Antibiofilm Peptides and Peptidomimetics with Focus on Surface Immobilization



Abstract
1. Introduction
2. Antibiofilm Peptides
2.1. Antibiofilm Peptides Tested In Vitro
2.2. Structure-Activity Relationship Studies of Antibiofilm Peptides
2.3. Mechanism of Action
2.4. Antibiofilm Peptides Tested In Vivo
3. Antibiofilm Peptidomimetics
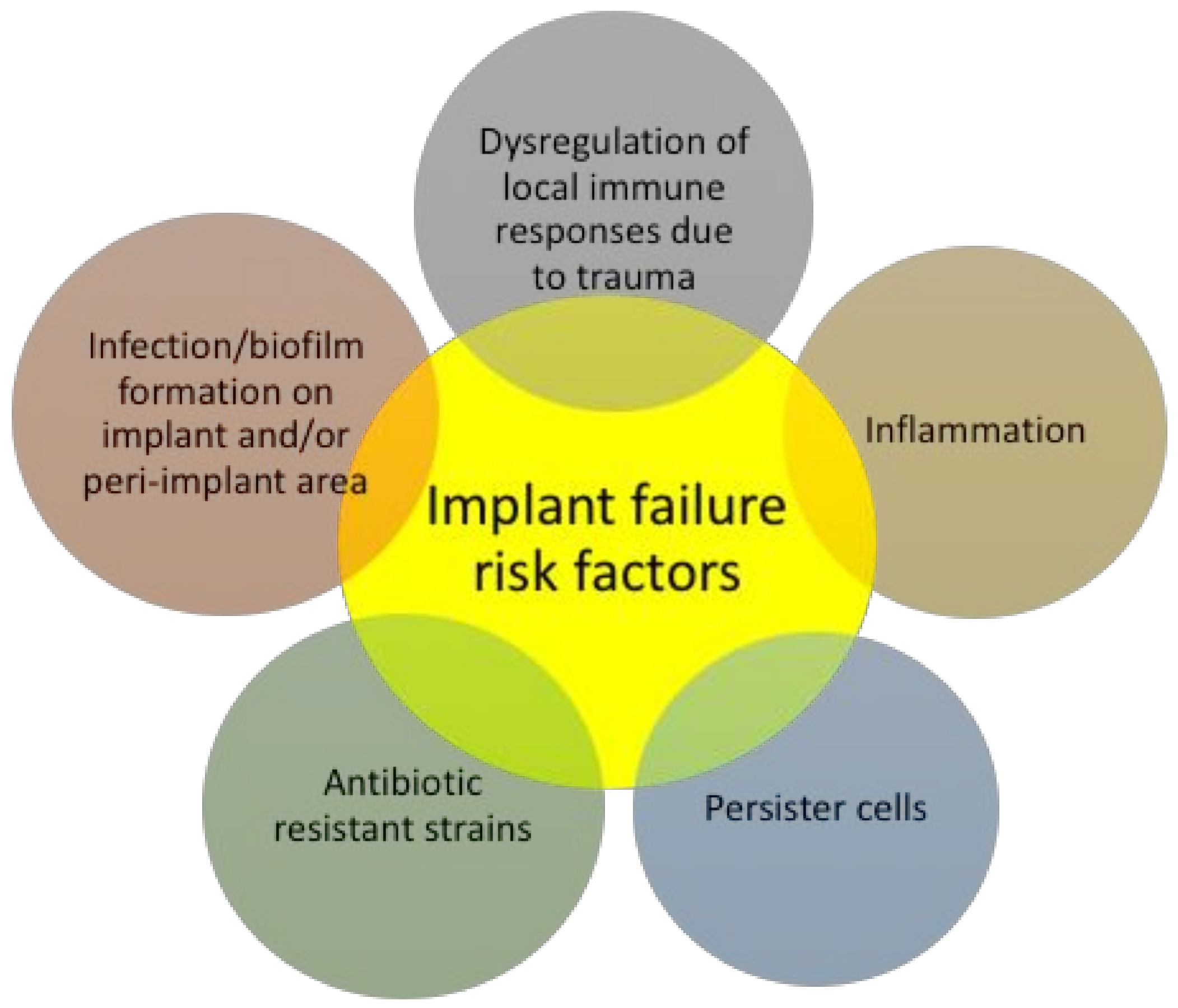
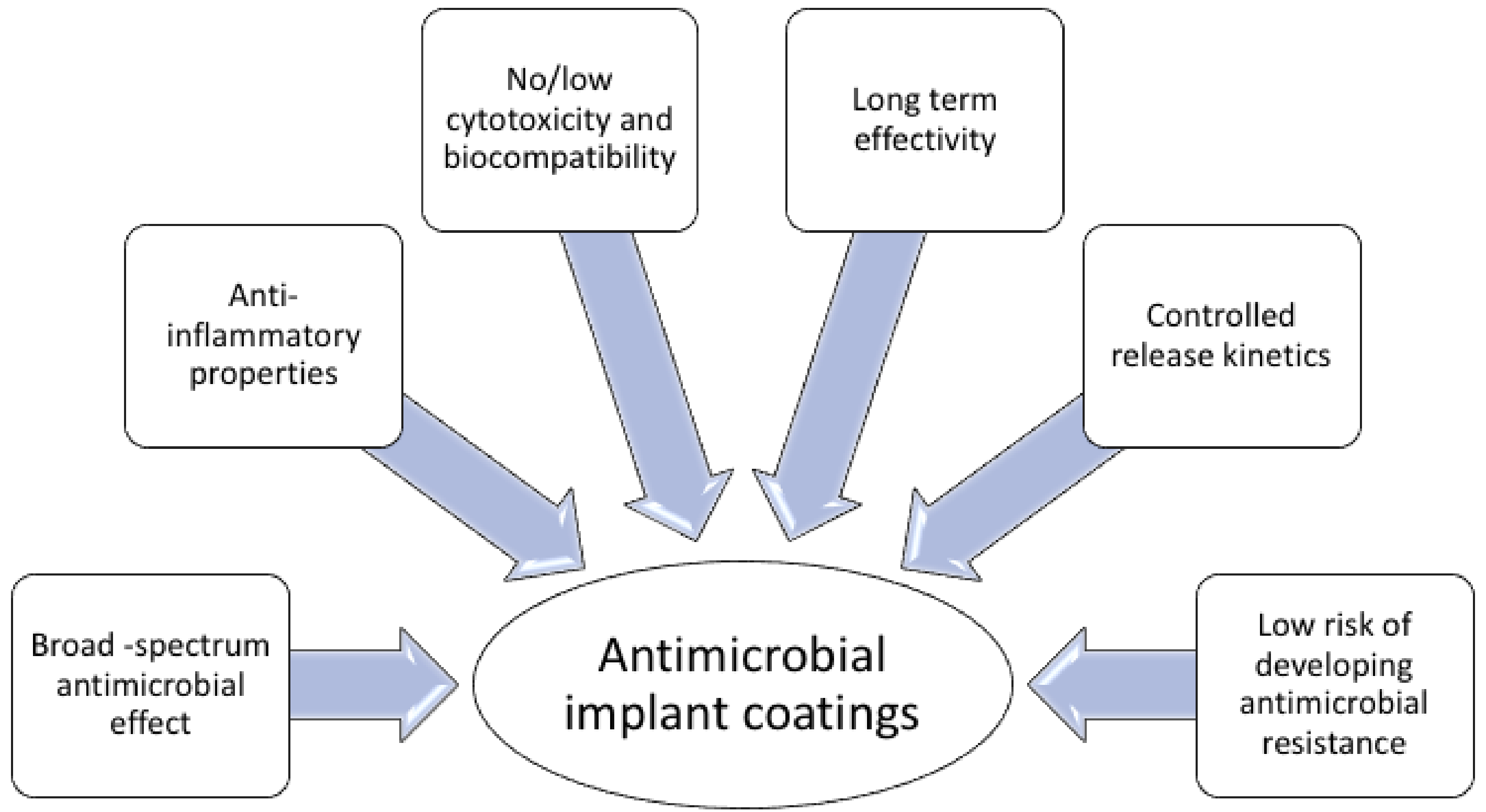
4. Surface-Immobilized Peptides on Medical Implants
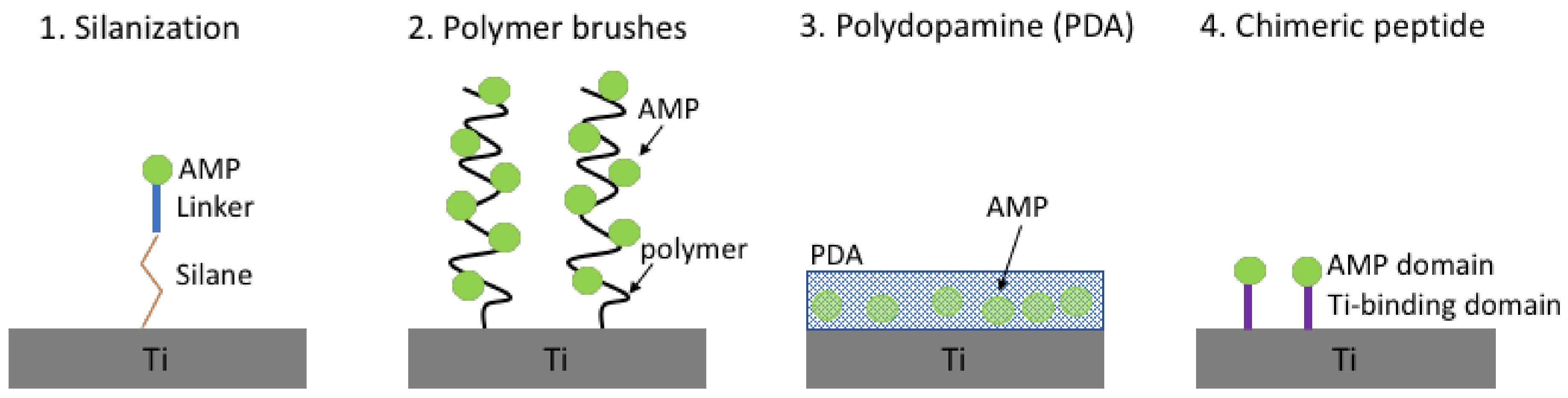
4.1. Antimicrobial Peptides Immobilised on Titanium
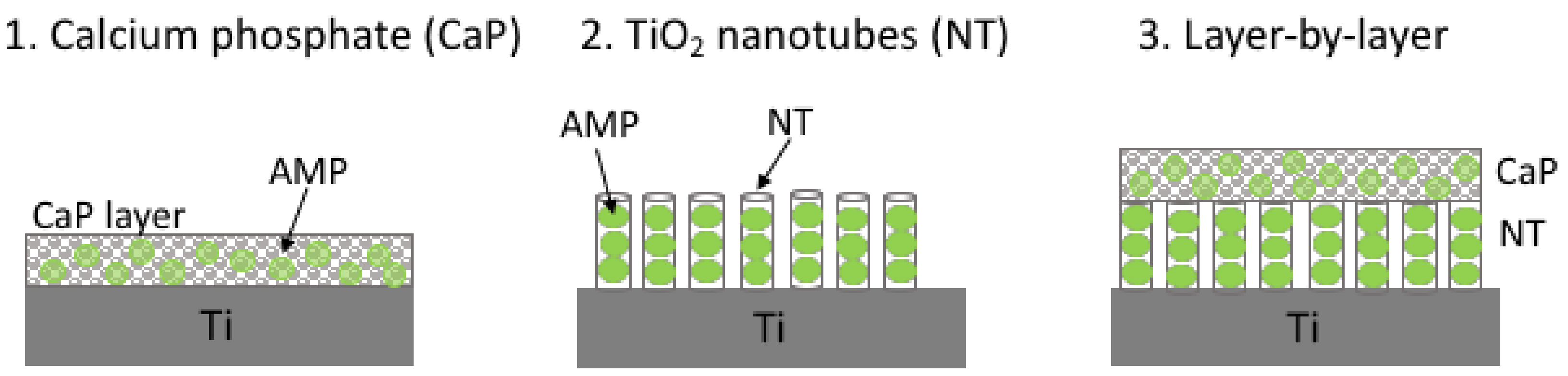
4.2. Antimicrobial Peptides Released from Titanium
4.3. Other Materials
5. Surface-Immobilized Peptidomimetics
6. Conclusions
Acknowledgments
Conflicts of Interest
References
- Costerton, J.W.; Stewart, P.S.; Greenberg, E.P. Bacterial biofilms: A common cause of persistent infections. Science 1999, 284, 1318–1322. [Google Scholar] [CrossRef] [PubMed]
- Satpathy, S.; Sen, S.K.; Pattanaik, S.; Raut, S. Review on bacterial biofilm: An universal cause of contamination. Biocatal. Agric. Biotechnol. 2016, 7, 56–66. [Google Scholar] [CrossRef]
- Flemming, H.C.; Wingender, J. The biofilm matrix. Nat. Rev. Microbiol. 2010, 8, 623–633. [Google Scholar] [CrossRef] [PubMed]
- Shrout, J.D.; Tolker-Nielsen, T.; Givskov, M.; Parsek, M.R. The contribution of cell-cell signaling and motility to bacterial biofilm formation. MRS Bull. 2011, 36, 367–373. [Google Scholar] [CrossRef] [PubMed]
- Fuqua, C.; Greenberg, E.P. Listening in on bacteria: Acyl-homoserine lactone signalling. Nat. Rev. Mol. Cell Biol. 2002, 3, 685–695. [Google Scholar] [CrossRef] [PubMed]
- Taga, M.E.; Bassler, B.L. Chemical communication among bacteria. Proc. Natl. Acad. Sci. USA 2003, 100, 14549–14554. [Google Scholar] [CrossRef] [PubMed]
- Lewis, K. Riddle of biofilm resistance. Antimicrob. Agents Chemother. 2001, 45, 999–1007. [Google Scholar] [CrossRef] [PubMed]
- Lewis, K. Programmed death in bacteria. Microbiol. Mol. Biol. Rev. 2000, 64, 503–514. [Google Scholar] [CrossRef] [PubMed]
- Costerton, W.; Veeh, R.; Shirtliff, M.; Pasmore, M.; Post, C.; Ehrlich, G. The application of biofilm science to the study and control of chronic bacterial infections. J. Clin. Investig. 2003, 112, 1466–1477. [Google Scholar] [CrossRef] [PubMed]
- Römling, U.; Balsalobre, C. Biofilm infections, their resilience to therapy and innovative treatment strategies. J. Intern. Med. 2012, 272, 541–561. [Google Scholar] [CrossRef] [PubMed]
- Claret, L.; Miquel, S.; Vieille, N.; Ryjenkov, D.A.; Gomelsky, M.; Darfeuille-Michaud, A. The flagellar sigma factor FliA regulates adhesion and invasion of Crohn disease-associated Escherichia coli via a cyclic dimeric GMP-dependent pathway. J. Biol. Chem. 2007, 282, 33275–33283. [Google Scholar] [CrossRef] [PubMed]
- Do, T.; Devine, D.; Marsh, P.D. Oral biofilms: Molecular analysis, challenges, and future prospects in dental diagnostics. Clin. Cosmet. Investig. Dent. 2013, 5, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Chenoweth, C.; Saint, S. Preventing catheter-associated urinary tract infections in the intensive care unit. Crit. Care Clin. 2013, 29, 19–32. [Google Scholar] [CrossRef] [PubMed]
- Geipel, U. Pathogenic organisms in hip joint infections. Int. J. Med. Sci. 2009, 6, 234–240. [Google Scholar] [CrossRef] [PubMed]
- Campoccia, D.; Montanaro, L.; Arciola, C.R. The significance of infection related to orthopedic devices and issues of antibiotic resistance. Biomaterials 2006, 27, 2331–2339. [Google Scholar] [CrossRef] [PubMed]
- Magill, S.S.; Edwards, J.R.; Bamberg, W.; Beldavs, Z.G.; Dumyati, G.; Kainer, M.A.; Lynfield, R.; Maloney, M.; McAllister-Hollod, L.; Nadle, J.; et al. Multistate point-prevalence survey of health care–associated infections. N. Engl. J. Med. 2014, 370, 1198–1208. [Google Scholar] [CrossRef] [PubMed]
- Chenoweth, C.E.; Saint, S. Urinary tract infections. Infect. Dis. Clin. N. Am. 2011, 25, 103–115. [Google Scholar] [CrossRef] [PubMed]
- Szaff, M.; Høiby, N.; Flensborg, E.W. Frequent antibiotic therapy improves survival of cystic fibrosis patients with chronic Pseudomonas aeruginosa infection. Acta Paediatr. Scand. 1983, 72, 651–657. [Google Scholar] [CrossRef] [PubMed]
- Høiby, N.; Ciofu, O.; Bjarnsholt, T. Pseudomonas aeruginosa biofilms in cystic fibrosis. Future Microbiol. 2010, 5, 1663–1674. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Chu, P.K.; Zhang, Y.; Wu, Z. Antibacterial coatings on titanium implants. J. Biomed. Mater. Res. B Appl. Biomater. 2009, 91, 470–480. [Google Scholar] [CrossRef] [PubMed]
- Sia, I.G.; Berbari, E.F.; Karchmer, A.W. Prosthetic joint infections. Infect. Dis. Clin. N. Am. 2005, 19, 885–914. [Google Scholar] [CrossRef] [PubMed]
- Stewart, P.S.; Costerton, J.W. Antibiotic resistance of bacteria in biofilms. Lancet 2001, 358, 135–138. [Google Scholar] [CrossRef]
- Shukla, A.; Fleming, K.E.; Chuang, H.F.; Chau, T.M.; Loose, C.R.; Stephanopoulos, G.N.; Hammond, P.T. Controlling the release of peptide antimicrobial agents from surfaces. Biomaterials 2010, 31, 2348–2357. [Google Scholar] [CrossRef] [PubMed]
- Trampuz, A.; Zimmerli, W. Antimicrobial agents in orthopaedic surgery: Prophylaxis and treatment. Drugs 2006, 66, 1089–1105. [Google Scholar] [CrossRef] [PubMed]
- Zilberman, M.; Elsner, J. Antibiotic-eluting medical devices for various applications. J. Control. Release 2008, 130, 202–215. [Google Scholar] [CrossRef] [PubMed]
- Hancock, R.E.W.; Diamond, G. The role of cationic antimicrobial peptides in innate host defences. Trends Microbiol. 2000, 8, 402–410. [Google Scholar] [CrossRef]
- Haney, E.F.; Mansour, S.C.; Hancock, R.E.W. Antimicrobial peptides: An introduction. Methods Mol. Biol. 2017, 1548, 3–22. [Google Scholar] [CrossRef] [PubMed]
- Fan, L.; Sun, J.; Zhou, M.; Zhou, J.; Lao, X.; Zheng, H.; Xu, H. DRAMP: A comprehensive data repository of antimicrobial peptides. Sci. Rep. 2016, 6, 24482. [Google Scholar] [CrossRef] [PubMed]
- Yin, L.M.; Edwards, M.A.; Li, J.; Yip, C.M.; Deber, C.M. Roles of hydrophobicity and charge distribution of cationic antimicrobial peptides in peptide-membrane interactions. J. Biol. Chem. 2012, 287, 7738–7745. [Google Scholar] [CrossRef] [PubMed]
- Bechinger, B.; Gorr, S.U. Antimicrobial peptides: Mechanisms of action and resistance. J. Dent. Res. 2017, 96, 254–260. [Google Scholar] [CrossRef] [PubMed]
- Ehrenstein, G.; Lecar, H. Electrically gated ionic channels in lipid bilayers. Q. Rev. Biophys. 1977, 10, 1–34. [Google Scholar] [CrossRef] [PubMed]
- Yeaman, M.R.; Yount, N.Y. Mechanisms of antimicrobial peptide action and resistance. Pharmacol. Rev. 2003, 55, 27–55. [Google Scholar] [CrossRef] [PubMed]
- Rotem, S.; Mor, A. Antimicrobial peptide mimics for improved therapeutic properties. Biochim. Biophys. Acta 2009, 1788, 1582–1592. [Google Scholar] [CrossRef] [PubMed]
- Straus, S.K.; Hancock, R.E.W. Mode of action of the new antibiotic for Gram-positive pathogens daptomycin: Comparison with cationic antimicrobial peptides and lipopeptides. Biochim. Biophys. Acta 2006, 1758, 1215–1223. [Google Scholar] [CrossRef] [PubMed]
