Redesigning TOR Kinase to Explore the Structural Basis for TORC1 and TORC2 Assembly



Abstract
1. Introduction
2. Results and Discussion
2.1. Experimental Approach
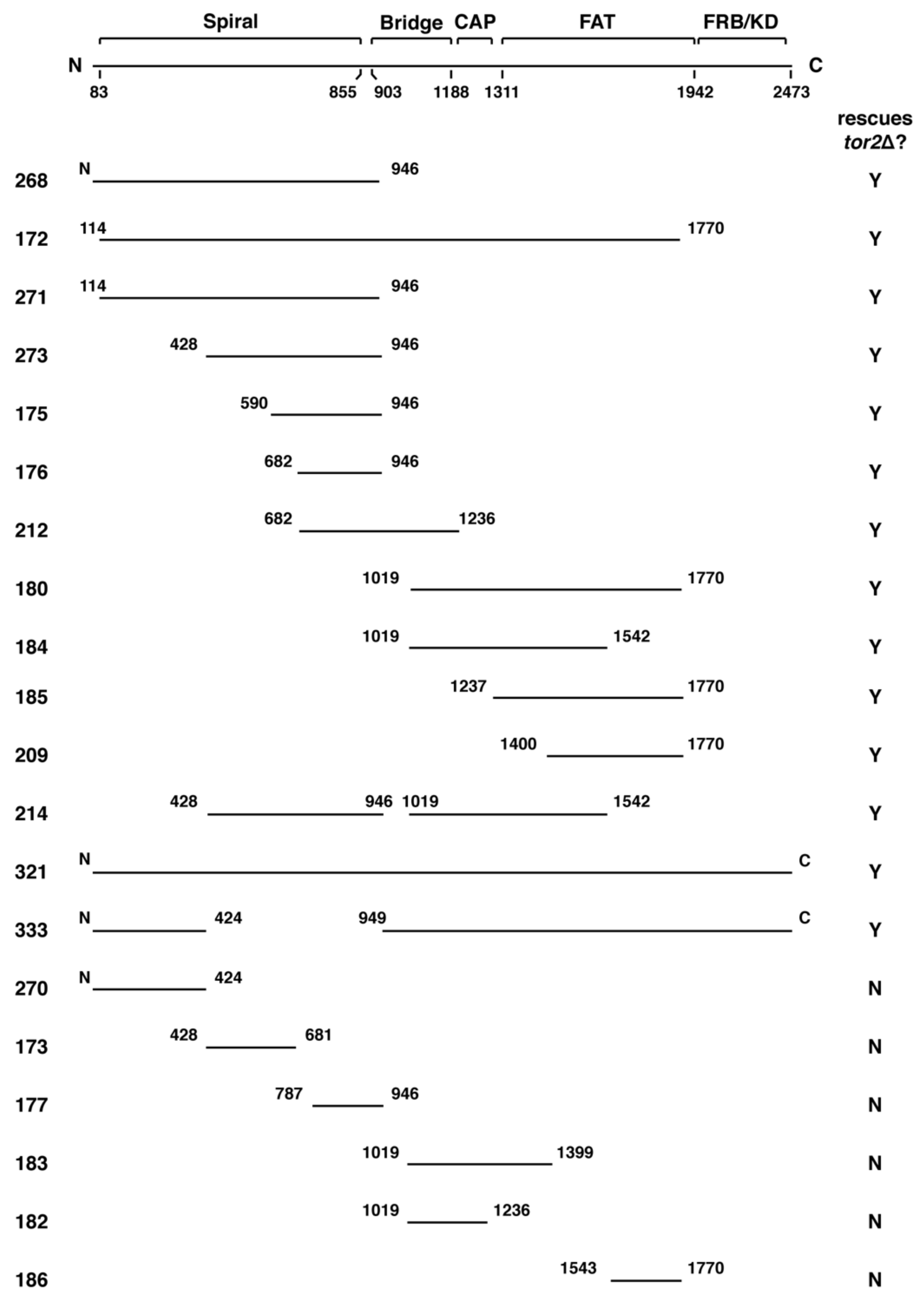
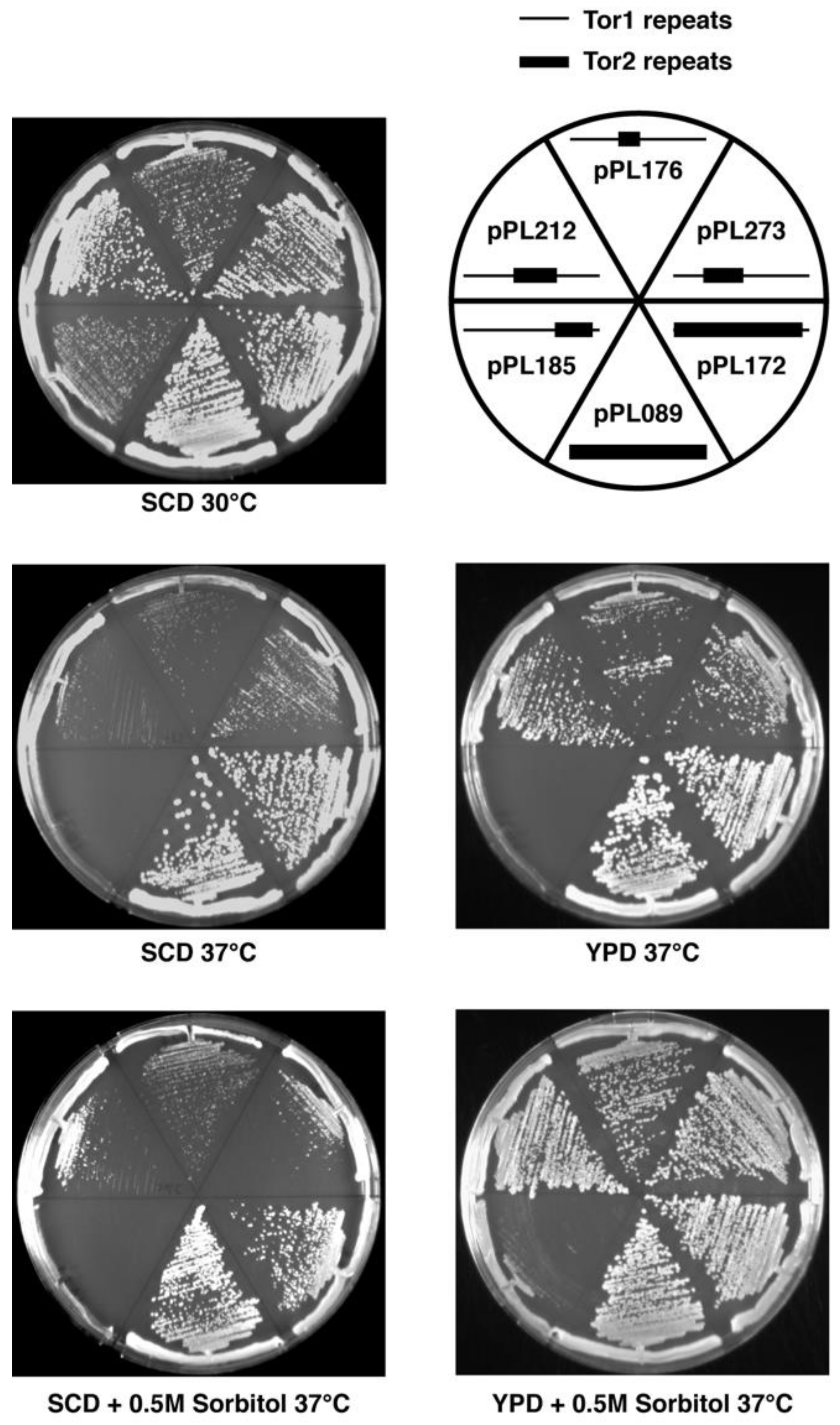
2.2. Genetic Analyses of TOR2–TOR1 Chimeras
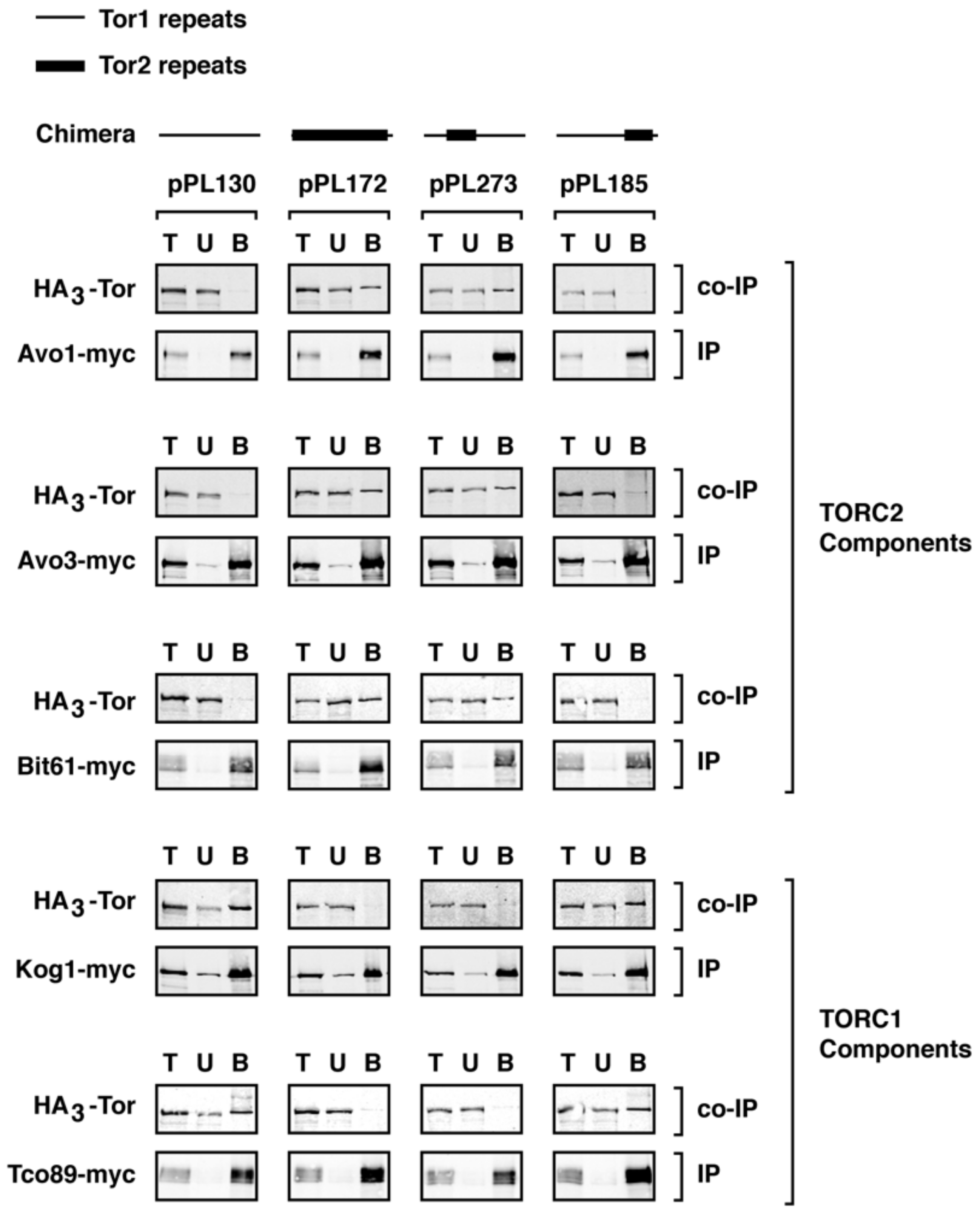
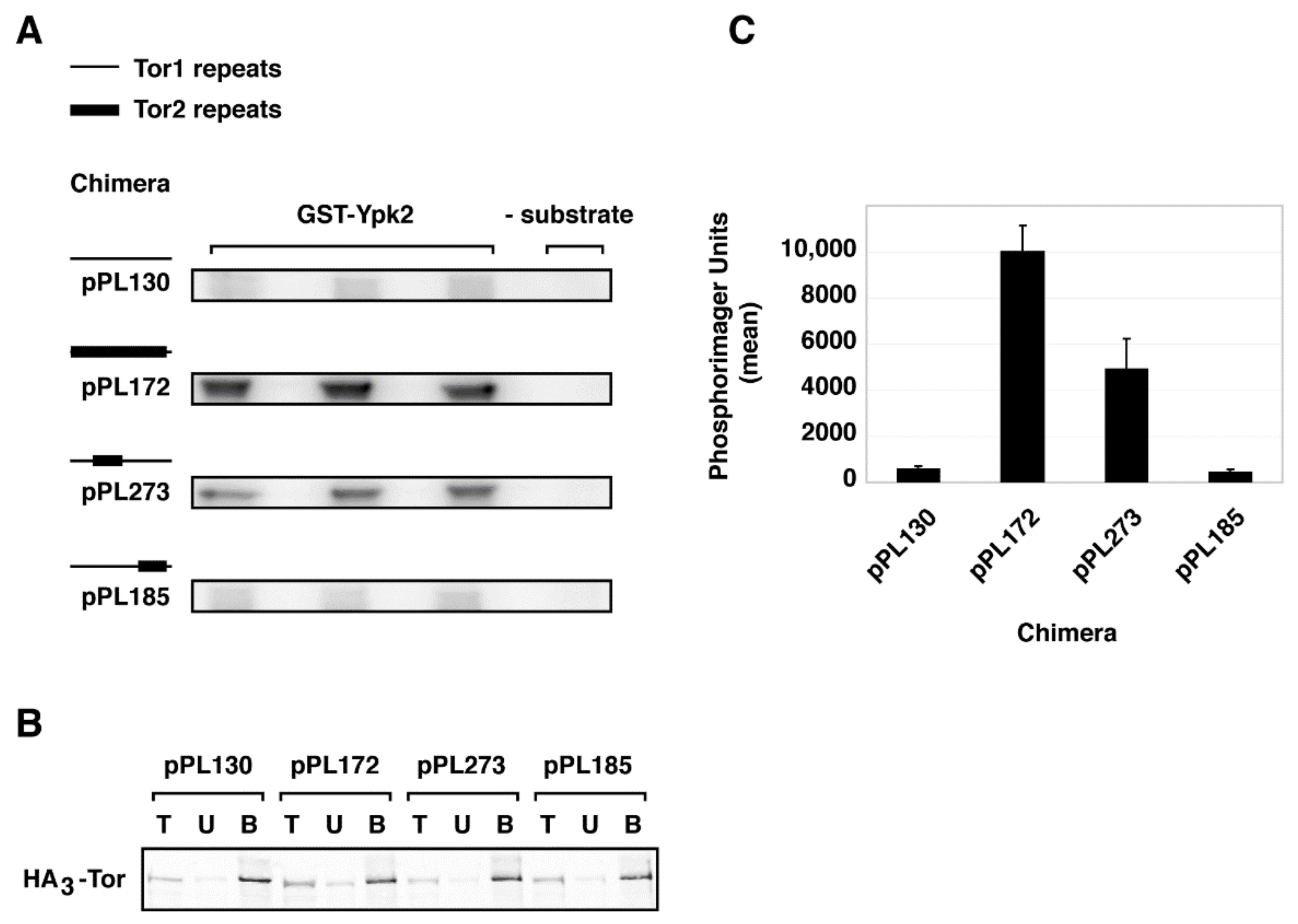
2.3. Monitoring Assembly of Chimeras into TORC1 and TORC2
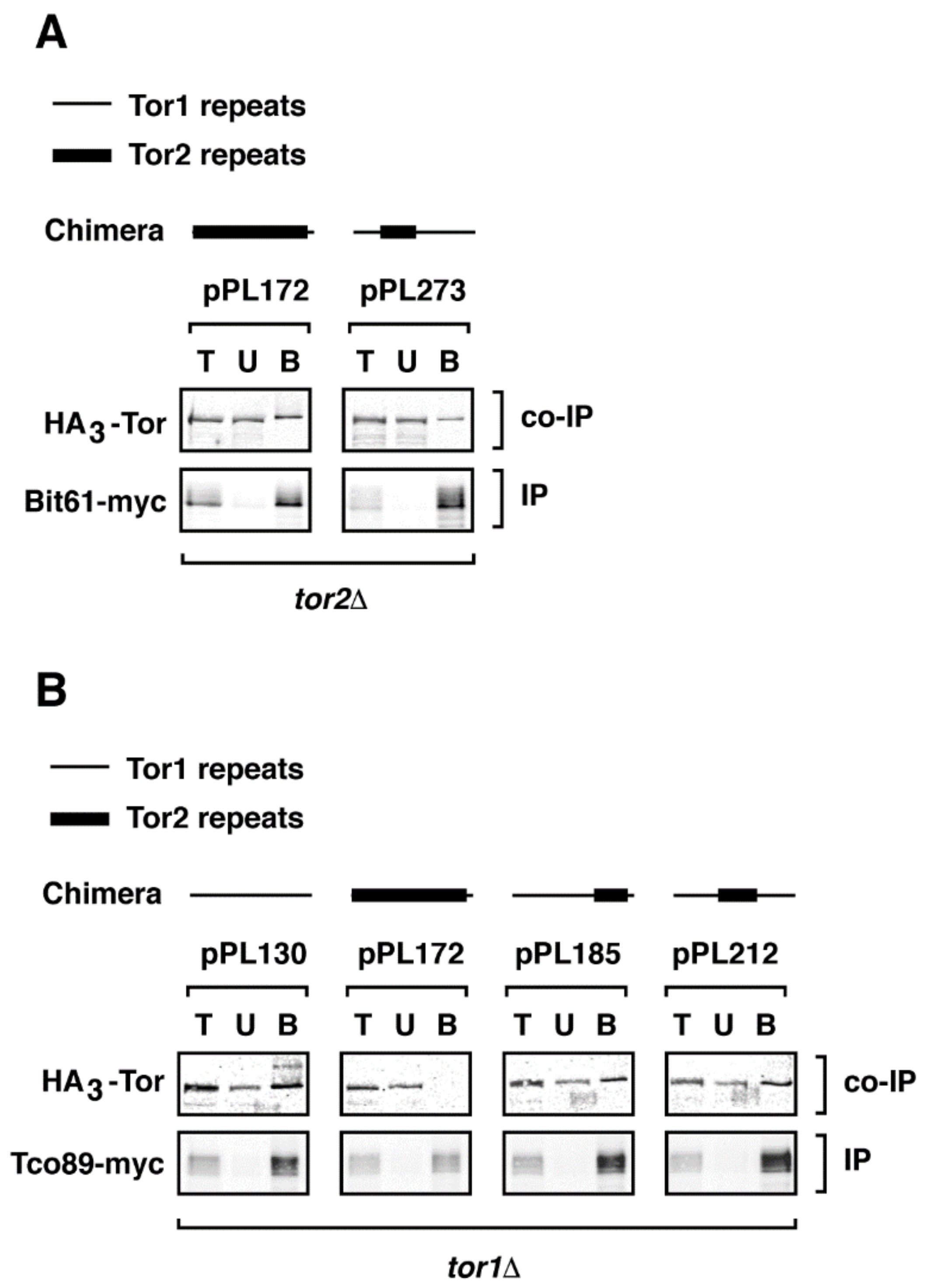
2.4. A Major Assembly SpecificityDomain in TOR
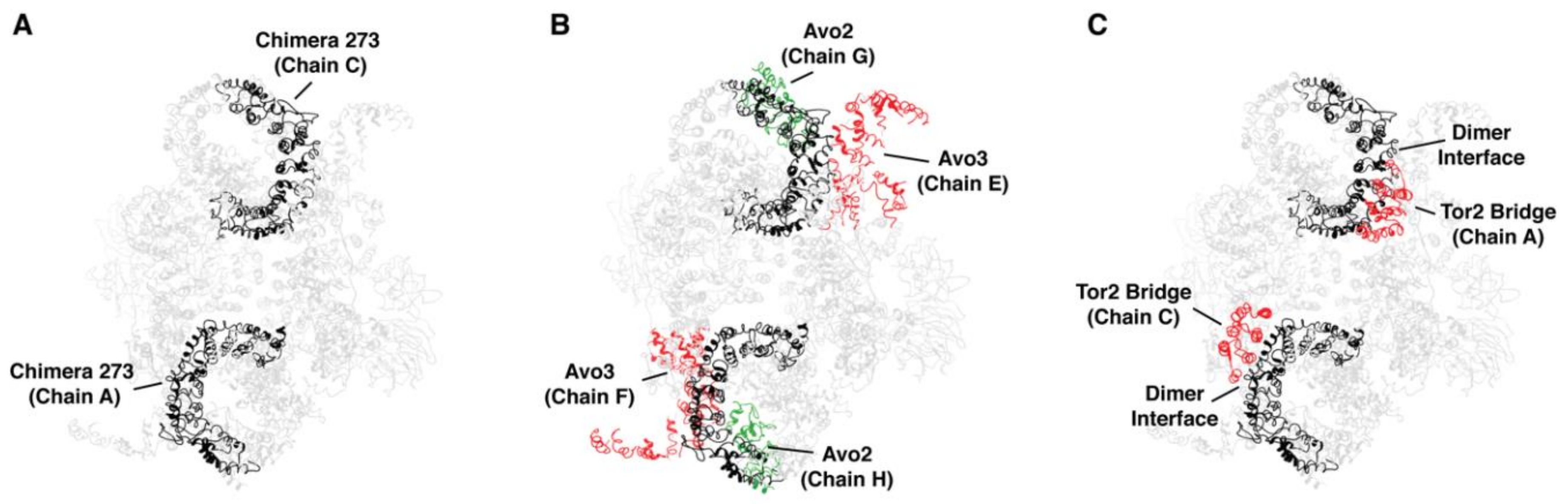
2.5. Toward Understanding Quaternary Interactions Important for TORC1 and TORC2 Assembly
3. Materials and Methods
3.1. Strains, Media, General Methods
3.2. Plasmid Construction
3.3. Immunoaffinity Purification of TOR and TOR-Complex Binding Partners
3.4. Immune-Complex In Vitro Kinase Assay
3.5. Actin Staining and Fluorescence Microscopy
3.6. Molecular Modeling
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Zeke, A.; Lukacs, M.; Lim, W.A.; Remenyi, A. Scaffolds: Interaction platforms for cellular signalling circuits. Trends Cell Biol. 2009, 19, 364–374. [Google Scholar] [CrossRef] [PubMed]
- Good, M.C.; Zalatan, J.G.; Lim, W.A. Scaffold proteins: Hubs for controlling the flow of cellular information. Science 2011, 332, 680–686. [Google Scholar] [CrossRef] [PubMed]
- Whitmarsh, A.J.; Davis, R.J. Structural organization of map-kinase signaling modules by scaffold proteins in yeast and mammals. Trends Biochem. Sci. 1998, 23, 481–485. [Google Scholar] [CrossRef]
- Witzel, F.; Maddison, L.; Bluthgen, N. How scaffolds shape MAPK signaling: What we know and opportunities for systems approaches. Front. Physiol. 2012, 3, 475. [Google Scholar] [CrossRef] [PubMed]
- Park, S.H.; Zarrinpar, A.; Lim, W.A. Rewiring MAP kinase pathways using alternative scaffold assembly mechanisms. Science 2003, 299, 1061–1064. [Google Scholar] [CrossRef] [PubMed]
- Wullschleger, S.; Loewith, R.; Hall, M.N. TOR signaling in growth and metabolism. Cell 2006, 124, 471–484. [Google Scholar] [CrossRef] [PubMed]
