A Marine Diterpenoid Modulates the Proteasome Activity in Murine Macrophages Stimulated with LPS

 , , and
, , and
Abstract
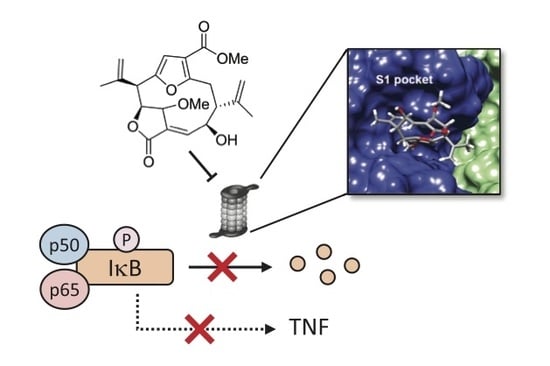
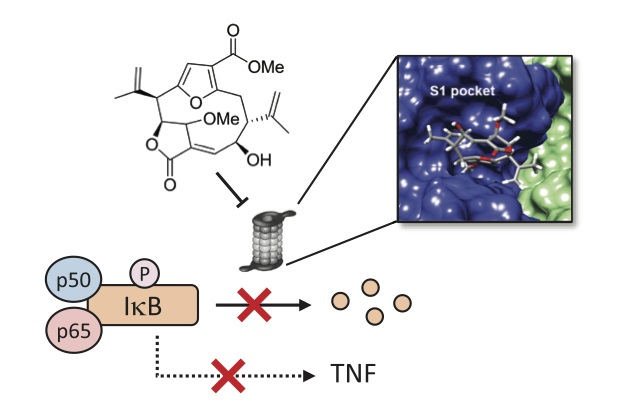{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Mice
2.2. Acute Pulmonary Inflammation
2.3. Cell Culture and Proteasome Activity Assay
2.4. Western Blot Analysis
2.5. Major Histocompatibility Complex Class I Flow Cytometry Analysis
2.6. Quantitative Real Time Polymerase Chain Reaction
2.7. Purified Proteasome Activity Assay
2.8. Cytokine Measurements
2.9. Molecular Docking Simulation
2.10. Statistical Analysis
3. Results
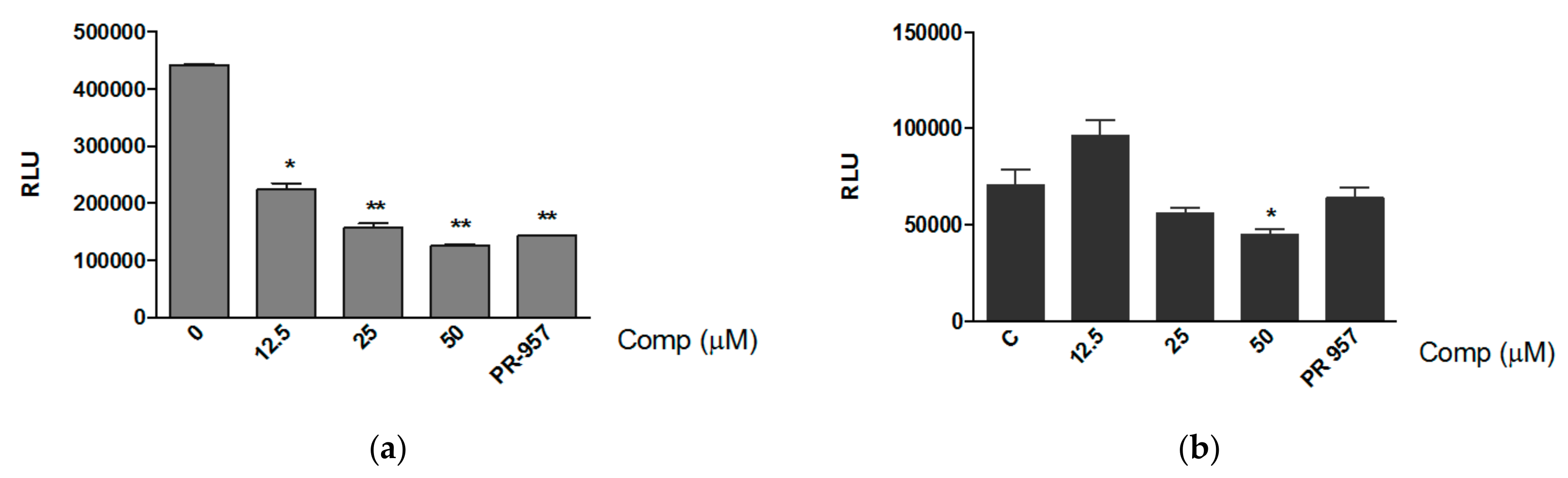
3.1. Compound 1 Inhibits the Activity of the Proteasome with the Consequent Accumulation of Ubiquitinated Proteins
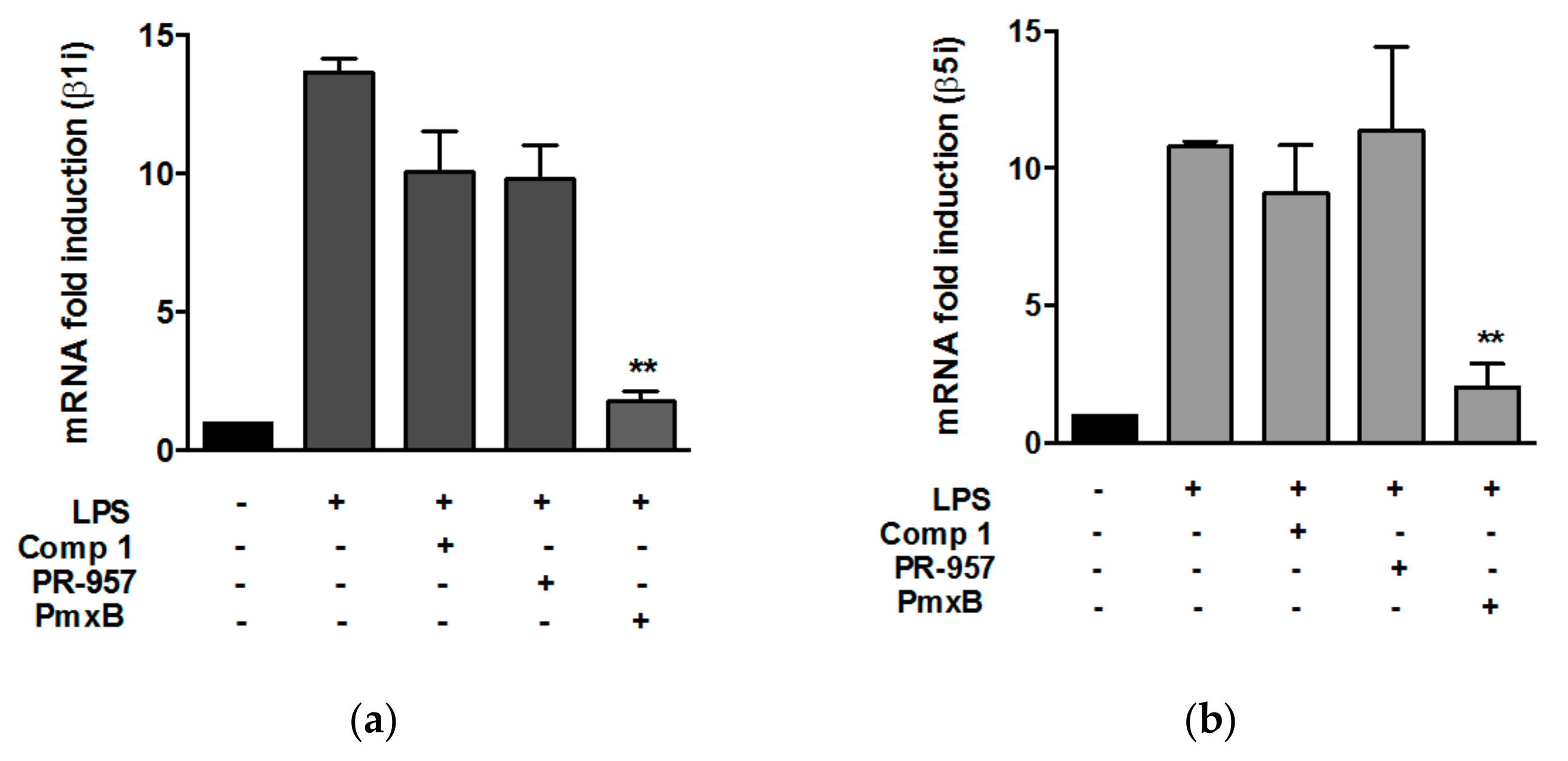
3.2. Compound 1 did not Affect the Expression of β1i and β5i Subunits in Peritoneal Macrophages Stimulated with Lypopolisaccharide
3.3. Compound 1 Inhibits the Chemotrypsin-like Activity of Purified Proteasomes
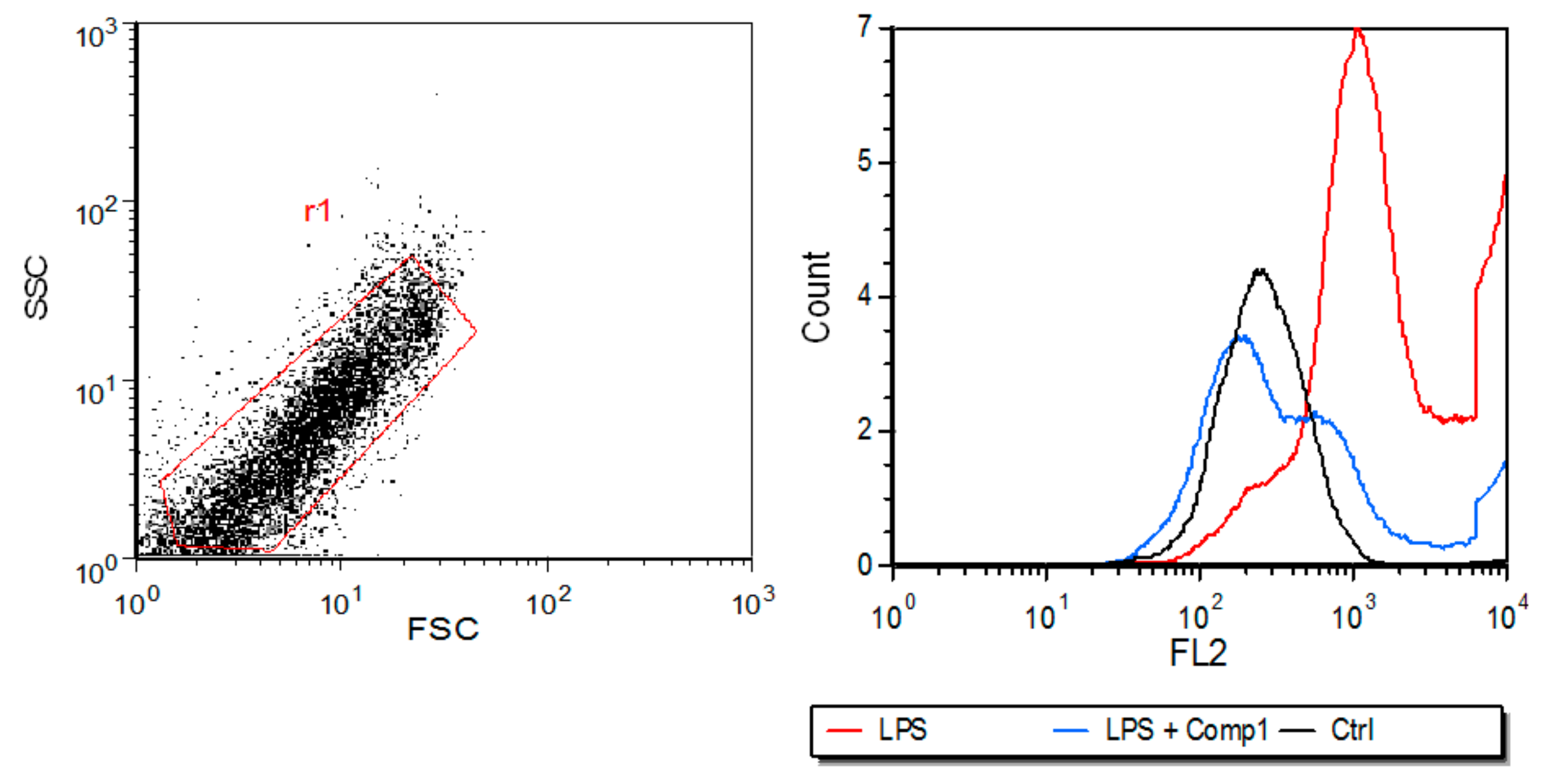
3.4. Compound 1 Inhibits the Expression of MHC-I In Vitro and the Production of Pro-Inflammatory Mediators In Vivo
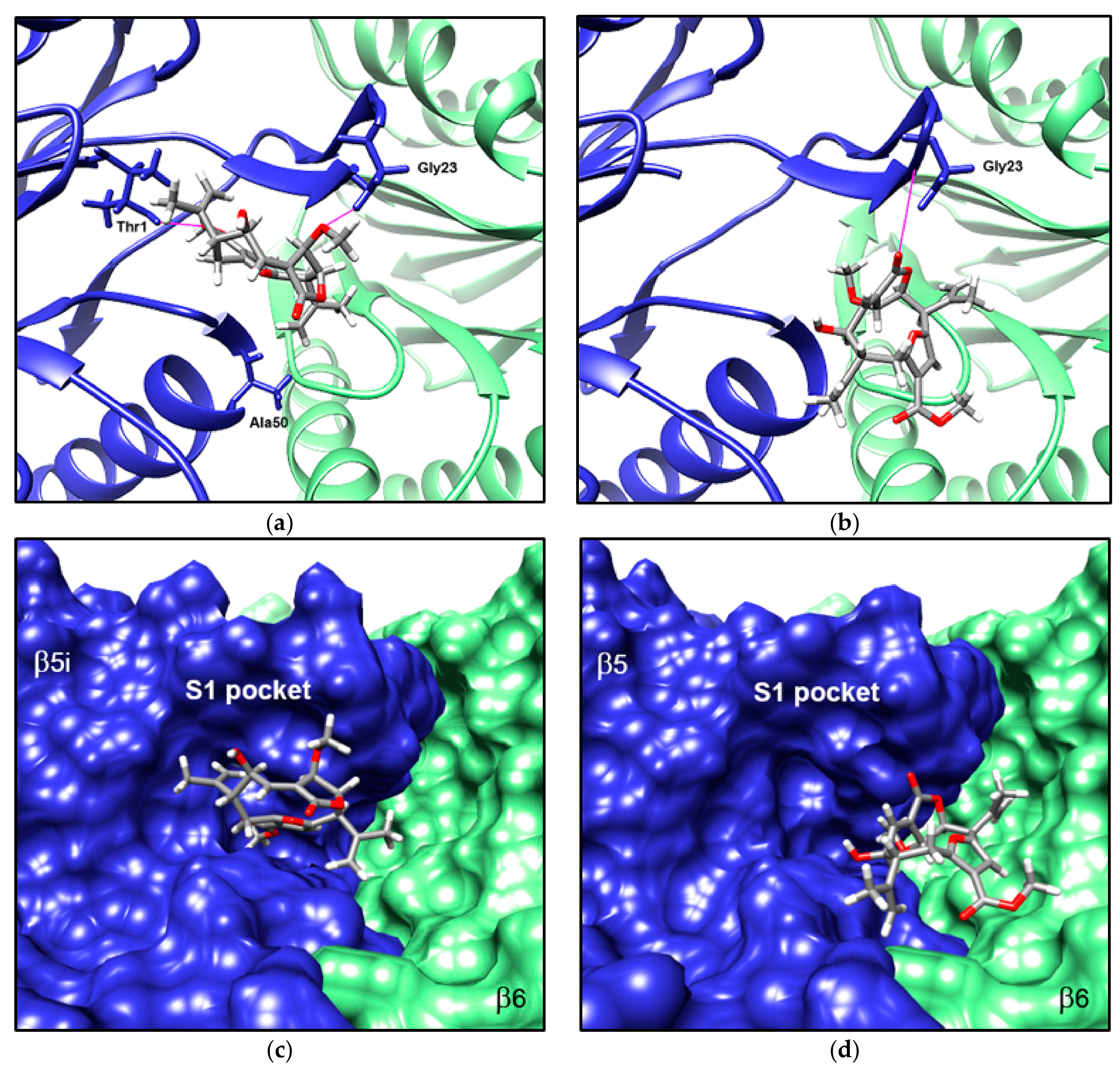
3.5. Compound 1 Appears to Bind to the Catalytic Pocket of β5i Subunit of the Immunoproteasome
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Brown, M.G.; Driscoll, J.; Monaco, J.J. Structural and serological similarity of MHC-linked LMP and proteasome (multicatalytic proteinase) complexes. Nature 1991, 353, 355–357. [Google Scholar] [CrossRef] [PubMed]
- Fehling, H.J.; Swat, W.; Laplace, C.; Kühn, R.; Rajewsky, K.; Müller, U.; von Boehmer, H. MHC class I expression in mice lacking the proteasome subunit LMP-7. Science 1994, 265, 1234–1237. [Google Scholar] [CrossRef] [PubMed]