- Guilhelmelli, F.; Vilela, N.; Albuquerque, P.; Derengowski, L.d.S.; Silva-Pereira, I.; Kyaw, C.M. Antibiotic development challenges: The various mechanisms of action of antimicrobial peptides and of bacterial resistance. Front. Microbiol. 2013, 4, 353. [Google Scholar] [CrossRef] [PubMed]
- Molchanova, N.; Hansen, P.R.; Franzyk, H. Advances in development of antimicrobial peptidomimetics as potential drugs. Molecules 2017, 22, 1430. [Google Scholar] [CrossRef] [PubMed]
- Marr, A.K.; Gooderham, W.J.; Hancock, R.E.W. Antibacterial peptides for therapeutic use: Obstacles and realistic outlook. Curr. Opin. Pharmacol. 2006, 6, 468–472. [Google Scholar] [CrossRef] [PubMed]
- Batoni, G.; Maisetta, G.; Esin, S. Antimicrobial peptides and their interaction with biofilms of medically relevant bacteria. Biochim. Biophys. Acta 2016, 1858, 1044–1060. [Google Scholar] [CrossRef] [PubMed]
- De La Fuente-Núñez, C.; Cardoso, M.H.; De Souza Cândido, E.; Franco, O.L.; Hancock, R.E.W. Synthetic antibiofilm peptides. Biochim. Biophys. Acta 2016, 1858, 1061–1069. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Shen, Y.; Haapasalo, M. Antibiofilm peptides against oral biofilms. J. Oral Microbiol. 2017, 9, 1327308. [Google Scholar] [CrossRef] [PubMed]
- Chung, P.Y.; Khanum, R. Antimicrobial peptides as potential anti-biofilm agents against multidrug- resistant bacteria. J. Microbiol. Immunol. Infect. 2017, 50, 405–410. [Google Scholar] [CrossRef] [PubMed]
- Jacobsen, A.S.; Jenssen, H. Human cathelicidin LL-37 prevents bacterial biofilm formation. Future Med. Chem. 2012, 4, 1587–1599. [Google Scholar] [CrossRef] [PubMed]
- Overhage, J.; Campisano, A.; Bains, M.; Torfs, E.C.W.; Rehm, B.H.A.; Hancock, R.E.W. Human host defense peptide LL-37 prevents bacterial biofilm formation. Infect. Immun. 2008, 76, 4176–4182. [Google Scholar] [CrossRef] [PubMed]
- Nagant, C.; Pitts, B.; Nazmi, K.; Vandenbranden, M.; Bolscher, J.G.; Stewart, P.S.; Dehaye, J.P. Identification of peptides derived from the human antimicrobial peptide LL-37 active against biofilms formed by Pseudomonas aeruginosa using a library of truncated fragments. Antimicrob. Agents Chemother. 2012, 56, 5698–5708. [Google Scholar] [CrossRef] [PubMed]
- De La Fuente-Núñez, C.; Korolik, V.; Bains, M.; Nguyen, U.; Breidenstein, E.B.M.; Horsman, S.; Lewenza, S.; Burrows, L.; Hancock, R.E.W. Inhibition of bacterial biofilm formation and swarming motility by a small synthetic cationic peptide. Antimicrob. Agents Chemother. 2012, 56, 2696–2704. [Google Scholar] [CrossRef] [PubMed]
- Haisma, E.M.; de Breij, A.; Chan, H.; van Dissel, J.T.; Drijfhout, J.W.; Hiemstra, P.S.; El Ghalbzouri, A.; Nibbering, P.H. LL-37-derived peptides eradicate multidrug-resistant Staphylococcus aureus from thermally wounded human skin equivalents. Antimicrob. Agents Chemother. 2014, 58, 4411–4419. [Google Scholar] [CrossRef] [PubMed]
- Butts, A.; Krysan, D.J. Antifungal drug discovery: Something old and something new. PLoS Pathog. 2012, 8, E1002870. [Google Scholar] [CrossRef] [PubMed]
- De Brucker, K.; Delattin, N.; Robijns, S.; Steenackers, H.; Verstraeten, N.; Landuyt, B.; Luyten, W.; Schoofs, L.; Dovgan, B.; Froḧlich, M.; et al. Derivatives of the mouse cathelicidin-related antimicrobial peptide (CRAMP) inhibit fungal and bacterial biofilm formation. Antimicrob. Agents Chemother. 2014, 58, 5395–5404. [Google Scholar] [CrossRef] [PubMed]
- Brancatisano, F.L.; Maisetta, G.; Di Luca, M.; Esin, S.; Bottai, D.; Bizzarri, R.; Campa, M.; Batoni, G. Inhibitory effect of the human liver-derived antimicrobial peptide hepcidin 20 on biofilms of polysaccharide intercellular adhesin (PIA)-positive and PIA-negative strains of Staphylococcus epidermidis. Biofouling 2014, 30, 435–446. [Google Scholar] [CrossRef] [PubMed]
- Mansour, S.C.; de la Fuente-Núñez, C.; Hancock, R.E.W. Peptide IDR-1018: Modulating the immune system and targeting bacterial biofilms to treat antibiotic-resistant bacterial infections. J. Pept. Sci. 2015, 21, 323–329. [Google Scholar] [CrossRef] [PubMed]
- De La Fuente-Núñez, C.; Reffuveille, F.; Mansour, S.C.; Reckseidler-Zenteno, S.L.; Hernández, D.; Brackman, G.; Coenye, T.; Hancock, R.E.W. d-enantiomeric peptides that eradicate wild-type and multidrug-resistant biofilms and protect against lethal Pseudomonas aeruginosa infections. Chem. Biol. 2015, 22, 196–205. [Google Scholar] [CrossRef] [PubMed]
- Mataraci, E.; Dosler, S. In vitro activities of antibiotics and antimicrobial cationic peptides alone and in combination against methicillin-resistant Staphylococcus aureus biofilms. Antimicrob. Agents Chemother. 2012, 56, 6366–6371. [Google Scholar] [CrossRef] [PubMed]
- Anunthawan, T.; de la Fuente-Nunez, C.; Hancock, R.E.W.; Klaynongsruang, S. Cationic amphipathic peptides KT2 and RT2 are taken up into bacterial cells and kill planktonic and biofilm bacteria. Biochim. Biophys. Acta 2015, 1848, 1352–1358. [Google Scholar] [CrossRef] [PubMed]
- Almaaytah, A.; Qaoud, M.T.; Mohammed, G.K.; Abualhaijaa, A.; Knappe, D.; Hoffmann, R.; Al-Balas, Q. Antimicrobial and antibiofilm activity of UP-5, an ultrashort antimicrobial peptide designed using only arginine and biphenylalanine. Pharmaceuticals 2018, 11, 3. [Google Scholar] [CrossRef] [PubMed]
- Di Luca, M.; Maccari, G.; Nifosí, R. Treatment of microbial biofilms in the post-antibiotic era: Prophylactic and therapeutic use of antimicrobial peptides and their design by bioinformatics tools. Pathog. Dis. 2014, 70, 257–270. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Baeder, D.Y.; Regoes, R.R.; Rolff, J. Combination effects of antimicrobial peptides. Antimicrob. Agents Chemother. 2016, 60, 1717–1724. [Google Scholar] [CrossRef] [PubMed]
- Cassone, M.; Otvos, L. Synergy among antibacterial peptides and between peptides and small-molecule antibiotics. Expert Rev. Anti-Infect. Ther. 2010, 8, 703–716. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Moser, C.; Wang, H.Z.; Høiby, N.; Song, Z.J. Strategies for combating bacterial biofilm infections. Int. J. Oral Sci. 2015, 7, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Reffuveille, F.; de La Fuente-Nú̃nez, C.; Mansour, S.; Hancock, R.E.W. A broad-spectrum antibiofilm peptide enhances antibiotic action against bacterial biofilms. Antimicrob. Agents Chemother. 2014, 58, 5363–5371. [Google Scholar] [CrossRef] [PubMed]
- Di Luca, M.; Maccari, G.; Maisetta, G.; Batoni, G. BaAMPs: The database of biofilm-active antimicrobial peptides. Biofouling 2015, 31, 193–199. [Google Scholar] [CrossRef] [PubMed]
- De la Fuente-Núñez, C.; Reffuveille, F.; Haney, E.F.; Straus, S.K.; Hancock, R.E.W. Broad-spectrum anti-biofilm peptide that targets a cellular stress response. PLoS Pathog. 2014, 10, E1004152. [Google Scholar] [CrossRef] [PubMed]
- Haney, E.F.; Brito-Sanchez, Y.; Trimble, M.J.; Mansour, S.C.; Cherkasov, A.; Hancock, R.E.W. Computer-aided discovery of peptides that specifically attack bacterial biofilms. Sci. Rep. 2018, 8, 1871. [Google Scholar] [CrossRef] [PubMed]