- Eltschinger, S.; Loewith, R. TOR complexes and the maintenance of cellular homeostasis. Trends Cell Biol. 2016, 26, 148–159. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, A.; Hall, M.N. Nutrient sensing and TOR signaling in yeast and mammals. EMBO J. 2017, 36, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Saxton, R.A.; Sabatini, D.M. mTOR signaling in growth, metabolism, and disease. Cell 2017, 169, 361–371. [Google Scholar] [CrossRef] [PubMed]
- Bosotti, R.; Isacchi, A.; Sonnhammer, E.L. Fat: A novel domain in PIK-related kinases. Trends Biochem. Sci. 2000, 25, 225–227. [Google Scholar] [CrossRef]
- Loewith, R.; Jacinto, E.; Wullschleger, S.; Lorberg, A.; Crespo, J.L.; Bonenfant, D.; Oppliger, W.; Jenoe, P.; Hall, M.N. Two TOR complexes, only one of which is rapamycin sensitive, have distinct roles in cell growth control. Mol. Cell 2002, 10, 457–468. [Google Scholar] [CrossRef]
- Laplante, M.; Sabatini, D.M. mTOR signaling at a glance. J. Cell Sci. 2009, 122, 3589–3594. [Google Scholar] [CrossRef] [PubMed]
- Shertz, C.A.; Bastidas, R.J.; Li, W.; Heitman, J.; Cardenas, M.E. Conservation, duplication, and loss of the TOR signaling pathway in the fungal kingdom. BMC Genomics 2010, 11, 510. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H.; Sarbassov, D.D.; Ali, S.M.; King, J.E.; Latek, R.R.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. mTOR interacts with Raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell 2002, 110, 163–175. [Google Scholar] [CrossRef]
- Sarbassov, D.D.; Ali, S.M.; Kim, D.H.; Guertin, D.A.; Latek, R.R.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and Raptor-independent pathway that regulates the cytoskeleton. Curr. Biol. 2004, 14, 1296–1302. [Google Scholar] [CrossRef] [PubMed]
- Wedaman, K.P.; Reinke, A.; Anderson, S.; Yates, J., 3rd; McCaffery, J.M.; Powers, T. TOR kinases are in distinct membrane-associated protein complexes in Saccharomyces cerevisiae. Mol. Biol. Cell 2003, 14, 1204–1220. [Google Scholar] [CrossRef] [PubMed]
- Helliwell, S.B.; Howald, I.; Barbet, N.; Hall, M.N. TOR2 is part of two related signaling pathways coordinating cell growth in Saccharomyces cerevisiae. Genetics 1998, 148, 99–112. [Google Scholar] [PubMed]
