Role of Nutrition in Alcoholic Liver Disease: Summary of the Symposium at the ESBRA 2017 Congress

 ,
, 


Abstract
:1. Introduction
2. Summary of Presentations at the Symposium
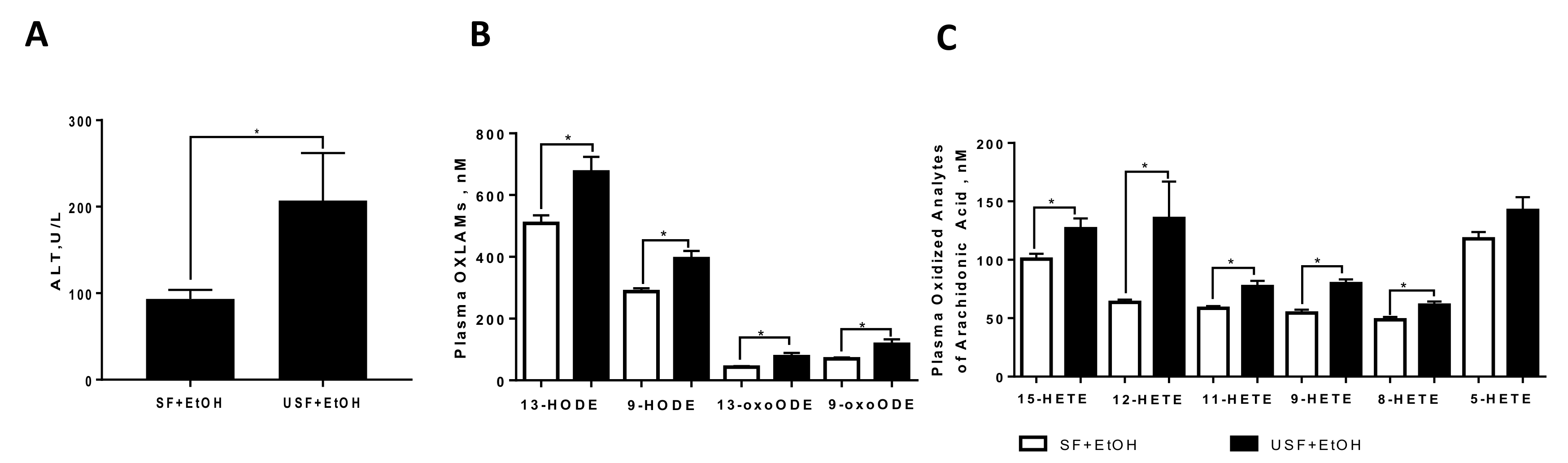
2.1. Dietary Linoleic Acid and Its Oxidized Metabolites Exacerbate Liver Injury Caused by Ethanol via Induction of Hepatic Pro-Inflammatory Response
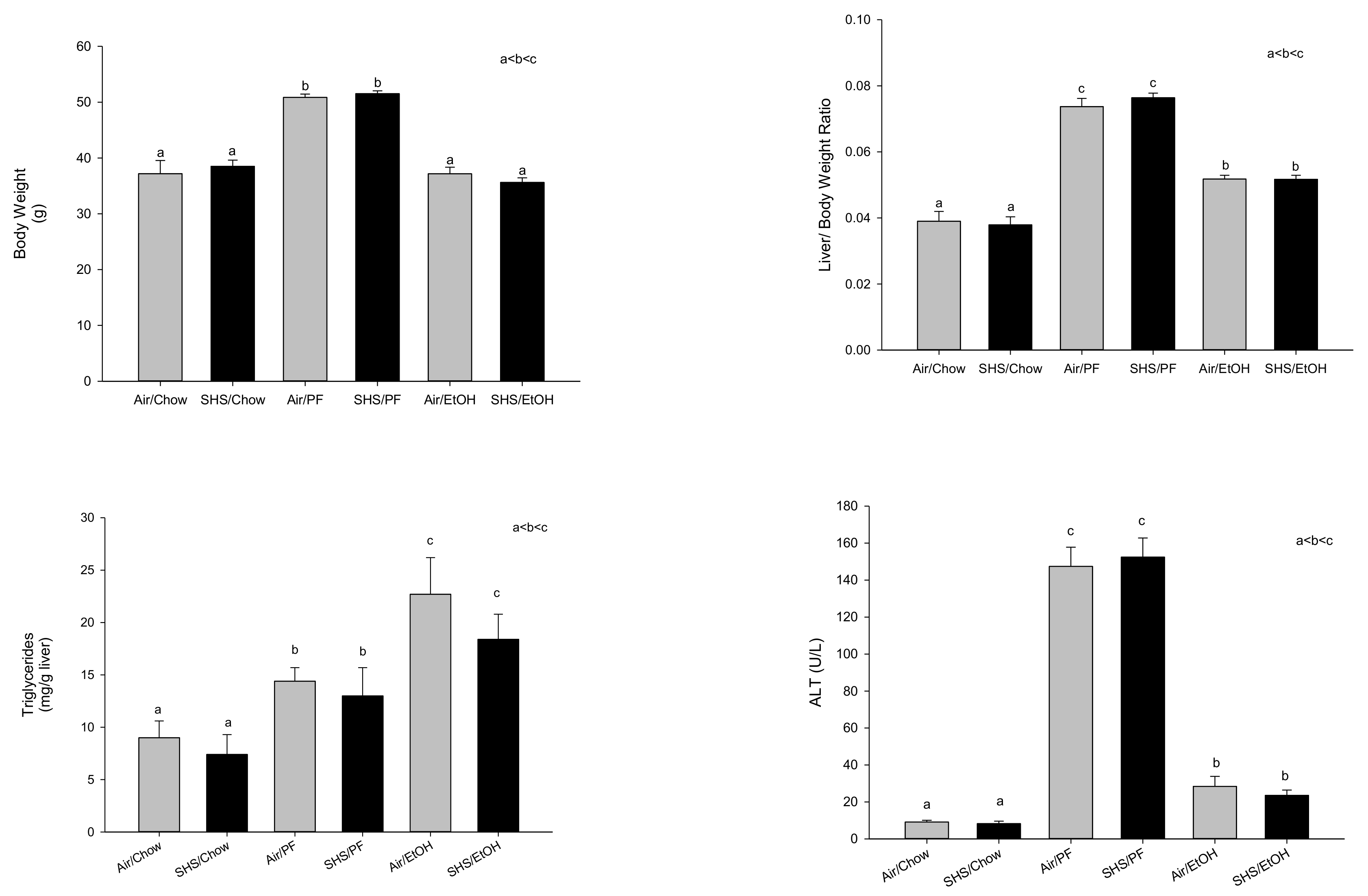
2.2. Role of Fat/Carbohydrate Ratio and Dietary Fat Type in Development of Alcoholic Liver Pathology
2.3. Betaine: A Promising Therapeutic in the Treatment of Alcohol-Induced Liver Injury
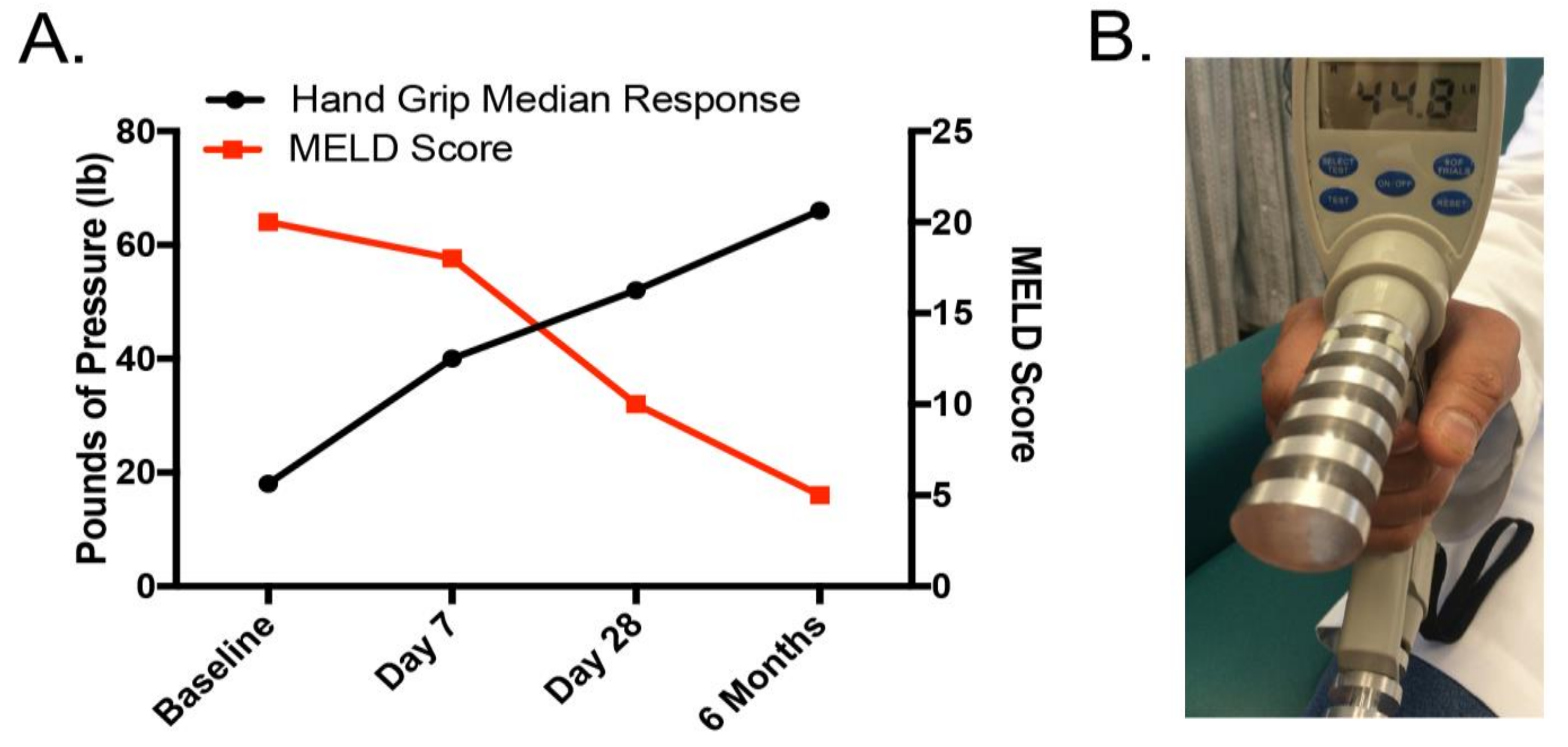
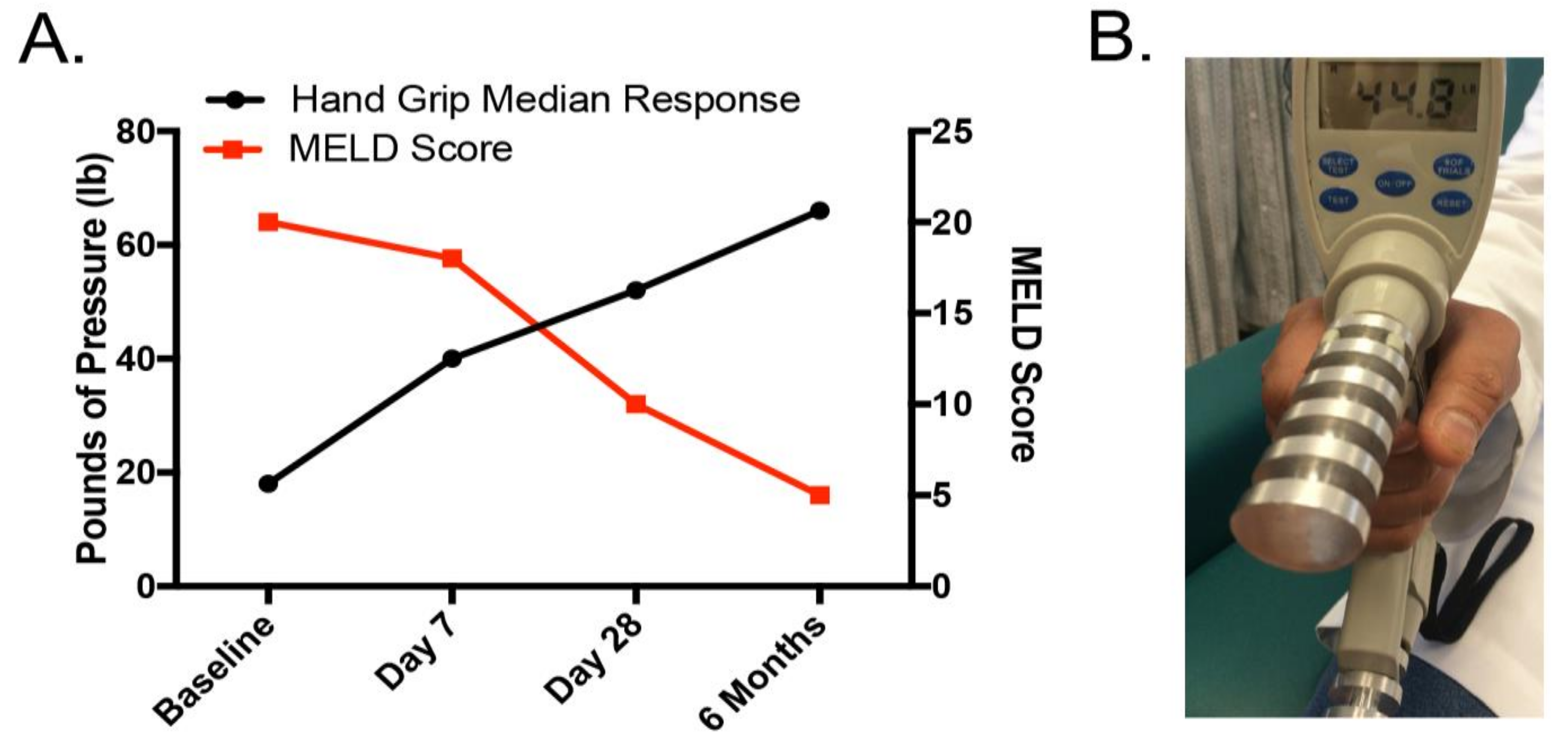
2.4. Malnutrition and Nutrition Support in Alcoholic Liver Disease: Clinical Relevance
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| AA | arachidonic acid |
| ALD | alcoholic liver disease |
| AH | alcoholic hepatitis |
| BIA | bioelectrical impedance |
| BHMT | betaine-homocysteine methyltransferase |
| BMI | body mass index |
| ChREBP | carbohydrate response element binding protein |
| HETEs | hydroxyeicosatetraenoic acids |
| HODEs | hydroxyoctadecadienoic acids |
| LA | linoleic acid |
| NASH | non-alcoholic steatohepatitis |
| 4-HNE | 4-hydroxynonenal |
| OXLAMs | oxidized linoleic acid metabolites |
| PUFA | polyunsaturated fatty acid |
| ROS | reactive oxygen species |
| SAH | S-adenosylhomocysteine |
| SAM | S-adenosylmethionine |
| SREBP-1c | steroid regulatory element binding protein 1c |
| SGA | Subjective global assessment |
| WAT | white adipose tissue |
| USF | unsaturated fat |
References
- De Minicis, S.; Brenner, D.A. Oxidative Stress in Alcoholic Liver Disease: Role of NADPH Oxidase Complex. J. Gastroenterol. Hepatol. 2008, 23 (Suppl. 1), S98–S103. [Google Scholar] [CrossRef] [PubMed]
- Ambade, A.; Mandrekar, P. Oxidative stress and inflammation: Essential partners in alcoholic liver disease. Int. J. Hepatol. 2012, 2012, 853175. [Google Scholar] [CrossRef] [PubMed]
- Dey, A.; Cederbaum, A.I. Alcohol and oxidative liver injury. Hepatology 2006, 43 (Suppl. 1), S63–S74. [Google Scholar] [CrossRef] [PubMed]
- Purohit, V.; Abdelmalek, M.F.; Barve, S.; Benevenga, N.J.; Halsted, C.H.; Kaplowitz, N.; Kharbanda, K.K.; Liu, Q.Y.; Lu, S.C.; McClain, C.J.; et al. Role of S-adenosylmethionine, folate, and betaine in the treatment of alcoholic liver disease: Summary of a symposium. Am. J. Clin. Nutr. 2007, 86, 14–24. [Google Scholar] [CrossRef] [PubMed]
- Kharbanda, K.K.; Mailliard, M.E.; Baldwin, C.R.; Beckenhauer, H.C.; Sorrell, M.F.; Tuma, D.J. Betaine attenuates alcoholic steatosis by restoring phosphatidylcholine generation via the phosphatidylethanolamine methyltransferase pathway. J. Hepatol. 2007, 46, 314–321. [Google Scholar] [CrossRef] [PubMed]
