Semantic Multi-Classifier Systems Identify Predictive Processes in Heart Failure Models across Species

 ,
,
Abstract
1. Introduction
2. Results and Discussion
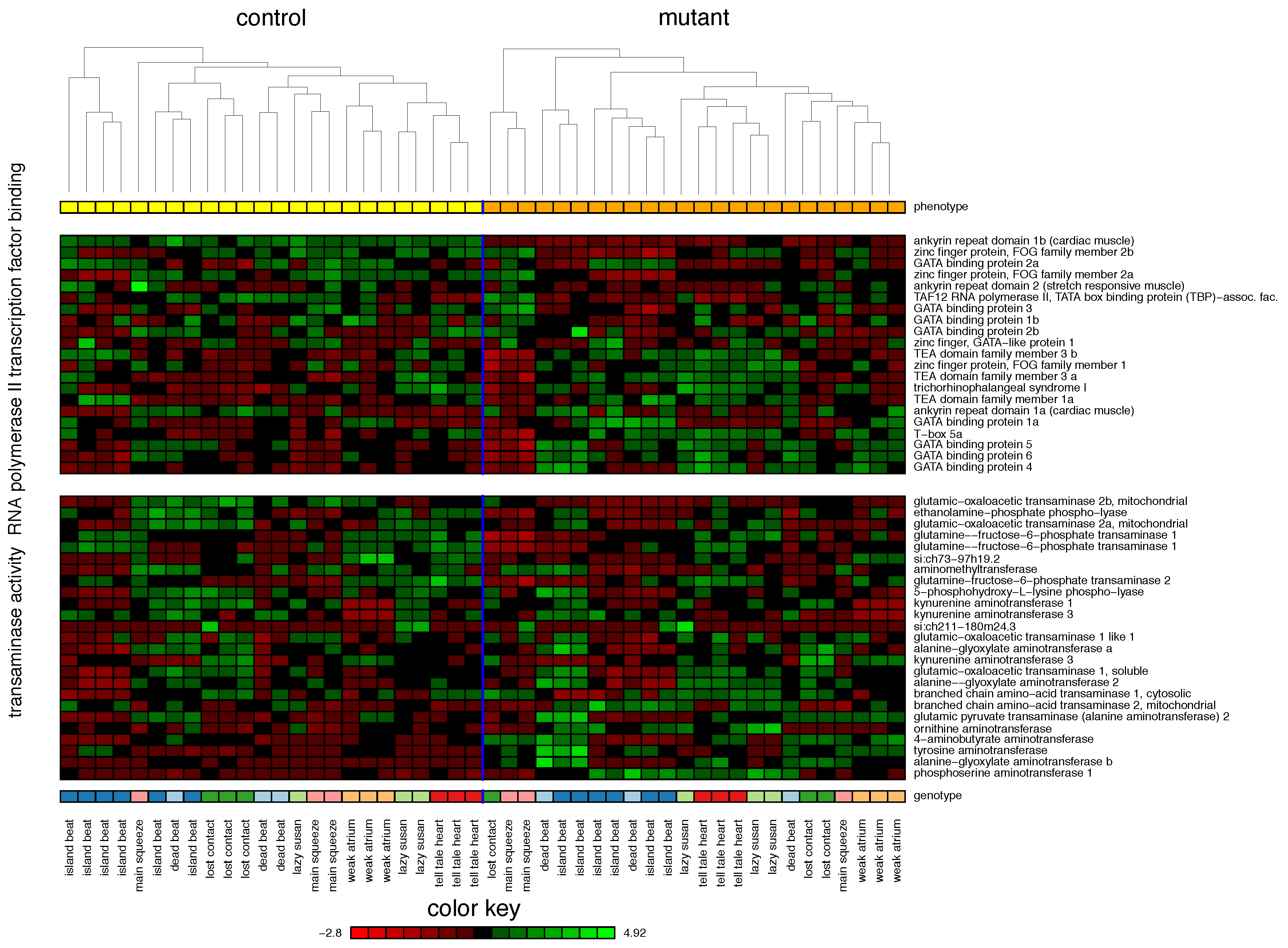
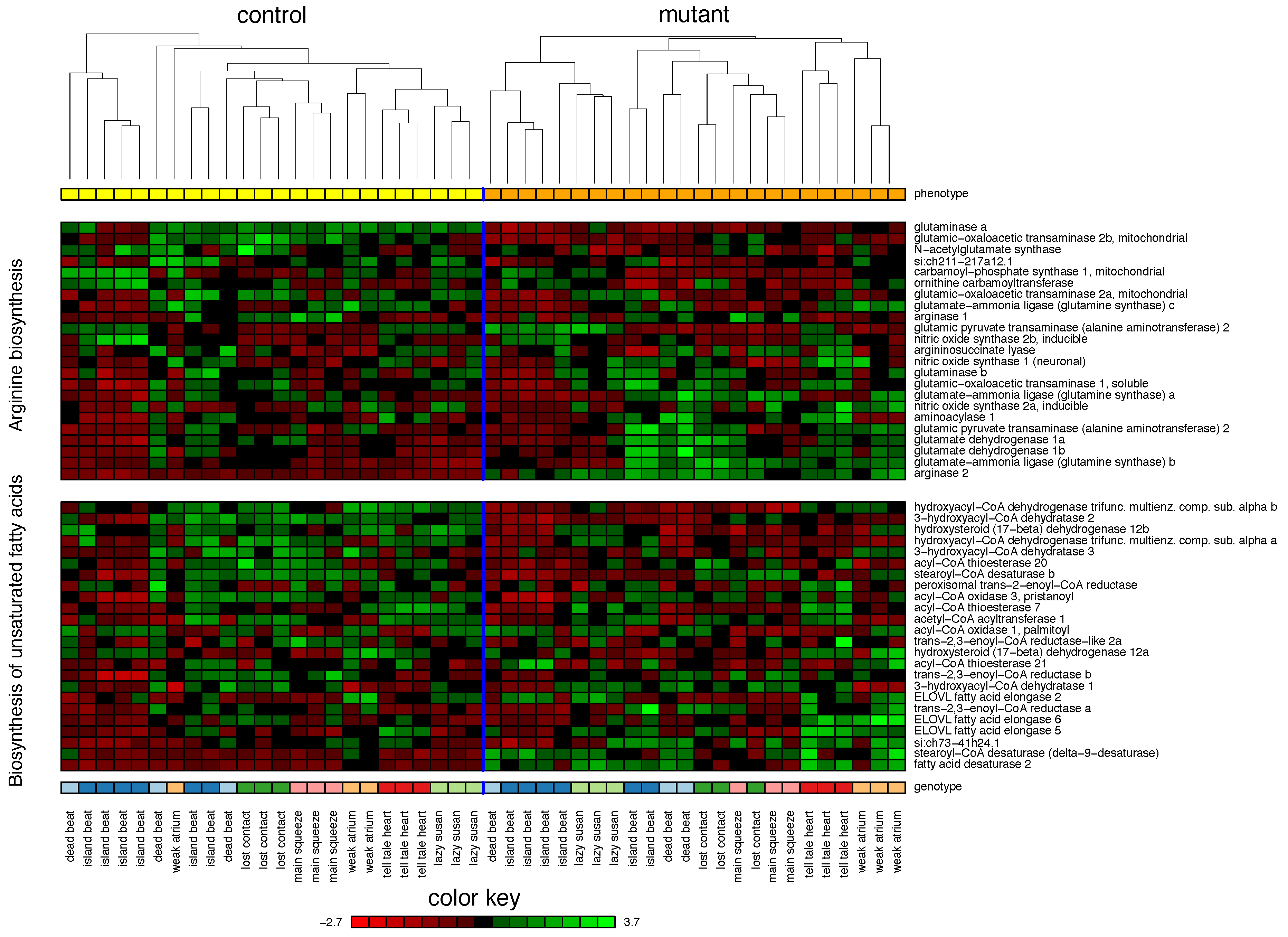
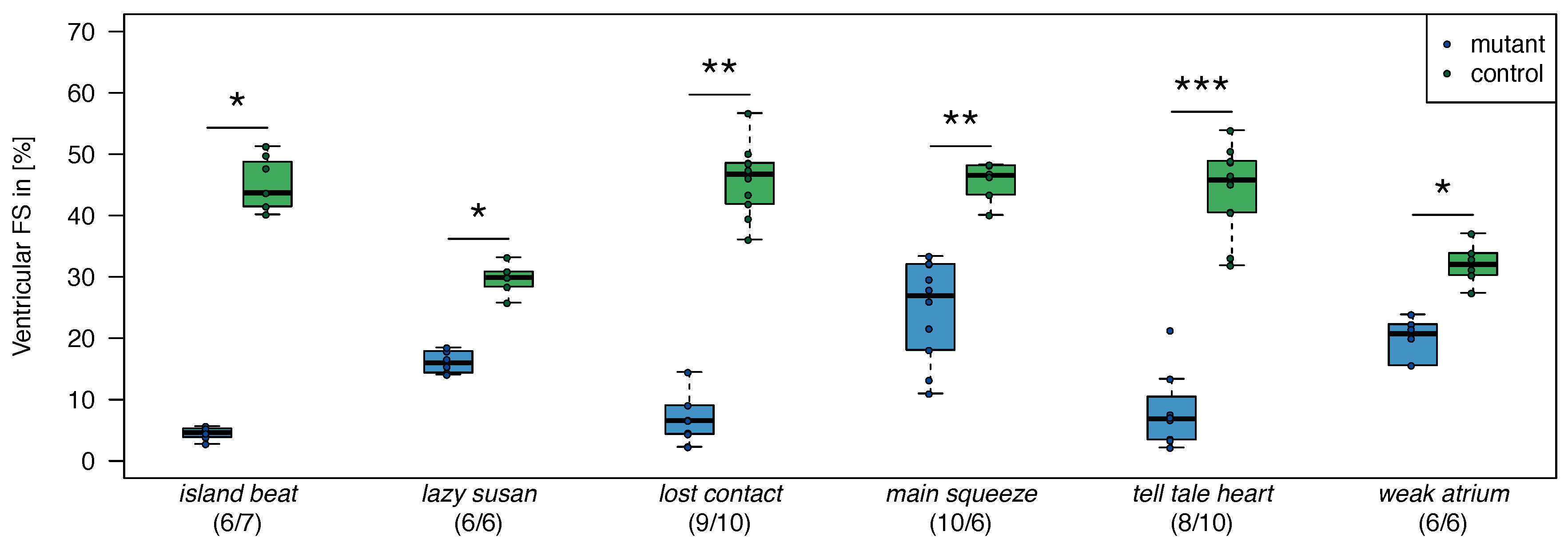
2.1. Zebrafish
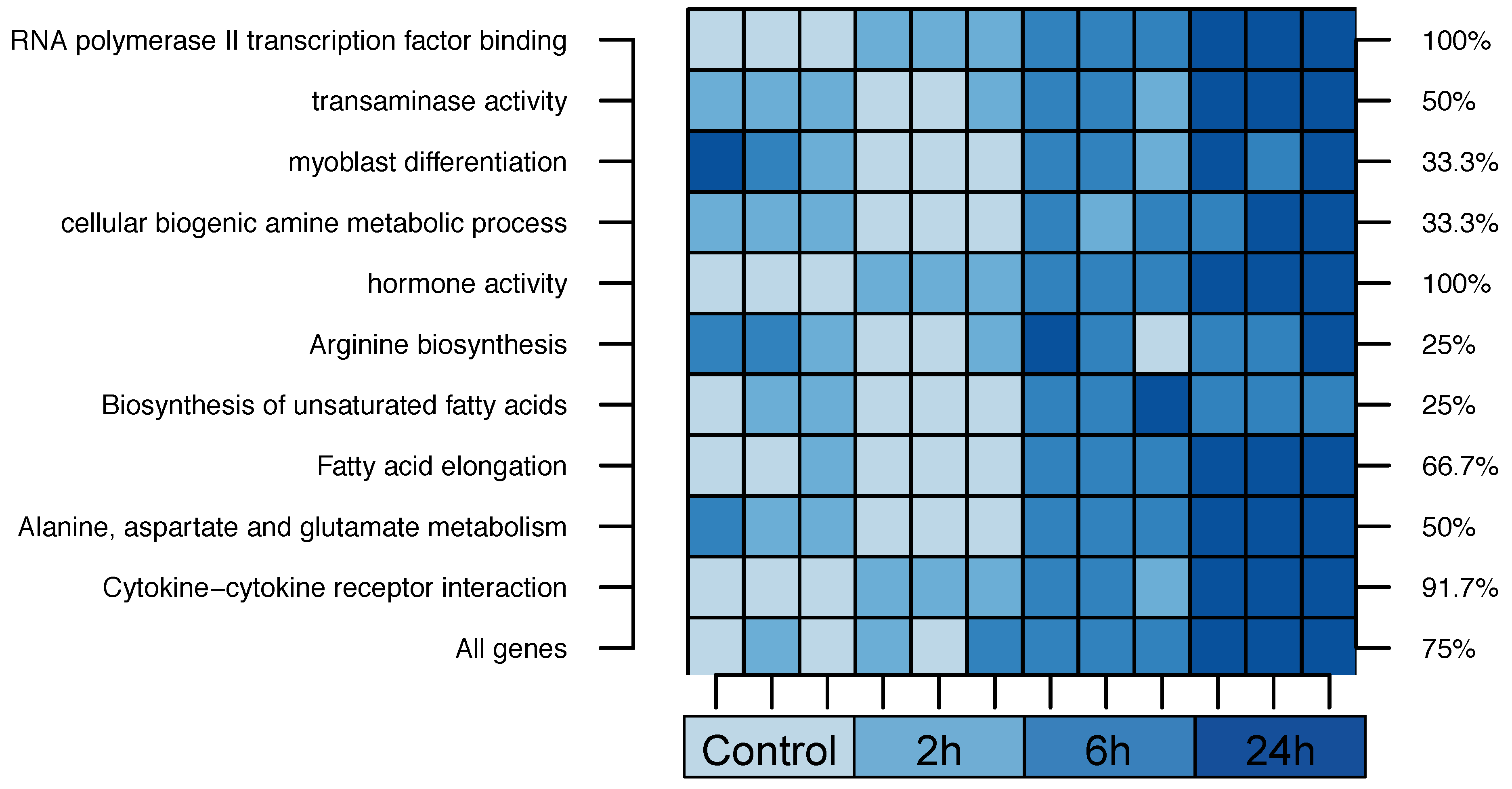
2.2. Cross-Species Experiment: Wistar Rats
3. Conclusions
4. Materials and Methods
4.1. Zebrafish Strains, Fractional Shortening Measurements, RNA Isolation and RNA Sequencing
4.2. Stretch Experiments in Neonatal Rat Ventricular Cardiomyocytes (NRVCMs)
4.3. Classification
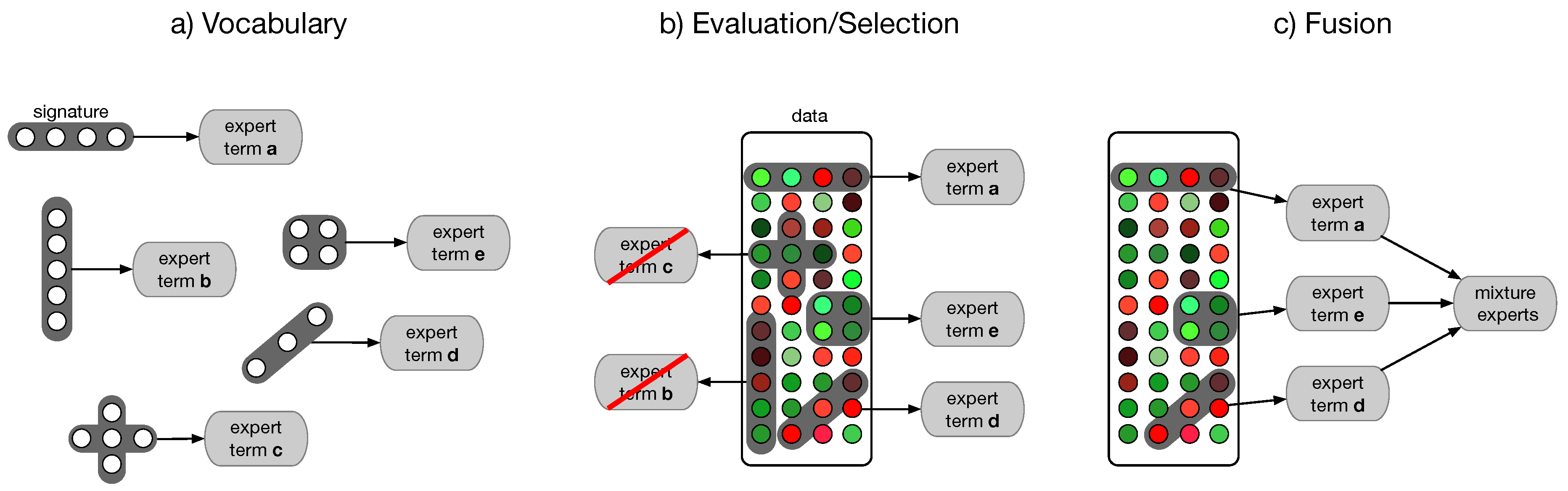
4.4. Semantic Multi-Classifier Systems (S-MCS)
4.5. Nearest Neighbor Classification
4.6. Pathways from Kyoto Encyclopedia of Genes and Genomes
4.7. Gene Ontology Terms
Author Contributions
Funding
Conflicts of Interest
References
- Cook, C.; Cole, G.; Asaria, P.; Jabbour, R.; Francis, D.P. The annual global economic burden of heart failure. Int. J. Cardiol. 2014, 171, 368–376. [Google Scholar] [CrossRef] [PubMed]
- Vigen, R.; Maddox, T.; Allen, L. Aging of the United States Population: Impact on Heart Failure. Curr. Heart Failure Rep. 2012, 9, 369–374. [Google Scholar] [CrossRef] [PubMed]
- Stainier, D.Y.; Fouquet, B.; Chen, J.N.; Warren, K.S.; Weinstein, B.M.; Meiler, S.E.; Mohideen, M.A.; Neuhauss, S.C.; Solnica-Krezel, L.; Schier, A.F.; et al. Mutations affecting the formation and function of the cardiovascular system in the zebrafish embryo. Development 1996, 123, 285–292. [Google Scholar] [PubMed]
- Rottbauer, W.; Wessels, G.; Dahme, T.; Just, S.; Trano, N.; Hassel, D.; Burns, C.; Katus, H.; Fishman, M. Cardiac myosin light chain-2: A novel essential component of thick-myofilament assembly and contractility of the heart. Circ. Res. 2006, 99, 323–331. [Google Scholar] [CrossRef] [PubMed]
- Bendig, G.; Grimmler, M.; Huttner, I.; Wessels, G.; Dahme, T.; Just, S.; Trano, N.; Katus, H.; Fishman, M.; Rottbauer, W. Integrin-linked kinase, a novel component of the cardiac mechanical stretch sensor, controls contractility in the zebrafish heart. Genes Dev. 2006, 20, 2361–2372. [Google Scholar] [CrossRef] [PubMed]
- Meder, B.; Huttner, I.; Sedaghat-Hamedani, F.; Just, S.; Dahme, T.; Frese, K.; Vogel, B.; Köhler, D.; Kloos, W.; Rudloff, J.; et al. PINCH proteins regulate cardiac contractility by modulating integrin-linked kinase-protein kinase B signaling. Mol. Cell. Biol. 2011, 31, 3424–3435. [Google Scholar] [CrossRef] [PubMed]
