In Silico Studies on Compounds Derived from Calceolaria: Phenylethanoid Glycosides as Potential Multitarget Inhibitors for the Development of Pesticides



Abstract
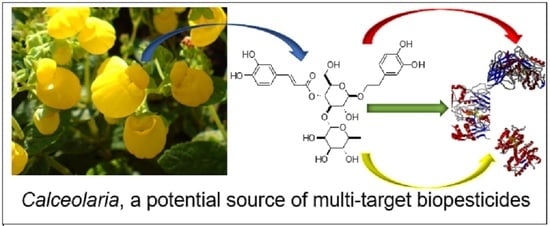
1. Introduction
2. Materials and Methods
2.1. Ligand Construction
2.2. Molecular Docking Studies
2.3. Molecular Dynamics Simulations
2.4. Construction of the Virtual Multitarget Index and the Weighed Multi-Target Index
3. Results
3.1. Docking Studies Results
3.2. Molecular Dynamics Studies on Complexes of Verbascoside with DmAChE and hAChE
3.3. Construction of the Virtual Multitarget Index and the Weighed Multitarget Index
4. Discussion
4.1. Docking Studies on Ecdysone Receptor
4.2. Docking Studies on Prophenoloxidase
4.3. Docking Studies and Molecular Dynamics Simulations on Drosophila and Human Acetylcholinesterase
4.4. Virtual Multitarget Index and Weighed Multitarget Index
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Panagiotakopulu, E.; Buckland, P.C.; Day, P.M. Natural Insecticides and Insect Repellents in Antiquity: A Review of the Evidence. J. Archeol. Sci. 1995, 22, 705–710. [Google Scholar] [CrossRef]
- Sporleder, M.; Lacey, L.A. Biopesticides. In Insect Pests of Potato; Elsevier: Amsterdam, The Netherlands, 2013; pp. 463–497. ISBN 9780123868954. [Google Scholar]
- Isman, M.B. Bridging the gap: Moving botanical insecticides from the laboratory to the farm. Ind. Crops Prod. 2017, 110, 10–14. [Google Scholar] [CrossRef]
- War, A.R.; Paulraj, M.G.; Ahmad, T.; Buhroo, A.A.; Hussain, B.; Ignacimuthu, S.; Sharma, H.C. Mechanisms of Plant Defense against Insect Herbivores. Plant Signal. Behav. 2012, 7, 1306–1320. [Google Scholar] [CrossRef] [PubMed]
- Miresmailli, S.; Isman, M.B. Botanical insecticides inspired by plant-herbivore chemical interactions. Trends Plant Sci. 2014, 19, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Isman, M.B.; Grieneisen, M.L. Botanical insecticide research: Many publications, limited useful data. Trends Plant Sci. 2014, 19, 140–145. [Google Scholar] [CrossRef] [PubMed]
- Schrader, K.K.; Andolfi, A.; Cantrell, C.L.; Cimmino, A.; Duke, S.O.; Osbrink, W.; Wedge, D.E.; Evidente, A. A survey of phytotoxic microbial and plant metabolites as potential natural products for pest management. Chem. Biodivers. 2010, 7, 2261–2280. [Google Scholar] [CrossRef] [PubMed]
- Cespedes, C.L.; Aqueveque, P.M.; Avila, J.G.; Alarcon, J.; Kubo, I. New advances in chemical defenses of plants: Researches in calceolariaceae. Phytochem. Rev. 2015, 14, 367–380. [Google Scholar] [CrossRef]
- Muñoz, E.; Escalona, D.; Salazar, J.R.; Alarcon, J.; Céspedes, C.L. Insect growth regulatory effects by diterpenes from Calceolaria talcana Grau & Ehrhart (Calceolariaceae: Scrophulariaceae) against Spodoptera frugiperda and Drosophila melanogaster. Ind. Crops Prod. 2013, 45, 283–292. [Google Scholar] [CrossRef]
- Muñoz, E.; Avila, J.G.; Alarcón, J.; Kubo, I.; Werner, E.; Céspedes, C.L. Tyrosinase inhibitors from Calceolaria integrifolia s.l.: Calceolaria talcana aerial parts. J. Agric. Food Chem. 2013, 61, 4336–4343. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, T.; Yang, Q.; Li, Z.; Qian, X. A Modeling Study for Structure Features of β-N-acetyl-d-hexosaminidase from Ostrinia furnacalis and its Novel Inhibitor Allosamidin: Species Selectivity and Multi-Target Characteristics. Chem. Biol. Drug Des. 2012, 79, 572–582. [Google Scholar] [CrossRef] [PubMed]