- Kirpich, I.A.; Feng, W.; Wang, Y.; Liu, Y.; Barker, D.F.; Barve, S.S.; McClain, C.J. The type of dietary fat modulates intestinal tight junction integrity, gut permeability, and hepatic toll-like receptor expression in a mouse model of alcoholic liver disease. Alcohol. Clin. Exp. Res. 2012, 36, 835–846. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Torralba, M.; Tan, J.; Embree, M.; Zengler, K.; Starkel, P.; van Pijkeren, J.P.; DePew, J.; Loomba, R.; Ho, S.B.; et al. Supplementation of saturated long-chain fatty acids maintains intestinal eubiosis and reduces ethanol-induced liver injury in mice. Gastroenterology 2015, 148, 203–214. [Google Scholar] [CrossRef] [PubMed]
- Calvey, H.; Davis, M.; Williams, R. Controlled trial of nutritional supplementation, with and without branched chain amino acid enrichment, in treatment of acute alcoholic hepatitis. J. Hepatol. 1985, 1, 141–151. [Google Scholar] [CrossRef]
- Moreno, C.; Langlet, P.; Hittelet, A.; Lasser, L.; Degre, D.; Evrard, S.; Colle, I.; Lemmers, A.; Deviere, J.; Le Moine, O. Enteral nutrition with or without N-acetylcysteine in the treatment of severe acute alcoholic hepatitis: A randomized multicenter controlled trial. J. Hepatol. 2010, 53, 1117–1122. [Google Scholar] [CrossRef] [PubMed]
- Mendenhall, C.L.; Anderson, S.; Weesner, R.E.; Goldberg, S.J.; Crolic, K.A. Protein-calorie malnutrition associated with alcoholic hepatitis. Veterans Administration Cooperative Study Group on Alcoholic Hepatitis. Am. J. Med. 1984, 76, 211–222. [Google Scholar] [CrossRef]
- Dasarathy, S. Nutrition and Alcoholic Liver Disease: Effects of alcoholism on nutrition, effects of nutrition on alcoholic liver disease, and nutritional therapies for alcoholic liver disease. Clin. Liver Dis. 2016, 20, 535–550. [Google Scholar] [CrossRef] [PubMed]
- Kirpich, I.A.; Miller, M.E.; Cave, M.C.; Joshi-Barve, S.; McClain, C.J. Alcoholic Liver Disease: Update on the Role of Dietary Fat. Biomolecules 2016, 6, 1. [Google Scholar] [CrossRef] [PubMed]
- Ronis, M.J.; Korourian, S.; Zipperman, M.; Hakkak, R.; Badger, T.M. Dietary saturated fat reduces alcoholic hepatotoxicity in rats by altering fatty acid metabolism and membrane composition. J. Nutr. 2004, 134, 904–912. [Google Scholar] [CrossRef] [PubMed]
- Zhong, W.; Li, Q.; Xie, G.; Sun, X.; Tan, X.; Sun, X.; Jia, W.; Zhou, Z. Dietary fat sources differentially modulate intestinal barrier and hepatic inflammation in alcohol-induced liver injury in rats. Am. J. Physiol. Gastrointest. Liver Physiol. 2013, 305, G919–G932. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Latchoumycandane, C.; McMullen, M.R.; Pratt, B.T.; Zhang, R.; Papouchado, B.G.; Nagy, L.E.; Feldstein, A.E.; McIntyre, T.M. Chronic alcohol exposure increases circulating bioactive oxidized phospholipids. J. Biol. Chem. 2010, 285, 22211–22220. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Zhong, W.; Sun, Q.; Sun, X.; Zhou, Z. Hepatic overproduction of 13-HODE due to ALOX15 upregulation contributes to alcohol-induced liver injury in mice. Sci. Rep. 2017, 7, 8976. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.; Gu, J.; Vandenhoff, G.E.; Liu, X.; Nadler, J.L. Role of 12/15-lipoxygenase in the expression of MCP-1 in mouse macrophages. Am. J. Physiol. Heart Circ. Physiol. 2008, 294, H1933–H1938. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.; Gu, J.; Chakrabarti, S.K.; Aylor, K.; Marshall, J.; Takahashi, Y.; Yoshimoto, T.; Nadler, J.L. The role of 12/15-lipoxygenase in the expression of interleukin-6 and tumor necrosis factor-alpha in macrophages. Endocrinology 2007, 148, 1313–1322. [Google Scholar] [CrossRef] [PubMed]
- Raszeja-Wyszomirska, J.; Safranow, K.; Milkiewicz, M.; Milkiewicz, P.; Szynkowska, A.; Stachowska, E. Lipidic last breath of life in patients with alcoholic liver disease. Prostag. Other Lipid Mediat. 2012, 99, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Obinata, H.; Hattori, T.; Nakane, S.; Tatei, K.; Izumi, T. Identification of 9-hydroxyoctadecadienoic acid and other oxidized free fatty acids as ligands of the G protein-coupled receptor G2A. J. Biol. Chem. 2005, 280, 40676–40683. [Google Scholar] [CrossRef] [PubMed]
- Patwardhan, A.M.; Scotland, P.E.; Akopian, A.N.; Hargreaves, K.M. Activation of TRPV1 in the spinal cord by oxidized linoleic acid metabolites contributes to inflammatory hyperalgesia. Proc. Natl. Acad. Sci. USA 2009, 106, 18820–18824. [Google Scholar] [CrossRef] [PubMed]
- Itoh, T.; Fairall, L.; Amin, K.; Inaba, Y.; Szanto, A.; Balint, B.L.; Nagy, L.; Yamamoto, K.; Schwabe, J.W. Structural basis for the activation of PPARgamma by oxidized fatty acids. Nat. Struct. Mol. Biol. 2008, 15, 924–931. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.W.; Lee, S.T.; Wu, W.T.; Fu, W.M.; Ho, F.M.; Lin, W.W. Signal transduction for inhibition of inducible nitric oxide synthase and cyclooxygenase-2 induction by capsaicin and related analogs in macrophages. Br. J. Pharmacol. 2003, 140, 1077–1087. [Google Scholar] [CrossRef] [PubMed]
- Ricote, M.; Li, A.C.; Willson, T.M.; Kelly, C.J.; Glass, C.K. The peroxisome proliferator-activated receptor-gamma is a negative regulator of macrophage activation. Nature 1998, 391, 79–82. [Google Scholar] [CrossRef] [PubMed]
- Ronis, M.J.; Mercer, K.; Suva, L.J.; Vantrease, J.; Ferguson, M.; Hogue, W.R.; Sharma, N.; Cleves, M.A.; Blackburn, M.L.; Badger, T.M. Influence of fat/carbohydrate ratio on progression of fatty liver disease and on development of osteopenia in male rats fed alcohol via total enteral nutrition (TEN). Alcohol 2014, 48, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Shen, Z.; Liang, X.; Rogers, C.Q.; Rideout, D.; You, M. Involvement of adiponectin-SIRT1-AMPK signaling in the protective action of rosiglitazone against alcoholic fatty liver in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2010, 298, G364–G374. [Google Scholar] [CrossRef] [PubMed]