- Griffin, T.A.; Nandi, D.; Cruz, M.; Fehling, H.J.; Kaer, L. V; Monaco, J.J.; Colbert, R.A. Immunoproteasome assembly: cooperative incorporation of interferon gamma (IFN-γ)-inducible subunits. J. Exp. Med. 1998, 187, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Qureshi, N.; Perera, P.; Shen, J.; Zhang, G.; Lenschat, A.; Splitter, G.; Morrison, D.C.; Vogel, S.N. The proteasome as a lipopolysaccharide-binding protein in macrophages: differential effects of proteasome inhibition on lipopolysaccharide-induced signaling events. J. Immunol. 2003, 171, 1515–1525. [Google Scholar] [CrossRef] [PubMed]
- Reis, J.; Guan, X.Q.; Kisselev, A.F.; Papasian, C.J.; Qureshi, A.A.; Morrison, D.C.; van Way, C.W.; Vogel, S.N.; Qureshi, N. LPS-Induced Formation of Immunoproteasomes: TNF-α and Nitric Oxide Production are Regulated by Altered Composition of Proteasome-Active Sites. Cell. Biochem. Biophys. 2011, 60, 77–88. [Google Scholar] [CrossRef] [PubMed]
- Reidlinger, J.; Pike, A.M.; Savory, P.J.; Murray, R.Z.; Rivett, A.J. Catalytic properties of 26 S and 20 S proteasomes and radiolabeling of MB1, LMP7, and C7 subunits associated with trypsin-like and chymotrypsin- like activities. J. Biol. Chem. 1997, 272, 24899–24905. [Google Scholar] [CrossRef] [PubMed]
- Ho, Y.K.; Bargagna-Mohan, P.; Wehenkel, M.; Mohan, R.; Kim, K.B. LMP2-Specific Inhibitors: Chemical Genetic Tools for Proteasome Biology. Chem. Biol. 2007, 14, 419–430. [Google Scholar] [CrossRef] [PubMed]
- Palombella, V.J.; Rando, O.J.; Goldberg, A.L.; Maniatis, T. The ubiquitin-proteasome pathway is required for processing the NF-κB1 precursor protein and the activation of NF-κB. Cell. 1994, 78, 773–785. [Google Scholar] [CrossRef]
- Karin, M.; Ben-Neriah, Y. Phosphorylation Meets Ubiquitination: The Control of NF-κB Activity. Annu. Rev. Immunol. 2000, 18, 621–663. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T.; Faustman, D. NOD mice are defective in proteasome production and activation of NF-κB. Mol. Cell. Biol. 1999, 19, 8646–8659. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T.; Faustman, D. Essential role of human leukocyte antigen-encoded proteasome subunits in NF-κB activation and prevention of tumor necrosis factor-α-induced apoptosis. J. Biol. Chem. 2000, 275, 5238. [Google Scholar] [CrossRef] [PubMed]