- Speck-Planche, A.; Kleandrova, V.V.; Scotti, M.T. Fragment-based approach for the in silico discovery of multi-target insecticides. Chemom. Intell. Lab. Syst. 2012, 111, 39–45. [Google Scholar] [CrossRef]
- Cavasotto, C.N.; Orry, A.J.W. Ligand docking and structure-based virtual screening in drug discovery. Curr. Top. Med. Chem. 2007, 7, 1006–1014. [Google Scholar] [CrossRef] [PubMed]
- Tuccinardi, T. Docking-based virtual screening: Recent developments. Comb. Chem. High Throughput Screen. 2009, 12, 303–314. [Google Scholar] [CrossRef] [PubMed]
- Cheng, T.; Li, Q.; Zhou, Z.; Wang, Y.; Bryant, S.H. Structure-Based Virtual Screening for Drug Discovery: A Problem-Centric Review. AAPS J. 2012, 14, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Kontoyianni, M. Docking and Virtual Screening in Drug Discovery. Methods Mol. Biol. 2017, 1647, 255–266. [Google Scholar] [PubMed]
- Toledo Warshaviak, D.; Golan, G.; Borrelli, K.W.; Zhu, K.; Kalid, O. Structure-Based Virtual Screening Approach for Discovery of Covalently Bound Ligands. J. Chem. Inf. Model. 2014, 54, 1941–1950. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Gu, Q.; Zheng, X.; Ye, J.; Liu, Z.; Li, J.; Hu, X.; Hagler, A.; Xu, J. Discovery of New Selective Human Aldose Reductase Inhibitors through Virtual Screening Multiple Binding Pocket Conformations. J. Chem. Inf. Model. 2013, 53, 2409–2422. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, F.F.; Mendonca Junior, F.J.B.; Ghasemi, J.B.; Ishiki, H.M.; Scotti, M.T.; Scotti, L. Docking of Natural Products against Neurodegenerative Diseases: General Concepts. Comb. Chem. High Throughput Screen. 2018, 21, 152–160. [Google Scholar] [CrossRef] [PubMed]
- Saldivar-Gonzalez, F.; Gómez-García, A.; Sánchez-Cruz, N.; Ruiz-Rios, J.; Pilón-Jiménez, B.; Medina-Franco, J. Computational Approaches to Identify Natural Products as Inhibitors of DNA Methyltransferases. Preprints 2018. [Google Scholar] [CrossRef]
- Singh, P.; Bast, F. Multitargeted molecular docking study of plant-derived natural products on phosphoinositide-3 kinase pathway components. Med. Chem. Res. 2013, 23. [Google Scholar] [CrossRef]
- Ambure, P.; Bhat, J.; Puzyn, T.; Roy, K. Identifying natural compounds as multi-target-directed ligands against Alzheimer’s disease: An in silico approach. J. Biomol. Struct. Dyn. 2018, 23, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-H.; Ha, K.B.; Park, D.H.; Fang, Y.; Kim, J.H.; Park, M.G.; Woo, R.M.; Kim, W.J.; Park, I.-K.; Choi, J.Y.; et al. Plant-derived compounds regulate formation of the insect juvenile hormone receptor complex. Pestic. Biochem. Physiol. 2018, 150, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Jankowska, M.; Rogalska, J.; Wyszkowska, J.; Stankiewicz, M.; Jankowska, M.; Rogalska, J.; Wyszkowska, J.; Stankiewicz, M. Molecular Targets for Components of Essential Oils in the Insect Nervous System—A Review. Molecules 2017, 23, 34. [Google Scholar] [CrossRef] [PubMed]
