Flavonoids as Putative Epi-Modulators: Insight into Their Binding Mode with BRD4 Bromodomains Using Molecular Docking and Dynamics


Abstract

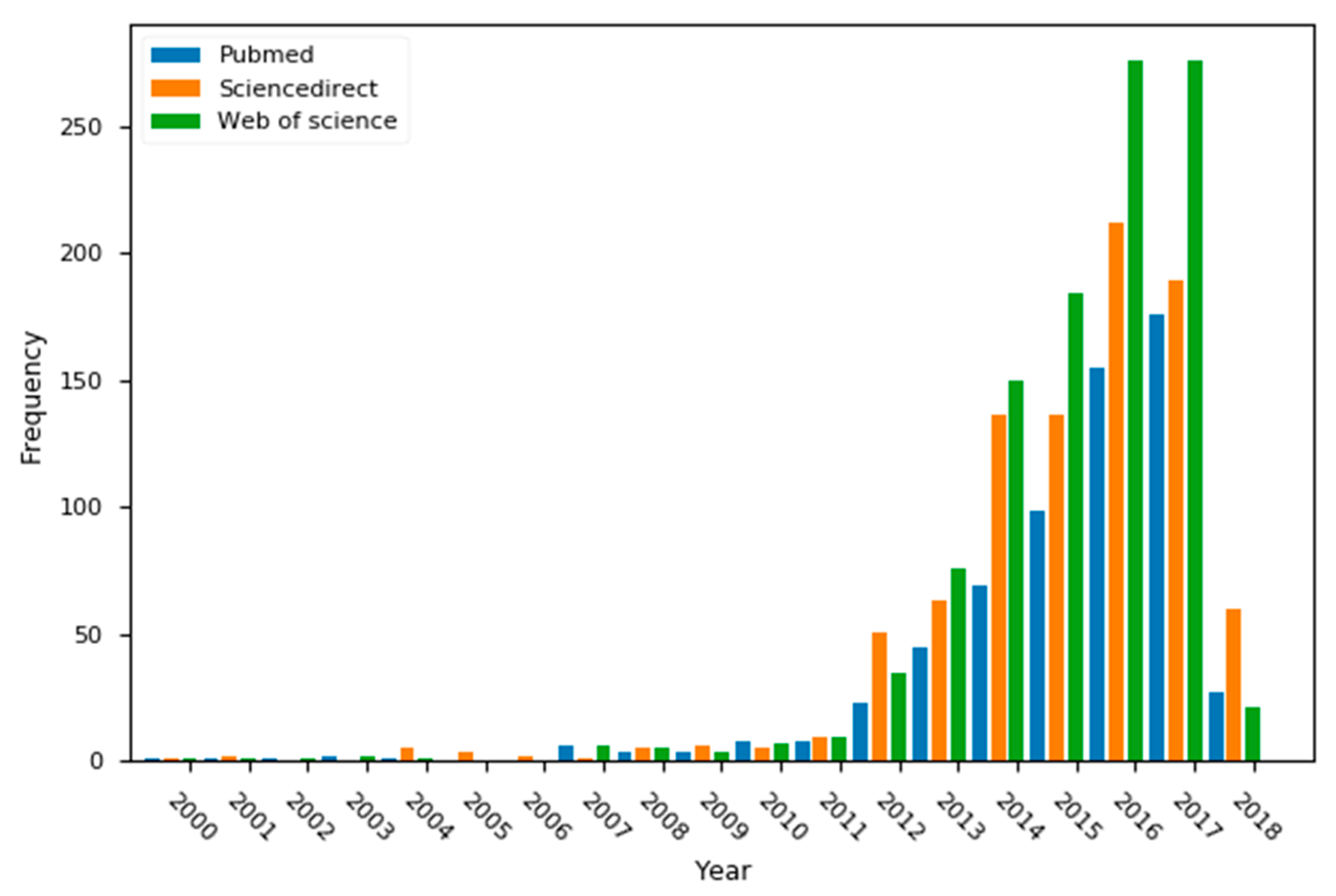
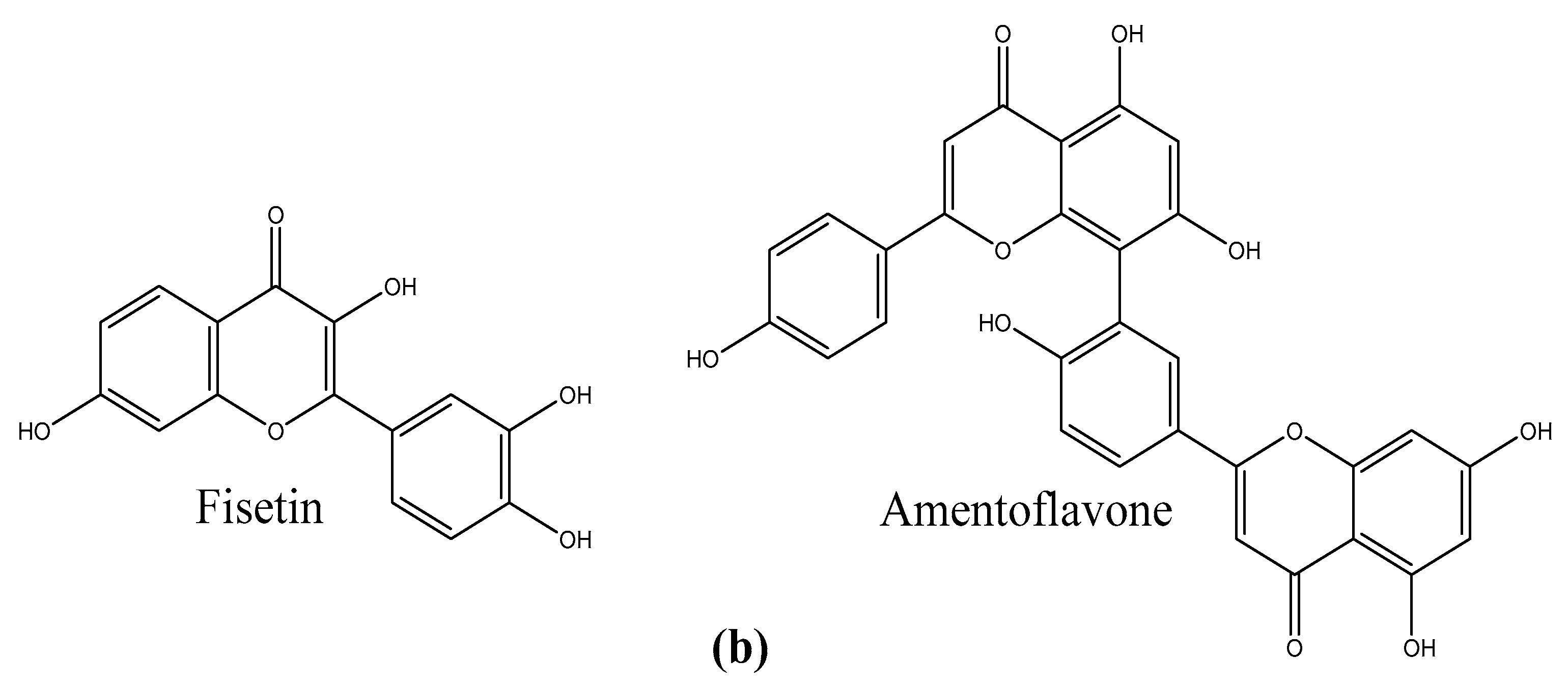
1. Introduction
2. Materials and Methods
2.1. Protein Preparation
2.2. Molecular Docking
2.3. Molecular Dynamics
2.4. Experimental Testing of Amentoflavone
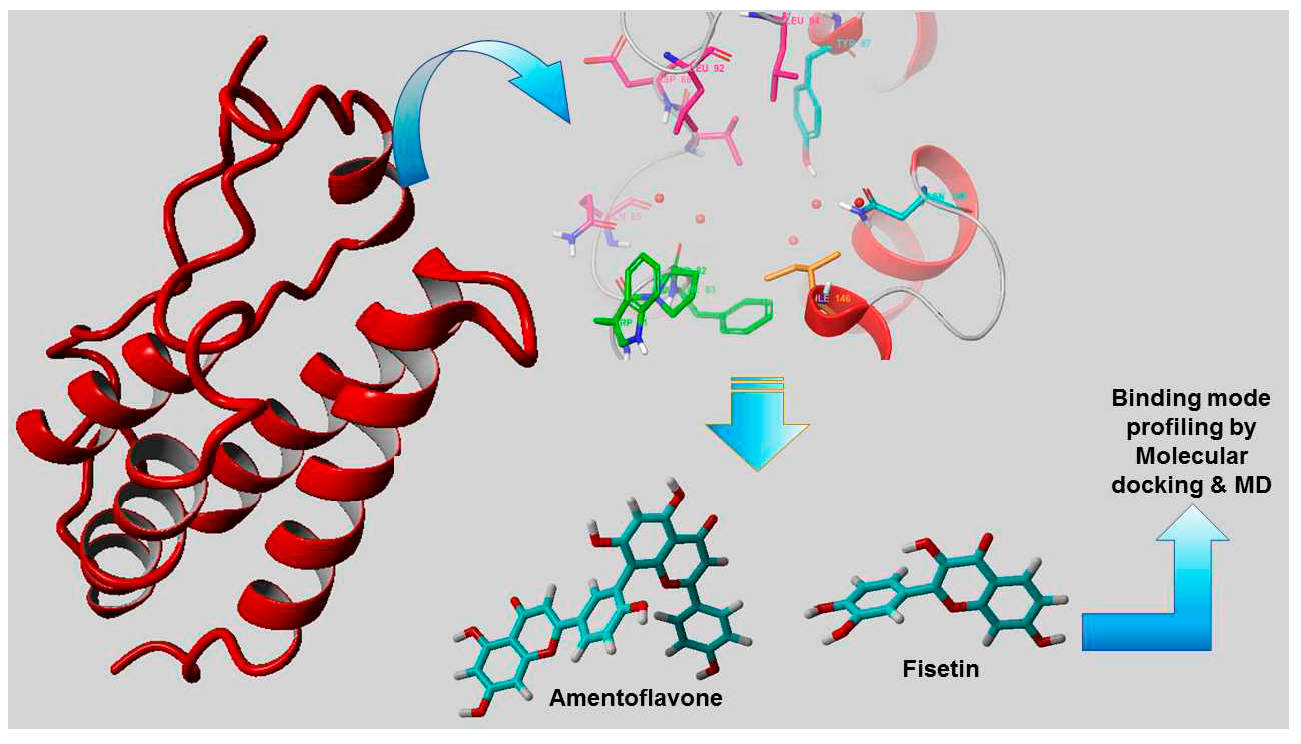
3. Results
3.1. Molecular Docking
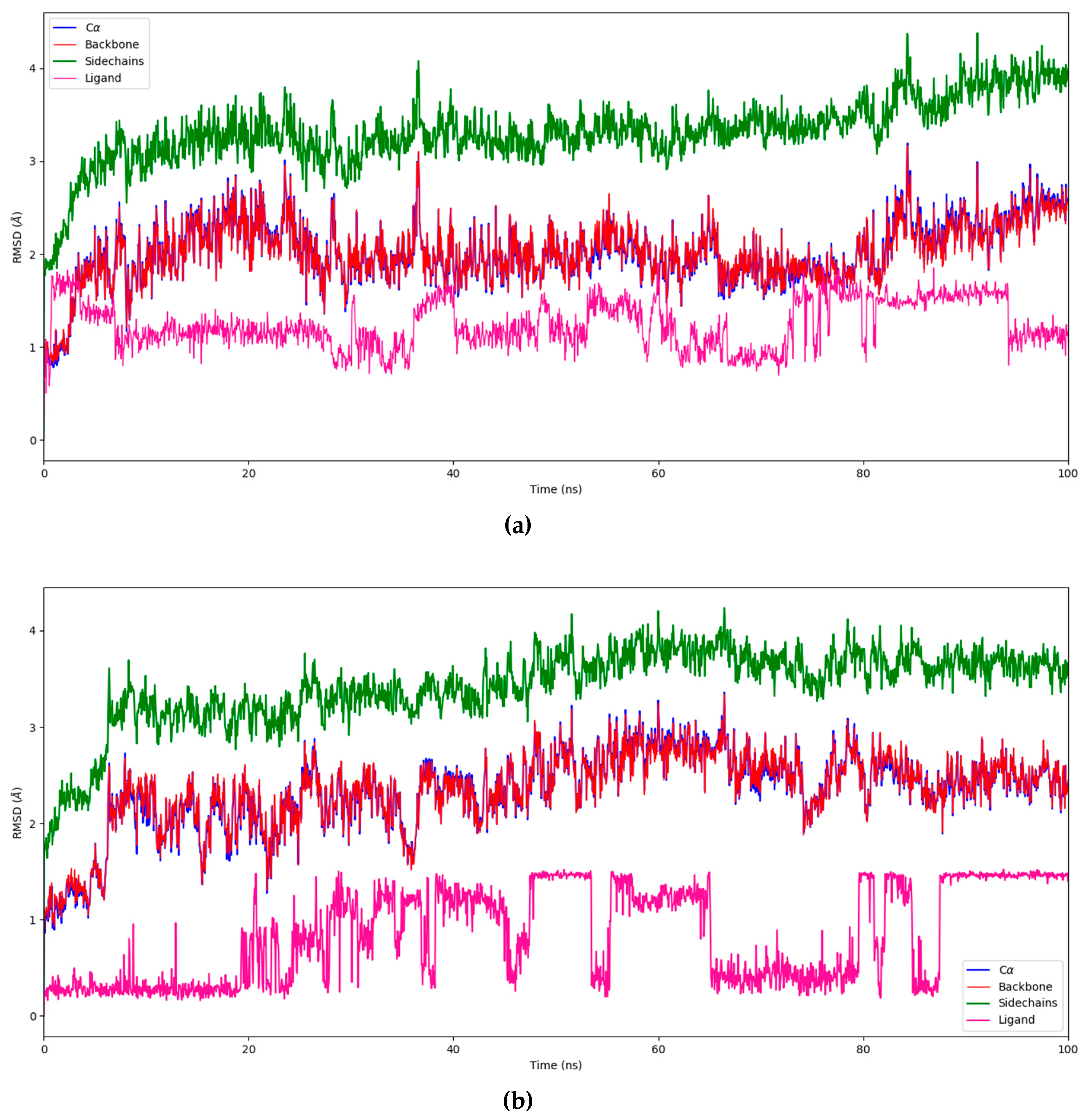
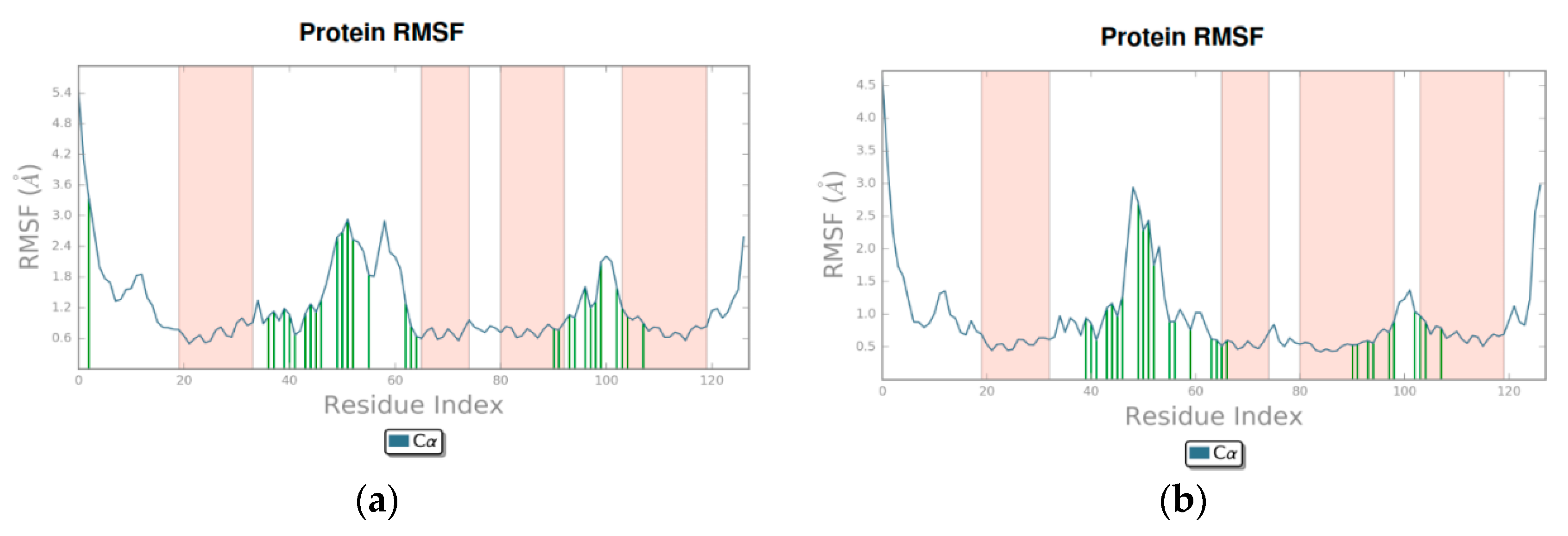
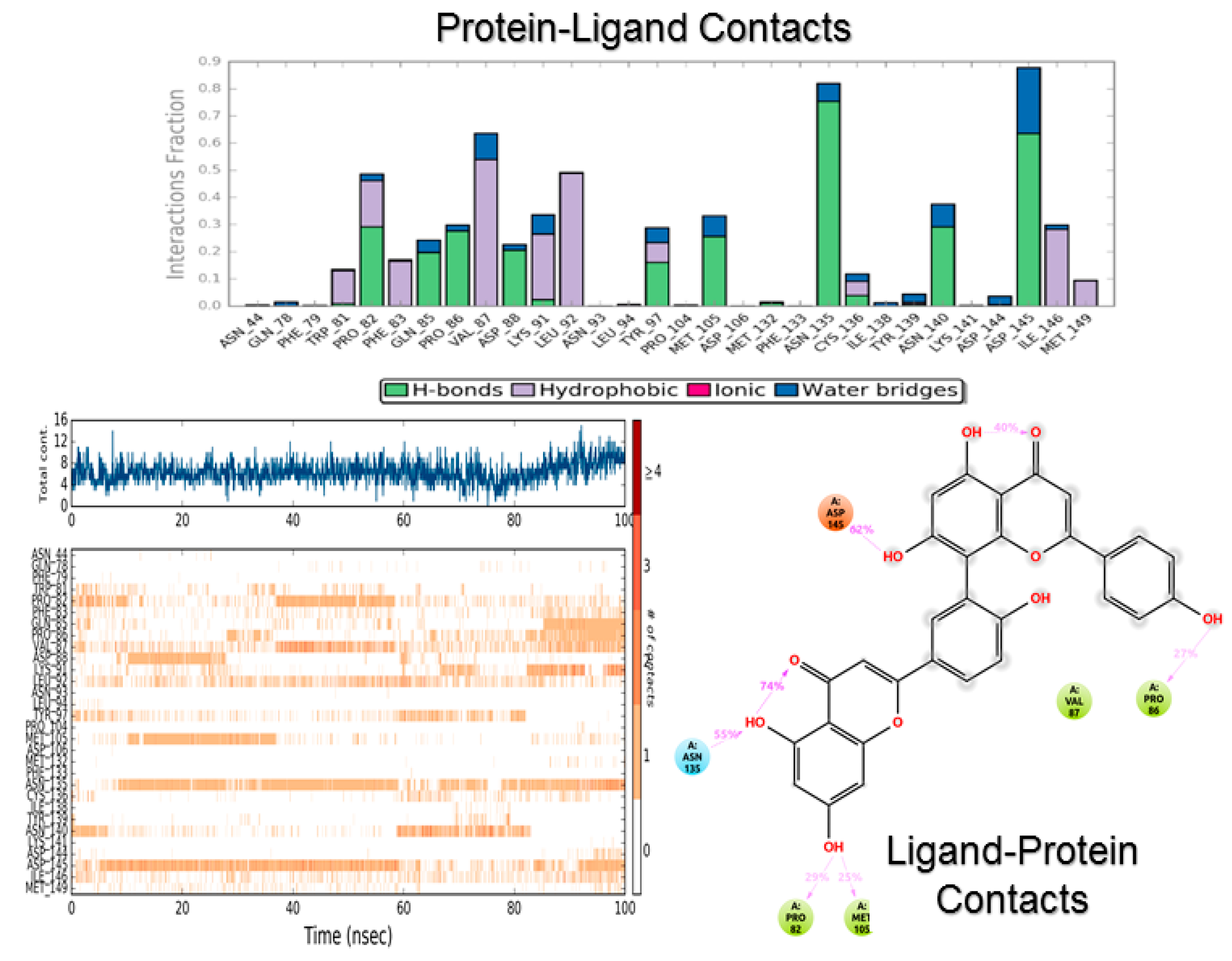
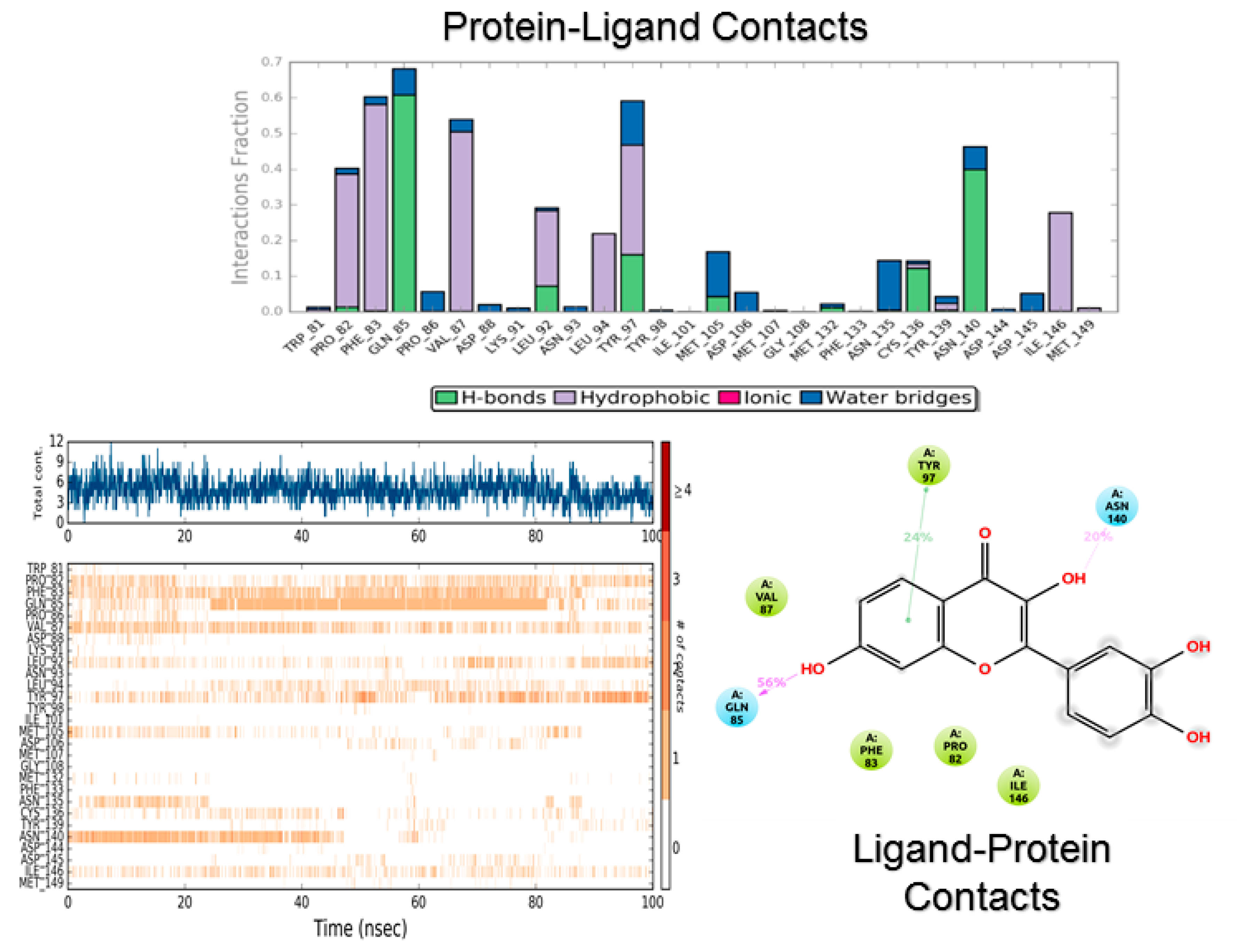
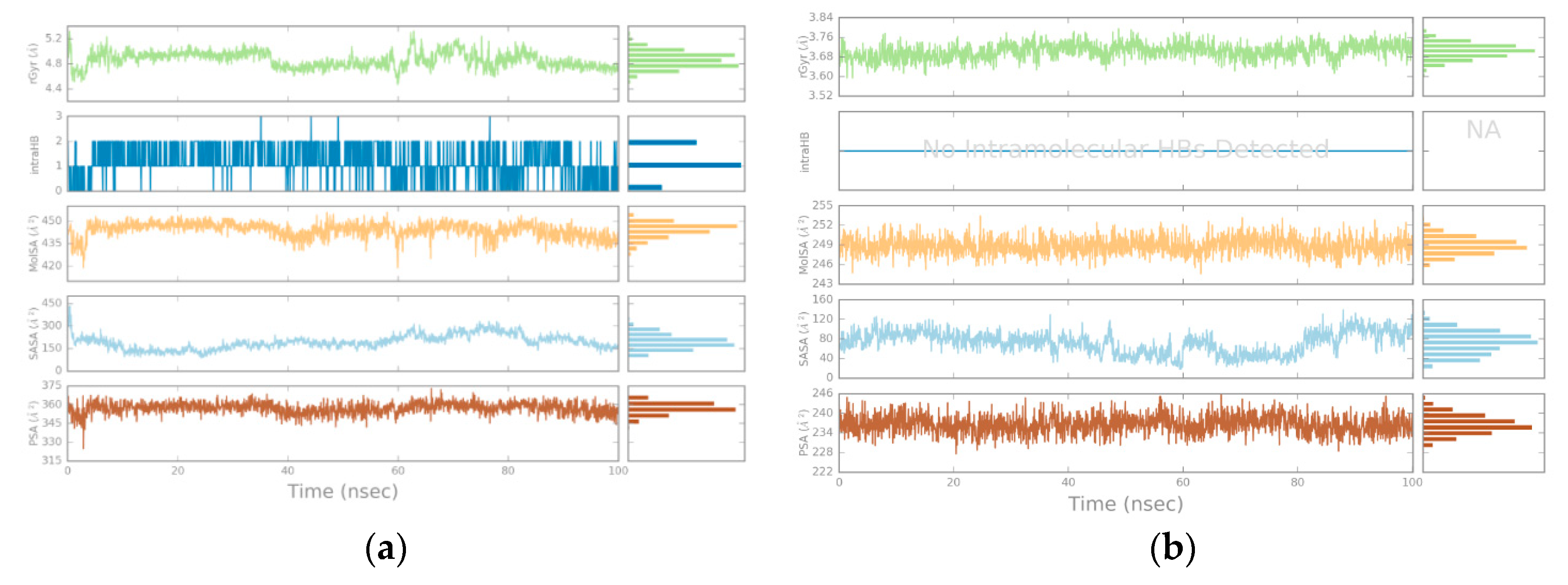
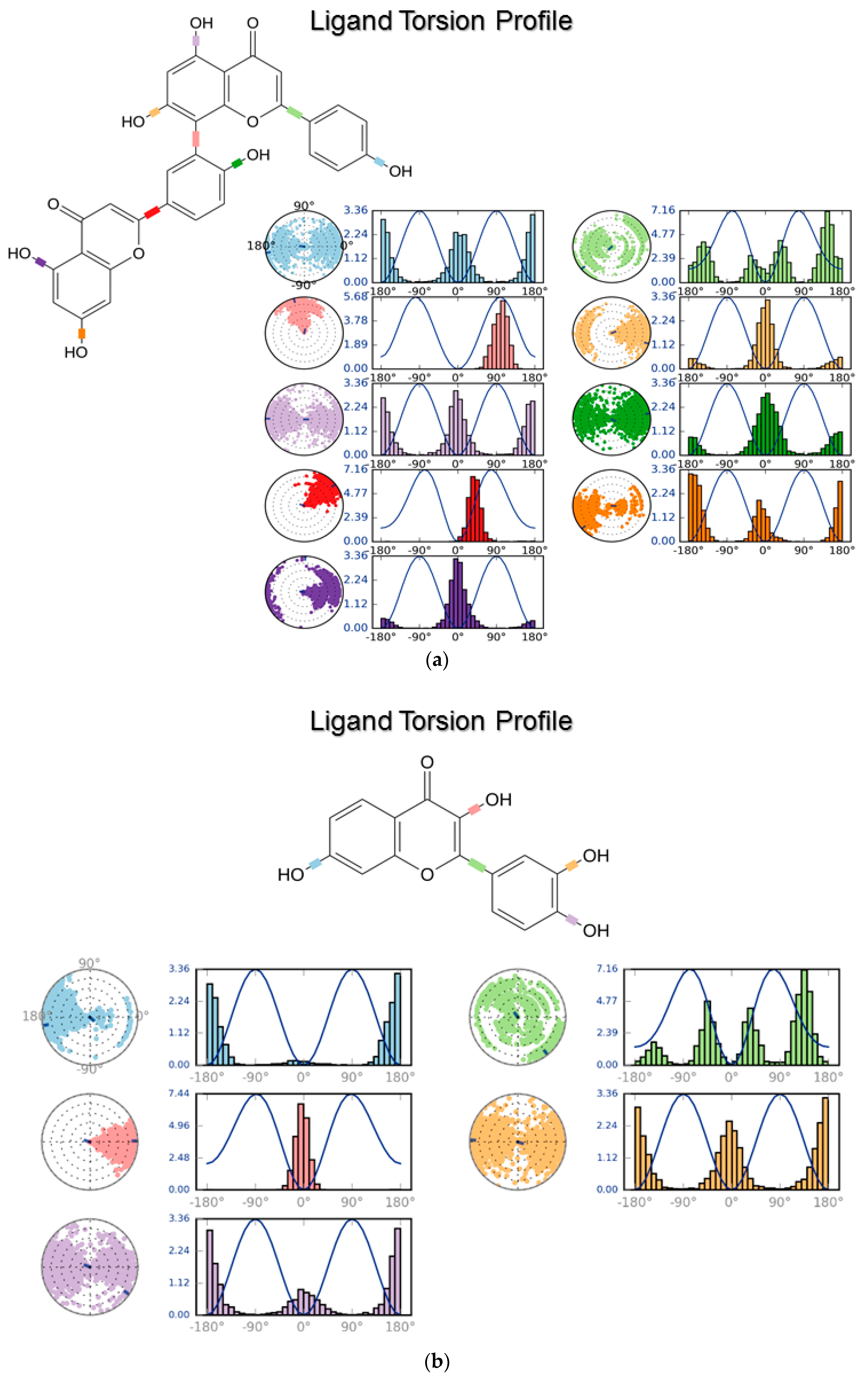
3.2. Molecular Dynamics
3.3. Binding Assay of Amentoflavone
4. Discussion
4.1. Molecular Docking
- Autodock VINA: It has a well-established performance against several protein families; additionally, its empirical scoring function has a significant correlation with experimental values [66]. Finally, its hybrid search algorithm optimized by local search allows a better sampling of the free-energy landscape [67].
- LeDock: Its search algorithm based on simulated annealing provides a significant clustering of poses. In addition, it was implemented successfully in virtual screening campaigns for BET bromodomains [52].
- MOE: Its docking algorithm allows for induced-fit search. Furthermore, its force-field-based scoring function (using AMBER parameters with generalized Born/volume integral (GB/VI) solvation) considers the solvation contributions to ligand binding [68].
- PLANTS: It provides a notable sampling of side-chain flexibility. Also, its searching algorithm (based on metaheuristics) and its empirical scoring function have a well-established performance [69].
4.2. Molecular Dynamics
4.3. Experimental Evaluation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cabaye, A.; Nguyen, K.T.; Liu, L.; Pande, V.; Schapira, M. Structural diversity of the epigenetics pocketome. Proteins Struct. Funct. Bioinform. 2015, 83, 1316–1326. [Google Scholar] [CrossRef] [PubMed]
- Eberharter, A.; Becker, P.B. Histone acetylation: A switch between repressive and permissive chromatin. Second in review on chromatin dynamics. EMBO Rep. 2002, 3, 224–229. [Google Scholar] [CrossRef] [PubMed]
- Kalyaanamoorthy, S.; Chen, Y.P.P. Energy based pharmacophore mapping of HDAC inhibitors against class i HDAC enzymes. Biochim. Biophys. Acta Proteins Proteomics 2013, 1834, 317–328. [Google Scholar] [CrossRef]
- Ortore, G.; Colo, F.D.; Martinelli, A. Docking of Hydroxamic Acids into HDAC1 and HDAC8: A Rationalization of Activity Trends and Selectivities. J. Chem. Inf. Model. 2009, 49, 2774–2785. [Google Scholar] [CrossRef] [PubMed]