- Knöll, R.; Postel, R.; Wang, J.; Krätzner, R.; Hennecke, G.; Vacaru, A.; Vakeel, P.; Schubert, C.; Murthy, K.; Rana, B.; et al. Laminin-alpha4 and integrin-linked kinase mutations cause human cardiomyopathy via simultaneous defects in cardiomyocytes and endothelial cells. Circulation 2007, 116, 515–525. [Google Scholar] [CrossRef]
- Lausser, L.; Müssel, C.; Kestler, H.A. Measuring and Visualizing the Stability of Biomarker Selection Techniques. Comput. Stat. 2013, 28, 51–65. [Google Scholar] [CrossRef]
- Schirra, L.R.; Lausser, L.; Kestler, H.A. Selection Stability as a Means of Biomarker Discovery in Classification. In Analysis of Large and Complex Data; Wilhelm, A.F.X., Kestler, H.A., Eds.; Studies in Classification, Data Analysis, and Knowledge Organization; Springer: Cham, Switzerland, 2016; pp. 79–89. [Google Scholar]
- Lausser, L.; Szekely, R.; Schirra, L.R.; Kestler, H.A. The Influence of Multi-class Feature Selection on the Prediction of Diagnostic Phenotypes. Neural Process. Lett. 2018, 48, 863–880. [Google Scholar] [CrossRef]
- Gress, T.M.; Lausser, L.; Schirra, L.R.; Ortmüller, L.; Diels, R.; Kong, B.; Michalski, C.W.; Hackert, T.; Strobel, O.; Giese, N.A.; et al. Combined microRNA and mRNA microfluidic TaqMan array cards for the diagnosis of malignancy of multiple types of pancreatico-biliary tumors in fine-needle aspiration material. Oncotarget 2017, 8, 108223–1082370. [Google Scholar] [CrossRef] [PubMed]
- Hesse, R.; Lausser, L.; Gummert, P.; Schmid, F.; Wahler, A.; Schnack, C.; Kroker, K.S.; Otto, M.; Tumani, H.; Kestler, H.A.; et al. Reduced cGMP levels in CSF of AD patients correlate with severity of dementia and current depression. Alzheimer’s Res. Therapy 2017, 9, 17. [Google Scholar] [CrossRef] [PubMed]
- Völkel, G.; Lausser, L.; Schmid, F.; Kraus, J.M.; Kestler, H.A. Sputnik: Ad hoc distributed computation. Bioinformatics 2015, 31, 1298–1301. [Google Scholar] [CrossRef] [PubMed]
- Lausser, L.; Schmid, F.; Platzer, M.; Sillanpää, M.J.; Kestler, H.A. Semantic Multi-classifier Systems for the Analysis of Gene Expression Profiles. Arch. Data Sci. Ser. A (Online First) 2016, 1, 157–176. [Google Scholar]
- Taudien, S.; Lausser, L.; Giamarellos-Bourboulis, E.J.; Sponholz, C.; Schöneweck, F.; Felder, M.; Schirra, L.R.; Schmid, F.; Gogos, C.; Groth, S.; et al. Genetic Factors of the Disease Course After Sepsis: Rare Deleterious Variants Are Predictive. EBioMedicine 2016, 12, 227–238. [Google Scholar] [CrossRef] [PubMed]
- Skrzynia, C.; Berg, J.S.; Willis, M.S.; Jensen, B.C. Genetics and Heart Failure: A Concise Guide for the Clinician. Curr. Cardiol. Rev. 2011, 11, 10–17. [Google Scholar] [CrossRef]
- Morita, H.; Seidman, J.; Seidman, C.E. Genetic causes of human heart failure. J. Clin. Investig. 2005, 115, 518–526. [Google Scholar] [CrossRef] [PubMed]