- Kunz, J.; Henriquez, R.; Schneider, U.; Deuter-Reinhard, M.; Movva, N.R.; Hall, M.N. Target of rapamycin in yeast, TOR2, is an essential phosphatidylinositol kinase homolog required for G1 progression. Cell 1993, 73, 585–596. [Google Scholar] [CrossRef]
- Helliwell, S.B.; Wagner, P.; Kunz, J.; Deuter-Reinhard, M.; Henriquez, R.; Hall, M.N. TOR1 and TOR2 are structurally and functionally similar but not identical phosphatidylinositol kinase homologues in yeast. Mol. Biol. Cell 1994, 5, 105–118. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.F.; Florentino, D.; Chen, J.; Crabtree, G.R.; Schreiber, S.L. TOR kinase domains are required for two distinct functions, only one of which is inhibited by rapamycin. Cell 1995, 82, 121–130. [Google Scholar] [CrossRef]
- Reinke, A.; Anderson, S.; McCaffery, J.M.; Yates, J., 3rd; Aronova, S.; Chu, S.; Fairclough, S.; Iverson, C.; Wedaman, K.P.; Powers, T. TOR complex 1 includes a novel component, Tco89p (YPL180w), and cooperates with Ssd1p to maintain cellular integrity in Saccharomyces cerevisiae. J. Biol. Chem. 2004, 279, 14752–14762. [Google Scholar] [CrossRef] [PubMed]
- Andrade, M.A.; Bork, P. HEAT repeats in the huntington’s disease protein. Nat. Genet. 1995, 11, 115–116. [Google Scholar] [CrossRef] [PubMed]
- Knutson, B.A. Insights into the domain and repeat architecture of target of rapamycin. J. Struct. Biol. 2010, 170, 354–363. [Google Scholar] [CrossRef] [PubMed]
- Andrade, M.A.; Petosa, C.; O′Donoghue, S.I.; Muller, C.W.; Bork, P. Comparison of ARM and HEAT protein repeats. J. Mol. Biol. 2001, 309, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Heitman, J.; Movva, N.R.; Hall, M.N. Targets for cell cycle arrest by the immunosuppressant rapamycin in yeast. Science 1991, 253, 905–909. [Google Scholar] [CrossRef] [PubMed]
- Cafferkey, R.; Young, P.R.; McLaughlin, M.M.; Bergsma, D.J.; Koltin, Y.; Sathe, G.M.; Faucette, L.; Eng, W.K.; Johnson, R.K.; Livi, G.P. Dominant missense mutations in a novel yeast protein related to mammalian phosphatidylinositol 3-kinase and VPS34 abrogate rapamycin cytotoxicity. Mol. Cell. Biol. 1993, 13, 6012–6023. [Google Scholar] [CrossRef] [PubMed]