- Von Borowski, R.G.; Macedo, A.J.; Gnoatto, S.C.B. Peptides as a strategy against biofilm-forming microorganisms: Structure-activity relationship perspectives. Eur. J. Pharm. Sci. 2018, 114, 114–137. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Tan, H.; Cheng, T.; Shen, H.; Shao, J.; Guo, Y.; Shi, S.; Zhang, X. Human β-defensin 3 inhibits antibiotic-resistant Staphylococcus biofilm formation. J. Surg. Res. 2013, 183, 204–213. [Google Scholar] [CrossRef] [PubMed]
- Pletzer, D.; Wolfmeier, H.; Bains, M.; Hancock, R.E.W. Synthetic peptides to target stringent response-controlled virulence in a Pseudomonas aeruginosa murine cutaneous infection model. Front. Microbiol. 2017, 8, 1867. [Google Scholar] [CrossRef] [PubMed]
- Lebeaux, D.; Chauhan, A.; Rendueles, O.; Beloin, C. From in vitro to in vivo models of bacterial biofilm-related infections. Pathogens 2013, 2, 288–356. [Google Scholar] [CrossRef] [PubMed]
- Sieprawska-Lupa, M.; Mydel, P.; Krawczyk, K.; Wójcik, K.; Puklo, M.; Lupa, B.; Suder, P.; Silberring, J.; Reed, M.; Pohl, J.; et al. Degradation of human antimicrobial peptide LL-37 by Staphylococcus aureus-derived proteinases. Antimicrob. Agents Chemother. 2004, 48, 4673–4679. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, M.F.; Brezden, A.; Mohammad, H.; Chmielewski, J.; Seleem, M.N. A short d-enantiomeric antimicrobial peptide with potent immunomodulatory and antibiofilm activity against multidrug-resistant Pseudomonas aeruginosa and Acinetobacter baumannii. Sci. Rep. 2017, 7, 6953. [Google Scholar] [CrossRef] [PubMed]
- Edwards, S.; Kjellerup, B.V. Exploring the applications of invertebrate host-pathogen models for in vivo biofilm infections. FEMS Immunol. Med. Microbiol. 2012, 65, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Glavis-Bloom, J.; Muhammed, M.; Mylonakis, E. Of model hosts and man: Using Caenorhabditis elegans, Drosophila melanogaster and Galleria mellonella as model hosts for infectious disease research. Adv. Exp. Med. Biol. 2012, 710, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Breij, A.D.; Riool, M.; Cordfunke, R.A.; Malanovic, N.; Boer, L.D.; Koning, R.I.; Ravensbergen, E.; Franken, M.; Heijde, T.V.D.; Boekema, B.K.; et al. The antimicrobial peptide SAAP-148 combats drug-resistant bacteria and biofilms. Sci. Transl. Med. 2018, 10, Eaan4044. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Han, J.; Chang, B.; Gao, L.; Lu, Z.; Lu, F.; Zhao, H.; Zhang, C.; Bie, X. Membrane-active amphipathic peptide WRL3 with in vitro antibiofilm capability and in vivo efficacy in treating methicillin-resistant Staphylococcus aureus burn wound infections. ACS Infect. Dis. 2017, 3, 820–832. [Google Scholar] [CrossRef] [PubMed]
- Azeredo, J.; Azevedo, N.F.; Briandet, R.; Cerca, N.; Coenye, T.; Costa, A.R.; Desvaux, M.; Di Bonaventura, G.; Hébraud, M.; Jaglic, Z.; et al. Critical review on biofilm methods. Crit. Rev. Microbiol. 2017, 43, 313–351. [Google Scholar] [CrossRef] [PubMed]
- Shin, H.M.; Kang, C.M.; Yoon, M.H.; Seo, J. Peptoid helicity modulation: Precise control of peptoid secondary structures via position-specific placement of chiral monomers. Chem. Commun. 2014, 50, 4465–4468. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, R.; Wadman, M.W.; Dohm, M.T.; Czyzewski, A.M.; Spormann, A.M.; Barron, A.E. Antimicrobial peptoids are effective against Pseudomonas aeruginosa biofilms. Antimicrob. Agents Chemother. 2011, 55, 3054–3057. [Google Scholar] [CrossRef] [PubMed]
- Padhee, S.; Li, Y.; Cai, J. Activity of lipo-cyclic γ-AApeptides against biofilms of Staphylococcus epidermidis and Pseudomonas aeruginosa. Bioorg. Med. Chem. Lett. 2015, 25, 2565–2569. [Google Scholar] [CrossRef] [PubMed]
- Jahnsen, R.D.; Haney, E.F.; Franzyk, H.; Hancock, R.E.W. Characterization of a proteolytically stable multifunctional host defense peptidomimetic. Chem. Biol. 2013, 20, 1286–1295. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Knapp, K.M.; Yang, L.; Molin, S.; Franzyk, H.; Folkesson, A. High in vitro antimicrobial activity of β-peptoid-peptide hybrid oligomers against planktonic and biofilm cultures of Staphylococcus epidermidis. Int. J. Antimicrob. Agents 2013, 41, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Ahn, M.; Gunasekaran, P.; Rajasekaran, G.; Kim, E.Y.; Lee, S.J.; Bang, G.; Cho, K.; Hyun, J.K.; Lee, H.J.; Jeon, Y.H.; et al. Pyrazole derived ultra-short antimicrobial peptidomimetics with potent anti-biofilm activity. Eur. J. Med. Chem. 2017, 125, 551–564. [Google Scholar] [CrossRef] [PubMed]
- Meir, O.; Zaknoon, F.; Cogan, U.; Mor, A. A broad-spectrum bactericidal lipopeptide with anti-biofilm properties. Sci. Rep. 2017, 7, 2198. [Google Scholar] [CrossRef] [PubMed]
- Kuppusamy, R.; Yasir, M.; Berry, T.; Cranfield, C.G.; Nizalapur, S.; Yee, E.; Kimyon, O.; Taunk, A.; Ho, K.K.K.; Cornell, B.; et al. Design and synthesis of short amphiphilic cationic peptidomimetics based on biphenyl backbone as antibacterial agents. Eur. J. Med. Chem. 2018, 143, 1702–1722. [Google Scholar] [CrossRef] [PubMed]
- Dewangan, R.P.; Joshi, S.; Kumari, S.; Gautam, H.; Yar, M.S.; Pasha, S. N-terminally modified linear and branched spermine backbone dipeptidomimetics against planktonic and sessile methicillin-resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 2014, 58, 5435–5447. [Google Scholar] [CrossRef] [PubMed]
- Zhang, E.; Bai, P.Y.; Cui, D.Y.; Chu, W.C.; Hua, Y.G.; Liu, Q.; Yin, H.Y.; Zhang, Y.J.; Qin, S.; Liu, H.M. Synthesis and bioactivities study of new antibacterial peptide mimics: The dialkyl cationic amphiphiles. Eur. J. Med. Chem. 2018, 143, 1489–1509. [Google Scholar] [CrossRef] [PubMed]
- Neut, D.; van de Belt, H.; van Horn, J.R.; van der Mei, H.C.; Busscher, H.J. Residual gentamicin-release from antibiotic-loaded polymethylmethacrylate beads after 5 years of implantation. Biomaterials 2003, 24, 1829–1831. [Google Scholar] [CrossRef]
- Thomas, M.V.; Puleo, D.A. Infection, inflammation, and bone regeneration: A paradoxical relationship. J. Dent. Res. 2011, 90, 1052–1061. [Google Scholar] [CrossRef] [PubMed]
- Stanford, C.M. Surface modification of biomedical and dental implants and the processes of inflammation, wound healing and bone formation. Int. J. Mol. Sci. 2010, 11, 354–369. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Lin, Z.; Ding, J.; Huang, W.; Chen, J.; Wu, D. Inflammatory and biocompatibility evaluation of antimicrobial peptide GL13K immobilized onto titanium by silanization. Colloids Surf. B Biointerfaces 2017, 160, 581–588. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Xia, X.; Xu, L.; Wang, Y. Design of hybrid β-hairpin peptides with enhanced cell specificity and potent anti-inflammatory activity. Biomaterials 2013, 34, 237–250. [Google Scholar] [CrossRef] [PubMed]
- Capparelli, R.; De Chiara, F.; Nocerino, N.; Montella, R.C.; Iannaccone, M.; Fulgione, A.; Romanelli, A.; Avitabile, C.; Blaiotta, G.; Capuano, F. New perspectives for natural antimicrobial peptides: Application as antinflammatory drugs in a murine model. BMC Immunol. 2012, 13, 61. [Google Scholar] [CrossRef] [PubMed]