- Rottbauer, W.; Just, S.; Wessels, G.; Trano, N.; Most, P.; Katus, H.; Fishman, M. VEGF–PLCγ1 pathway controls cardiac contractility in the embryonic heart. Genes Dev. 2005, 19, 1624–1634. [Google Scholar] [CrossRef] [PubMed]
- Rottbauer, W.; Baker, K.; Wo, Z.; Mohideen, M.A.P.; Cantiello, H.F.; Fishman, M.C. Growth and Function of the Embryonic Heart Depend upon the Cardiac-Specific l-Type Calcium Channel α1 Subunit. Dev. Cell 2001, 1, 265–275. [Google Scholar] [CrossRef]
- Berdougo, E.; Coleman, H.; Lee, D.H.; Stainier, D.Y.R.; Yelon, D. Mutation of weak atrium/atrial myosin heavy chain disrupts atrial function and influences ventricular morphogenesis in zebrafish. Development 2003, 130, 6121–6129. [Google Scholar] [CrossRef]
- Meder, B.; Laufer, C.; Hassel, D.; Just, S.; Marquart, S.; Vogel, B.; Hess, A.; Fishman, M.; Katus, H.; Rottbauer, W. A single serine in the carboxyl terminus of cardiac essential myosin light chain-1 controls cardiomyocyte contractility in vivo. Circ. Res. 2009, 104, 650–659. [Google Scholar] [CrossRef]
- Iwaki, H.; Sasaki, S.; Matsushita, A.; Ohba, K.; Matsunaga, H.; Misawa, H.; Oki, Y.; Ishizuka, K.; Nakamura, H.; Suda, T. Essential Role of TEA Domain Transcription Factors in the Negative Regulation of the MYH 7 Gene by Thyroid Hormone and Its Receptors. PLoS ONE 2014, 9, e88610. [Google Scholar] [CrossRef] [PubMed]
- Kohli, S.; Ahuja, S.; Rani, W. Transcription Factors in Heart: Promising Therapeutic Targets in Cardiac Hypertrophy. Curr. Cardiol. Rev. 2011, 7, 262–271. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Lee, J.; Kim, B.S.; Wang, Q.; Buxton, S.K.; Balasubramanyam, N.; Kim, J.J.; Dong, J.; Zhang, A.; Li, S.; et al. Tead1 is required for maintaining adult cardiomyocyte function, and its loss results in lethal dilated cardiomyopathy. JCI Insight 2017, 2, e93343. [Google Scholar] [CrossRef] [PubMed]
- Fosset, N.; Zhang, Q.; Gajewski, K.; Choi, C.Y.; Kim, Y.; Schulz, R.A. The multitype zinc-finger protein U-shaped functions in heart cell specification in the Drosophila embryo. Proc. Natl. Acad. Sci. USA 2000, 97, 7348–7353. [Google Scholar] [CrossRef]
- Zhou, B.; Ma, Q.; Kong, S.W.; Hu, Y.; Campbell, P.H.; McGowan, F.X.; Ackerman, K.G.; Wu, B.; Bin, Z.; Tevosian, S.G.; et al. Fog2 is critical for cardiac function and maintenance of coronary vasculature in the adult mouse heart. J. Clin. Investig. 2009, 119, 1462–1476. [Google Scholar] [CrossRef] [PubMed]