- Cespedes, C.L.; Muñoz, E.; Salazar, J.R.; Yamaguchi, L.; Werner, E.; Alarcon, J.; Kubo, I. Inhibition of cholinesterase activity by extracts, fractions and compounds from Calceolaria talcana and C. integrifolia (Calceolariaceae: Scrophulariaceae). Food Chem. Toxicol. 2013, 62, 919–926. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, E.; Lamilla, C.; Marin, J.C.; Alarcon, J.; Cespedes, C.L. Antifeedant, insect growth regulatory and insecticidal effects of Calceolaria talcana (Calceolariaceae) on Drosophila melanogaster and Spodoptera frugiperda. Ind. Crops Prod. 2013, 42, 137–144. [Google Scholar] [CrossRef]
- Céspedes, C.L.; Salazar, J.R.; Alarcon, J. Chemistry and biological activities of Calceolaria spp. (Calceolariaceae: Scrophulariaceae). Phytochem. Rev. 2013, 12, 733–749. [Google Scholar] [CrossRef]
- Thomsen, R.; Christensen, M.H. MolDock: A New Technique for High-Accuracy Molecular Docking. J. Med. Chem. 2006, 49, 3315–3321. [Google Scholar] [CrossRef] [PubMed]
- Harel, M.; Kryger, G.; Rosenberry, T.; Mallender, W.; Lewis, T.; Fletcher, R.; Guss, J.; Silman, I.; Sussman, J.L. Three-Dimensional Structures of Drosophila melanogaster Acetylcholinesterase and of its Complexes with Two Potent Inhibitors. Protein Sci. 2000, 9, 1063–1072. [Google Scholar] [CrossRef] [PubMed]
- Moras, D.; Billas, I.M.; Browning, C. Adaptability of the ecdysone receptor bound to synthetic ligands. [CrossRef]
- Browning, C.; Martin, E.; Loch, C.; Wurtz, J.-M.; Moras, D.; Stote, R.H.; Dejaegere, A.P.; Billas, I.M.L. Critical role of desolvation in the binding of 20-hydroxyecdysone to the ecdysone receptor. J. Biol. Chem. 2007, 282, 32924–32934. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, Y.; Jiang, H.; Deng, J. Crystal structure of Manduca sexta prophenoloxidase provides insights into the mechanism of type 3 copper enzymes. Proc. Natl. Acad. Sci. USA 2009, 106, 17002–17006. [Google Scholar] [CrossRef] [PubMed]
- Cheung, J.; Rudolph, M.J.; Burshteyn, F.; Cassidy, M.S.; Gary, E.N.; Love, J.; Franklin, M.C.; Height, J.J. Structures of human acetylcholinesterase in complex with pharmacologically important ligands. J. Med. Chem. 2012, 55, 10282–10286. [Google Scholar] [CrossRef] [PubMed]
- Cheung, J.; Gary, E.N.; Shiomi, K.; Rosenberry, T.L. Structures of Human Acetylcholinesterase Bound to Dihydrotanshinone I and Territrem B Show Peripheral Site Flexibility. ACS Med. Chem. Lett. 2013, 4, 1091–1096. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Ogungbe, I.V.; Erwin, W.R.; Setzer, W.N. Antileishmanial phytochemical phenolics: Molecular docking to potential protein targets. J. Mol. Graph. Model. 2014, 48, 105–117. [Google Scholar] [CrossRef] [PubMed]
- Loza-Mejía, M.A.; Salazar, J.R. Sterols and triterpenoids as potential anti-inflammatories: Molecular docking studies for binding to some enzymes involved in inflammatory pathways. J. Mol. Graph. Model. 2015, 62, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Krieger, E.; Vriend, G. YASARA View—Molecular graphics for all devices—From smartphones to workstations. Bioinformatics 2014, 30, 2981–2982. [Google Scholar] [CrossRef] [PubMed]
- Yasara Dynamics. Available online: www.yasara.org (accessed on 23 October 2018).