- Hassanzadeh, M.; Bagherzadeh, K.; Amanlou, M. A comparative study based on docking and molecular dynamics simulations over HDAC-tubulin dual inhibitors. J. Mol. Graph. Model. 2016, 70, 170–180. [Google Scholar] [CrossRef] [PubMed]
- VanderMolen, K.M.; McCulloch, W.; Pearce, C.J.; Oberlies, N.H. Romidepsin (Istodax, NSC 630176, FR901228, FK228, depsipeptide): A natural product recently approved for cutaneous T-cell lymphoma. J. Antibiot. (Tokyo) 2011, 64, 525–531. [Google Scholar] [CrossRef] [PubMed]
- Arrowsmith, C.H.; Bountra, C.; Fish, P.V.; Lee, K.; Schapira, M. Epigenetic protein families: a new frontier for drug discovery. Nat. Rev. Drug Discov. 2012, 11, 384–400. [Google Scholar] [CrossRef] [PubMed]
- Berkovits, B.D.; Wolgemuth, D.J. The Role of the Double Bromodomain-Containing BET Genes During Mammalian Spermatogenesis. Curr. Top. Dev. Biol. 2013, 102, 293–326. [Google Scholar] [CrossRef] [PubMed]
- Hewings, D.S.; Rooney, T.P.C.; Jennings, L.E.; Hay, D.A.; Schofield, C.J.; Brennan, P.E.; Knapp, S.; Conway, S.J. Progress in the Development and Application of Small Molecule Inhibitors of Bromodomain–Acetyl-lysine Interactions. J. Med. Chem. 2012, 55, 9393–9413. [Google Scholar] [CrossRef] [PubMed]
- Ferri, E.; Petosa, C.; McKenna, C.E. Bromodomains: Structure, function and pharmacology of inhibition. Biochem. Pharmacol. 2016, 106, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Brand, M.; Measures, A.M.; Wilson, B.G.; Cortopassi, W.A.; Alexander, R.; Höss, M.; Hewings, D.S.; Rooney, T.P.C.; Paton, R.S.; Conway, S.J. Small Molecule Inhibitors of Bromodomain–Acetyl-lysine Interactions. ACS Chem. Biol. 2015, 10, 22–39. [Google Scholar] [CrossRef] [PubMed]
- Ladbury, J.E. Just add water! The effect of water on the specificity of protein-ligand binding sites and its potential application to drug design. Chem. Biol. 1996, 3, 973–980. [Google Scholar] [CrossRef]
- Plumridge, T.H.; Waigh, R.D. Water structure theory and some implications for drug design. J. Pharm. Pharmacol. 2002, 54, 1155–1179. [Google Scholar] [CrossRef] [PubMed]
- Crawford, T.D.; Tsui, V.; Flynn, E.M.; Wang, S.; Taylor, A.M.; Côté, A.; Audia, J.E.; Beresini, M.H.; Burdick, D.J.; Cummings, R.; et al. Diving into the Water: Inducible Binding Conformations for BRD4, TAF1(2), BRD9, and CECR2 Bromodomains. J. Med. Chem. 2016, 59, 5391–5402. [Google Scholar] [CrossRef] [PubMed]
- Pan, A.C.; Borhani, D.W.; Dror, R.O.; Shaw, D.E. Molecular determinants of drug-receptor binding kinetics. Drug Discov. Today 2013, 18, 667–673. [Google Scholar] [CrossRef] [PubMed]
- Huggins, D.J.; Sherman, W.; Tidor, B. Rational approaches to improving selectivity in drug design. J. Med. Chem. 2012, 55, 1424–1444. [Google Scholar] [CrossRef] [PubMed]