- Pikkarainen, S.; Tokola, H.; Kerkelä, R.; Ruskoaho, H. GATA transcription factors in the developing and adult heart. Cardiovasc. Res. 2004, 63, 196–207. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y.J. Cell signaling pathways for the regulation of GATA4 transcription factor: Implications for cell growth and apoptosis. Cell. Signal. 2011, 23, 1094–1099. [Google Scholar] [CrossRef] [PubMed]
- Pan, H.; Qin, K.; Guo, Z.; Ma, Y.; April, C.; Gao, X.; Andrews, T.G.; Bokov, A.; Zhang, J.; Chen, Y.; et al. Negative Elongation Factor Controls Energy Homeostasis in Cardiomyocytes. Cell Rep. 2014, 7, 79–85. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.X.; Qian, L.X.; Yu, Z.; Jiang, Q.; Dong, Y.X.; Liu, X.F.; Yang, X.Y.; Zhong, T.P.; Song, H.Y. Requirements of myocyte-specific enhancer factor 2A in zebrafish cardiac contractility. FEBS Lett. 2005, 579, 4843–4850. [Google Scholar] [CrossRef]
- Yilbas, A.; Hamilton, A.; Wang, Y.; Mach, H.; Lacroix, N.; Davis, D.; Chen, J.; Li, Q. Activation of GATA4 gene expression at the early stage of cardiac specification. Front. Chem. 2014, 2, 12. [Google Scholar] [CrossRef]
- Bang, M.L.; Gu, Y.; Dalton, N.D.; Peterson, K.L.; Chien, K.R.; Chen, J. The Muscle Ankyrin Repeat Proteins CARP, Ankrd2, and DARP Are Not Essential for Normal Cardiac Development and Function at Basal Conditions and in Response to Pressure Overload. PLoS ONE 2014, 9, e93638. [Google Scholar] [CrossRef]
- Miano, J.M.; Long, X.; Fujiwara, K. Serum response factor: Master regulator of the actin cytoskeleton and contractile apparatus. Am. J. Physiol. Cell Physiol. 2007, 292, C70–C81. [Google Scholar] [CrossRef] [PubMed]
- Fowler, T.; Sen, R.; Roy, A.L. Regulation of Primary Response Genes. Mol. Cell 2011, 44, 348–360. [Google Scholar] [CrossRef] [PubMed]
- Venturini, A.; Ascione, R.; Lin, H.; Polesel, E.; Angelini, G.D.; Suleiman, M.S. The importance of myocardial amino acids during ischemia and reperfusion in dilated left ventricle of patients with degenerative mitral valve disease. Mol. Cell. Biochem. 2009, 330. [Google Scholar] [CrossRef] [PubMed]
- Drake, K.J.; Sidorov, V.Y.; Mcguinness, O.P.; Wasserman, D.H.; Wikswo, J.P. Amino acids as metabolic substrates during cardiac ischemia. Exp. Biol. Med. 2012, 237, 1369–1378. [Google Scholar] [CrossRef] [PubMed]
- Williams, H.; King, N.; Griffiths, E.J.; Suleiman, M.S. Glutamate-loading Stimulates Metabolic Flux and Improves Cell Recovery Following Chemical Hypoxia in Isolated Cardiomyocyte. J. Mol. Cell. Cardiol. 2001, 33, 2109–2119. [Google Scholar] [CrossRef] [PubMed]