- Perry, J.; Kleckner, N. The ATRs, ATMs, and TORs are giant HEAT repeat proteins. Cell 2003, 112, 151–155. [Google Scholar] [CrossRef]
- Gaubitz, C.; Oliveira, T.M.; Prouteau, M.; Leitner, A.; Karuppasamy, M.; Konstantinidou, G.; Rispal, D.; Eltschinger, S.; Robinson, G.C.; Thore, S.; et al. Molecular basis of the rapamycin insensitivity of Target of Rapamycin complex 2. Mol. Cell 2015, 58, 977–988. [Google Scholar] [CrossRef] [PubMed]
- Baretic, D.; Berndt, A.; Ohashi, Y.; Johnson, C.M.; Williams, R.L. TOR forms a dimer through an N-terminal helical solenoid with a complex topology. Nat. Commun. 2016, 7, 11016. [Google Scholar] [CrossRef] [PubMed]
- Karuppasamy, M.; Kusmider, B.; Oliveira, T.M.; Gaubitz, C.; Prouteau, M.; Loewith, R.; Schaffitzel, C. Cryo-EM structure of Saccharomyces cerevisiae Target of Rapamycin complex 2. Nat. Commun. 2017, 8, 1729. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Rudge, D.G.; Koos, J.D.; Vaidialingam, B.; Yang, H.J.; Pavletich, N.P. mTOR kinase structure, mechanism and regulation. Nature 2013, 497, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Aylett, C.H.; Sauer, E.; Imseng, S.; Boehringer, D.; Hall, M.N.; Ban, N.; Maier, T. Architecture of human mTOR complex 1. Science 2016, 351, 48–52. [Google Scholar] [CrossRef] [PubMed]
- Stuttfeld, E.; Aylett, C.H.; Imseng, S.; Boehringer, D.; Scaiola, A.; Sauer, E.; Hall, M.N.; Maier, T.; Ban, N. Architecture of the human mTORC2 core complex. Elife 2018, 7, e33101. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Liu, M.; Tian, Y.; Li, J.; Qi, Y.; Zhao, D.; Wu, Z.; Huang, M.; Wong, C.C.L.; Wang, H.W.; et al. Cryo-EM structure of human mTOR complex 2. Cell Res. 2018, 28, 518. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H.; Sarbassov, D.D.; Ali, S.M.; Latek, R.R.; Guntur, K.V.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. GbetaL, a positive regulator of the rapamycin-sensitive pathway required for the nutrient-sensitive interaction between raptor and mTOR. Mol. Cell 2003, 11, 895–904. [Google Scholar] [CrossRef]
