High Glucose Represses the Anti-Proliferative and Pro-Apoptotic Effect of Metformin in Triple Negative Breast Cancer Cells

 , ,
, ,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Chemicals, Biochemicals, Reagents, and Antibodies
2.2. Cell Culture
2.3. Cell Treatments
2.4. Cell Proliferation Assay
2.5. Apoptosis by Flow Cytometry
2.6. Cell Cycle Analysis Using Propidium Iodide Staining by Flow Cytometry
2.7. Protein Isolation and Total Protein Estimation
2.8. Immunoblotting
2.9. Statistical Analysis
3. Results
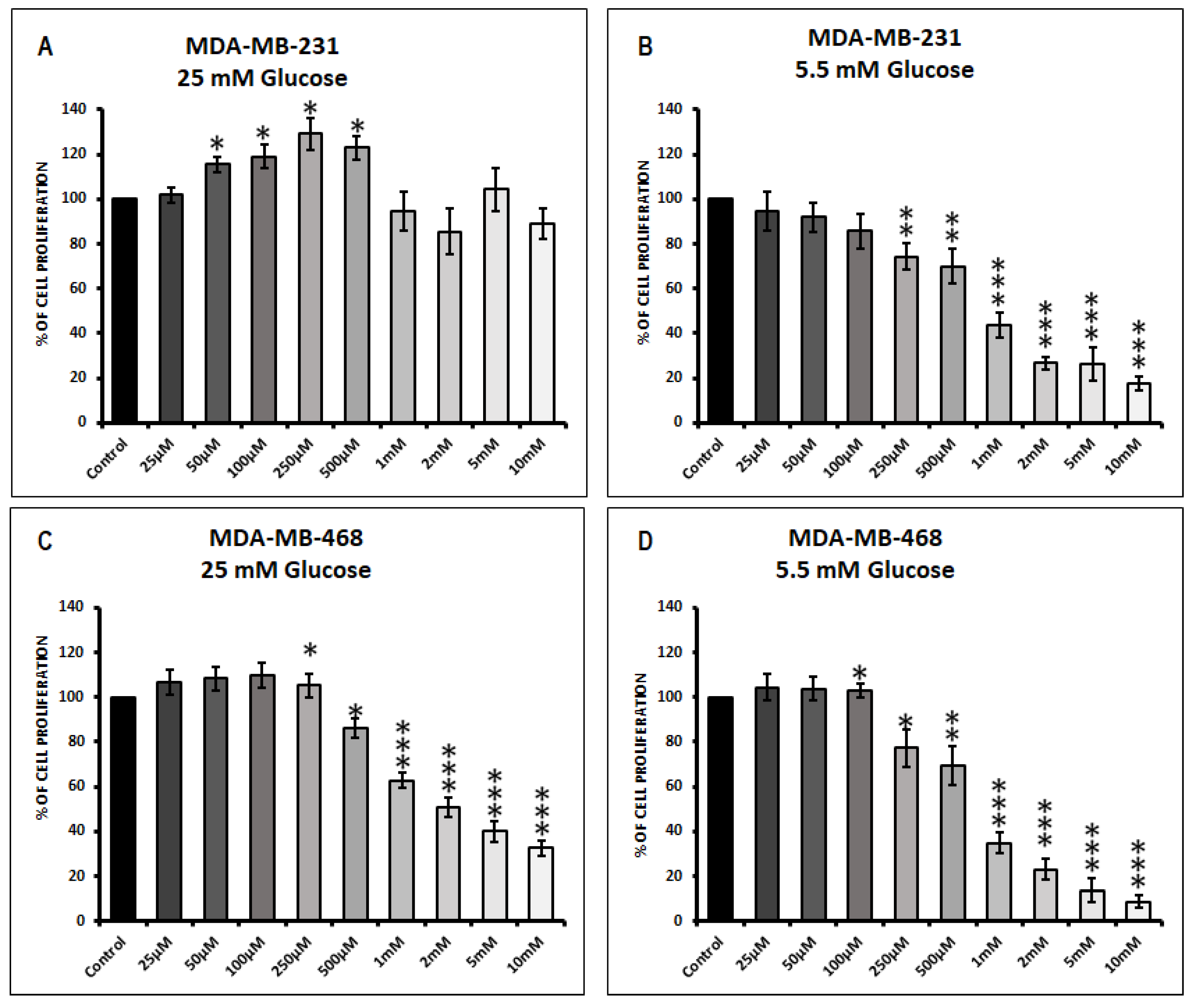
3.1. Effect of Metformin on Cell Proliferation in MDA-MB-231 and MDA-MB-468 Cells Exposed to 25 mM Glucose and 5.5 mM Glucose Conditions
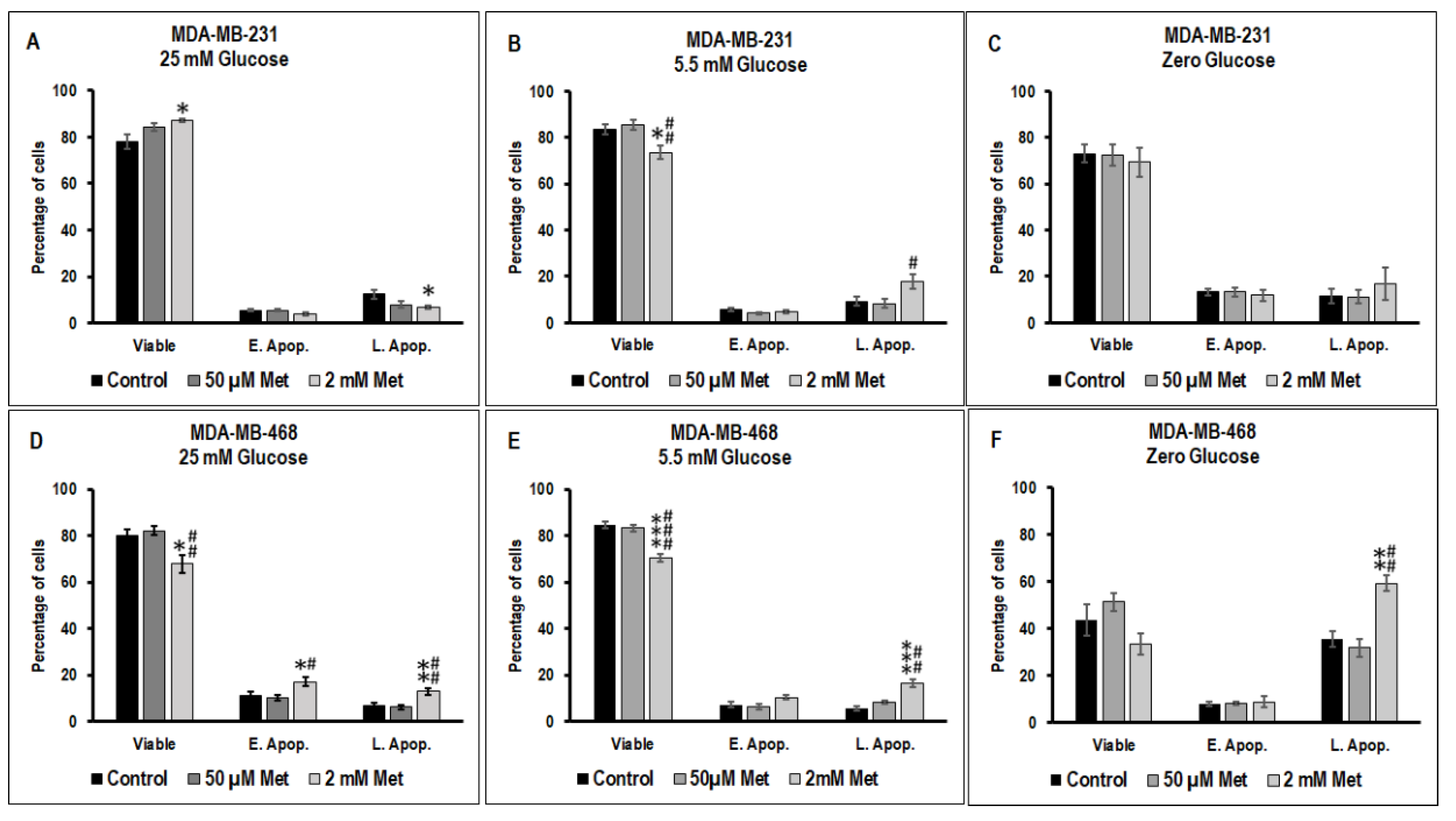
3.2. Effect of Metformin on Apoptotic Cell Death in MDA-MB-231 and MDA-MB-468 Cells Exposed to Different Glucose (25 mM, 5.5 mM, and Zero Glucose/Glucose-Starved) Conditions
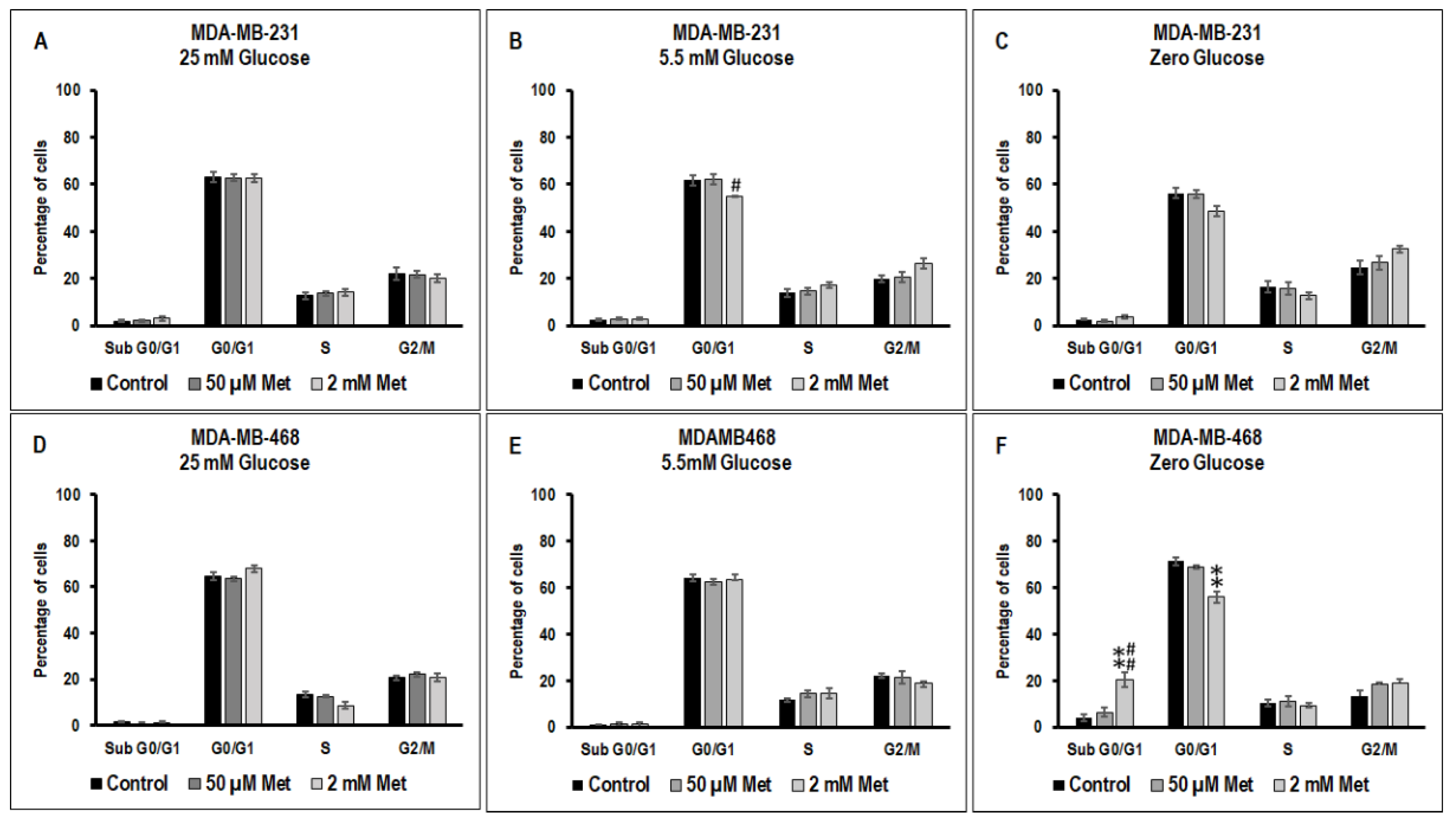
3.3. Effect of Metformin on Cell Cycle in MDA-MB-231 and MDA-MB-468 Cells Exposed to Different Glucose (25 mM, 5.5 mM, and Zero Glucose/Glucose-Starved) Conditions
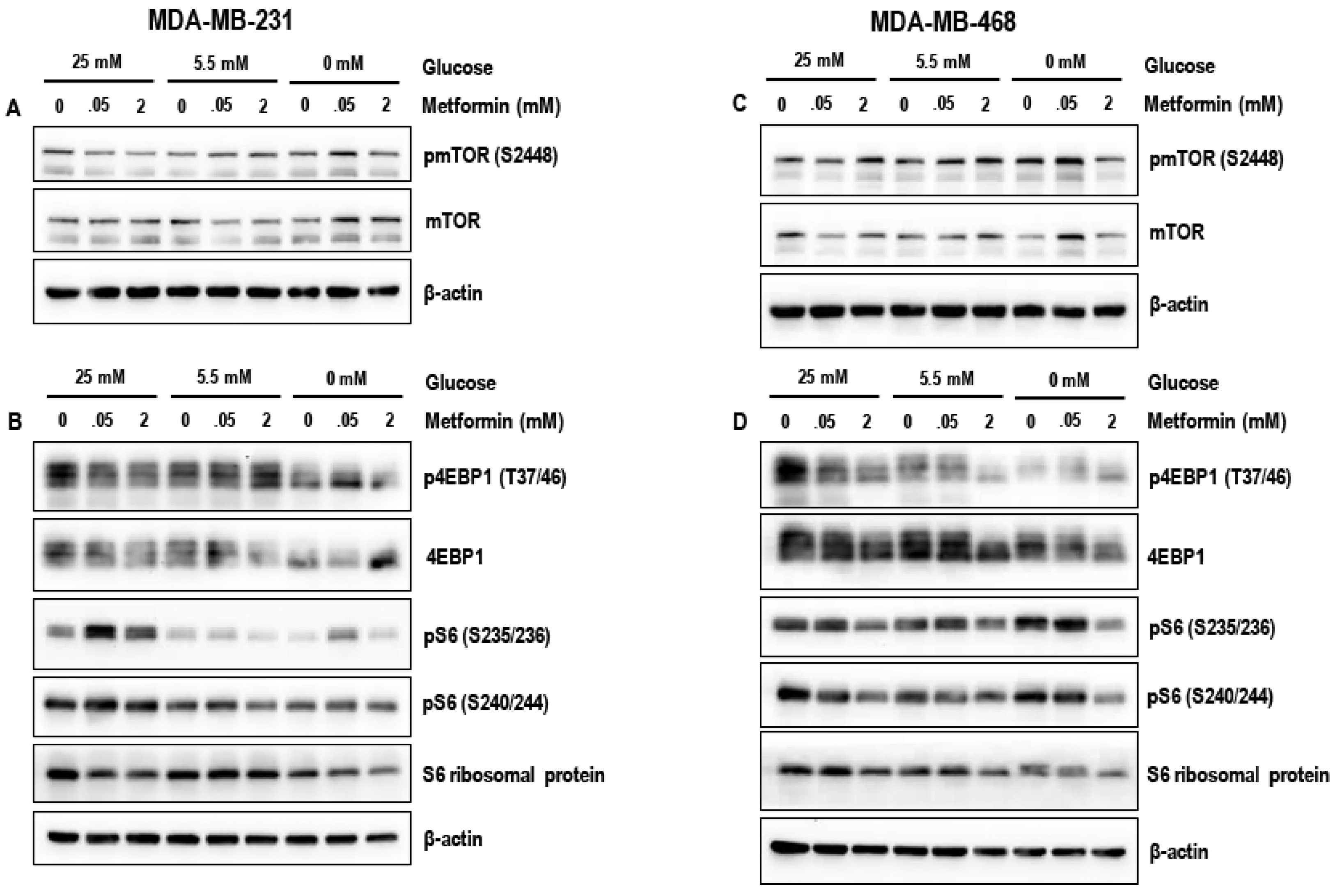
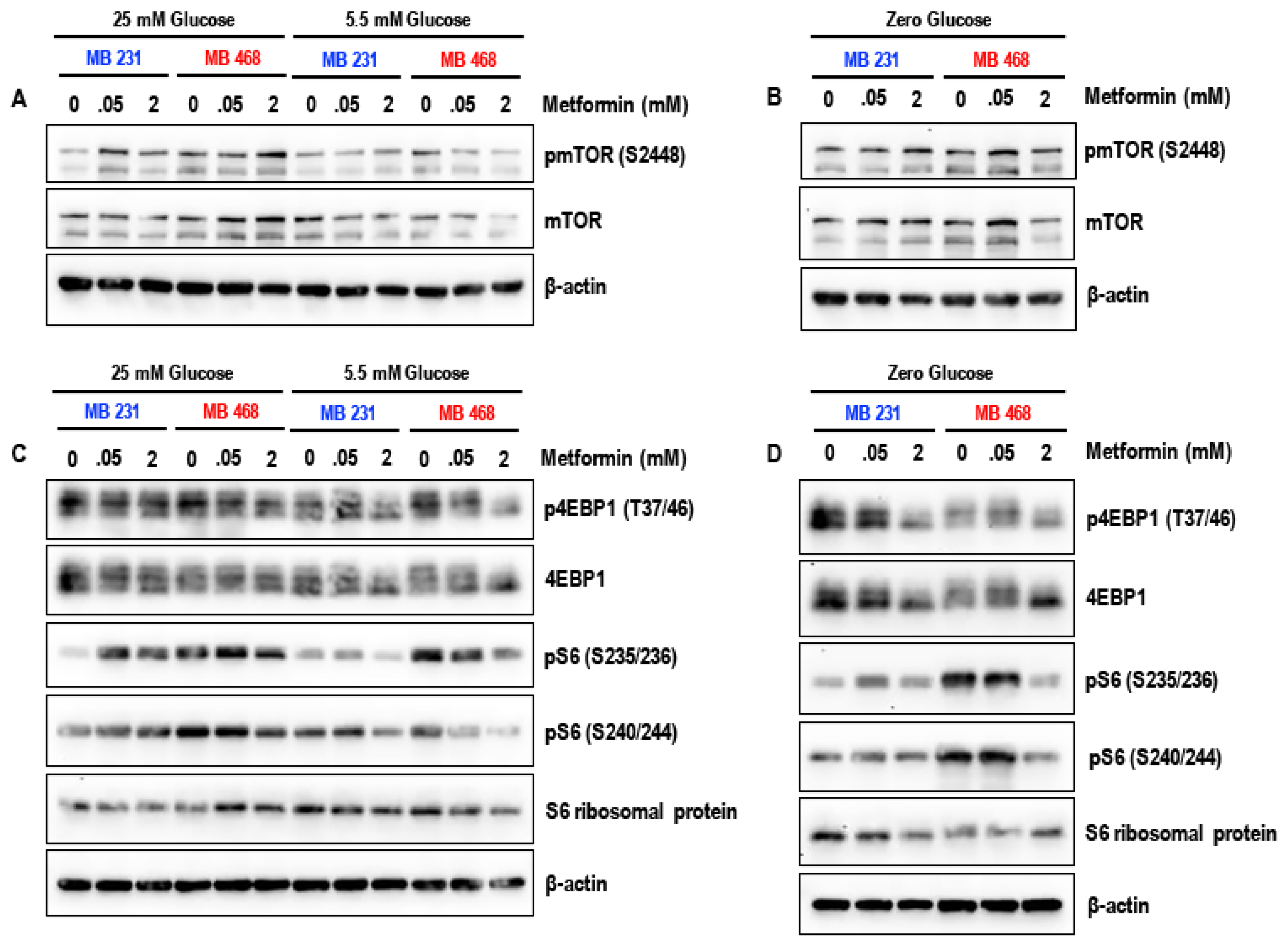
3.4. Effect of Metformin on the mTOR Pathway in MDA-MB-231 and MDA-MB-468 Cells Exposed to Different Glucose (25 mM, 5.5 mM, and Zero Glucose/Glucose-Starved) Conditions
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Hamanaka, R.B.; Chandel, N.S. Targeting glucose metabolism for cancer therapy. J. Exp. Med. 2012, 209, 211–215. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. On the origin of cancer cells on the origin of cance. Source Sci. New Ser. 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Samuel, S.M.; Varghese, E.; Varghese, S.; Busselberg, D. Challenges and perspectives in the treatment of diabetes associated breast cancer. Cancer Treat. Rev. 2018, 70, 98–111. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, M.S.; Sharp, P.A. Pyruvate kinase m2-specific siRNA induces apoptosis and tumor regression. J. Exp. Med. 2012, 209, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Jae, H.J.; Chung, J.W.; Park, H.S.; Lee, M.J.; Lee, K.C.; Kim, H.C.; Yoon, J.H.; Chung, H.; Park, J.H. The antitumor effect and hepatotoxicity of a hexokinase II inhibitor 3-bromopyruvate: In vivo investigation of intraarterial administration in a rabbit VX2 hepatoma model. Korean J. Radiol. 2009, 10, 596–603. [Google Scholar] [CrossRef]
- Basha, B.; Samuel, S.M.; Triggle, C.R.; Ding, H. Endothelial dysfunction in diabetes mellitus: Possible involvement of endoplasmic reticulum stress? Exp. Diabetes Res. 2012, 2012, 481840. [Google Scholar] [CrossRef]
- Alimova, I.N.; Liu, B.; Fan, Z.; Edgerton, S.M.; Dillon, T.; Lind, S.E.; Thor, A.D. Metformin inhibits breast cancer cell growth, colony formation and induces cell cycle arrest in vitro. Cell Cycle 2009, 8, 909–915. [Google Scholar] [CrossRef]
- Mallik, R.; Chowdhury, T.A. Metformin in cancer. Diabetes Res. Clin. Pract. 2018, 143, 409–419. [Google Scholar] [CrossRef]
- Starup-Linde, J.; Karlstad, O.; Eriksen, S.A.; Vestergaard, P.; Bronsveld, H.K.; de Vries, F.; Andersen, M.; Auvinen, A.; Haukka, J.; Hjellvik, V.; et al. Caring (cancer risk and insulin analogues): The association of diabetes mellitus and cancer risk with focus on possible determinants-a systematic review and a meta-analysis. Curr. Drug Saf. 2013, 8, 296–332. [Google Scholar] [CrossRef]
- Ali Kaplan, M.; Pekkolay, Z.; Kucukoner, M.; Urakci, Z.; Ertugrul, H.; Akdogan, R.; Firat, U.; Yildiz, S.; Isikdogan, A. Type 2 diabetes mellitus and prognosis in early stage breast cancer women. Med. Oncol. 2012, 29, 1576–1580. [Google Scholar] [CrossRef]
- Boyle, P.; Boniol, M.; Koechlin, A.; Robertson, C.; Valentini, F.; Coppens, K.; Fairley, L.L.; Boniol, M.; Zheng, T.; Zhang, Y.; et al. Diabetes and breast cancer risk: A meta-analysis. Br. J. Cancer 2012, 107, 1608–1617. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Virnig, B.; Hendryx, M.; Wen, S.; Chelebowski, R.; Chen, C.; Rohan, T.; Tinker, L.; Wactawski-Wende, J.; Lessin, L.; et al. Diabetes, diabetes treatment and breast cancer prognosis. Breast Cancer Res. Treat. 