- Duan, Y.; Wu, C.; Chowdhury, S.; Lee, M.C.; Xiong, G.; Zhang, W.; Yang, R.; Cieplak, P.; Luo, R.; Lee, T.; et al. A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J. Comput. Chem. 2003, 24, 1999–2012. [Google Scholar] [CrossRef] [PubMed]
- Gan, R.; Zhao, L.; Sun, Q.; Tang, P.; Zhang, S.; Yang, H.; He, J.; Li, H. Binding behavior of trelagliptin and human serum albumin: Molecular docking, dynamical simulation, and multi-spectroscopy. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2018, 202, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.; Suo, Z.; Sun, Q.; Gan, R.; Tang, P.; Hou, Q.; Wu, D.; Li, H. Study of the interaction of broad-spectrum antimicrobial drug sitafloxacin with human serum albumin using spectroscopic methods, molecular docking, and molecular dynamics simulation. J. Pharm. Biomed. Anal. 2018, 160, 397–403. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Srivastava, G.; Negi, A.S.; Sharma, A. Docking, molecular dynamics, binding energy-MM-PBSA studies of naphthofuran derivatives to identify potential dual inhibitors against BACE-1 and GSK-3β. J. Biomol. Struct. Dyn. 2018, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Schwedes, C.; Tulsiani, S.; Carney, G.E. Ecdysone receptor expression and activity in adult Drosophila melanogaster. J. Insect Physiol. 2011, 57, 899–907. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Lazaridis, T. The Effect of Water Displacement on Binding Thermodynamics: Concanavalin A. J. Phys. Chem. B 2005, 109, 662–670. [Google Scholar] [CrossRef] [PubMed]
- Whitehill, J.; Rigsby, C.; Cipollini, D.; Herms, D.A.; Bonello, P. Decreased emergence of emerald ash borer from ash treated with methyl jasmonate is associated with induction of general defense traits and the toxic phenolic compound verbascoside. Oecologia 2014, 176, 1047–1059. [Google Scholar] [CrossRef] [PubMed]
- Harmatha, J.; Dinan, L. Biological activities of lignans and stilbenoids associated with plant-insect chemical interactions. Phytochem. Rev. 2003, 2, 321–330. [Google Scholar] [CrossRef]
- Dinan, L.; Hormann, R.E. Comprehensive Molecular Insect Science; Elsevier: Amsterdam, The Netherlands, 2005; ISBN 9780444519245. [Google Scholar]
- Jiang, H.; Wang, Y.; Kanost, M.R. Pro-phenol oxidase activating proteinase from an insect, Manduca sexta: A bacteria-inducible protein similar to Drosophila easter. Proc. Natl. Acad. Sci. USA 1998, 95, 12220–12225. [Google Scholar] [CrossRef] [PubMed]
- Sugumaran, M.; Barek, H. Critical Analysis of the Melanogenic Pathway in Insects and Higher Animals. Int. J. Mol. Sci. 2016, 17, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Aloui, S.; Raboudi, F.; Ghazouani, T.; Salghi, R.; Hamdaoui, M.H.; Fattouch, S. Use of molecular and in silico bioinformatic tools to investigate pesticide binding to insect (Lepidoptera) phenoloxidases (PO): Insights to toxicological aspects. J. Environ. Sci. Health B 2014, 49, 654–660. [Google Scholar] [CrossRef] [PubMed]