- Kanellakopoulou, K.; Giamarellos-Bourboulis, E.J. Carrier systems for the local delivery of antibiotics in bone infections. Drugs 2000, 59, 1223–1232. [Google Scholar] [CrossRef] [PubMed]
- Eltorai, A.E.M.; Haglin, J.; Perera, S.; Brea, B.A.; Ruttiman, R.; Garcia, D.R.; Born, C.T.; Daniels, A.H. Antimicrobial technology in orthopedic and spinal implants. World J. Orthop. 2016, 7, 361–369. [Google Scholar] [CrossRef] [PubMed]
- Stigter, M.; Bezemer, J.; de Groot, K.; Layrolle, P. Incorporation of different antibiotics into carbonated hydroxyapatite coatings on titanium implants, release and antibiotic efficacy. J. Control. Release 2004, 99, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Price, J.S.; Tencer, A.F.; Arm, D.M.; Bohach, G.A. Controlled release of antibiotics from coated orthopedic implants. J. Biomed. Mater. Res. 1996, 30, 281–286. [Google Scholar] [CrossRef]
- Willcox, M.D.P.; Hume, E.B.H.; Aliwarga, Y.; Kumar, N.; Cole, N. A novel cationic-peptide coating for the prevention of microbial colonization on contact lenses. J. Appl. Microbiol. 2008, 105, 1817–1825. [Google Scholar] [CrossRef] [PubMed]
- Rathbone, C.R.; Cross, J.D.; Brown, K.V.; Murray, C.K.; Wenke, J.C. Effect of various concentrations of antibiotics on osteogenic cell viability and activity. J. Orthop. Res. 2011, 29, 1070–1074. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Willcox, M.D.P.; Ka, K.; Ho, K.; Smyth, D.; Kumar, N. Antimicrobial peptide melimine coating for titanium and its in vivo antibacterial activity in rodent subcutaneous infection models. Biomaterials 2016, 85, 142–151. [Google Scholar] [CrossRef] [PubMed]
- Gao, G.; Yu, K.; Kindrachuk, J.; Brooks, D.E.; Hancock, R.E.W.; Kizhakkedathu, J.N. Antibacterial surfaces based on polymer brushes: Investigation on the influence of brush properties on antimicrobial peptide immobilization and antimicrobial activity. Biomacromolecules 2011, 12, 3715–3727. [Google Scholar] [CrossRef] [PubMed]
- Gao, G.; Lange, D.; Hilpert, K.; Kindrachuk, J.; Zou, Y.; Cheng, J.T.J.; Kazemzadeh-Narbat, M.; Yu, K.; Wang, R.; Straus, S.K.; et al. The biocompatibility and biofilm resistance of implant coatings based on hydrophilic polymer brushes conjugated with antimicrobial peptides. Biomaterials 2011, 32, 3899–3909. [Google Scholar] [CrossRef] [PubMed]
- Costa, F.M.T.A.; Maia, S.R.; Gomes, P.A.C.; Cristina, M.; Martins, L. Dhvar5 antimicrobial peptide (AMP) chemoselective covalent immobilization results on higher antiadherence effect than simple physical adsorption. Biomaterials 2015, 52, 531–538. [Google Scholar] [CrossRef] [PubMed]
- Veenstra, D.L.; Saint, S.; Saha, S.; Lumley, T.; Sullivan, S.D. Efficacy of antiseptic-impregnated central venous catheters in preventing catheter-related bloodstream infection. JAMA 1999, 281, 261–267. [Google Scholar] [CrossRef] [PubMed]
- Mishra, B.; Basu, A.; Chua, R.R.Y.; Saravanan, R.; Tambyah, P.A.; Ho, B.; Chang, M.W.; Leong, S.S.J. Site specific immobilization of a potent antimicrobial peptide onto silicone catheters: Evaluation against urinary tract infection pathogens. J. Mater. Chem. B 2014, 2, 1706. [Google Scholar] [CrossRef]
- Lim, K.; Chua, R.R.Y.; Ho, B.; Tambyah, P.A.; Hadinoto, K.; Leong, S.S.J. Development of a catheter functionalized by a polydopamine peptide coating with antimicrobial and antibiofilm properties. Acta Biomater. 2015, 15, 127–138. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Li, P.; Saravanan, R.; Basu, A.; Mishra, B.; Lim, S.H.; Su, X.; Tambyah, P.A.; Leong, S.S.J.; Su, S.; et al. Antimicrobial functionalization of silicone surfaces with engineered short peptides having broad spectrum antimicrobial and salt-resistant properties. Acta Biomater. 2014, 10, 258–266. [Google Scholar] [CrossRef] [PubMed]
- Goddard, J.M.; Hotchkiss, J.H. Polymer surface modification for the attachment of bioactive compounds. Prog. Polym. Sci. 2007, 32, 698–725. [Google Scholar] [CrossRef]
- Hilpert, K.; Elliott, M.; Jenssen, H.; Kindrachuk, J.; Fjell, C.D.; Körner, J.; Winkler, D.F.H.; Weaver, L.L.; Henklein, P.; Ulrich, A.S.; et al. Screening and characterization of surface-tethered cationic peptides for antimicrobial activity. Chem. Biol. 2009, 16, 58–69. [Google Scholar] [CrossRef] [PubMed]
- Koon, Q.; Neoh, G.; Hu, X.; Zheng, D.; Kang, E.T. Balancing osteoblast functions and bacterial adhesion on functionalized titanium surfaces. Biomaterials 2012, 33, 2813–2822. [Google Scholar] [CrossRef]
- Zimmerli, W.; Sendi, P. Pathogenesis of implant-associated infection: The role of the host. Semin. Immunopathol. 2011, 33, 295–306. [Google Scholar] [CrossRef] [PubMed]
- Broekhuizen, C.A.N.; Sta, M.; Vandenbroucke-Grauls, C.M.J.E.; Zaat, S.A.J. Microscopic detection of viable Staphylococcus epidermidis in peri-implant tissue in experimental biomaterial-associated infection, identified by bromodeoxyuridine incorporation. Infect. Immun. 2010, 78, 954–962. [Google Scholar] [CrossRef] [PubMed]
- Boelens, J.J.; van der Poll, T.; Zaat, S.A.; Murk, J.L.; Weening, J.J.; Dankert, J. Interleukin-1 receptor type I gene-deficient mice are less susceptible to Staphylococcus epidermidis biomaterial-associated infection than are wild-type mice. Infect. Immun. 2000, 68, 6924–6931. [Google Scholar] [CrossRef] [PubMed]
- Boelens, J.; van der Poll, T.S.; Dankert, J.F.; Zaat, S.A.J. Interferon-γ protects against biomaterial-associated Staphylococcus epidermidis infection in mice. J. Infect. Dis. 2000, 181, 1167–1171. [Google Scholar] [CrossRef] [PubMed]
- Boelens, J.J.; Dankert, J.; Murk, J.L.; Weening, J.J.; van der Poll, T.; Dingemans, K.P.; Koole, L.; Laman, J.D.; Zaat, S.A.J. Biomaterial-associated persistence of Staphylococcus epidermidis in pericatheter macrophages. J. Infect. Dis. 2000, 181, 1337–1349. [Google Scholar] [CrossRef] [PubMed]
- Riool, M.; de Breij, A.; Drijfhout, J.W.; Nibbering, P.H.; Zaat, S.A.J. Antimicrobial peptides in biomedical device manufacturing. Front. Chem. 2017, 5, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Nie, B.E.; Ao, H.; Long, T.; Zhou, J.; Tang, T.; Yue, B. Immobilizing bacitracin on titanium for prophylaxis of infections and for improving osteoinductivity: An in vivo study. Colloids Surf. B Biointerfaces 2017, 150, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Tan, X.W.; Goh, T.W.; Saraswathi, P.; Nyein, C.L.; Setiawan, M.; Riau, A.; Lakshminarayanan, R.; Liu, S.; Tan, D.; Beuerman, R.W.; et al. Effectiveness of antimicrobial peptide immobilization for preventing perioperative cornea implant-associated bacterial infection. Antimicrob. Agents Chemother. 2014, 58, 5229–5238. [Google Scholar] [CrossRef] [PubMed]