- Liangpunsakul, S.; Ross, R.A.; Crabb, D.W. Activation of carbohydrate response element-binding protein by ethanol. J. Investig. Med. 2013, 61, 270–277. [Google Scholar] [CrossRef] [PubMed]
- Korourian, S.; Hakkak, R.; Ronis, M.J.; Shelnutt, S.R.; Waldron, J.; Ingelman-Sundberg, M.; Badger, T.M. Diet and risk of ethanol-induced hepatotoxicity: Carbohydrate-fat relationships in rats. Toxicol. Sci. 1999, 47, 110–117. [Google Scholar] [CrossRef] [PubMed]
- Ronis, M.J.; Mercer, K.E.; Gannon, B.; Engi, B.; Zimniak, P.; Shearn, C.T.; Orlicky, D.J.; Albano, E.; Badger, T.M.; Petersen, D.R. Increased 4-hydroxynonenal protein adducts in male GSTA4-4/PPAR-alpha double knockout mice enhance injury during early stages of alcoholic liver disease. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 308, G403–G415. [Google Scholar] [CrossRef] [PubMed]
- Smathers, R.L.; Galligan, J.J.; Shearn, C.T.; Fritz, K.S.; Mercer, K.; Ronis, M.; Orlicky, D.J.; Davidson, N.O.; Petersen, D.R. Susceptibility of L-FABP-/- mice to oxidative stress in early-stage alcoholic liver. J. Lipid Res. 2013, 54, 1335–1345. [Google Scholar] [CrossRef] [PubMed]
- Ronis, M.J.; Baumgardner, J.N.; Marecki, J.C.; Hennings, L.; Wu, X.; Shankar, K.; Cleves, M.A.; Gomez-Acevedo, H.; Badger, T.M. Dietary fat source alters hepatic gene expression profile and determines the type of liver pathology in rats overfed via total enteral nutrition. Physiol. Genomics 2012, 44, 1073–1089. [Google Scholar] [CrossRef] [PubMed]
- Ronis, M.; Mercer, K.; Engi, B.; Pulliam, C.; Zimniak, P.; Hennings, L.; Shearn, C.; Badger, T.; Petersen, D. Global Deletion of Glutathione S-Transferase A4 Exacerbates Developmental Nonalcoholic Steatohepatitis. Am. J. Pathol. 2017, 187, 418–430. [Google Scholar] [CrossRef] [PubMed]
- Altamirano, J.; Bataller, R. Alcoholic liver disease: Pathogenesis and new targets for therapy. Nat. Rev. Gastroenterol. Hepatol. 2011, 8, 491–501. [Google Scholar] [CrossRef] [PubMed]
- Gao, B.; Bataller, R. Alcoholic liver disease: Pathogenesis and new therapeutic targets. Gastroenterology 2011, 141, 1572–1585. [Google Scholar] [CrossRef] [PubMed]
- Orman, E.S.; Odena, G.; Bataller, R. Alcoholic liver disease: Pathogenesis, management, and novel targets for therapy. J. Gastroenterol. Hepatol. 2013, 28 (Suppl. 1), 77–84. [Google Scholar] [CrossRef] [PubMed]
- Stickel, F.; Datz, C.; Hampe, J.; Bataller, R. Pathophysiology and Management of Alcoholic Liver Disease: Update 2016. Gut Liver 2017, 11, 173–188. [Google Scholar] [CrossRef] [PubMed]
- Song, Z.; Zhou, Z.; Deaciuc, I.; Chen, T.; McClain, C.J. Inhibition of adiponectin production by homocysteine: A potential mechanism for alcoholic liver disease. Hepatology 2008, 47, 867–879. [Google Scholar] [CrossRef] [PubMed]