- Kanteev, M.; Goldfeder, M.; Fishman, A. Structure–function correlations in tyrosinases. Protein Sci. 2015, 24, 1360–1369. [Google Scholar] [CrossRef] [PubMed]
- Karioti, A.; Protopappa, A.; Megoulas, N.; Skaltsa, H. Identification of tyrosinase inhibitors from Marrubium velutinum and Marrubium cylleneum. Bioorg. Med. Chem. 2007, 15, 2708–2714. [Google Scholar] [CrossRef] [PubMed]
- Yoshimori, A.; Oyama, T.; Takahashi, S.; Abe, H.; Kamiya, T.; Abe, T.; Tanuma, S.I. Structure-activity relationships of the thujaplicins for inhibition of human tyrosinase. Bioorg. Med. Chem. 2014, 22, 6193–6200. [Google Scholar] [CrossRef] [PubMed]
- Tan, X.; Song, Y.H.; Park, C.; Lee, K.W.; Kim, J.Y.; Kim, D.W.; Kim, K.D.; Lee, K.W.; Curtis-Long, M.J.; Park, K.H. Highly potent tyrosinase inhibitor, neorauflavane from Campylotropis hirtella and inhibitory mechanism with molecular docking. Bioorg. Med. Chem. 2016, 24, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Houghton, P.J.; Ren, Y.; Howes, M.-J. Acetylcholinesterase inhibitors from plants and fungi. Nat. Prod. Rep. 2006, 23, 181–199. [Google Scholar] [CrossRef] [PubMed]
- Thapa, S.L.; Xu, H. Acetylcholinesterase: A Primary Target for Drugs and Insecticides. Mini Rev. Med. Chem. 2017, 17, 1665–1676. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, H.; Suzuki, T.; Akahori, F.; Satoh, T. Acetylcholinesterase and Acetylcholine Receptors: Brain Regional Heterogeneity. Anticholinesterase Pestic. Metab. Neurotox. Epidemiol. 2011, 3–18. [Google Scholar] [CrossRef]
- Pang, Y.; Brimijoin, S.; Ragsdale, D.W.; Zhu, K.Y.; Suranyi, R.; Gormley, M.; Company, K. Novel and Viable Acetylcholinesterase Target Site for Developing Effective and Environmentally Safe Insecticides. Curr. Drug Targets 2012, 13, 471–482. [Google Scholar] [CrossRef] [PubMed]
- Wiesner, J.; Kříž, Z.; Kuča, K.; Jun, D.; Koča, J. Acetylcholinesterases—The structural similarities and differences. J. Enzyme Inhib. Med. Chem. 2007, 22, 417–424. [Google Scholar] [CrossRef] [PubMed]
- Katselou, M.G.; Matralis, A.N.; Kourounakis, A.P. Multi-target drug design approaches for multifactorial diseases: From neurodegenerative to cardiovascular applications. Curr. Med. Chem. 2014, 21, 2743–2787. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.J.; Pan, W.; Hu, Y.J.; Wang, Y.T. Multi-target drugs: The trend of drug research and development. PLoS ONE 2012, 7, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Lavecchia, A.; Cerchia, C. In silico methods to address polypharmacology: Current status, applications and future perspectives. Drug Discov. Today 2016, 21, 288–298. [Google Scholar] [CrossRef] [PubMed]
- Prado-Prado, F.; García-Mera, X.; Abeijón, P.; Alonso, N.; Caamaño, O.; Yáñez, M.; Gárate, T.; Mezo, M.; González-Warleta, M.; Muiño, L.; et al. Using entropy of drug and protein graphs to predict FDA drug-target network: Theoretic-experimental study of MAO inhibitors and hemoglobin peptides from Fasciola hepatica. Eur. J. Med. Chem. 2011, 46, 1074–1094. [Google Scholar] [CrossRef] [PubMed]