- Wullschleger, S.; Loewith, R.; Oppliger, W.; Hall, M.N. Molecular organization of target of rapamycin complex 2. J. Biol. Chem. 2005, 280, 30697–30704. [Google Scholar] [CrossRef] [PubMed]
- Alarcon, C.M.; Cardenas, M.E.; Heitman, J. Mammalian RAFT1 kinase domain provides rapamycin-sensitive TOR function in yeast. Genes Dev. 1996, 10, 279–288. [Google Scholar] [CrossRef] [PubMed]
- Kamada, Y.; Fujioka, Y.; Suzuki, N.N.; Inagaki, F.; Wullschleger, S.; Loewith, R.; Hall, M.N.; Ohsumi, Y. TOR2 directly phosphorylates the AGC kinase Ypk2 to regulate actin polarization. Mol. Cell Biol. 2005, 25, 7239–7248. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, A.; Kunz, J.; Hall, M.N. TOR2 is required for organization of the actin cytoskeleton in yeast. Proc. Natl. Acad. Sci. USA 1996, 93, 13780–13785. [Google Scholar] [CrossRef] [PubMed]
- Prouteau, M.; Desfosses, A.; Sieben, C.; Bourgoint, C.; Lydia Mozaffari, N.; Demurtas, D.; Mitra, A.K.; Guichard, P.; Manley, S.; Loewith, R. TORC1 organized in inhibited domains (Toroids) regulate TORC1 activity. Nature 2017, 550, 265–269. [Google Scholar] [CrossRef] [PubMed]
- Niles, B.J.; Mogri, H.; Hill, A.; Vlahakis, A.; Powers, T. Plasma membrane recruitment and activation of the AGC kinase Ypk1 is mediated by target of rapamycin complex 2 (TORC2) and its effector proteins Slm1 and Slm2. Proc. Natl. Acad. Sci. USA 2012, 109, 1536–1541. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Weisman, R. The fission yeast TOR proteins and the rapamycin response: An unexpected tale. Curr. Top. Microbiol. Immunol. 2004, 279, 85–95. [Google Scholar] [PubMed]
- Hayashi, T.; Hatanaka, M.; Nagao, K.; Nakaseko, Y.; Kanoh, J.; Kokubu, A.; Ebe, M.; Yanagida, M. Rapamycin sensitivity of the Schizosaccharomyces pombe TOR2 mutant and organization of two highly phosphorylated TOR complexes by specific and common subunits. Genes Cells 2007, 12, 1357–1370. [Google Scholar] [CrossRef] [PubMed]
- Sherman, F. Getting started with yeast. Methods Enzymol. 1991, 194, 3–21. [Google Scholar] [PubMed]