2014, 148, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Marshall, S.M. 60 years of metformin use: A glance at the past and a look to the future. Diabetologia 2017, 60, 1561–1565. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Massey, S.; Story, D.; Li, L.; Zhou, J.; Massey, S.; Story, D.; Li, L. Metformin: An old drug with new applications. Int. J. Mol. Sci. 2018, 19, 2863. [Google Scholar] [CrossRef]
- Ning, H.-H.; Le, J.; Wang, Q.; Young, C.A.; Deng, B.; Gao, P.-X.; Zhang, H.-Q.; Qin, S.-L. The effects of metformin on simple obesity: A meta-analysis. Endocrine 2018, 1–7. [Google Scholar] [CrossRef]
- Xu, H.; Chen, K.; Jia, X.; Tian, Y.; Dai, Y.; Li, D.; Xie, J.; Tao, M.; Mao, Y. Metformin use is associated with better survival of breast cancer patients with diabetes: A meta-analysis. Oncologist 2015, 20, 1236–1244. [Google Scholar] [CrossRef]
- Zhang, J.; Li, G.; Chen, Y.; Fang, L.; Guan, C.; Bai, F.; Ma, M.; Lyu, J.; Meng, Q.H. Metformin inhibits tumorigenesis and tumor growth of breast cancer cells by upregulating miR-200c but downregulating AKT2 expression. J. Cancer 2017, 8, 1849–1864. [Google Scholar] [CrossRef]
- Zakikhani, M.; Dowling, R.; Fantus, I.G.; Sonenberg, N.; Pollak, M. Metformin is an AMP kinase-dependent growth inhibitor for breast cancer cells. Cancer Res. 2006, 66, 10269–10273. [Google Scholar] [CrossRef]
- Sahra, I.B.; Laurent, K.; Loubat, A.; Giorgetti-Peraldi, S.; Colosetti, P.; Auberger, P.; Tanti, J.F.; Le Marchand-Brustel, Y.; Bost, F. The antidiabetic drug metformin exerts an antitumoral effect in vitro and in vivo through a decrease of cyclin D1 level. Oncogene 2008, 27, 3576–3586. [Google Scholar] [CrossRef]
- Grossmann, M.E.; Yang, D.Q.; Guo, Z.; Potter, D.A.; Cleary, M.P. Metformin treatment for the prevention and/or treatment of breast/mammary tumorigenesis. Curr. Pharmacol. Rep. 2015, 1, 312–323. [Google Scholar] [CrossRef] [PubMed]
- Abotaleb, M.; Kubatka, P.; Caprnda, M.; Varghese, E.; Zolakova, B.; Zubor, P.; Opatrilova, R.; Kruzliak, P.; Stefanicka, P.; Büsselberg, D. Chemotherapeutic agents for the treatment of metastatic breast cancer: An update. Biomed. Pharmacother. 2018, 101, 458–477. [Google Scholar] [CrossRef] [PubMed]