- Prado-Prado, F.J.; García, I.; García-Mera, X.; González-Díaz, H. Entropy multi-target QSAR model for prediction of antiviral drug complex networks. Chemom. Intell. Lab. Syst. 2011, 107, 227–233. [Google Scholar] [CrossRef]
- Speck-Planche, A.; Kleandrova, V.; Scotti, M.; Cordeiro, M. 3D-QSAR Methodologies and Molecular Modeling in Bioinformatics for the Search of Novel Anti-HIV Therapies: Rational Design of Entry Inhibitors. Curr. Bioinform. 2013, 8, 452–464. [Google Scholar] [CrossRef]
- Speck-Planche, A.; Kleandrova, V.; Luan, F.; Natalia, D.S.; Cordeiro, M. Multi-Target Inhibitors for Proteins Associated with Alzheimer: In Silico Discovery using Fragment-Based Descriptors. Curr. Alzheimer Res. 2013, 10, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Lackey, K.E. The Discovery of Lapatinib. In Designing Multi-Target Drugs; Morphy, J.R., Harris, J.C., Eds.; Royal Society of Chemistry: Cambridge, UK, 2012; pp. 181–205. [Google Scholar]
- Alipieva, K.; Korkina, L.; Orhan, I.E.; Georgiev, M.I. Verbascoside—A review of its occurrence, (bio)synthesis and pharmacological significance. Biotechnol. Adv. 2014, 32, 1065–1076. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ligand | Skeleton Type | Compound Name | PDB: 3IXP | PDB: 2R40 | Average MolDock Scores |
|---|---|---|---|---|---|
| 88 | PEG | Calceolarioside C | −214.0 | −207.2 | −210.6 |
| 90 | PEG | Forsythoside A | −202.8 | −213.5 | −208.2 |
| 89 | PEG | Calceolarioside E | −184.7 | −212.5 | −198.6 |
| 92 | PEG | Isoarenarioside | −183.0 | −205.5 | −194.2 |
| 87 | PEG | Verbascoside | −197.4 | −184.1 | −190.8 |
| 86 | PEG | Calceolarioside A | −174.2 | −183.6 | −178.9 |
| 91 | PEG | Calceolarioside B | −162.1 | −176.7 | −169.4 |
| 93 | PEG | Calceolarioside D | −160.2 | −169.3 | −164.7 |
| 68 | Scopadulane | 3-Isovaleroyl-7-malonyloxy-thyrsiflorane | −147.2 | −157.1 | −152.2 |
| 45 | Isopimarane | 3-β-Isovaleroyl-18-hydroxy-7-α-malonyloxyent-isopimara-9(11), 15-diene | −150.2 | −151.5 | −150.8 |
| Ligand | Skeleton | Compound Name | MolDock Score |
|---|---|---|---|
| 86 | PEG | Calceolarioside A | −161.187 |
| 110 | Flavonoid | Kaempferol-7-methyl ether | −142.825 |
| 93 | PEG | Calceolarioside D | −142.017 |
| 109 | Flavonoid | Gossypetin-7,8,3′-trimethyl ether | −140.618 |
| 108 | Flavonoid | Herbacetin-8,4′-dimethyl ether | −138.595 |
| 88 | PEG | Calceolarioside C | −137.969 |
| 111 | Flavonoid | Kaempferol-4′-methyl ether | −137.519 |
| 104 | Flavonoid | Naringenin-4′-methyl ether | −137.451 |
| 107 | Flavonoid | Isoscutellarein-8,4′-dimethyl ether | −137.188 |
| DmAChE MolDock Scores | hAChE MolDock Scores | |||||||
|---|---|---|---|---|---|---|---|---|
| Ligand | Compound Name | PDB: 1DX4 | PDB: 1QON | Average Score | PDB: 4EY7 | PDB: 4M0E | Average Score | SR 1 |
| 90 | Forsythoside A | −171.3 | −254.5 | −212.9 | −177.7 | −247.6 | −212.6 | 1.00 |
