603
Thaljeh LF, Rothschild JA, Naderi M, Coghill LM, Brown JM, Brylinski M. Hinge Region in DNA Packaging Terminase pUL15 of Herpes Simplex Virus: A Potential Allosteric Target for Antiviral Drugs. Biomolecules. 2019; 9(10):603. https://doi.org/10.3390/biom9100603
Chicago/Turabian StyleThaljeh, Lana F., J. Ainsley Rothschild, Misagh Naderi, Lyndon M. Coghill, Jeremy M. Brown, and Michal Brylinski. 2019. "Hinge Region in DNA Packaging Terminase pUL15 of Herpes Simplex Virus: A Potential Allosteric Target for Antiviral Drugs" Biomolecules 9, no. 10: 603. https://doi.org/10.3390/biom9100603
APA StyleThaljeh, L. F., Rothschild, J. A., Naderi, M., Coghill, L. M., Brown, J. M., & Brylinski, M. (2019). Hinge Region in DNA Packaging Terminase pUL15 of Herpes Simplex Virus: A Potential Allosteric Target for Antiviral Drugs. Biomolecules, 9(10), 603. https://doi.org/10.3390/biom9100603