- Løfgren, B.; Povlsen, J.A.; Rasmussen, L.E.; Støttrup, N.B.; Solskov, L.; Krarup, P.; Kristiansen, S.B.; Bøtker, H.E.; Nielsen, T.T. Amino acid transamination is crucial for ischaemic cardioprotection in normal and preconditioned isolated rat hearts—Focus on l-glutamate. Exp. Physiol. 2010, 95, 140–152. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Morris, S.M. Arginine metabolism: Nitric oxide and beyond. Biochem. J. 1998, 336, 1–17. [Google Scholar] [CrossRef]
- Tousoulis, D.; Antoniades, C.; Tentolouris, C.; Goumas, G.; Stefanadis, C.; Toutouzas, P. L-Arginine in cardiovascular disease: Dream or reality? Vasc. Med. 2002, 7, 203–211. [Google Scholar] [CrossRef] [PubMed]
- Jung, A.S.; Kubo, H.; Wilson, R.; Houser, S.R.; Margulies, K.B. Modulation of contractility by myocyte-derived arginase in normal and hypertrophied feline myocardium. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H1756–H1762. [Google Scholar] [CrossRef] [PubMed]
- Cotton, J.M.; Kearney, M.T.; Shah, A.M. Nitric oxide and myocardial function in heart failure: Friend or foe? Heart 2002, 88, 564–566. [Google Scholar] [CrossRef] [PubMed]
- Steppan, J.; Ryoo, S.; Schuleri, K.H.; Gregg, C.; Hasan, R.K.; White, A.R.; Bugaj, L.J.; Khan, M.; Santhanam, L.; Nyhan, D.; et al. Arginase modulates myocardial contractility by a nitric oxide synthase 1-dependent mechanism. Proc. Natl. Acad. Sci. USA 2006, 103, 4759–4764. [Google Scholar] [CrossRef] [PubMed]
- Ziolo, M.T.; Kohr, M.J.; Wang, H. Nitric oxide signaling and the regulation of myocardial function. J. Mol. Cell. Cardiol. 2008, 45, 625–632. [Google Scholar] [CrossRef] [PubMed]
- Heidorn, M.; Frodermann, T.; Böning, A.; Schreckenberg, R.; Schlüter, K.D. Citrulline Improves Early Post-Ischemic Recovery or Rat Hearts In Vitro by Shifting Arginine Metabolism From Polyamine to Nitric Oxide Formation. Clin. Med. Insights Cardiol. 2018, 12. [Google Scholar] [CrossRef] [PubMed]
- Dyck, J.R.B.; Lopaschuk, G.D. Malonyl CoA Control of Fatty Acid Oxidation in the Ischemic Heart. J. Mol. Cell. Cardiol. 2002, 34, 1099–1109. [Google Scholar] [CrossRef] [PubMed]
- Wende, A.R.; Brahma, M.K.; McGinnis, G.R.; Young, M.E. Metabolic Origins of Heart Failure. JACC Basic Transl. Sci. 2017, 2, 297–310. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Leskinen, H.; Liimatta, E.; Sormunen, R.T.; Miinalainen, I.J.; Hassinen, I.E.; Hiltunen, J.K. Myocardial Overexpression of Mecr, a Gene of Mitochondrial FAS II Leads to Cardiac Dysfunction in Mouse. PLoS ONE 2009, 4, e5589. [Google Scholar] [CrossRef]
- Guertl, B.; Noehammer, C.; Hoefler, G. Metabolic cardiomyopathies. Int. J. Exp. Pathol. 2000, 81, 349–372. [Google Scholar] [CrossRef]