- Ross, G.A.; Morris, G.M.; Biggin, P.C. Rapid and accurate prediction and scoring of water molecules in protein binding sites. PLoS ONE 2012, 7, e32036. [Google Scholar] [CrossRef] [PubMed]
- García-Sosa, A.T.; Firth-Clark, S.; Mancera, R.L. Including tightly-bound water molecules in de novo drug design. exemplification through the in silico generation of poly(ADP-ribose)polymerase ligands. J. Chem. Inf. Model. 2005, 45, 624–633. [Google Scholar] [CrossRef] [PubMed]
- Shadrick, W.R.; Slavish, P.J.; Chai, S.C.; Waddell, B.; Connelly, M.; Low, J.A.; Tallant, C.; Young, B.M.; Bharatham, N.; Knapp, S.; et al. Exploiting a water network to achieve enthalpy-driven, bromodomain-selective BET inhibitors. Bioorgan. Med. Chem. 2018, 26, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Bharatham, N.; Slavish, P.J.; Young, B.M.; Shelat, A.A. The role of ZA channel water-mediated interactions in the design of bromodomain-selective BET inhibitors. J. Mol. Graph. Model. 2018, 81, 197–210. [Google Scholar] [CrossRef] [PubMed]
- Geist, L.; Mayer, M.; Cockcroft, X.L.; Wolkerstorfer, B.; Kessler, D.; Engelhardt, H.; McConnell, D.B.; Konrat, R. Direct NMR Probing of Hydration Shells of Protein Ligand Interfaces and Its Application to Drug Design. J. Med. Chem. 2017, 60, 8708–8715. [Google Scholar] [CrossRef] [PubMed]
- Prieto-Martínez, F.D.; Fernandez-de Gortari, E.; Méndez-Lucio, O.; Medina-Franco, J.L. A chemical space odyssey of inhibitors of histone deacetylases and bromodomains. RSC Adv. 2016, 6, 56225–56239. [Google Scholar] [CrossRef]
- Galdeano, C.; Ciulli, A. Selectivity on-target of bromodomain chemical probes by structure-guided medicinal chemistry and chemical biology. Future Med. Chem. 2016, 8, 1655–1680. [Google Scholar] [CrossRef] [PubMed]
- Kharenko, O.A.; Gesner, E.M.; Patel, R.G.; Norek, K.; White, A.; Fontano, E.; Suto, R.K.; Young, P.R.; McLure, K.G.; Hansen, H.C. RVX-297-a novel BD2 selective inhibitor of BET bromodomains. Biochem. Biophys. Res. Commun. 2016, 477, 62–67. [Google Scholar] [CrossRef] [PubMed]
- Ember, S.W.J.; Zhu, J.Y.; Olesen, S.H.; Martin, M.P.; Becker, A.; Berndt, N.; Georg, G.I.; Schonbrunn, E. Acetyl-lysine binding site of bromodomain-containing protein 4 (BRD4) interacts with diverse kinase inhibitors. ACS Chem. Biol. 2014, 9, 1160–1171. [Google Scholar] [CrossRef] [PubMed]
- Dhananjayan, K. Molecular Docking Study Characterization of Rare Flavonoids at the Nac-Binding Site of the First Bromodomain of BRD4 (BRD4 BD1). J. Cancer Res. 2015, 2015, 1–15. [Google Scholar] [CrossRef]
- Singh, M.; Kaur, M.; Silakari, O. Flavones: An important scaffold for medicinal chemistry. Eur. J. Med. Chem. 2014, 84, 206–239. [Google Scholar] [CrossRef] [PubMed]
- Perry, N.S.L.; Bollen, C.; Perry, E.K.; Ballard, C. Salvia for dementia therapy: Review of pharmacological activity and pilot tolerability clinical trial. Pharmacol. Biochem. Behav. 2003, 75, 651–659. [Google Scholar] [CrossRef]