- Zhong, W.; Zhao, Y.; Tang, Y.; Wei, X.; Shi, X.; Sun, W.; Sun, X.; Yin, X.; Kim, S.; McClain, C.J.; et al. Chronic alcohol exposure stimulates adipose tissue lipolysis in mice: Role of reverse triglyceride transport in the pathogenesis of alcoholic steatosis. Am. J. Pathol. 2012, 180, 998–1007. [Google Scholar] [CrossRef] [PubMed]
- Dou, X.; Xia, Y.; Chen, J.; Qian, Y.; Li, S.; Zhang, X.; Song, Z. Rectification of impaired adipose tissue methylation status and lipolytic response contributes to hepatoprotective effect of betaine in a mouse model of alcoholic liver disease. Br. J. Pharmacol. 2014, 171, 4073–4086. [Google Scholar] [CrossRef] [PubMed]
- Steiner, J.L.; Lang, C.H. Alcohol, Adipose Tissue and Lipid Dysregulation. Biomolecules 2017, 7, 16. [Google Scholar] [CrossRef] [PubMed]
- Keshavarzian, A.; Fields, J. Alcoholic liver disease: Is it an “extraintestinal” complication of alcohol-induced intestinal injury? J. Lab. Clin. Med. 2003, 142, 285–287. [Google Scholar] [CrossRef]
- Bode, C.; Bode, J.C. Activation of the innate immune system and alcoholic liver disease: Effects of ethanol per se or enhanced intestinal translocation of bacterial toxins induced by ethanol? Alcohol. Clin. Exp. Res. 2005, 29, 166S–171S. [Google Scholar] [CrossRef] [PubMed]
- Zhong, W.; McClain, C.J.; Cave, M.; Kang, Y.J.; Zhou, Z. The role of zinc deficiency in alcohol-induced intestinal barrier dysfunction. Am. J. Physiol. Gastrointest. Liver Physiol. 2010, 298, G625–G633. [Google Scholar] [CrossRef] [PubMed]
- Bull-Otterson, L.; Feng, W.; Kirpich, I.; Wang, Y.; Qin, X.; Liu, Y.; Gobejishvili, L.; Joshi-Barve, S.; Ayvaz, T.; Petrosino, J.; et al. Metagenomic analyses of alcohol induced pathogenic alterations in the intestinal microbiome and the effect of Lactobacillus rhamnosus GG treatment. PLoS ONE 2013, 8, e53028. [Google Scholar] [CrossRef] [PubMed]
- Szabo, G. Gut-liver axis in alcoholic liver disease. Gastroenterology 2015, 148, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Starkel, P.; Schnabl, B. Bidirectional Communication between Liver and Gut during Alcoholic Liver Disease. Semin. Liver Dis. 2016, 36, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Ji, C.; Kaplowitz, N. Betaine decreases hyperhomocysteinemia, endoplasmic reticulum stress, and liver injury in alcohol-fed mice. Gastroenterology 2003, 124, 1488–1499. [Google Scholar] [CrossRef]
- Ji, C.; Kaplowitz, N. Hyperhomocysteinemia, endoplasmic reticulum stress, and alcoholic liver injury. World J. Gastroenterol. 2004, 10, 1699–1708. [Google Scholar] [CrossRef] [PubMed]
- Kharbanda, K.K.; Barak, A.J. Defects in methionine metabolism: Its role in ethanol-induced liver injury. In Comprehensive Handbook of Alcohol-Related Pathology; Preedy, V.R., Watson, R.R., Eds.; Elsevier Academic Press: San Diego, CA, USA, 2005; pp. 735–747. [Google Scholar]
- Kharbanda, K.K. Role of transmethylation reactions in alcoholic liver disease. World J. Gastroenterol. 2007, 13, 4947–4954. [Google Scholar] [CrossRef] [PubMed]