- Hensley, S.E.; Zanker, D.; Dolan, B.P.; David, A.; Hickman, H.D.; Embry, A.C.; Skon, C.N.; Grebe, K.M.; Griffin, T. a; Chen, W.; Bennink, J.R.; Yewdell, J.W. Unexpected role for the immunoproteasome subunit LMP2 in antiviral humoral and innate immune responses. J. Immunol. 2010, 184, 4115–4122. [Google Scholar] [CrossRef] [PubMed]
- Maldonado, M.; Kapphahn, R.J.; Terluk, M.R.; Heuss, N.D.; Yuan, C.; Gregerson, D.S.; Ferrington, D.A. Immunoproteasome Deficiency Modifies the Alternative Pathway of NFκB Signaling. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [PubMed]
- Kessler, B.M.; Lennon-Duménil, A.M.; Shinohara, M.L.; Lipes, M.A.; Ploegh, H.L. LMP2 expression and proteasome activity in NOD mice. Nat. Med. 2000, 6, 1064. [Google Scholar] [CrossRef] [PubMed]
- Visekruna, A.; Joeris, T.; Seidel, D.; Kroesen, A.; Loddenkemper, C.; Zeitz, M.; Kaufmann, S.H.E.; Schmidt-Ullrich, R.; Steinhoff, U. Proteasome-mediated degradation of IκBα and processing of p105 in Crohn disease and ulcerative colitis. J. Clin. Invest. 2006, 116, 3195–3203. [Google Scholar] [CrossRef] [PubMed]
- González, Y.; Doens, D.; Santamaría, R.; Ramos, M.; Restrepo, C.M.; Barros De Arruda, L.; Lleonart, R.; Gutiérrez, M.; Fernández, P.L. A pseudopterane diterpene isolated from the octocoral Pseudopterogorgia acerosa inhibits the inflammatory response mediated by TLR-ligands and TNF-α in macrophages. PLoS ONE 2013, 8, e84107. [Google Scholar] [CrossRef] [PubMed]
- Bitzer, A.; Basler, M.; Krappmann, D.; Groettrup, M. Immunoproteasome subunit deficiency has no influence on the canonical pathway of NF-κB activation. Mol. Immunol. 2017, 83, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Jang, E.R.; Lee, N.-R.; Han, S.; Wu, Y.; Sharma, L.K.; Carmony, K.C.; Marks, J.; Lee, D.-M.; Ban, J.-O.; Wehenkel, M.; et al. Revisiting the role of the immunoproteasome in the activation of the canonical NF-κB pathway. Mol. Biosyst. 2012, 8, 2295–2302. [Google Scholar] [CrossRef] [PubMed]