- Varghese, E.; Samuel, S.M.; Abotaleb, M.; Cheema, S.; Mamtani, R.; Busselberg, D. The “yin and yang” of natural compounds in anticancer therapy of triple-negative breast cancers. Cancers 2018, 10, 346. [Google Scholar] [CrossRef] [PubMed]
- Bianchini, G.; Balko, J.M.; Mayer, I.A.; Sanders, M.E.; Gianni, L. Triple-negative breast cancer: Challenges and opportunities of a heterogeneous disease. Nat. Rev. Clin. Oncol. 2016, 13, 674–690. [Google Scholar] [CrossRef] [PubMed]
- Creighton, C.J. A gene transcription signature of the Akt/mTOR pathway in clinical breast tumors. Oncogene 2007, 26, 4648–4655. [Google Scholar] [CrossRef] [PubMed]
- Hay, N. The Akt-mTOR tango and its relevance to cancer. Cancer Cell 2005, 8, 179–183. [Google Scholar] [CrossRef] [PubMed]
- Umemura, S.; Yoshida, S.; Ohta, Y.; Naito, K.; Osamura, R.Y.; Tokuda, Y. Increased phosphorylation of Akt in triple-negative breast cancers. Cancer Sci. 2007, 98, 1889–1892. [Google Scholar] [CrossRef] [PubMed]
- Shrestha, S.; Sorolla, A.; Fromont, J.; Blancafort, P.; Flematti, G.R. Aurantoside C targets and induces apoptosis in triple negative breast cancer cells. Mar. Drugs 2018, 16, 361. [Google Scholar] [CrossRef]
- Easton, J.B.; Houghton, P.J. mTOR and cancer therapy. Oncogene 2006, 25, 6436–6446. [Google Scholar] [CrossRef]
- Vivanco, I.; Sawyers, C.L. The phosphatidylinositol 3-kinase Akt pathway in human cancer. Nat. Rev. Cancer 2002, 2, 489–501. [Google Scholar] [CrossRef]
- Hennessy, B.T.; Smith, D.L.; Ram, P.T.; Lu, Y.; Mills, G.B. Exploiting the PI3K/AKT pathway for cancer drug discovery. Nat. Rev. Drug Discov. 2005, 4, 988–1004. [Google Scholar] [CrossRef] [PubMed]
- Viollet, B.; Guigas, B.; Garcia, N.S.; Leclerc, J.; Foretz, M.; Andreelli, F. Cellular and molecular mechanisms of metformin: An overview. Clin. Sci. 2012, 122, 253–270. [Google Scholar] [CrossRef] [PubMed]
- Samuel, S.M.; Ghosh, S.; Majeed, Y.; Arunachalam, G.; Emara, M.M.; Ding, H.; Triggle, C.R. Metformin represses glucose starvation induced autophagic response in microvascular endothelial cells and promotes cell death. Biochem. Pharmacol. 2017, 132, 118–132. [Google Scholar] [CrossRef] [PubMed]