- Kharbanda, K.K. Alcoholic liver disease and methionine metabolism. Sem. Liver Dis. 2009, 29, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Kharbanda, K.K. Methionine metabolic pathway in alcoholic liver injury. Curr. Opin. Clin. Nutr. Metab. Care 2013, 16, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Clarke, S.; Banfield, K. S-adenosylmethionine-dependent methyltransferases. In Homocysteine in Health and Disease; Carmel, R., Jacobsen, D.W., Eds.; Cambridge University Press: Cambridge, UK, 2001; pp. 63–78. [Google Scholar]
- Barak, A.J.; Beckenhauer, H.C.; Mailliard, M.E.; Kharbanda, K.K.; Tuma, D.J. Betaine lowers elevated S-adenosylhomocysteine levels in hepatocytes from ethanol-fed rats. J. Nutr. 2003, 133, 2845–2848. [Google Scholar] [CrossRef] [PubMed]
- Kharbanda, K.K.; Rogers, D.D., 2nd; Mailliard, M.E.; Siford, G.L.; Barak, A.J.; Beckenhauer, H.C.; Sorrell, M.F.; Tuma, D.J. A comparison of the effects of betaine and S-adenosylmethionine on ethanol-induced changes in methionine metabolism and steatosis in rat hepatocytes. J. Nutr. 2005, 135, 519–524. [Google Scholar] [CrossRef] [PubMed]
- Kharbanda, K.K.; Rogers, D.D., 2nd; Mailliard, M.E.; Siford, G.L.; Barak, A.J.; Beckenhauer, H.C.; Sorrell, M.F.; Tuma, D.J. Role of elevated S-adenosylhomocysteine in rat hepatocyte apoptosis: Protection by betaine. Biochem. Pharmacol. 2005, 70, 1883–1890. [Google Scholar] [CrossRef] [PubMed]
- Kharbanda, K.K.; Mailliard, M.E.; Baldwin, C.R.; Sorrell, M.F.; Tuma, D.J. Accumulation of proteins bearing atypical isoaspartyl residues in livers of alcohol-fed rats is prevented by betaine administration: Effects on protein-l-isoaspartyl methyltransferase activity. J. Hepatol. 2007, 46, 1119–1125. [Google Scholar] [CrossRef] [PubMed]
- Kharbanda, K.K.; Todero, S.L.; Moats, J.C.; Harris, R.M.; Osna, N.A.; Thomes, P.G.; Tuma, D.J. Alcohol consumption decreases rat hepatic creatine biosynthesis via altered guanidinoacetate methyltransferase activity. Alcohol. Clin. Exp. Res. 2014, 38, 641–648. [Google Scholar] [CrossRef] [PubMed]
- Kharbanda, K.K.; Todero, S.L.; Ward, B.W.; Cannella, J.J., 3rd.; Tuma, D.J. Betaine administration corrects ethanol-induced defective VLDL secretion. Mol. Cell. Biochem. 2009, 327, 75–78. [Google Scholar] [CrossRef] [PubMed]
- Carter, W.G.; Vigneswara, V.; Atkins, R.; Tuma, D.J.; Kharbanda, K.K. Proteomic characterization of both altered protein level and isoaspartate carboxyl methylation in a model of alcoholic liver disease. Alcohol. Clin. Exp. Res. 2008, 32, 343A. [Google Scholar]
- Osna, N.A.; Donohue, T.M.; White, R.L.; Beard, M.R.; Kharbanda, K.K. Ethanol and hepatic C viral proteins regulate interferon signaling in liver cells via impaired methylation of Stat1. Hepatology 2008, 48, 327A. [Google Scholar]