- Blondelle, J.; Ohno, Y.; Gache, V.; Guyot, S.; Storck, S.; Blanchard-Gutton, N.; Barthélémy, I.; Walmsley, G.; Rahier, A.; Gadin, S.; et al. HACD1, a regulator of membrane composition and fluidity, promotes myoblast fusion and skeletal muscle growth. J. Mol. Cell Biol. 2015, 7, 429–440. [Google Scholar] [CrossRef]
- Mulligan, C.M.; Le, C.H.; deMooy, A.B.; Nelson, C.B.; Chicco, A.J. Inhibition of Delta-6 Desaturase Reverses Cardiolipin Remodeling and Prevents Contractile Dysfunction in the Aged Mouse Heart Without Altering Mitochondrial Respiratory Function. J. Gerontol. Ser. A 2014, 69, 799–809. [Google Scholar] [CrossRef]
- Frank, D.; Kuhn, C.; Brors, B.; Hanselmann, C.; Lüdde, M.; Katus, H.A.; Frey, N. Gene expression pattern in biomechanically stretched cardiomyocytes: Evidence for a stretch-specific gene program. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2008, 51, 309–318. [Google Scholar] [CrossRef] [PubMed]
- Hill, J.A.; Olson, E.N. Cardiac Plasticity. N. Engl. J. Med. 2008, 358, 1370–1380. [Google Scholar] [CrossRef] [PubMed]
- Sîrbu, A.; Kerr, G.; Crane, M.; Ruskin, H.J. RNA-Seq vs Dual- and Single-Channel Microarray Data: Sensitivity Analysis for Differential Expression and Clustering. PLoS ONE 2012, 7, e50986. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Fung-Leung, W.P.; Bittner, A.; Ngo, K.; Liu, X. Comparison of RNA-Seq and Microarray in Transcriptome Profiling of Activated T Cells. PLoS ONE 2014, 9, e78644. [Google Scholar] [CrossRef] [PubMed]
- Lowe, R.; Shirley, N.; Bleackley, M.; Dolan, S.; Shafee, T. Transcriptomics technologies. PLoS ONE Comput. Biol. 2017, 13, e2005457. [Google Scholar] [CrossRef] [PubMed]
- Rai, M.F.; Tycksen, E.D.; Sandell, L.J.; Brophy, R.H. Advantages of RNA-seq compared to RNA microarrays for transcriptome profiling of anterior cruciate ligament tears. J. Orthop. Res. 2017, 36, 484–497. [Google Scholar] [CrossRef] [PubMed]
- Marioni, J.C.; Mason, C.E.; Mane, S.M.; Stephens, M.; Gilad, Y. RNA-seq: An assessment of technical reproducibility and comparison with gene expression arrays. Genome Res. 2008, 18, 1509–1517. [Google Scholar] [CrossRef]
- Nookaeq, I.; Papini, M.; Pornputtapong, N.; Scalcinati, G.; Fagerberg, L.; Uhlen, M.; Nielsen, J. A comprehensive comparison of RNA-Seq-based transcriptome analysis from reads to differential gene expression and cross-comparison with microarrays: A case study in Saccharomyces cerevisiae. Nucleic Acids Res. 2012, 40, 10084–10097. [Google Scholar] [CrossRef]
- Chen, L.; Sun, F.; Yang, X.; Jin, Y.; Shi, M.; Wang, L.; Shi, Y.; Zhan, C.; Wang, Q. Correlation between RNA-Seq and microarrays results using TCGA data. Gene 2017, 628, 200–204. [Google Scholar] [CrossRef]