- Hwang, S.-L.; Shih, P.-H.; Yen, G.-C. Citrus Flavonoids and Effects in Dementia and Age-Related Cognitive Decline. In Diet and Nutrition in Dementia and Cognitive Decline; Elsevier: Cambridge, MA, USA, 2015; pp. 869–878. ISBN 9780124079397. [Google Scholar]
- Fernández, S.P.; Wasowski, C.; Paladini, A.C.; Marder, M. Synergistic interaction between hesperidin, a natural flavonoid, and diazepam. Eur. J. Pharmacol. 2005, 512, 189–198. [Google Scholar] [CrossRef] [PubMed]
- Thilakarathna, W.W.; Langille, M.G.; Rupasinghe, H.V. Polyphenol-based prebiotics and synbiotics: potential for cancer chemoprevention. Curr. Opin. Food Sci. 2018, 20, 51–57. [Google Scholar] [CrossRef]
- Khan, N.; Syed, D.N.; Ahmad, N.; Mukhtar, H. Fisetin: A Dietary Antioxidant for Health Promotion. Antioxid. Redox Signal. 2013, 19, 151–162. [Google Scholar] [CrossRef] [PubMed]
- Vasantha Rupasinghe, H.P.; Nair, S.V.G.; Robinson, R.A. Chemopreventive Properties of Fruit Phenolic Compounds and Their Possible Mode of Actions, 1st ed.; Elsevier: Amsterdam, The Netherlands, 2014; Volume 42, ISBN 9780444632814. [Google Scholar]
- Sung, B.; Pandey, M.K.; Aggarwal, B.B. Fisetin, an Inhibitor of Cyclin-Dependent Kinase 6, Down-Regulates Nuclear Factor-B-Regulated Cell Proliferation, Antiapoptotic and Metastatic Gene Products through the Suppression of TAK-1 and Receptor-Interacting Protein-Regulated IκBα Kinase Activation. Mol. Pharmacol. 2007, 71, 1703–1714. [Google Scholar] [CrossRef] [PubMed]
- Syed, D.N.; Adhami, V.M.; Khan, N.; Khan, M.I.; Mukhtar, H. Exploring the molecular targets of dietary flavonoid fisetin in cancer. Semin. Cancer Biol. 2016, 40–41, 130–140. [Google Scholar] [CrossRef] [PubMed]
- Sundarraj, K.; Raghunath, A.; Perumal, E. A review on the chemotherapeutic potential of fisetin: In vitro evidences. Biomed. Pharmacother. 2018, 97, 928–940. [Google Scholar] [CrossRef] [PubMed]
- Kashyap, D.; Sharma, A.; Sak, K.; Tuli, H.S.; Buttar, H.S.; Bishayee, A. Fisetin: A bioactive phytochemical with potential for cancer prevention and pharmacotherapy. Life Sci. 2018, 194, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Rengarajan, T.; Yaacob, N.S. The flavonoid fisetin as an anticancer agent targeting the growth signaling pathways. Eur. J. Pharmacol. 2016, 789, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Nadeem, A.; Al-Harbi, N.O.; Al-Harbi, M.M.; El-Sherbeeny, A.M.; Ahmad, S.F.; Siddiqui, N.; Ansari, M.A.; Zoheir, K.M.A.; Attia, S.M.; Al-Hosaini, K.A.; et al. Imiquimod-induced psoriasis-like skin inflammation is suppressed by BET bromodomain inhibitor in mice through RORC/IL-17A pathway modulation. Pharmacol. Res. 2015, 99, 248–257. [Google Scholar] [CrossRef] [PubMed]