- Ben Sahra, I.; Regazzetti, C.; Robert, G.; Laurent, K.; Le Marchand-Brustel, Y.; Auberger, P.; Tanti, J.F.; Giorgetti-Peraldi, S.; Bost, F. Metformin, independent of AMPK, induces mTOR inhibition and cell-cycle arrest through REDD1. Cancer Res. 2011, 71, 4366–4372. [Google Scholar] [CrossRef] [PubMed]
- Daugan, M.; Dufaÿ Wojcicki, A.; D’Hayer, B.; Boudy, V. Metformin: An anti-diabetic drug to fight cancer. Pharmacol. Res. 2016, 113, 675–685. [Google Scholar] [CrossRef] [PubMed]
- Wahdan-Alaswad, R.S.; Edgerton, S.M.; Salem, H.S.; Thor, A.D. Metformin targets glucose metabolism in triple negative breast cancer. J. Oncol. Transl. Res. 2018, 4, 1–6. [Google Scholar] [CrossRef]
- Menendez, J.A.; Oliveras-Ferraros, C.; Cufi, S.; Corominas-Faja, B.; Joven, J.; Martin-Castillo, B.; Vazquez-Martin, A. Metformin is synthetically lethal with glucose withdrawal in cancer cells. Cell Cycle 2012, 11, 2782–2792. [Google Scholar] [CrossRef]
- Varghese, E.; Busselberg, D. Auranofin, an anti-rheumatic gold compound, modulates apoptosis by elevating the intracellular calcium concentration ([Ca2+]i) in MCF-7 breast cancer cells. Cancers 2014, 6, 2243–2258. [Google Scholar] [CrossRef]
- Arunachalam, G.; Samuel, S.M.; Marei, I.; Ding, H.; Triggle, C.R. Metformin modulates hyperglycaemia-induced endothelial senescence and apoptosis through SIRT1. Br. J. Pharmacol. 2014, 171, 523–535. [Google Scholar] [CrossRef] [PubMed]
- Keenan, M.M.; Chi, J.T. Alternative fuels for cancer cells. Cancer J. 2015, 21, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Evans, J.M.M.; Donnelly, L.A.; Emslie-Smith, A.M.; Alessi, D.R.; Morris, A.D. Metformin and reduced risk of cancer in diabetic patients. BMJ 2005, 330, 1304–1305. [Google Scholar] [CrossRef] [PubMed]
- Noto, H.; Goto, A.; Tsujimoto, T.; Noda, M. Cancer risk in diabetic patients treated with metformin: A systematic review and meta-analysis. PLoS ONE 2012, 7, e33411. [Google Scholar] [CrossRef] [PubMed]
- Safe, S.; Nair, V.; Karki, K. Metformin-induced anticancer activities: Recent insights. Biol. Chem. 2018, 399, 321–335. [Google Scholar] [CrossRef] [PubMed]