- Gietz, R.D.; Woods, R.A. Transformation of yeast by Lithium acetate/single-stranded carrier DNA/polyethylene glycol method. Methods Enzymol. 2002, 350, 87–96. [Google Scholar] [PubMed]
- Nasmyth, K.; Adolf, G.; Lydall, D.; Seddon, A. The identification of a second cell cycle control on the HO promoter in yeast: Cell cycle regulation of SW15 nuclear entry. Cell 1990, 62, 631–647. [Google Scholar] [CrossRef]
- Reinke, A.; Chen, J.C.; Aronova, S.; Powers, T. Caffeine targets TOR complex I and provides evidence for a regulatory link between the FRB and kinase domains of TOR1p. J. Biol. Chem. 2006, 281, 31616–31626. [Google Scholar] [CrossRef] [PubMed]
- Horton, R.M. PCR-mediated recombination and mutagenesis. SOEing together tailor-made genes. Mol. Biotechnol. 1995, 3, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Sikorski, R.S.; Hieter, P. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics 1989, 122, 19–27. [Google Scholar] [PubMed]
- Aronova, S.; Wedaman, K.; Anderson, S.; Yates, J., 3rd; Powers, T. Probing the membrane environment of the TOR kinases reveals functional interactions between TORC1, actin, and membrane trafficking in Saccharomyces cerevisiae. Mol. Biol. Cell 2007, 18, 2779–2794. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Plasmid | WT 30 °C + Rap | tor1Δ 30 °C + Rap | tor2Δ 30 °C | tor2Δ 37 °C | tor2Δ 37 °C + Sorbitol | tor2Δ 37 °C + YPD | tor2Δ 37 °C + YPD +Sorbitol |
|---|---|---|---|---|---|---|---|
| pRS315 | 0 | 0 | 0 | ND | ND | ND | ND |
| pPL130 | +++++ | +++++ | 0 | ND | ND | ND | ND |
| pPL132 2 | 0 | 0 | 0 | ND | ND | ND | ND |
| pPL089 2 | 0 | 0 | +++++ | +++++ | +++++ | +++++ | +++++ |
| pPL268 | +++++ | +++++ | +++++ | + | ++ | + | +++++ |
| pPL172 2 | ND | ND | +++++ | +++++ | +++++ | +++++ | +++++ |
| pPL271 | +++++ | +++++ | +++++ | 0 | ++ | ++ | +++++ |
| pPL273 | +++++ | +++++ | +++++ | + | ++ | ++++ | +++++ |
| pPL175 | +++++ | +++++ | +++++ | ++ | +++ | ++++ | +++++ |
| pPL176 | +++++ | +++++ | ++++ | + | + | ++ | +++++ |
| pPL212 | ++++ | +++++ | +++++ | + | ++ | ++++ | +++++ |
| pPL180 | +++++ | +++++ | ++++ | 0 | 0 | 0 | + |