- Coletta, D.K. Genetic and Epigenetics of Type 2 Diabetes. In Pathobiology of Human Disease; Elsevier: Amsterdam, The Netherlands, 2014; pp. 467–476. ISBN 9780123864567. [Google Scholar]
- Mele, D.A.; Salmeron, A.; Ghosh, S.; Huang, H.-R.; Bryant, B.M.; Lora, J.M. BET bromodomain inhibition suppresses TH17-mediated pathology. J. Exp. Med. 2013, 210, 2181–2190. [Google Scholar] [CrossRef] [PubMed]
- Burkard, M.; Leischner, C.; Lauer, U.M.; Busch, C.; Venturelli, S.; Frank, J. Dietary flavonoids and modulation of natural killer cells: implications in malignant and viral diseases. J. Nutr. Biochem. 2017, 46, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Catarino, M.D.; Talhi, O.; Rabahi, A.; Silva, A.M.S.; Cardoso, S.M. The Antiinflammatory Potential of Flavonoids. In Studies in Natural Products Chemistry; Elsevier: Amsterdam, The Netherlands, 2016; Volume 48, pp. 65–99. ISBN 9780444636027. [Google Scholar]
- Onawole, A.T.; Sulaiman, K.O.; Adegoke, R.O.; Kolapo, T.U. Identification of potential inhibitors against the Zika virus using consensus scoring. J. Mol. Graph. Model. 2017, 73, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Śledź, P.; Caflisch, A. Protein structure-based drug design: from docking to molecular dynamics. Curr. Opin. Struct. Biol. 2018, 48, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Raj, U.; Kumar, H.; Varadwaj, P.K. Molecular docking and dynamics simulation study of flavonoids as BET bromodomain inhibitors. J. Biomol. Struct. Dyn. 2016, 1102, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Prieto-Martínez, F.D.; Medina-Franco, J.L. Charting the Bromodomain BRD4: Towards the Identification of Novel Inhibitors with Molecular Similarity and Receptor Mapping. Lett. Drug Des. Discov. 2018, 15, 1–10. [Google Scholar] [CrossRef]
- Molecular Operating Environment (MOE), 2013.08; Chemical Computing Group ULC: Montreal, QC, Canada, 2018.
- Case, D.A.; Babin, V.; Berryman, J.; Betz, R.; Cai, Q.; Cerutti, D.S.; Cheatham, T.; Darden, T.; Duke, R.; Gohlke, H.; et al. AMBER 14, University of California: San Francisco, CA, USA, 2014.
- Gerber, P.R.; Müller, K. MAB, a generally applicable molecular force field for structure modelling in medicinal chemistry. J. Comput. Aided. Mol. Des. 1995, 9, 251–268. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2009, 31. [Google Scholar] [CrossRef] [PubMed]
- Unzue, A.; Zhao, H.; Lolli, G.; Dong, J.; Zhu, J.; Zechner, M.; Dolbois, A.; Caflisch, A.; Nevado, C. The “gatekeeper” Residue Influences the Mode of Binding of Acetyl Indoles to Bromodomains. J. Med. Chem. 2016, 59, 3087–3097. [Google Scholar] [CrossRef] [PubMed]
- Korb, O.; Stützle, T.; Exner, T.E. Empirical scoring functions for advanced Protein-Ligand docking with PLANTS. J. Chem. Inf. Model. 2009, 49, 84–96. [Google Scholar] [CrossRef] [PubMed]