- Tseng, C.-H. Metformin may reduce breast cancer risk in Taiwanese women with type 2 diabetes. Breast Cancer Res. Treat. 2014, 145, 785–790. [Google Scholar] [CrossRef] [PubMed]
- Chlebowski, R.T.; McTiernan, A.; Wactawski-Wende, J.; Manson, J.E.; Aragaki, A.K.; Rohan, T.; Ipp, E.; Kaklamani, V.G.; Vitolins, M.; Wallace, R.; et al. Diabetes, metformin, and breast cancer in postmenopausal women. J. Clin. Oncol. 2012, 30, 2844–2852. [Google Scholar] [CrossRef]
- Aksoy, S.; Sendur, M.A.N.; Altundag, K. Demographic and clinico-pathological characteristics in patients with invasive breast cancer receiving metformin. Med. Oncol. 2013, 30, 590. [Google Scholar] [CrossRef]
- Jiralerspong, S.; Palla, S.L.; Giordano, S.H.; Meric-Bernstam, F.; Liedtke, C.; Barnett, C.M.; Hsu, L.; Hung, M.C.; Hortobagyi, G.N.; Gonzalez-Angulo, A.M. Metformin and pathologic complete responses to neoadjuvant chemotherapy in diabetic patients with breast cancer. J. Clin. Oncol. 2009, 27, 3297–3302. [Google Scholar] [CrossRef]
- Wilcock, C.; Wyre, N.D.; Bailey, C.J. Subcellular distribution of metformin in rat liver. J. Pharm. Pharmacol. 1991, 43, 442–444. [Google Scholar] [CrossRef]
- Graham, G.G.; Punt, J.; Arora, M.; Day, R.O.; Doogue, M.P.; Duong, J.K.; Furlong, T.J.; Greenfield, J.R.; Greenup, L.C.; Kirkpatrick, C.M.; et al. Clinical pharmacokinetics of metformin. Clin. Pharmacokinet. 2011, 50, 81–98. [Google Scholar] [CrossRef]
- Wahdan-Alaswad, R.; Fan, Z.; Edgerton, S.M.; Liu, B.; Deng, X.S.; Arnadottir, S.S.; Richer, J.K.; Anderson, S.M.; Thor, A.D. Glucose promotes breast cancer aggression and reduces metformin efficacy. Cell Cycle 2013, 12, 3759–3769. [Google Scholar] [CrossRef]
- Liu, H.; Scholz, C.; Zang, C.; Schefe, J.H.; Habbel, P.; Regierer, A.C.; Schulz, C.O.; Possinger, K.; Eucker, J. Metformin and the mTOR inhibitor everolimus (RAD001) sensitize breast cancer cells to the cytotoxic effect of chemotherapeutic drugs in vitro. Anticancer Res. 2012, 32, 1627–1637. [Google Scholar] [PubMed]