- Thomes, P.G.; Osna, N.A.; Bligh, S.M.; Tuma, D.J.; Kharbanda, K.K. Role of defective methylation reactions in ethanol-induced dysregulation of intestinal barrier integrity. Biochem. Pharmacol. 2015, 96, 30–38. [Google Scholar] [CrossRef] [PubMed]
- McClain, C.J.; Barve, S.S.; Barve, A.; Marsano, L. Alcoholic liver disease and malnutrition. Alcohol. Clin. Exp. Res. 2011, 35, 815–820. [Google Scholar] [CrossRef] [PubMed]
- Patel, J.J.; McClain, C.J.; Sarav, M.; Hamilton-Reeves, J.; Hurt, R.T. Protein Requirements for Critically Ill Patients With Renal and Liver Failure. Nutr. Clin. Pract. 2017, 32, 101S–111S. [Google Scholar] [CrossRef] [PubMed]
- Johnson, T.M.; Overgard, E.B.; Cohen, A.E.; DiBaise, J.K. Nutrition assessment and management in advanced liver disease. Nutr. Clin. Pract. 2013, 28, 15–29. [Google Scholar] [CrossRef] [PubMed]
- Bharadwaj, S.; Ginoya, S.; Tandon, P.; Gohel, T.D.; Guirguis, J.; Vallabh, H.; Jevenn, A.; Hanouneh, I. Malnutrition: Laboratory markers vs. nutritional assessment. Gastroenterol. Rep. 2016, 4, 272–280. [Google Scholar] [CrossRef] [PubMed]
- Boosalis, M.G.; Ott, L.; Levine, A.S.; Slag, M.F.; Morley, J.E.; Young, B.; McClain, C.J. Relationship of visceral proteins to nutritional status in chronic and acute stress. Crit. Care Med. 1989, 17, 741–747. [Google Scholar] [CrossRef] [PubMed]
- Da Silva Fink, J.; Daniel de Mello, P.; Daniel de Mello, E. Subjective global assessment of nutritional status —A systematic review of the literature. Clin. Nutr. 2015, 34, 785–792. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.L.; Lin, X.H.; Daniels, L. Seven-Point Subjective Global Assessment Is More Time Sensitive Than Conventional Subjective Global Assessment in Detecting Nutrition Changes. J. Parenter. Enteral. Nutr. 2016, 40, 966–972. [Google Scholar] [CrossRef] [PubMed]
- Pirlich, M.; Schutz, T.; Spachos, T.; Ertl, S.; Weiss, M.L.; Lochs, H.; Plauth, M. Bioelectrical impedance analysis is a useful bedside technique to assess malnutrition in cirrhotic patients with and without ascites. Hepatology 2000, 32, 1208–1215. [Google Scholar] [CrossRef] [PubMed]
- Gaikwad, N.R.; Gupta, S.J.; Samarth, A.R.; Sankalecha, T.H. Handgrip dynamometry: A surrogate marker of malnutrition to predict the prognosis in alcoholic liver disease. Ann. Gastroenterol. 2016, 29, 509–514. [Google Scholar] [PubMed]
- Mendenhall, C.; Roselle, G.A.; Gartside, P.; Moritz, T. Relationship of protein calorie malnutrition to alcoholic liver disease: A reexamination of data from two Veterans Administration Cooperative Studies. Alcohol. Clin. Exp. Res. 1995, 19, 635–641. [Google Scholar] [CrossRef] [PubMed]
- Mendenhall, C.L.; Tosch, T.; Weesner, R.E.; Garcia-Pont, P.; Goldberg, S.J.; Kiernan, T.; Seeff, L.B.; Sorell, M.; Tamburro, C.; Zetterman, R.; et al. VA cooperative study on alcoholic hepatitis. II: Prognostic significance of protein-calorie malnutrition. Am. J. Clin. Nutr. 1986, 43, 213–218. [Google Scholar] [CrossRef] [PubMed]