- De Breij, A.; Riool, M.; Kwakman, P.H.S.; De Boer, L.; Cordfunke, R.A.; Drijfhout, J.W.; Cohen, O.; Emanuel, N.; Zaat, S.A.J.; Nibbering, P.H.; et al. Prevention of Staphylococcus aureus biomaterial-associated infections using a polymer-lipid coating containing the antimicrobial peptide OP-145. J. Control. Release 2016, 222, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Riool, M.; de Breij, A.; de Boer, L.; Kwakman, P.H.S.; Cordfunke, R.A.; Cohen, O.; Malanovic, N.; Emanuel, N.; Lohner, K.; Drijfhout, J.W.; et al. Controlled release of LL-37-derived synthetic antimicrobial and anti-biofilm peptides SAAP-145 and SAAP-276 prevents experimental biomaterial-associated Staphylococcus aureus infection. Adv. Funct. Mater. 2017, 27, 1606623. [Google Scholar] [CrossRef]
- Shi, J.; Liu, Y.; Wang, Y.; Zhang, J.; Zhao, S.; Yang, G. Biological and immunotoxicity evaluation of antimicrobial peptide-loaded coatings using a layer-by-layer process on titanium. Sci. Rep. 2015, 5, 16336. [Google Scholar] [CrossRef] [PubMed]
- Sawant, S.N.; Selvaraj, V.; Prabhawathi, V.; Doble, M. Antibiofilm properties of silver and gold incorporated PU, PCLm, PC and PMMA nanocomposites under two shear conditions. PLoS ONE 2013, 8, E63311. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Hirt, H.; Li, Y.; Gorr, S.U.; Aparicio, C. Antimicrobial GL13K peptide coatings killed and ruptured the wall of Streptococcus gordonii and prevented formation and growth of biofilms. PLoS ONE 2014, 9, e111579. [Google Scholar] [CrossRef] [PubMed]
- Tschernitschek, H.; Borchers, L.; Geurtsen, W. Nonalloyed titanium as a bioinert metal—A review. Quintessence Int. 1985, 36, 523–530. [Google Scholar] [CrossRef]
- Gabriel, M.; Nazmi, K.; Veerman, E.C.; Amerongen, A.V.N.; Zentner, A. Preparation of LL-37-grafted titanium surfaces with bactericidal activity. Bioconj. Chem. 2006, 17, 548–550. [Google Scholar] [CrossRef] [PubMed]
- Piehler, J.; Brecht, A.; Valiokas, R.; Liedberg, B.; Gauglitz, G. A high-density poly(ethylene glycol) polymer brush for immobilization on glass-type surfaces. Biosens. Bioelectron. 2000, 15, 473–481. [Google Scholar] [CrossRef]
- Godoy-Gallardo, M.; Mas-Moruno, C.; Fernández-Calderón, M.C.; Pérez-Giraldo, C.; Manero, J.M.; Albericio, F.; Gil, F.J.; Rodríguez, D. Covalent immobilization of hLf1-11 peptide on a titanium surface reduces bacterial adhesion and biofilm formation. Acta Biomater. 2014, 10, 3522–3534. [Google Scholar] [CrossRef] [PubMed]
- De Zoysa, G.H.; Sarojini, V. Feasibility study exploring the potential of novel battacin lipopeptides as antimicrobial coatings. ACS Appl. Mater. Interfaces 2017, 9, 1373–1383. [Google Scholar] [CrossRef] [PubMed]
- Godoy-Gallardo, M.; Mas-Moruno, C.; Yu, K.; Manero, J.M.; Gil, F.J.; Kizhakkedathu, J.N.; Rodriguez, D. Antibacterial properties of hLf1-11 peptide onto titanium surfaces: A comparison study between silanization and surface initiated polymerization. Biomacromolecules 2015, 16, 483–496. [Google Scholar] [CrossRef] [PubMed]
- Holmberg, K.V.; Abdolhosseini, M.; Li, Y.; Chen, X.; Gorr, S.-U.; Aparicio, C. Bio-inspired stable antimicrobial peptide coatings for dental applications. Acta Biomater. 2013, 9, 8224–8231. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Ma, S.; Duan, S.; Xuliang, D.; Sun, Y.; Zhang, X.; Xu, X.; Guan, B.; Wang, C.; Hu, M.; et al. Modification of titanium substrates with chimeric peptides comprising antimicrobial and titanium-binding motifs connected by linkers to inhibit biofilm formation. ACS Appl. Mater. Interfaces 2016, 8, 5124–5136. [Google Scholar] [CrossRef] [PubMed]
- Tan, X.W.; Lakshminarayanan, R.; Liu, S.P.; Goh, E.; Tan, D.; Beuerman, R.W.; Mehta, J.S. Dual functionalization of titanium with vascular endothelial growth factor and β-defensin analog for potential application in keratoprosthesis. J. Biomed. Mater. Res. B Appl. Biomater. 2012, 100, 2090–2100. [Google Scholar] [CrossRef] [PubMed]
- Yucesoy, D.T.; Hnilova, M.; Boone, K.; Arnold, P.M.; Snead, M.L.; Tamerler, C. Chimeric peptides as implant functionalization agents for titanium alloy implants with antimicrobial properties. JOM 2015, 67, 754–766. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Yue, K.; Kazemzadeh-Narbat, M.; Liu, Y.; Khalilpour, A.; Li, B.; Zhang, Y.S.; Annabi, N.; Khademhosseini, A. Mussel-inspired multifunctional hydrogel coating for prevention of infections and enhanced osteogenesis. ACS Appl. Mater. Interfaces 2017, 9, 11428–11439. [Google Scholar] [CrossRef] [PubMed]
- Kazemzadeh-Narbat, M.; Kindrachuk, J.; Duan, K.; Jenssen, H.; Hancock, R.E.W.; Wang, R. Antimicrobial peptides on calcium phosphate-coated titanium for the prevention of implant-associated infections. Biomaterials 2010, 31, 9519–9526. [Google Scholar] [CrossRef] [PubMed]
- Kazemzadeh-Narbat, M.; Lai, B.F.L.; Ding, C.; Kizhakkedathu, J.N.; Hancock, R.E.W.; Wang, R. Multilayered coating on titanium for controlled release of antimicrobial peptides for the prevention of implant-associated infections. Biomaterials 2013, 34, 5969–5977. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Wang, N.; Chen, S.; Lu, R.; Li, H.; Zhang, Z. Antibacterial activity and cytocompatibility of an implant coating consisting of TiO2 nanotubes combined with a GL13K antimicrobial peptide. Int. J. Nanomed. 2017, 12, 2995–3007. [Google Scholar] [CrossRef] [PubMed]
- Ma, M.; Kazemzadeh-Narbat, M.; Hui, Y.; Lu, S.; Ding, C.; Chen, D.D.Y.; Hancock, R.E.W.; Wang, R. Local delivery of antimicrobial peptides using self-organized TiO2 nanotube arrays for peri-implant infections. J. Biomed. Mater. Res. A 2012, 100, 278–285. [Google Scholar] [CrossRef] [PubMed]
- Mateescu, M.; Baixe, S.; Garnier, T.; Jierry, L.; Ball, V.; Haikel, Y.; Metz-Boutigue, M.H.; Nardin, M.; Schaaf, P.; Etienne, O.; et al. Antibacterial peptide-based gel for prevention of medical implanted-device infection. PLoS ONE 2015, 10, E0145143. [Google Scholar] [CrossRef] [PubMed]
- Whiting, G.L.; Farhan, T.; Huck, W.T.S. Polymer brushes: Towards applications. In Polymer Brushes; Advincula, R.C., Brittain, W.J., Caster, K.C., Rühe, J., Eds.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2005. [Google Scholar] [CrossRef]
- Edmondson, S.; Osborne, V.L.; Huck, W.T.S. Polymer brushes via surface-initiated polymerizations. Chem. Soc. Rev. 2004, 33, 14–22. [Google Scholar] [CrossRef] [PubMed]
- Nie, B.E.; Ao, H.; Zhou, J.; Tang, T.; Yue, B. Biofunctionalization of titanium with bacitracin immobilization shows potential for anti-bacteria, osteogenesis and reduction of macrophage inflammation. Colloids Surf. B Biointerfaces 2016, 145, 728–739. [Google Scholar] [CrossRef] [PubMed]
- Waite, J.H.; Qin, X. Polyphosphoprotein from the adhesive pads of Mytilus edulis. Biochemistry 2001, 40, 2887–2893. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Dellatore, S.M.; Miller, W.M.; Messersmith, P.B. Mussel-inspired surface chemistry for multifunctional coatings. Science 2007, 318, 426–430. [Google Scholar] [CrossRef] [PubMed]
- Waite, J.H. Evidence for a repeating 3,4-dihydroxyphenylalanine- and hydroxyproline-containing decapeptide in the adhesive protein of the mussel, Mytilus edulis L. J. Biol. Chem. 1983, 258, 2911–2915. [Google Scholar] [PubMed]