- Stewart, J.J.P. Application of the PM6 method to modeling the solid state. J. Mol. Model. 2008, 14, 499–535. [Google Scholar] [CrossRef] [PubMed]
- Hostaš, J.; Řezáč, J.; Hobza, P. On the performance of the semiempirical quantum mechanical PM6 and PM7 methods for noncovalent interactions. Chem. Phys. Lett. 2013, 568–569, 161–166. [Google Scholar] [CrossRef]
- Bowers, K.; Chow, E.; Xu, H.; Dror, R.; Eastwood, M.; Gregersen, B.; Klepeis, J.; Kolossvary, I.; Moraes, M.; Sacerdoti, F.; et al. Scalable Algorithms for Molecular Dynamics Simulations on Commodity Clusters. In Proceedings of the 2006 ACM/IEEE SC Conference on Supercomputing (SC’06), Tampa, FL, USA, 11–17 November 2006. [Google Scholar]
- Kräutler, V.; van Gusteren, W.F.; Hünenberger, P.H. A fast SHAKE algorithm to solve distance constraints for small molecules in molecular dynamics simulations. J. Comput. Chem. 2001, 22, 501–508. [Google Scholar] [CrossRef]
- Andrews, F.H.; Singh, A.R.; Joshi, S.; Smith, C.A.; Morales, G.A.; Garlich, J.R.; Durden, D.L.; Kutateladze, T.G. Dual-activity PI3K–BRD4 inhibitor for the orthogonal inhibition of MYC to block tumor growth and metastasis. Proc. Natl. Acad. Sci. USA 2017, 114, E1072–E1080. [Google Scholar] [CrossRef] [PubMed]
- Sargsyan, K.; Grauffel, C.; Lim, C. How Molecular Size Impacts RMSD Applications in Molecular Dynamics Simulations. J. Chem. Theory Comput. 2017, 13, 1518–1524. [Google Scholar] [CrossRef] [PubMed]
- Romero, F.A.; Taylor, A.M.; Crawford, T.D.; Tsui, V.; Côté, A.; Magnuson, S. Disrupting Acetyl-Lysine Recognition: Progress in the Development of Bromodomain Inhibitors. J. Med. Chem. 2016, 59, 1271–1298. [Google Scholar] [CrossRef] [PubMed]
- Ran, T.; Zhang, Z.; Liu, K.; Lu, Y.; Li, H.; Xu, J.; Xiong, X.; Zhang, Y.; Xu, A.; Lu, S.; et al. Insight into the key interactions of bromodomain inhibitors based on molecular docking, interaction fingerprinting, molecular dynamics and binding free energy calculation. Mol. Biosyst. 2015, 11, 1295–1304. [Google Scholar] [CrossRef] [PubMed]
- Korb, O.; Olsson, T.S.G.; Bowden, S.J.; Hall, R.J.; Verdonk, M.L.; Liebeschuetz, J.W.; Cole, J.C. Potential and Limitations of Ensemble Docking. J. Chem. Inf. Model. 2012, 1262–1274. [Google Scholar] [CrossRef] [PubMed]
- Evangelista, W.; Weir, R.L.; Ellingson, S.R.; Harris, J.B.; Kapoor, K.; Smith, J.C.; Baudry, J. Ensemble-based docking: From hit discovery to metabolism and toxicity predictions. Bioorganic Med. Chem. 2016, 24, 4928–4935. [Google Scholar] [CrossRef] [PubMed]
- Tuccinardi, T.; Poli, G.; Romboli, V.; Giordano, A.; Martinelli, A. Extensive consensus docking evaluation for ligand pose prediction and virtual screening studies. J. Chem. Inf. Model. 2014, 54, 2980–2986. [Google Scholar] [CrossRef] [PubMed]