- Teicher, B.A.; Tomaszewski, J.E. Proteasome inhibitors. Biochem. Pharmacol. 2015, 96, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.; Kim, K.B. The immunoproteasome: an emerging therapeutic target. Curr. Top. Med. Chem. 2011, 11, 2923–2930. [Google Scholar] [CrossRef] [PubMed]
- Miller, Z.; Ao, L.; Kim, K.B.; Lee, W. Inhibitors of the immunoproteasome: current status and future directions. Curr. Pharm. Des. 2013, 19, 4140–4151. [Google Scholar] [CrossRef] [PubMed]
- Sula Karreci, E.; Fan, H.; Uehara, M.; Mihali, A.B.; Singh, P.K.; Kurdi, A.T.; Solhjou, Z.; Riella, L.V.; Ghobrial, I.; Laragione, T.; et al. Brief treatment with a highly selective immunoproteasome inhibitor promotes long-term cardiac allograft acceptance in mice. Proc. Natl. Acad. Sci. 2016. [Google Scholar] [CrossRef] [PubMed]
- Sosič, I.; Gobec, M.; Brus, B.; Knez, D.; Živec, M.; Konc, J.; Lešnik, S.; Ogrizek, M.; Obreza, A.; Žigon, D. Nonpeptidic Selective Inhibitors of the Chymotrypsin-Like (β5 i) Subunit of the Immunoproteasome. Angew. Chemie. 2016. [Google Scholar] [CrossRef]
- Nam, S.; Smith, D.M.; Dou, Q.P. Ester Bond-containing Tea Polyphenols Potently Inhibit Proteasome Activity in Vitro and in Vivo. J Biol Chem. 2001, 20. [Google Scholar] [CrossRef]
- Cheng, X.; Shi, W.; Zhao, C.; Zhang, D.; Liang, P.; Wang, G.; Lu, L. Triptolide sensitizes human breast cancer cells to tumor necrosis factor-α-induced apoptosis by inhibiting activation of the nuclear factor-κB pathway. Mol. Med. Rep. 2016. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, K.; Yokota, K.; Kagawa, S.; Shimbara, N.; Tamura, T.; Akioka, H.; Nothwang, H.G.; Noda, C.; Tanaka, K.; Ichihara, A. cDNA cloning and interferon gamma down-regulation of proteasomal subunits X and Y. Science 1994, 265, 1231–1234. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeerschd, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 2012, 4. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, W.; Kollmann, P.; Case, D. Antechamber, An Accessory Software Package For Molecular Mechanical Calculation. J. Comput. Chem. 2005, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Allen, W.J.; Balius, T.E.; Mukherjee, S.; Brozell, S.R.; Moustakas, D.T.; Lang, P.T.; Case, D.A.; Kuntz, I.D.; Rizzo, R.C. DOCK 6: Impact of new features and current docking performance. J. Comput. Chem. 2015, 36, 1132–1156. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A Visualization System for Exploratory Research and Analysis. J. Comput. Chem 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple ligand-protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 2011, 51, 2778–2786. [Google Scholar] [CrossRef] [PubMed]
- Groettrup, M.; Kirk, C.J.; Basler, M. Proteasomes in immune cells: More than peptide producers? Nat. Rev. Immunol. 2010, 10, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Basler, M.; Dajee, M.; Moll, C.; Groettrup, M.; Kirk, C.J. Prevention of experimental colitis by a selective inhibitor of the immunoproteasome. J. Immunol. 2010, 185, 634–641. [Google Scholar] [CrossRef] [PubMed]
- Lei, B.; Abdul Hameed, M.; Hamza, A.; Wehenkel, M.; Muzyka, J.L.; Yao, X.J.; Kim, K.B.; Zhan, C.G. Molecular basis of the selectivity of the immunoproteasome catalytic subunit LMP2-specific inhibitor revealed by molecular modeling and dynamics simulations. J. Phys. Chem. B 2010, 114, 12333–12339. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Huber, E.M.; Basler, M.; Schwab, R.; Heinemeyer, W.; Kirk, C.J.; Groettrup, M.; Groll, M. Immuno- and constitutive proteasome crystal structures reveal differences in substrate and inhibitor specificity. Cell. 2012, 148, 727–738. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Reis, J.; Morrison, D.C.; Papasian, C.; Raghavakaimal, S.; Kolbert, C.; Qureshi, A.A.; Vogel, S.N.; Qureshi, N. Key inflammatory signaling pathways are regulated by the proteasome. Shock 2006, 25, 472–484. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Gao, J.J.; Zhang, G.; Tan, X.; Morrison, D.C.; Papasian, C.; Vogel, S.N.; Qureshi, N. Proteasome-mediated regulation of CpG DNA- and peptidoglycan-induced cytokines, inflammatory genes, and mitogen-activated protein kinase activation. Shock 2006, 25, 594–599. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Chen, D.; Qiuzhi, C.C.; Yuan, X.; Dou, Q.P. Celastrol, a triterpene extracted from the Chinese “Thunder of God Vine,” is a potent proteasome inhibitor and suppresses human prostate cancer growth in nude mice. Cancer Res. 2006, 66, 4758–4765. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Kanwar, J.; Schmitt, S.; Cui, Q.C.; Zhang, C.; Zhao, C.; Dou, Q.P. Inhibition of tumor cellular proteasome activity by triptolide extracted from the Chinese medicinal plant “thunder god vine”. Anticancer Res. 2011, 31, 1–10. [Google Scholar] [PubMed]
- Boes, B.; Hengel, H.; Ruppert, T.; Multhaup, G.; Koszinowski, U.H.; Kloetzel, P.M. Interferon γ stimulation modulates the proteolytic activity and cleavage site preference of 20S mouse proteasomes. J. Exp. Med. 1994, 179, 901–909. [Google Scholar] [CrossRef] [PubMed]
- Guillaume, B.; Chapiro, J.; Stroobant, V.; Colau, D.; Van Holle, B.; Parvizi, G.; Bousquet-Dubouch, M.-P.; Théate, I.; Parmentier, N.; Van den Eynde, B.J. Two abundant proteasome subtypes that uniquely process some antigens presented by HLA class I molecules. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 18599–18604. [Google Scholar] [CrossRef] [PubMed]
- Kloetzel, P.M. Antigen processing by the proteasome. Nat. Rev. Mol. Cell. Biol. 2001, 2, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Groettrup, M.; van den Broek, M.; Schwarz, K.; Macagno, A.; Khan, S.; de Giuli, R.; Schmidtke, G. Structural plasticity of the proteasome and its function in antigen processing. Crit. Rev. Immunol. 2001, 21, 339–358. [Google Scholar] [CrossRef] [PubMed]