- Lee, H.; Rho, J.; Messersmith, P.B. Facile conjugation of biomolecules onto surfaces via mussel adhesive protein inspired coatings. Adv. Mater. 2009, 21, 431–434. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Scherer, N.F.; Messersmith, P.B. Single-molecule mechanics of mussel adhesion. Proc. Natl. Acad. Sci. USA 2006, 103, 12999–13003. [Google Scholar] [CrossRef] [PubMed]
- Dalsin, J.L.; Hu, B.-H.; Lee, B.P.; Messersmith, P.B. Mussel adhesive protein mimetic polymers for the preparation of nonfouling surfaces. J. Am. Chem. Soc. 2003, 125, 4253–4258. [Google Scholar] [CrossRef] [PubMed]
- Dalsin, J.L.; Lin, L.; Tosatti, S.; Vörös, J.; Textor, M.; Messersmith, P.B. Protein resistance of titanium oxide surfaces modified by biologically inspired mPEG-DOPA. Langmuir 2004, 21, 640–646. [Google Scholar] [CrossRef] [PubMed]
- Baneyx, F.; Schwartz, D.T. Selection and analysis of solid-binding peptides. Curr. Opin. Biotechnol. 2007, 18, 312–317. [Google Scholar] [CrossRef] [PubMed]
- Sano, K.-I.; Shiba, K. A hexapeptide motif that electrostatically binds to the surface of titanium. J. Am. Chem. Soc. 2003, 125, 14234–14235. [Google Scholar] [CrossRef] [PubMed]
- Jones, F.H. Teeth and bones: Applications of surface science to dental materials and related biomaterials. Surf. Sci. Rep. 2001, 42, 75–205. [Google Scholar] [CrossRef]
- Hayashi, T.; Sano, K.-I.; Shiba, K.; Kumashiro, Y.; Iwahori, K.; Yamashita, I.; Hara, M. Mechanism underlying specificity of proteins targeting inorganic materials. Nano Lett. 2006, 6, 515–519. [Google Scholar] [CrossRef] [PubMed]
- Nilebäck, L.; Hedin, J.; Widhe, M.; Floderus, L.S.; Krona, A.; Bysell, H.; Hedhammar, M. Self-assembly of recombinant silk as a strategy for chemical-free formation of bioactive coatings: A real-time study. Biomacromolecules 2017, 18, 846–854. [Google Scholar] [CrossRef] [PubMed]
- Leon, B.; Jansen, J. (Eds.) Thin Calcium Phosphate Coatings for Medical Implants; Springer: New York, NY, USA, 2009. [Google Scholar]
- Narayanan, R.; Seshadri, S.K.; Kwon, T.Y.; Kim, K.H. Calcium phosphate-based coatings on titanium and its alloys. J. Biomed. Mater. Res. B Appl. Biomater. 2008, 85, 279–299. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.; Daraio, C.; Chen, L.-H.; Pisanic, T.R.; Fiñones, R.R.; Jin, S. Significantly accelerated osteoblast cell growth on aligned TiO2 nanotubes. J. Biomed. Mater. Res. A 2006, 78, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Von Wilmowsky, C.; Bauer, S.; Lutz, R.; Meisel, M.; Neukam, F.W.; Toyoshima, T.; Schmuki, P.; Nkenke, E.; Schlegel, K.A. In vivo evaluation of anodic TiO2 nanotubes: An experimental study in the pig. J. Biomed. Mater. Res. B Appl. Biomater. 2009, 89, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Brammer, K.S.; Oh, S.; Cobb, C.J.; Bjursten, L.M.; Heyde, H.v.d.; Jin, S. Improved bone-forming functionality on diameter-controlled TiO2 nanotube surface. Acta Biomater. 2009, 5, 3215–3223. [Google Scholar] [CrossRef] [PubMed]
- Bjursten, L.M.; Rasmusson, L.; Oh, S.; Smith, G.C.; Brammer, K.S.; Jin, S. Titanium dioxide nanotubes enhance bone bonding in vivo. J. Biomed. Mater. Res. A 2009, 92, 1218–1224. [Google Scholar] [CrossRef]
- Macak, J.M.; Tsuchiya, H.; Ghicov, A.; Yasuda, K.; Hahn, R.; Bauer, S.; Schmuki, P. TiO2 nanotubes: Self-organized electrochemical formation, properties and applications. Curr. Opin. Solid State Mater. Sci. 2007, 11, 3–18. [Google Scholar] [CrossRef]
- Vasilev, K.; Poh, Z.; Kant, K.; Chan, J.; Michelmore, A.; Losic, D. Tailoring the surface functionalities of titania nanotube arrays. Biomaterials 2010, 31, 532–540. [Google Scholar] [CrossRef] [PubMed]
- Balaur, E.; Macak, J.M.; Taveira, L.; Schmuki, P. Tailoring the wettability of TiO2 nanotube layers. Electrochem. Commun. 2005, 7, 1066–1070. [Google Scholar] [CrossRef]
- Losic, D.; Simovic, S. Self-ordered nanopore and nanotube platforms for drug delivery applications. Expert Opin. Drug Deliv. 2009, 6, 1363–1381. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.E.; Lim, J.H.; Lee, S.C.; Nam, S.-C.; Kang, H.-G.; Choi, J. Anodically nanostructured titanium oxides for implant applications. Electrochim. Acta 2008, 53, 4846–4851. [Google Scholar] [CrossRef]
- Cherkasov, A.; Hilpert, K.; Jenssen, H.; Fjell, C.D.; Waldbrook, M.; Mullaly, S.C.; Volkmer, R.; Hancock, R.E.W. Use of artificial intelligence in the design of small peptide antibiotics effective against a broad spectrum of highly antibiotic-resistant superbugs. ACS Chem. Biol. 2009, 4, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Konno, T.; Takai, M.; Ishihara, K. Regulation of cell proliferation by multi-layered phospholipid polymer hydrogel coatings through controlled release of paclitaxel. Biomaterials 2012, 33, 954–961. [Google Scholar] [CrossRef] [PubMed]
- Satsangi, A.; Satsangi, N.; Glover, R.; Satsangi, R.K.; Ong, J.L. Osteoblast response to phospholipid modified titanium surface. Biomaterials 2003, 24, 4585–4589. [Google Scholar] [CrossRef]
- Willumeit, R.; Schuster, A.; Iliev, P.; Linser, S.; Feyerabend, F. Phospholipids as implant coatings. J. Mater. Sci. Mater. Med. 2007, 18, 367–380. [Google Scholar] [CrossRef] [PubMed]
- Susin, C.; Qahash, M.; Hall, J.; Sennerby, L.; Wikesjö, U.M.E. Histological and biomechanical evaluation of phosphorylcholine-coated titanium implants. J. Clin. Periodontol. 2008, 35, 270–275. [Google Scholar] [CrossRef] [PubMed]
- Metsemakers, W.J.; Emanuel, N.; Cohen, O.; Reichart, M.; Potapova, I.; Schmid, T.; Segal, D.; Riool, M.; Kwakman, P.H.S.; De Boer, L.; et al. A doxycycline-loaded polymer-lipid encapsulation matrix coating for the prevention of implant-related osteomyelitis due to doxycycline-resistant methicillin-resistant Staphylococcus aureus. J. Control. Release 2015, 209, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Emanuel, N.; Rosenfeld, Y.; Cohen, O.; Applbaum, Y.H.; Segal, D.; Barenholz, Y. A lipid-and-polymer-based novel local drug delivery system—BonyPid™: From physicochemical aspects to therapy of bacterially infected bones. J. Control. Release 2012, 160, 353–361. [Google Scholar] [CrossRef] [PubMed]
- Steven, M.D.; Hotchkiss, J.H. Covalent immobilization of an antimicrobial peptide on poly(ethylene) film. J. Appl. Polym. Sci. 2008, 110, 2665–2670. [Google Scholar] [CrossRef]
- Mohorčič, M.; Jerman, I.; Zorko, M.; Butinar, L.; Orel, B.; Jerala, R.; Friedrich, J. Surface with antimicrobial activity obtained through silane coating with covalently bound polymyxin B. J. Mater. Sci. Mater. Med. 2010, 21, 2775–2782. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.R.; Johnston, B.D.; Kuskowski, M.A.; Pitout, J. In vitro activity of available antimicrobial coated Foley catheters against Escherichia coli, including strains resistant to extended spectrum cephalosporins. J. Urol. 2010, 184, 2572–2577. [Google Scholar] [CrossRef] [PubMed]
- Yu, K.; Lo, J.C.Y.; Yan, M.; Yang, X.; Brooks, D.E.; Hancock, R.E.W.; Lange, D.; Kizhakkedathu, J.N. Anti-adhesive antimicrobial peptide coating prevents catheter associated infection in a mouse urinary infection model. Biomaterials 2017, 116, 69–81. [Google Scholar] [CrossRef] [PubMed]