- Strekalova, E.; Malin, D.; Rajanala, H.; Cryns, V.L. Metformin sensitizes triple-negative breast cancer to proapoptotic TRAIL receptor agonists by suppressing XIAP expression. Breast Cancer Res. Treat. 2017, 163, 435–447. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, E.J.; Baldwin, L.A. Hormesis: The dose-response revolution. Annu. Rev. Pharmacol. Toxicol. 2003, 43, 175–197. [Google Scholar] [CrossRef] [PubMed]
- Arunachalam, G.; Lakshmanan, A.P.; Samuel, S.M.; Triggle, C.R.; Ding, H. Molecular interplay between microRNA-34a and Sirtuin1 in hyperglycemia-mediated impaired angiogenesis in endothelial cells: Effects of metformin. J. Pharmacol. Exp. Ther. 2016, 356, 314–323. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.C.; Singleton, B.A.; Llopis, S.D.; Skripnikova, E.V. Metformin induces a senescence-associated gene signature in breast cancer cells. J. Health Care Poor Underserved 2013, 24, 93–103. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.-Y.; Liu, Z.; Bi, M.-H.; Zhang, J.-J.; Han, Z.-Q.; Han, X.; Wang, H.-Y.; Sun, G.-P.; Liu, H. Metformin induces apoptosis via a mitochondria-mediated pathway in human breast cancer cells in vitro. Exp. Ther. Med. 2016, 11, 1700–1706. [Google Scholar] [CrossRef]
- Rajh, M.; Dolinar, K.; Miš, K.; Pavlin, M.; Pirkmajer, S. Medium renewal blocks anti-proliferative effects of metformin in cultured MDA-MB-231 breast cancer cells. PLoS ONE 2016, 11, e0154747. [Google Scholar] [CrossRef]
- Jaune, E.; Rocchi, S. Metformin: Focus on melanoma. Front. Endocrinol. 2018, 9, 472. [Google Scholar] [CrossRef]
- Li, K.; Zhang, T.T.; Wang, F.; Cui, B.; Zhao, C.X.; Yu, J.J.; Lv, X.X.; Zhang, X.W.; Yang, Z.N.; Huang, B.; et al. Metformin suppresses melanoma progression by inhibiting KAT5-mediated SMAD3 acetylation, transcriptional activity and TRIB3 expression. Oncogene 2018, 37, 2967–2981. [Google Scholar] [CrossRef]
- Lehmann, B.D.; Bauer, J.A.; Chen, X.; Sanders, M.E.; Chakravarthy, A.B.; Shyr, Y.; Pietenpol, J.A. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J. Clin. Investig. 2011, 121, 2750–2767. [Google Scholar] [CrossRef]