- MacAry, P.A.; Lindsay, M.; Scott, M.A.; Craig, J.I.; Luzio, J.P.; Lehner, P.J. Mobilization of MHC class I molecules from late endosomes to the cell surface following activation of CD34-derived human Langerhans cells. Proc. Natl. Acad. Sci. USA 2001, 98, 3982–3987. [Google Scholar] [CrossRef] [PubMed]
- Boehm, U.; Klamp, T.; Groot, M.; Howard, J.C. Cellular responses to interferon-γ. Annu. Rev. Immunol. 1997, 15, 749–795. [Google Scholar] [CrossRef] [PubMed]
- Muchamuel, T.; Basler, M.; Aujay, M.; Suzuki, E.; Kalim, K.W.; Lauer, C.; Sylvain, C.; Ring, E.R.; Shields, J.; Jiang, J.; et al. A selective inhibitor of the immunoproteasome subunit LMP7 blocks cytokine production and attenuates progression of experimental arthritis. Nat. Med. 2009, 15, 781–787. [Google Scholar] [CrossRef] [PubMed]
- Van Kaer, L.; Ashton-Rickardt, P.G.; Eichelberger, M.; Gaczynska, M.; Nagashima, K.; Rock, K.L.; Goldberg, A.L.; Doherty, P.C.; Tonegawa, S. Altered peptidase and viral-specific T cell response in LMP2 mutant mice. Immunity 1994, 1, 533–541. [Google Scholar] [CrossRef]
- Basler, M.; Mundt, S.; Muchamuel, T.; Moll, C.; Jiang, J.; Groettrup, M.; Kirk, C.J. Inhibition of the immunoproteasome ameliorates experimental autoimmune encephalomyelitis. EMBO Mol. Med. 2014, 6, 226–238. [Google Scholar] [CrossRef] [PubMed]
- Keller, I.E.; Vosyka, O.; Takenaka, S.; Kloß, A.; Dahlmann, B.; Willems, L.I.; Verdoes, M.; Overkleeft, H.S.; Marcos, E.; Adnot, S.; et al. Regulation of Immunoproteasome Function in the Lung. Sci. Rep. 2015, 5, 10230. [Google Scholar] [CrossRef] [PubMed]
- Löwe, J.; Stock, D.; Jap, B.; Zwickl, P.; Baumeister, W.; Huber, R. Crystal structure of the 20S proteasome from the archaeon T. acidophilum at 3.4 A resolution. Science 1995, 268, 533–539. [Google Scholar] [CrossRef] [PubMed]
- Groll, M.; Ditzel, L.; Löwe, J.; Stock, D.; Bochtler, M.; Bartunik, H.D.; Huber, R. Structure of 20S proteasome from yeast at 2.4 A resolution. Nature 1997, 386, 463–471. [Google Scholar] [CrossRef] [PubMed]