- Bottomly, D.; Walter, N.A.R.; Hunter, J.E.; Darakjian, P.; Kawane, S.; Buck, K.J.; Searles, R.P.; Mooney, M.; McWeeney, S.K.; Hitzemann, R. Evaluating Gene Expression in C57BL/6J and DBA/2J Mouse Striatum Using RNA-Seq and Microarrays. PLoS ONE 2011, 6, e17820. [Google Scholar] [CrossRef]
- Etard, C.; Armant, O.; Roostalu, U.; Gourain, V.; Ferg, M.; Strähle, U. Loss of function of myosin chaperones triggers Hsf1-mediated transcriptional response in skeletal muscle cells. Genome Biol. 2015, 16, 267. [Google Scholar] [CrossRef] [PubMed]
- Kajimura, S.; Aida, K.; Duan, C. Insulin-like growth factor-binding protein-1 (IGFBP-1) mediates hypoxia-induced embryonic growth and developmental retardation. Proc. Natl. Acad. Sci. USA 2005, 102, 1240–1245. [Google Scholar] [CrossRef] [PubMed]
- Kustermann, M.; Manta, L.; Paone, C.; Kustermann, J.; Lausser, L.; Wiesner, C.; Eichinger, L.; Clemen, C.; Schröder, R.; Kestler, H.; et al. Loss of the novel Vcp (valosin containing protein) interactor Washc4 interferes with autophagy-mediated proteostasis in striated muscle and leads to myopathy in vivo. Autophagy 2018, 14, 1911–1927. [Google Scholar] [CrossRef] [PubMed]
- Hirth, S.; Bühler, A.; Bührdel, J.; Rudeck, S.; Dahme, T.; Rottbauer, W.; Just, S. Paxillin and Focal Adhesion Kinase (FAK) Regulate Cardiac Contractility in the Zebrafish Heart. PLoS ONE 2016, 11, e0150323. [Google Scholar] [CrossRef] [PubMed]
- Rangrez, A.Y.; Pott, J.; Kluge, A.; Frauen, R.; Stiebeling, K.; Hoppe, P.; Sossalla, S.; Frey, N.; Frank, D. Myeloid leukemia factor-1 is a novel modulator of neonatal rat cardiomyocyte proliferation. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2017, 1864, 634–644. [Google Scholar] [CrossRef] [PubMed]
- Japkowicz, N.; Shah, M. Evaluating Learning Algorithms: A Classification Perspective; Cambridge University Press: New York, NY, USA, 2011. [Google Scholar]
- Müssel, C.; Lausser, L.; Maucher, M.; Kestler, H.A. Multi-objective Parameter Selection for Classifiers. J. Stat. Softw. 2012, 46, 1–27. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Ashburner, M.; Ball, C.; Blake, J.; Botstein, D.; Butler, H.; Cherry, J.; Davis, A.; Dolinski, K.; Dwight, S.; Eppig, J.; et al. Gene Ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Fix, E.; Hodges, J.L. Discriminatory Analysis: Nonparametric Discrimination: Consistency Properties; Technical Report Project 21-49-004, Report Number 4; USAF School of Aviation Medicine, Randolf Field: Universal City, TX, USA, 1951. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cross-validation performance ( cross-validation (CV)): | ||||
|---|---|---|---|---|