- Kitchen, D.B.; Decornez, H.; Furr, J.R.; Bajorath, J. Docking and scoring in virtual screening for drug discovery: methods and applications. Nat. Rev. Drug Discov. 2004, 3, 935–949. [Google Scholar] [CrossRef] [PubMed]
- Palestro, P.H.; Gavernet, L.; Estiu, G.L.; Bruno Blanch, L.E. Docking Applied to the Prediction of the Affinity of Compounds to P-Glycoprotein. Biomed Res. Int. 2014, 2014, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, L.; dos Santos, R.; Oliva, G.; Andricopulo, A. Molecular Docking and Structure-Based Drug Design Strategies. Molecules 2015, 20, 13384–13421. [Google Scholar] [CrossRef] [PubMed]
- Naïm, M.; Bhat, S.; Rankin, K.N.; Dennis, S.; Chowdhury, S.F.; Siddiqi, I.; Drabik, P.; Sulea, T.; Bayly, C.I.; Jakalian, A.; et al. Solvated Interaction Energy (SIE) for scoring protein-ligand binding affinities. 1. Exploring the parameter space. J. Chem. Inf. Model. 2007, 47, 122–133. [Google Scholar] [CrossRef] [PubMed]
- Richter, L.; de Graaf, C.; Sieghart, W.; Varagic, Z.; Mörzinger, M.; de Esch, I.J.P.; Ecker, G.F.; Ernst, M. Diazepam-bound GABAA receptor models identify new benzodiazepine binding-site ligands. Nat. Chem. Biol. 2012, 8, 455–464. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Sun, H.; Yao, X.; Li, D.; Xu, L.; Li, Y.; Tian, S.; Hou, T. Comprehensive evaluation of ten docking programs on a diverse set of protein-ligand complexes: The prediction accuracy of sampling power and scoring power. Phys. Chem. Chem. Phys. 2016, 18, 12964–12975. [Google Scholar] [CrossRef] [PubMed]
- Hoffer, L.; Voitovich, Y.V.; Raux, B.; Carrasco, K.; Muller, C.; Fedorov, A.Y.; Derviaux, C.; Amouric, A.; Betzi, S.; Horvath, D.; et al. Integrated Strategy for Lead Optimization Based on Fragment Growing: The Diversity-Oriented-Target-Focused-Synthesis Approach. J. Med. Chem. 2018. [Google Scholar] [CrossRef]
- Waterman, M.J.; Nugraha, A.S.; Hendra, R.; Ball, G.E.; Robinson, S.A.; Keller, P.A. Antarctic Moss Biflavonoids Show High Antioxidant and Ultraviolet-Screening Activity. J. Nat. Prod. 2017, 80, 2224–2231. [Google Scholar] [CrossRef] [PubMed]
- Glunz, P.W. Recent encounters with atropisomerism in drug discovery. Bioorgan. Med. Chem. Lett. 2018, 28, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Jung, M.; Philpott, M.; Müller, S.; Schulze, J.; Badock, V.; Eberspächer, U.; Moosmayer, D.; Bader, B.; Schmees, N.; Fernández-Montalván, A.; Haendler, B. Affinity map of bromodomain protein 4 (BRD4) interactions with the histone H4 tail and the small molecule inhibitor JQ1. J. Biol. Chem. 2014, 289, 9304–9319. [Google Scholar] [CrossRef] [PubMed]