- Mendenhall, C.L.; Moritz, T.E.; Roselle, G.A.; Morgan, T.R.; Nemchausky, B.A.; Tamburro, C.H.; Schiff, E.R.; McClain, C.J.; Marsano, L.S.; Allen, J.I.; et al. Protein energy malnutrition in severe alcoholic hepatitis: Diagnosis and response to treatment. The VA Cooperative Study Group #275. J. Parenter. Enteral. Nutr. 1995, 19, 258–265. [Google Scholar]
- Mendenhall, C.L.; Moritz, T.E.; Roselle, G.A.; Morgan, T.R.; Nemchausky, B.A.; Tamburro, C.H.; Schiff, E.R.; McClain, C.J.; Marsano, L.S.; Allen, J.I.; et al. A study of oral nutritional support with oxandrolone in malnourished patients with alcoholic hepatitis: Results of a Department of Veterans Affairs cooperative study. Hepatology 1993, 17, 564–576. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, M.K.; Zhou, Z.; Cave, M.; Barve, A.; McClain, C.J. Zinc and liver disease. Nutr. Clin. Pract. 2012, 27, 8–20. [Google Scholar] [CrossRef] [PubMed]
- Kirpich, I.A.; McClain, C.J.; Vatsalya, V.; Schwandt, M.; Phillips, M.; Falkner, K.C.; Zhang, L.; Harwell, C.; George, D.T.; Umhau, J.C. Liver injury and endotoxemia in male and female alcohol-dependent individuals admitted to an alcohol treatment program. Alcohol. Clin. Exp. Res. 2017, 41, 747–757. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Methods | |
|---|---|
| 1 | Anthropometry |
| 2 | Biologic Indicators |
| 3 | Creatinine Height Index |
| 4 | Muscle Strength |
| 5 | Bioelectrical Impedance |
| 6 | Air Displacement, Plethysmography |
| 7 | Imaging (DEXA, MRI, CT, etc.) |
| 8 | Subjective Global Assessment |
| 9 | Energy Balance |
| 10 | Metabolomics |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kharbanda, K.K.; Ronis, M.J.J.; Shearn, C.T.; Petersen, D.R.; Zakhari, S.; Warner, D.R.; Feldstein, A.E.; McClain, C.J.; Kirpich, I.A. Role of Nutrition in Alcoholic Liver Disease: Summary of the Symposium at the ESBRA 2017 Congress. Biomolecules 2018, 8, 16. https://doi.org/10.3390/biom8020016
Kharbanda KK, Ronis MJJ, Shearn CT, Petersen DR, Zakhari S, Warner DR, Feldstein AE, McClain CJ, Kirpich IA. Role of Nutrition in Alcoholic Liver Disease: Summary of the Symposium at the ESBRA 2017 Congress. Biomolecules. 2018; 8(2):16. https://doi.org/10.3390/biom8020016
Chicago/Turabian StyleKharbanda, Kusum K., Martin J. J. Ronis, Colin T. Shearn, Dennis R. Petersen, Samir Zakhari, Dennis R. Warner, Ariel E. Feldstein, Craig J. McClain, and Irina A. Kirpich. 2018. "Role of Nutrition in Alcoholic Liver Disease: Summary of the Symposium at the ESBRA 2017 Congress" Biomolecules 8, no. 2: 16. https://doi.org/10.3390/biom8020016
APA StyleKharbanda, K. K., Ronis, M. J. J., Shearn, C. T., Petersen, D. R., Zakhari, S., Warner, D. R., Feldstein, A. E., McClain, C. J., & Kirpich, I. A. (2018). Role of Nutrition in Alcoholic Liver Disease: Summary of the Symposium at the ESBRA 2017 Congress. Biomolecules, 8(2), 16. https://doi.org/10.3390/biom8020016