| 88 | Calceolarioside C | −174.0 | −251.6 | −212.8 | −145.7 | −217.7 | −181.7 | 1.17 |
| 87 | Verbascoside | −178.8 | −233.5 | −206.1 | −152.6 | −200.8 | −176.7 | 1.17 |
| 89 | Calceolarioside E | −169.0 | −239.1 | −204.0 | −116.7 | −209.0 | −162.8 | 1.25 |
| 93 | Calceolarioside D | −162.3 | −227.7 | −195.0 | −147.9 | −188.4 | −168.2 | 1.16 |
| 92 | Isoarenarioside | −141.1 | −244.8 | −193.0 | −165.1 | −208.0 | −186.6 | 1.03 |
| 86 | Calceolarioside A | −156.2 | −212.8 | −184.5 | −164.6 | −189.6 | −177.1 | 1.04 |
| 91 | Calceolarioside B | −128.0 | −210.5 | −169.2 | −137.9 | −183.9 | −160.9 | 1.05 |
| 44 | Isopimarane | −119.9 | −180.3 | −150.1 | −137.0 | −159.7 | −148.4 | 1.01 |
| 43 | Isopimarane | −127.3 | −164.5 | −145.9 | −150.2 | −154.6 | −152.4 | 0.96 |
| Ligand | Skeleton | Compound Name | vMTi | wMTi |
|---|---|---|---|---|
| 88 | PEG | Calceolarioside C | 2.86 | 0.60 |
| 89 | PEG | Calceolarioside E | 2.67 | 0.57 |
| 86 | PEG | Calceolarioside A | 2.72 | 0.56 |
| 87 | PEG | Verbascoside | 2.67 | 0.55 |
| 93 | PEG | Calceolarioside D | 2.58 | 0.54 |
| 90 | PEG | Forsythoside A | 2.77 | 0.53 |
| 92 | PEG | Isoarenarioside | 2.64 | 0.53 |
| 91 | PEG | Calceolarioside B | 2.39 | 0.49 |
| 109 | Flavonoid | Gossypetin-7,8,3′-trimethyl ether | 2.05 | 0.43 |
| 110 | Flavonoid | Kaempferol-7-methyl ether | 2.03 | 0.43 |
| 77 | Labdane | 19-Malonyloxy-9-epi-ent-labda- 8(17), 12 Z, 14-triene | 2.04 | 0.42 |
| 45 | Isopimarane | 3-β-Isovaleroyl-18-hydroxy-7-α-malonyloxyent-isopimara-9(11), 15-diene | 2.13 | 0.42 |
| 3 | Abietane | 19-Malonyloxy-dehydroabietinol | 1.99 | 0.42 |
| 57 | Stemarane | 17-Acetoxy-19-malonyloxy-ent-stemar-13(14)-ene | 2.03 | 0.41 |
| 108 | Flavonoid | Herbacetin-8,4′-dimethyl ether | 1.92 | 0.40 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Loza-Mejía, M.A.; Salazar, J.R.; Sánchez-Tejeda, J.F. In Silico Studies on Compounds Derived from Calceolaria: Phenylethanoid Glycosides as Potential Multitarget Inhibitors for the Development of Pesticides. Biomolecules 2018, 8, 121. https://doi.org/10.3390/biom8040121
Loza-Mejía MA, Salazar JR, Sánchez-Tejeda JF. In Silico Studies on Compounds Derived from Calceolaria: Phenylethanoid Glycosides as Potential Multitarget Inhibitors for the Development of Pesticides. Biomolecules. 2018; 8(4):121. https://doi.org/10.3390/biom8040121
Chicago/Turabian StyleLoza-Mejía, Marco A., Juan Rodrigo Salazar, and Juan Francisco Sánchez-Tejeda. 2018. "In Silico Studies on Compounds Derived from Calceolaria: Phenylethanoid Glycosides as Potential Multitarget Inhibitors for the Development of Pesticides" Biomolecules 8, no. 4: 121. https://doi.org/10.3390/biom8040121
APA StyleLoza-Mejía, M. A., Salazar, J. R., & Sánchez-Tejeda, J. F. (2018). In Silico Studies on Compounds Derived from Calceolaria: Phenylethanoid Glycosides as Potential Multitarget Inhibitors for the Development of Pesticides. Biomolecules, 8(4), 121. https://doi.org/10.3390/biom8040121