- Lehmann, B.D.; Pietenpol, J.A. Identification and use of biomarkers in treatment strategies for triple-negative breast cancer subtypes. J. Pathol. 2014, 232, 142–150. [Google Scholar] [CrossRef] [PubMed]
- Ben Sahra, I.; Laurent, K.; Giuliano, S.; Larbret, F.; Ponzio, G.; Gounon, P.; Le Marchand-Brustel, Y.; Giorgetti-Peraldi, S.; Cormont, M.; Bertolotto, C.; et al. Targeting cancer cell metabolism: The combination of metformin and 2-deoxyglucose induces p53-dependent apoptosis in prostate cancer cells. Cancer Res. 2010, 70, 2465–2475. [Google Scholar] [CrossRef] [PubMed]
- Ben Sahra, I.; Tanti, J.F.; Bost, F. The combination of metformin and 2-deoxyglucose inhibits autophagy and induces AMPK-dependent apoptosis in prostate cancer cells. Autophagy 2010, 6, 670–671. [Google Scholar] [CrossRef] [PubMed]
- Sui, X.; Xu, Y.; Wang, X.; Han, W.; Pan, H.; Xiao, M. Metformin: A novel but controversial drug in cancer prevention and treatment. Mol. Pharm. 2015, 12, 3783–3791. [Google Scholar] [CrossRef] [PubMed]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Varghese, S.; Samuel, S.M.; Varghese, E.; Kubatka, P.; Büsselberg, D. High Glucose Represses the Anti-Proliferative and Pro-Apoptotic Effect of Metformin in Triple Negative Breast Cancer Cells. Biomolecules 2019, 9, 16. https://doi.org/10.3390/biom9010016
Varghese S, Samuel SM, Varghese E, Kubatka P, Büsselberg D. High Glucose Represses the Anti-Proliferative and Pro-Apoptotic Effect of Metformin in Triple Negative Breast Cancer Cells. Biomolecules. 2019; 9(1):16. https://doi.org/10.3390/biom9010016
Chicago/Turabian StyleVarghese, Sharon, Samson Mathews Samuel, Elizabeth Varghese, Peter Kubatka, and Dietrich Büsselberg. 2019. "High Glucose Represses the Anti-Proliferative and Pro-Apoptotic Effect of Metformin in Triple Negative Breast Cancer Cells" Biomolecules 9, no. 1: 16. https://doi.org/10.3390/biom9010016
APA StyleVarghese, S., Samuel, S. M., Varghese, E., Kubatka, P., & Büsselberg, D. (2019). High Glucose Represses the Anti-Proliferative and Pro-Apoptotic Effect of Metformin in Triple Negative Breast Cancer Cells. Biomolecules, 9(1), 16. https://doi.org/10.3390/biom9010016