- Mishra, B.; Lushnikova, T.; Golla, R.M.; Wang, X.; Wang, G. Design and surface immobilization of short anti-biofilm peptides. Acta Biomater. 2017, 49, 316–328. [Google Scholar] [CrossRef] [PubMed]
- Cao, P.; Yang, Y.; Uche, F.I.; Hart, S.R.; Li, W.W.; Yuan, C. Coupling plant-derived cyclotides to metal surfaces: An antibacterial and antibiofilm study. Int. J. Mol. Sci. 2018, 9, pii: E793. [Google Scholar] [CrossRef] [PubMed]
- Bumgardner, J.D.; Chesnutt, B.M.; Yuan, Y.; Yang, Y.; Appleford, M.; Oh, S.; McLaughlin, R.; Elder, S.H.; Ong, J.L. The integration of chitosan-coated titanium in bone: An in vivo study in rabbits. Implant Dent. 2007, 16, 66–79. [Google Scholar] [CrossRef] [PubMed]
- Moutzouri, A.G.; Athanassiou, G.M. Attachment, spreading, and adhesion strength of human bone marrow cells on chitosan. Ann. Biomed. Eng. 2011, 39, 730–741. [Google Scholar] [CrossRef] [PubMed]
- Carlson, R.P.; Taffs, R.; Davison, W.M.; Stewart, P.S. Anti-biofilm properties of chitosan-coated surfaces. J. Biomater. Sci. Polym. Ed. 2008, 19, 1035–1046. [Google Scholar] [CrossRef] [PubMed]
- Kong, M.; Chen, X.G.; Xing, K.; Park, H.J. Antimicrobial properties of chitosan and mode of action: A state of the art review. Int. J. Food. Microbiol. 2010, 144, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Costa, F.; Maia, S.; Gomes, J.; Gomes, P.; Martins, M.C.L. Characterization of hLf1-11 immobilization onto chitosan ultrathin films, and its effects on antimicrobial activity. Acta Biomater. 2014, 10, 3513–3521. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.M.; Simon, R.J.; Ng, S.; Zuckermann, R.N.; Kerr, J.M.; Moos, W.H. Comparison of the proteolytic susceptibilities of homologous l-amino acid, d-amino acid, and N-substituted glycine peptide and peptoid oligomers. Drug Dev. Res. 1995, 35, 20–32. [Google Scholar] [CrossRef]
- Zuckermann, R.N.; Kerr, J.M.; Kent, S.B.H.; Moos, W.H. Efficient method for the preparation of peptoids [oligo(N-substituted glycines)] by submonomer solid-phase synthesis. J. Am. Chem. Soc. 1992, 114, 10647–10649. [Google Scholar] [CrossRef]
- Luxenhofer, R.; Fetsch, C.; Grossmann, A. Polypeptoids: A perfect match for molecular definition and macromolecular engineering? J. Polym. Sci. Part A Polym. Chem. 2013, 51, 2731–2752. [Google Scholar] [CrossRef]
- Secker, C.; Brosnan, S.M.; Luxenhofer, R.; Schlaad, H. Poly(α-peptoid)s revisited: Synthesis, properties, and use as biomaterial. Macromol. Biosci. 2015, 15, 881–891. [Google Scholar] [CrossRef] [PubMed]
- Statz, A.R.; Meagher, R.J.; Barron, A.E.; Messersmith, P.B. New peptidomimetic polymers for antifouling surfaces. J. Am. Chem. Soc. 2005, 127, 7972–7973. [Google Scholar] [CrossRef] [PubMed]
- Ostuni, E.; Chapman, R.G.; Holmlin, R.E.; Takayama, S.; Whitesides, G.M. A survey of structure-property relationships of surfaces that resist the adsorption of protein. Langmuir 2001, 17, 5605–5620. [Google Scholar] [CrossRef]
- Statz, A.R.; Barron, A.E.; Messersmith, P.B. Protein, cell and bacterial fouling resistance of polypeptoid-modified surfaces: Effect of side-chain chemistry. Soft Matter 2008, 4, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Statz, A.R.; Park, J.P.; Chongsiriwatana, N.P.; Barron, A.E.; Messersmith, P.B. Surface-immobilized antimicrobial peptoids. Biofouling 2009, 24, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Lau, K.H.A.; Ren, C.; Sileika, T.S.; Park, S.H.; Szleifer, I.; Messersmith, P.B. Surface-grafted polysarcosine as a peptoid antifouling polymer brush. Langmuir 2012, 28, 16099–16107. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.; Cao, Z. Ultralow-fouling, functionalizable, and hydrolyzable zwitterionic materials and their derivatives for biological applications. Adv. Mater. 2010, 22, 920–932. [Google Scholar] [CrossRef] [PubMed]
- Blaszykowski, C.; Sheikh, S.; Thompson, M. Surface chemistry to minimize fouling from blood-based fluids. Chem. Soc. Rev. 2012, 41, 5599. [Google Scholar] [CrossRef] [PubMed]
- Lau, K.H.A.; Sileika, T.S.; Park, S.H.; Sousa, A.M.L.; Burch, P.; Szleifer, I.; Messersmith, P.B. Molecular design of antifouling polymer brushes using sequence-specific peptoids. Adv. Mater. Interfaces 2015, 2, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Patterson, A.L.; Wenning, B.; Rizis, G.; Calabrese, D.R.; Finlay, J.A.; Franco, S.C.; Zuckermann, R.N.; Clare, A.S.; Kramer, E.J.; Ober, C.K.; et al. Role of backbone chemistry and monomer sequence in amphiphilic oligopeptide- and oligopeptoid-functionalized PDMS- and PEO-based block copolymers for marine antifouling and fouling release coatings. Macromolecules 2017, 50, 2656–2667. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| AMP | Sequence | Bacteria | Activity | Ref |
|---|---|---|---|---|
| LL-37 | LLGDFFRKSKEKIGKEFKRIVQRIKDFLRNLVPRTES | P. aeruginosa | Inhibition | [43] |
| AS10 | KLKKIAQKIKNFFQKLVP | C. albicans | Inhibition | [48] |
| P. aeruginosa | ||||
| E. coli | ||||
| IDR-1018 | VRLIVAVRIWRR-NH2 | P. aeruginosa | Inhibition/Eradication | [50] |
| E. coli | ||||
| A. baumannii | ||||
| K. pneumoniae | ||||
| S. enterica | ||||
| MRSA | ||||
| DJK-5 | vqwrairvrvir ** | P. aeruginosa E. coli A. baumannii K. pneumonia S. enterica | Inhibition/Eradication | [51] |
| DJK-6 | vqwrrirvwvir ** | [51] | ||
| KT2 | NGVQPKYKWWKWWKKWW-NH2 | E. coli | Inhibition/Eradication | [53] |
| RT2 | NGVQPKYRWWRWWRRWW-NH2 | [53] | ||
| CAMA | KWKLFKKIGIGKFLQSAKKF-NH2 | MRSA | Inhibition | [52] |
| P10 | LAREYKKIVEKLKRWLRQVLRTLR | MDR S. aureus | Inhibition/Eradication | [46] |
| UP-5 | RBRBR * | MRSA | Inhibition | [54] |
| hep20 | ICIFCCGCCHRSHCGMCCKT | S. epidermidis | Inhibition | [49] |
| AMP | Sequence | Bacteria | In Vivo Model | Ref |
|---|---|---|---|---|
| SAAP-148 | LKRVWKRVFKLLKRYWRQLKKPVR | A. baumannii | A mouse wound skin model | [71] |
| MRSA | ||||
| 3002 | ILVRWIRWRIQW-NH2 | MRSA | A mouse cutaneous abscess model | [62] |
| IDR-1018 | VRLIVAVRIWRR-NH2 | |||
| DJK-5 | vqwrairvrvir * | P. aeruginosa | Caenorhabditis elegans nematodes Galleria mellonella larvae A mouse cutaneous abscess model | [51,65] |
| DJK-6 | vqwrrirvwvir * | |||
| WRL3 | WLRAFRRLVRRLARGLRRNH2 | MRSA | An infected burn mouse wound model | [72] |
| D-RR4 | wlrrikawlrrika-NH2 * | P. aeruginosa | C. elegans model | [68] |
| A. baumannii |
| Compound | Structure | Bacteria | Activity | Ref. |
|---|---|---|---|---|
| 1 |  | P. aeruginosa | Inhibiotion/Prevention | [75] |
| 1-C134mer |  | P. aeruginosa | Inhibition/Prevention | [75] |
| Y-36 |  | P. aeruginosa | Inhibition/Prevention | [76] |
| MRSE | ||||
| HDM-4 |  | E. coli | Inhibition | [77] |
| P. aeruginosa | ||||
| S. enterica Typhimurium | ||||
| A. baumannii | ||||
| K. pneumoniae | ||||
| 4d |  | S. epidermidis | Inhibition | [78] |
| Py11 |  | P. aeruginosa | Inhibition | [79] |
| C14KKc12K |  | Streptococcus mutans | Inhibition | [80] |
| 23b |  | S. aureus | Inhibition | [81] |
| E. coli | ||||
| 4g |  | S. aureus | Inhibition | [83] |
| E. coli | ||||