| Accuracy(Acc): | Sensitivity(Sens): | Specificity (Spec): | ||
| S-MCS (GO) | 95.2% | 95.8% | 94.6% | |
| S-MCS (KEGG) | 91.5% | 87.5% | 95.4% | |
| 1-NN (all genes) | 74.8% | 79.2% | 70.4% | |
| Most Frequent GO Terms (%): | ||||
| 1. RNA Polymerase II Transcription Factor Binding | 55% | |||
| 2. Transaminase Activity | 42% | |||
| 3. Myoblast Differentiation | 23% | |||
| 4. Cellular Biogenic Amine Metabolic Process | 19% | |||
| 5. Hormone Activity | 16% | |||
| Most Frequent KEGG Pathways (%): | ||||
| 1. Arginine biosynthesis | 75% | |||
| 2. Biosynthesis of unsaturated fatty acids | 55% | |||
| 3. Fatty acid elongation | 37% | |||
| 4. Alanine, aspartate and glutamate metabolism | 29% | |||
| 5. Cytokine-cytokine receptor interaction | 29% | |||
| Random Vocabularies (100 repetitions, cross-validation (CV)): | ||||
| Acc: | Sens: | Spec: | ||
| S-MCS (100 × 15 rand. genes) | median | 91.6% | 91.7% | 92.9% |
| IQR | [88.8–94.6%] | [87.1–94.6%] | [88.3–95.5%] | |
| S-MCS (100 × 20 rand. genes) | median | 92.6% | 91.3% | 93.5% |
| IQR | [89.1–95.4%] | [87.8–95.0%] | [91.2–96.7%] | |
| 1-NN (100 rand. genes) | median | 78.5% | 78.5% | 78.8% |
| IQR | [67.2–86.1%] | [66.6–87.5%] | [70.0–87.2%] | |
| No. | Genotype | Samples (mut/crt) |
|---|---|---|
| 1. | dead beat (m582) | (3/3) |
| 2. | island beat (m458) | (6/6) |
| 3. | lazy susan (m647) | (3/3) |
| 4. | lost contact (hu801) | (3/3) |
| 5. | main squeeze (m347) | (3/3) |
| 6. | tell-tale heart (m225) | (3/3) |
| 7. | weak atrium (m229) | (3/3) |
| summary | (24/24) |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lausser, L.; Siegle, L.; Rottbauer, W.; Frank, D.; Just, S.; Kestler, H.A. Semantic Multi-Classifier Systems Identify Predictive Processes in Heart Failure Models across Species. Biomolecules 2018, 8, 158. https://doi.org/10.3390/biom8040158
Lausser L, Siegle L, Rottbauer W, Frank D, Just S, Kestler HA. Semantic Multi-Classifier Systems Identify Predictive Processes in Heart Failure Models across Species. Biomolecules. 2018; 8(4):158. https://doi.org/10.3390/biom8040158
Chicago/Turabian StyleLausser, Ludwig, Lea Siegle, Wolfgang Rottbauer, Derk Frank, Steffen Just, and Hans A. Kestler. 2018. "Semantic Multi-Classifier Systems Identify Predictive Processes in Heart Failure Models across Species" Biomolecules 8, no. 4: 158. https://doi.org/10.3390/biom8040158
APA StyleLausser, L., Siegle, L., Rottbauer, W., Frank, D., Just, S., & Kestler, H. A. (2018). Semantic Multi-Classifier Systems Identify Predictive Processes in Heart Failure Models across Species. Biomolecules, 8(4), 158. https://doi.org/10.3390/biom8040158