- Scotti, L.; Bezerra Mendonca, F.J.; Ribeiro, F.F.; Tavares, J.F.; da Silva, M.S.; Barbosa Filho, J.M.; Scotti, M.T. Natural Product Inhibitors of Topoisomerases: Review and Docking Study. Curr. Protein Pept. Sci. 2018, 19, 275–291. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, C.R.P.; de Syllos Cólus, I.M.; Bernardi, C.C.; Sannomiya, M.; Vilegas, W.; Varanda, E.A. Mutagenic activity promoted by amentoflavone and methanolic extract of Byrsonima crassa Niedenzu. Toxicology 2006, 225, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.-J.; Shin, S.-Y.; Lee, J.-Y.; Lee, S.-J.; Kim, J.-K.; Yoon, D.-Y.; Woo, E.-R.; Kim, Y.-M. Cytotoxic Activities of Amentoflavone against Human Breast and Cervical Cancers are Mediated by Increasing of PTEN Expression Levels due to Peroxisome Proliferator-Activated Receptor γ Activation. Bull. Korean Chem. Soc. 2012, 33, 2219–2223. [Google Scholar] [CrossRef]
- Grynberg, N.F.; Carvalho, M.G.; Velandia, J.R.; Oliveira, M.C.; Moreira, I.C.; Braz- Filho, R.; Echevarria, A. DNA topoisomerase inhibitors: Biflavonoids from Ouratea species. Braz. J. Med. Biol. Res. 2002, 35, 819–822. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecule | Summary Stats * | Autodock VINA (kcal/mol) | LeDock (kcal/mol) | MOE (kcal/mol) | PLANTS |
|---|---|---|---|---|---|
| Amentoflavone | Min | −10.5 | −7.9 | −9.0 | −102.1 |
| 1Q | −9.5 | −7.3 | −7.9 | −89.4 | |
| Avg | −9.2 | −7.0 | −7.6 | −86.9 | |
| 3Q | −9.0 | −6.8 | −7.2 | −84.0 | |
| Max | −8.2 | −6.3 | −6.4 | −77.4 | |
| SD | 0.46 | 0.34 | 0.54 | 4.6 | |
| Fisetin | Min | −8.6 | −6.0 | −7.4 | −79.6 |
| 1Q | −8.2 | −5.6 | −6.5 | −73.2 | |
| Avg | −7.9 | −5.4 | −6.2 | −71.0 | |
| 3Q | −7.7 | −5.3 | −6.0 | −68.9 | |
| Max | −7.1 | −4.7 | −5.6 | −65.0 | |
| SD | 0.31 | 0.24 | 0.39 | 2.94 |
| DATA 1 | DATA 2 | |
|---|---|---|
| IC50 (μM) | 36.1 | 30.4 |
| Hill slope | −2.5 | −1.9 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Prieto-Martínez, F.D.; Medina-Franco, J.L. Flavonoids as Putative Epi-Modulators: Insight into Their Binding Mode with BRD4 Bromodomains Using Molecular Docking and Dynamics. Biomolecules 2018, 8, 61. https://doi.org/10.3390/biom8030061
Prieto-Martínez FD, Medina-Franco JL. Flavonoids as Putative Epi-Modulators: Insight into Their Binding Mode with BRD4 Bromodomains Using Molecular Docking and Dynamics. Biomolecules. 2018; 8(3):61. https://doi.org/10.3390/biom8030061
Chicago/Turabian StylePrieto-Martínez, Fernando D., and José L. Medina-Franco. 2018. "Flavonoids as Putative Epi-Modulators: Insight into Their Binding Mode with BRD4 Bromodomains Using Molecular Docking and Dynamics" Biomolecules 8, no. 3: 61. https://doi.org/10.3390/biom8030061
APA StylePrieto-Martínez, F. D., & Medina-Franco, J. L. (2018). Flavonoids as Putative Epi-Modulators: Insight into Their Binding Mode with BRD4 Bromodomains Using Molecular Docking and Dynamics. Biomolecules, 8(3), 61. https://doi.org/10.3390/biom8030061