| pPL184 | +++ | ++++ | ++++ | 0 | <+ | <+ | ++++ |
| pPL185 | +++++ | +++++ | +++ | 0 | 0 | 0 | <+ |
| pPL209 | +++++ | +++++ | +++ | 0 | 0 | 0 | ++++ |
| pPL214 | ++++ | ++++ | +++++ | 0 | + | 0 | ++++ |
| pPL321 2 | ND | ND | +++++ | +++++ | +++++ | +++++ | +++++ |
| pPL333 2 | ND | ND | +++++ | +++ | ++++ | ++++ | +++++ |
| pPL270 | +++++ | +++++ | 0 | ND | ND | ND | ND |
| pPL173 | +++++ | +++++ | 0 | ND | ND | ND | ND |
| pPL177 | ++++ | +++ | 0 | ND | ND | ND | ND |
| pPL183 | +++ | ++++ | 0 | ND | ND | ND | ND |
| pPL182 | ++++ | ++++ | 0 | ND | ND | ND | ND |
| pPL186 | +++++ | +++++ | 0 | ND | ND | ND | ND |
| Chimera 172 | Chimera 273 | Chimera 212 | Chimera 185 | Chimera 333 | |
|---|---|---|---|---|---|
| Tor2: Intramolecular | 28,174 | 8877 | 9107 | 9667 | 32,603 |
| Tor2: Intermolecular | 193 | 193 | 193 | 0 | 206 |
| Lst8 | 0 | 0 | 0 | 0 | 140 |
| Avo1 | 0 | 0 | 0 | 0 | 143 |
| Avo2 | 86 | 65 | 0 | 21 | 21 |
| Avo3 | 69 | 55 | 48 | 0 | 36 |
| Observed Stable Complex | TORC2 | TORC2 | TORC1 | TORC1 | TORC1/2 |
| Strain | Genotype | Source |
|---|---|---|
| PLY061 | W303a (leu2-3,-112; his3-11,-15; trp1-1; ura3-1; ade2-1; can1-100; ssd1-d) | [47] |
| PLY314 | W303a/α tor2::HIS/TOR2 | [48] |
| PLY497 | W303a tor1::TRP | This study |
| PLY577 | W303a AVO1-13MYC:TRP1 | This study |
| PLY671 | W303a KOG1-13MYC:TRP1 | This study |
| PLY699 | W303a tor2::HIS3 [pPL089] | This study |
| PLY718 | W303a AVO3-13MYC:TRP1 | This study |
| PLY737 | W303a tor2::HIS3 [pPL273] | This study |
| PLY738 | W303α tor2::HIS3 [pPL273] | This study |
| PLY820 | W303α tor2::HIS3 [pPL172] | This study |
| PLY862 | W303α tor2::HIS3 [pPL176] | This study |
| PLY1020 | W303a tor2::HIS3 [pPL212] | This study |
| PLY1029 | W303α tor2::His3 [pPL185] | This study |
| PLY1164 | W303a BIT61-13MYC:TRP1 | This study |
| PLY1283 | W303a tor2::HIS3 [pPL321] | This study |
| PLY1285 | W303a tor2::HIS3 [pPL333] | This study |
| PLY1416 | W303a TCO89-13MYC:TRP1 TOR1::HIS3 | This study |
| PLY1417 | W303a BIT61-13MYC:TRP1 TOR2::HIS3 [pPL172] | This study |
| PLY1418 | W303a BIT61-13MYC:TRP1 TOR2::HIS3 [pPL273] | This study |