- Huber, E.M.; Heinemeyer, W.; Li, X.; Arendt, C.S.; Hochstrasser, M.; Groll, M. A unified mechanism for proteolysis and autocatalytic activation in the 20S proteasome. Nat. Commun. 2016, 7, 10900. [Google Scholar] [CrossRef] [PubMed]
- Kisselev, A.F.; Callard, A.; Goldberg, A.L. Importance of the different proteolytic sites of the proteasome and the efficacy of inhibitors varies with the protein substrate. J. Biol. Chem. 2006, 281, 8582–8590. [Google Scholar] [CrossRef] [PubMed]
- Dahlmann, B.; Ruppert, T.; Kuehn, L.; Merforth, S.; Kloetzel, P.M. Different proteasome subtypes in a single tissue exhibit different enzymatic properties. J. Mol. Biol. 2000, 303, 643–653. [Google Scholar] [CrossRef] [PubMed]
- Yewdell, J.W. Immunoproteasomes: Regulating the regulator. Proc. Natl. Acad. Sci. 2005, 102, 9089–9090. [Google Scholar] [CrossRef] [PubMed]







© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
González, Y.; Doens, D.; Cruz, H.; Santamaría, R.; Gutiérrez, M.; Llanes, A.; Fernández, P.L. A Marine Diterpenoid Modulates the Proteasome Activity in Murine Macrophages Stimulated with LPS. Biomolecules 2018, 8, 109. https://doi.org/10.3390/biom8040109
González Y, Doens D, Cruz H, Santamaría R, Gutiérrez M, Llanes A, Fernández PL. A Marine Diterpenoid Modulates the Proteasome Activity in Murine Macrophages Stimulated with LPS. Biomolecules. 2018; 8(4):109. https://doi.org/10.3390/biom8040109
Chicago/Turabian StyleGonzález, Yisett, Deborah Doens, Héctor Cruz, Ricardo Santamaría, Marcelino Gutiérrez, Alejandro Llanes, and Patricia L. Fernández. 2018. "A Marine Diterpenoid Modulates the Proteasome Activity in Murine Macrophages Stimulated with LPS" Biomolecules 8, no. 4: 109. https://doi.org/10.3390/biom8040109
APA StyleGonzález, Y., Doens, D., Cruz, H., Santamaría, R., Gutiérrez, M., Llanes, A., & Fernández, P. L. (2018). A Marine Diterpenoid Modulates the Proteasome Activity in Murine Macrophages Stimulated with LPS. Biomolecules, 8(4), 109. https://doi.org/10.3390/biom8040109