| 1c, 1d |  | S. aureus | Inhibition | [82] |
| Biomaterial | AMP | Sequence | Coating Method | Bacteria | In Vitro Testing | In Vivo Testing | Biocompatibility Tested on | Ref. |
|---|---|---|---|---|---|---|---|---|
| Immobilised | ||||||||
| Titanium, disks or hollow round casings | Melimine | CTLISWIKNKRKQRPRVSRRRRRRGGRRRR | Three-step:
| S. aureus strain 38 P. aeruginosa PAO1 | Bacterial adhesion via fluorescence microscopy | Mouse and rat subcutaneous infection models. CFU determination. | n/a | [96] |
| Titanium, commercially pure Grade II discs | GL13K | GKIIKLKASLKLL-NH2 | Two-step:
| Streptococcus gordonii strain ML-5 | Drip Flow Bioreactor Culture CFU assay, ATP Assay, L/D staining BacLight, SEM | n/a | n/a | [119] |
| Titanium | GZ3.163 | 4-methylhexanoyl-Cys-d-Dab-Dab-Dab-Leu-d-Phe-Dab-Dab-Leu-NH2 | Three-step:
| E. coli DH5α P. aeruginosa ATCC 27853 S. aureus 10 | CFU assay, L/D staining BacLight, SEM | n/a | Mouse blood cells lysis assay | [124] |
| Titanium, platelets | LL-37 | CLLGDFFRKSKEKIGKEFKRIVQRIKDFLRNLVPRTES | Three-step:
| E. coli strain K12 | Bacterial killing assay (Propidium iodide staining) | n/a | n/a | [121] |
| Titanium, deposited on silicon wafer | Tet213 | KRWWKWWRRC | Three-step:
| P. aeruginosa PA01 (luxCDABE) | CFU assay, luminescence | n/a | n/a | [97] |
| Titanium, deposited on silicon wafer | Tet-20 | KRWRIRVRVIRKC |
| P. aeruginosa PA01 (luxCDABE) | CFU assay, luminescence, SEM | Rat subcutaneous infection model | MG-63 human osteoblast-like cells, Platelet activation, Complement activation analysis | [98] |
| Titanium, commercially pure Grade II | hLF1-11 | MPA-Ahx-Ahx-Ahx-GRRRRSVQWCA-NH2 6 | Three-step:
| S. sanguinis 10 L. salivarius 10 | CFU assay, L/D staining BackLight, CLSM, BacTiter-Glo Reagent for biofilm | n/a | Human foreskin fibroblasts | [125] |
| Titanium, commercially pure Grade II | hLF1-11 | MPA-Ahx-Ahx-Ahx-GRRRRSVQWCA-NH2 6 | Three-step:
| S. sanguinis 11 L. salivarius 11 | CFU assay, SEM, luminescence BacTiter-Glo Reagent for biofilm | n/a | Human foreskin fibroblasts | [123] |
| Titanium, commercially pure Grade II | GL13K | GKIIKLKASLKLL-NH2 | Two-step:
| Porphyromonas gingivalis ATCC 33277 | ATP assay, CFU assay | n/a | Human gingival fibroblasts (HGF) and MC3T3-E1 murine osteoblasts | [126] |
| Titanium foils 99.2% pure | Ti-binding- linker-JPH8194 | RKLPDA-PAPAP-KRLFRRWQWRMKKY | Chimeric peptide, with titanium-binding domain. | S. gordonii ATCC 51656, S. sanguis ATCC 10556 | L/D staining BacLight, CLSM | n/a | MC3T3-E1 Osteoblasts Culture | [127] |
| Titanium alloy, Ti6AL4V | Bacitracin | Ile-Cys-Leu-d-Glu-Ile-cy(Lys-d-Orn-Ile-d-Phe-His-d-Asp-Asp) | Polydopamine | S. aureus ATCC 25923, MRSA | n/a | Rat model, Ti rods were implanted into the femurs. CFU on the implant and at the peri-implant tissues. | Histopathology evaluation of the bone tissue around the Ti rod implant. nephrotoxicity of bacitracin-modified Ti in vivo | [113] |
| Titanium | SESB2V | [(RGRKVVRR)2K]2KK | Polydopamine | S. aureus ATCC 29213 P. aeruginosa ATCC 9027 | L/D staining BacLight | rabbit keratitis model, CFU/cornea | n/a | [114] |
| Titanium alloy, Ti6Al4V | SESB2V | [(RGRKVVRR)2K]2KK | Polydopamine | B. cereus ATCC 14579 E. coli ATCC 35218 | L/D staining, fluorescent microscopy | n/a | Human corneal stroma cells from donors tissue | [128] |
| Titanium, grade V powder | AMP1 | LKLLKKLLKLLKKL | Chimeric peptide, with titanium-binding domain. | E. coli ATCC 2592 S. mutans ATCC 25175 S. epidermidis ATCC 29886 | SYTO9 green fluorescent nucleic acid stain fluorescent microscopy | n/a | n/a | [129] |
| AMP2 | KWKRWWWWR | |||||||
| Release | ||||||||
| Titanium | HHC-36 | KRWWKWWRR-NH2 | hydrogel, cathehol functionalised, addition of AMP | P. aeruginosa E. coli S. aureus S. epidermidis | CFU assay, SEM | n/a | human mesenchymal stem cells | [130] |
| Titanium | OP-145 | Ac-IGKEFKRIVERIKRFLRELVRPLR-NH2 | PLEX 8 coating, mixed with peptide. Immersion for in vitro testing, spraying for in vivo. | S. aureus clinical strain JAR060131 | CFU assay, Crystal violet | Mouse subcutaneous and Rabbit intramedullary nail infection models. Biopsy fom skin, subcutaneous tissue and implant. | n/a | [115] |
| Titanium | Tet213 | KRWWKWWRRC | Calcium phosphate by electrolytic deposition, soaking in the AMP solution. | P. aeruginosa H1001: lux-CDABE S. aureus ATCC 25293 | CFU assay, luminescence | n/a | MG-63 human osteoblast-like cells | [131] |
| Titanium | HHC-36 | KRWWKWWRR-NH2 | LBL 9 coating. Three layers of vertically oriented TiO2 nanotubes, a thin layer of calcium phosphate coating and a phospholipid. | P. aeruginosa H1001: lux-CDABE S. aureus ATCC 25293 | CFU assay, SEM | n/a | MG-63 human osteoblast-like cells Platelet activation Red blood cell (RBC) haemolysis assay | [132] |
| Titanium | GL13K | GKIIKLKASLKLL-NH2 | TiO2 nanotubes. | F. nucleatum ATCC 25586 P. gingivalis ATCC 33277 | CFU assay | n/a | MC3T3-E1 cells, a clonal mouse preosteoblastic cell line, J774A.1 mouse macrophage | [133] |
| Titanium | HHC36 | KRWWKWWRR | TiO2 nanotubes, adsorption via a simple vacuum-assisted physical adsorption method. | S. aureus ATCC 25293 | CFU assay, SEM | n/a | n/a | [134] |
| Titanium alloy, Ti6Al4V | Cateslytin | RSMRLSFRARGYGFR | Hydrogel made of natural polysaccharide, sodium alginate, modified by catechol groups along the polymer chain. | P. gingivalis ATCC 33277 | Alamar Blue cell viability assay CFU assay | n/a | Gingival fibroblasts HGF-1 | [135] |
| Titanium, solid medical grade implants | SAAP-145 | Ac-LKRLYKRLAKLIKRLYRYLKKPVR-NH2 | Biodegradable PLEX was mixed with peptide. | S. aureus JAR060131, MDR S. aureus LUH15101 | Propidium iodine fluorescence | mouse model of subcutaneous biomaterial-associated infection. CFU on the implant and at the peri-implant area. Biopsies. | n/a | [116] |
| Titanium plaHNUtes | Tet213 | KRWWKWWRRC | layer-by-layer assembly, chitosan, hyalouronic acid. AMP was covalently linked to free amines of collagen IV | S. aureus ATCC 25923, P. gingivalis ATCC 33277 | CFU and fluorescent microscopy | mice, intraperitoneal administration | Cytotoxicity Assay. HaCaT cells Human erythrocytes, haemolysis assay In vivo immunotoxicity assay. | [117] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Andrea, A.; Molchanova, N.; Jenssen, H. Antibiofilm Peptides and Peptidomimetics with Focus on Surface Immobilization. Biomolecules 2018, 8, 27. https://doi.org/10.3390/biom8020027
Andrea A, Molchanova N, Jenssen H. Antibiofilm Peptides and Peptidomimetics with Focus on Surface Immobilization. Biomolecules. 2018; 8(2):27. https://doi.org/10.3390/biom8020027
Chicago/Turabian StyleAndrea, Athina, Natalia Molchanova, and Håvard Jenssen. 2018. "Antibiofilm Peptides and Peptidomimetics with Focus on Surface Immobilization" Biomolecules 8, no. 2: 27. https://doi.org/10.3390/biom8020027
APA StyleAndrea, A., Molchanova, N., & Jenssen, H. (2018). Antibiofilm Peptides and Peptidomimetics with Focus on Surface Immobilization. Biomolecules, 8(2), 27. https://doi.org/10.3390/biom8020027