| Plasmid | Description | Source |
|---|---|---|
| pRS315 | LEU2 CEN/ARS | [50] |
| pPL089 | LEU2 CEN/ARS TOR2 | [17] |
| pPL130 | LEU2 CEN/ARS TOR1-1 | [48] |
| pPL132 | LEU2 CEN/ARS TOR1 | [48] |
| pPL172 | pPL132, Tor2 114-1770 | This study |
| pPL173 | pPL130 Tor2 428-681 | This study |
| pPL175 | pPL130, Tor2 590-946 | This study |
| pPL176 | pPL130, Tor2 682-946 | This study |
| pPL177 | pPL130, Tor2 787-946 | This study |
| pPL180 | pPL130, Tor2 1019-1770 | This study |
| pPL182 | pPL130, Tor2 1019-1236 | This study |
| pPL183 | pPL130, Tor2 1019-1399 | This study |
| pPL184 | pPL130, Tor2 1019-1542 | This study |
| pPL185 | pPL130, Tor2 1237-1770 | This study |
| pPL186 | pPL130, Tor2 1543-1770 | This study |
| pPL209 | pPL130, Tor2 1400-1770 | This study |
| pPL212 | pPL130, Tor2 682-1236 | This study |
| pPL214 | pPL130, Tor2 428-946 + 1019-1542 | This study |
| pPL268 | pPL130, Tor2 N-946 | This study |
| pPL270 | pPL130, Tor2 N-424 | This study |
| pPL271 | pPL130, Tor2 114-946 | This study |
| pPL273 | pPL130, Tor2 428-946 | This study |
| pPL321 | pPL130, TOR2 | This study |
| pPL333 | pPL321, Tor2 N-424 + 949-C | This study |
| pYE352 | URA3 2-micron YPK2-D239A | [38] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hill, A.; Niles, B.; Cuyegkeng, A.; Powers, T. Redesigning TOR Kinase to Explore the Structural Basis for TORC1 and TORC2 Assembly. Biomolecules 2018, 8, 36. https://doi.org/10.3390/biom8020036
Hill A, Niles B, Cuyegkeng A, Powers T. Redesigning TOR Kinase to Explore the Structural Basis for TORC1 and TORC2 Assembly. Biomolecules. 2018; 8(2):36. https://doi.org/10.3390/biom8020036
Chicago/Turabian StyleHill, Andrew, Brad Niles, Andrew Cuyegkeng, and Ted Powers. 2018. "Redesigning TOR Kinase to Explore the Structural Basis for TORC1 and TORC2 Assembly" Biomolecules 8, no. 2: 36. https://doi.org/10.3390/biom8020036
APA StyleHill, A., Niles, B., Cuyegkeng, A., & Powers, T. (2018). Redesigning TOR Kinase to Explore the Structural Basis for TORC1 and TORC2 Assembly. Biomolecules, 8(2), 36. https://doi.org/10.3390/biom8020036