p53-Mediated Molecular Control of Autophagy in Tumor Cells



Abstract
1. Introduction: Autophagy and Tumorigenesis
2. The Cellular Mechanism of Autophagy
3. Autophagosome Formation and Its Molecular Control
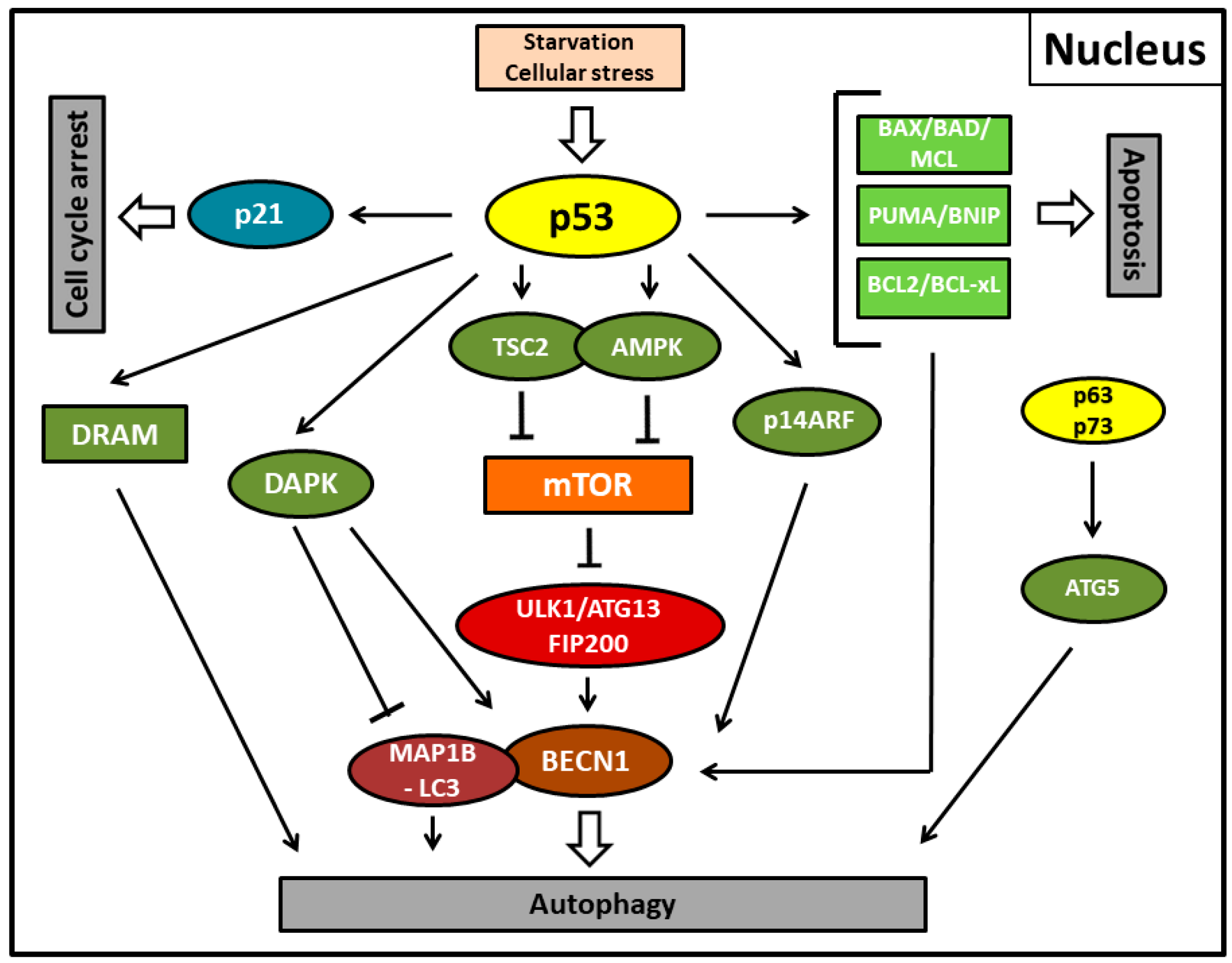
4. Positive Regulation of p53-Mediated Autophagy
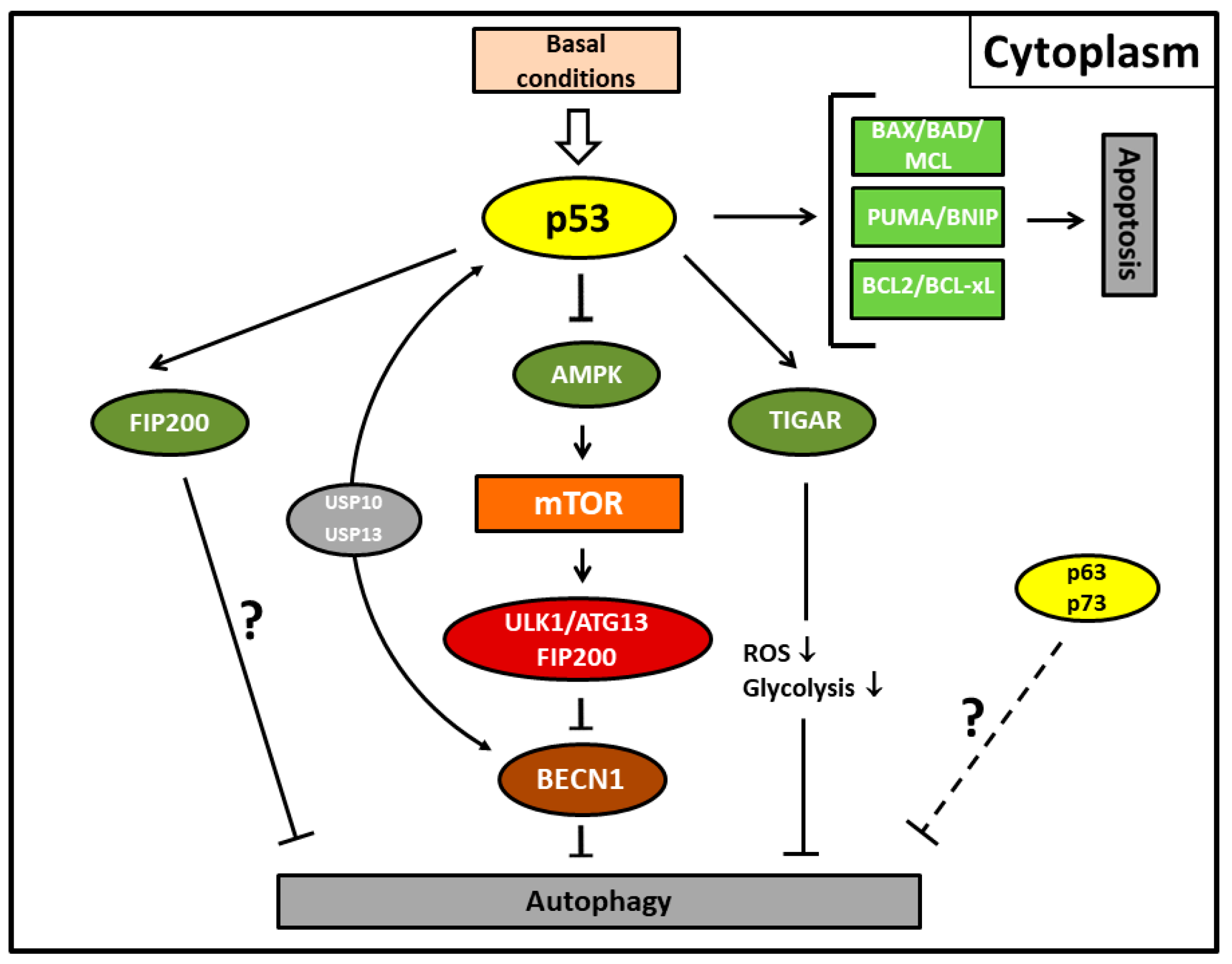
5. Negative Regulation of p53-Mediated Autophagy
6. Regulation of Autophagy in p53-Inactivated Cells
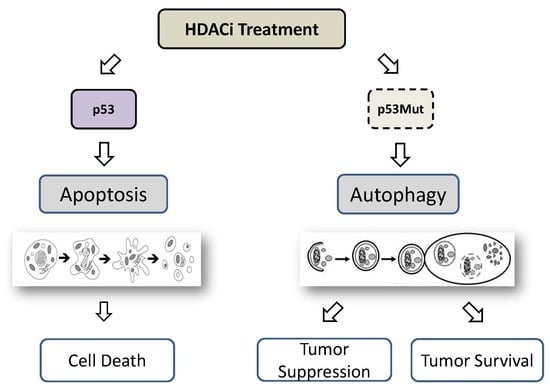
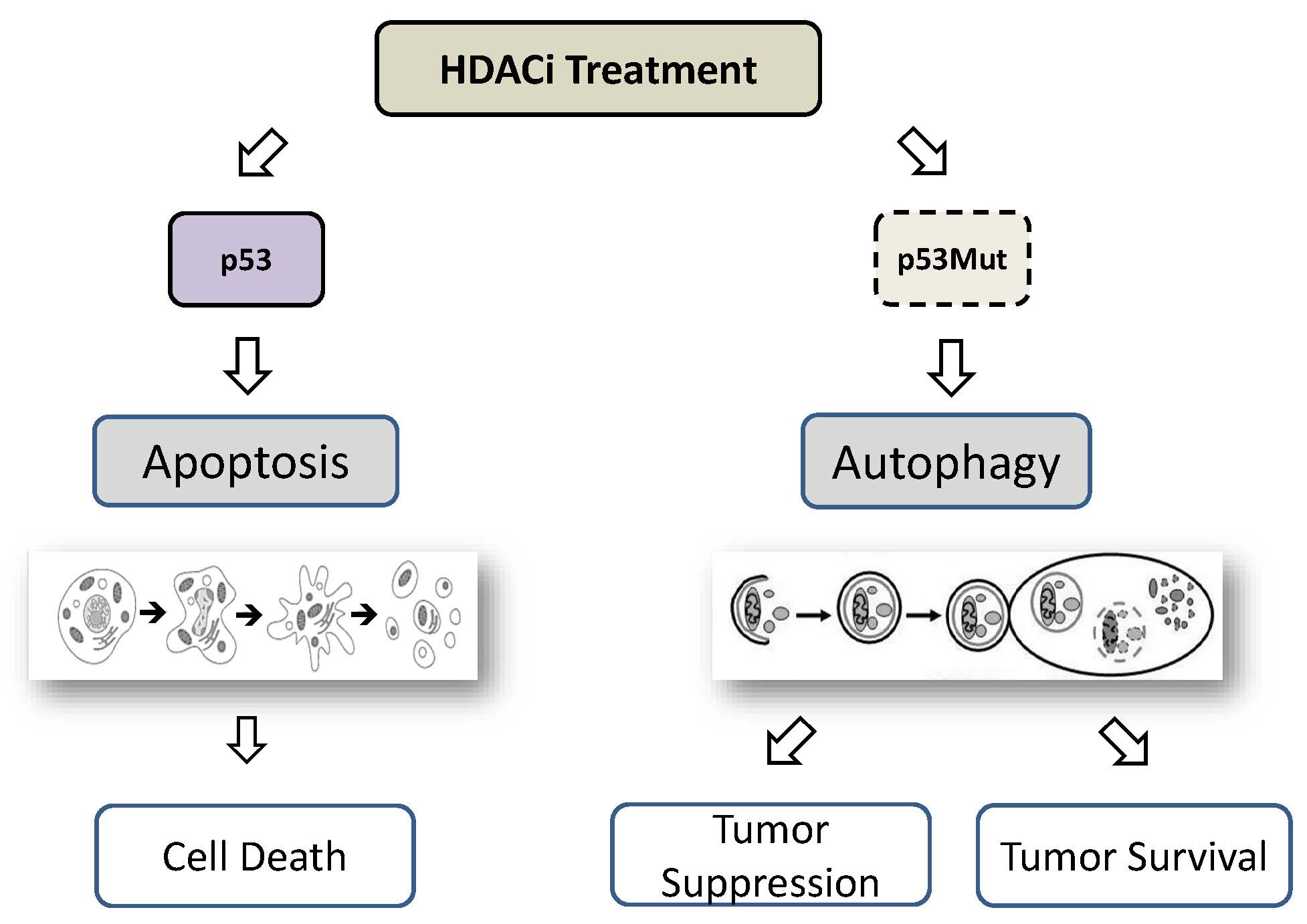
7. HDACi-Induced Autophagy Mediated by p53
8. Conclusions and Outlook
Acknowledgments
Conflicts of Interest
References
- Mizushima, N.; Noda, T.; Yoshimori, T.; Tanaka, Y.; Ishii, T.; George, M.D.; Klionsky, D.J.; Ohsumi, M.; Ohsumi, Y. A protein conjugation system essential for autophagy. Nature 1998, 395, 395–398. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Klionsky, D.J. Mammalian autophagy: Core molecular machinery and signaling regulation. Curr. Opin. Cell Biol. 2010, 22, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of cells and tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef] [PubMed]
- Li, W.W.; Li, J.; Bao, J. Microautophagy: lesser-known self-eating. Cell. Mol. Life Sci. 2012, 69, 1125–1136. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, S.; Cuervo, A.M. Chaperone-mediated autophagy: A unique way to enter the lysosome world. Trends Cell Biol. 2012, 22, 305–309. [Google Scholar] [CrossRef] [PubMed]
- Youle, R.; Narendra, D. Mechanisms of mitophagy. Nat. Rev. Mol. Cell Biol. 2011, 12, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Mizushima, N.; Virgin, H.W. Autophagy in immunity and inflammation. Nature 2011, 469, 323–335. [Google Scholar] [CrossRef] [PubMed]
- Jung, C.H.; Ro, S.-H.; Cao, J.; Otto, N.M.; Kim, D.-H. mTOR regulation of autophagy. FEBS Lett. 2010, 584, 1287–1295. [Google Scholar] [CrossRef] [PubMed]
- Takeshige, K.; Baba, M.; Tsuboi, S.; Noda, T.; Ohsumi, Y. Autophagy in yeast demonstrated with proteinase-deficient mutants and conditions for its induction. J. Cell Biol. 1992, 119, 301–311. [Google Scholar] [CrossRef] [PubMed]
- Tsukada, M.; Ohsumi, Y. Isolation and characterization of autophagy-defective mutants of Saccharomyces cerevisiae. FEBS Lett. 1992, 333, 169–174. [Google Scholar] [CrossRef]
- Ichimura, Y.; Kirisako, T.; Takao, T.; Satomi, Y.; Shimonishi, Y.; Ishihara, N.; Mizushima, N.; Tanida, I.; Kominami, E.; Ohsumi, M.; et al. A ubiquitin-like system mediates protein lipidation. Nature 2000, 408, 488–492. [Google Scholar] [PubMed]
- Harding, T.; Morano, K.; Scott, S.; Klionsky, D. Isolation and characterization of yeast mutants in the cytoplasm to vacuole protein targeting pathway. J. Cell Biol. 1995, 131, 591–602. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J. Autophagy revisited: A conversation with Christian de Duve. Autophagy 2008, 4, 740–743. [Google Scholar] [CrossRef] [PubMed]
- Kraft, C.; Martens, S. Mechanisms and regulation of autophagosome formation. Curr. Opin. Cell Biol. 2012, 24, 496–501. [Google Scholar] [CrossRef] [PubMed]
- Choi, A.M.; Ryter, S.W.; Levine, B. Autophagy in human health and disease. N. Engl. J. Med. 2013, 368, 651–662. [Google Scholar] [CrossRef] [PubMed]
- Mariño, G.; Niso-Santano, M.; Baehrecke, E.H.; Kroemer, G. Self consumption: The interplay between autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 2014, 15, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Hou, W.; Goldstein, L.A.; Lu, C.; Stolz, D.B.; Yin, X.-M.; Rabinowich, H. Involvement of protective autophagy in TRAIL resistance of apoptosis-defective tumor cells. J. Biol. Chem. 2008, 283, 19665–19677. [Google Scholar] [CrossRef] [PubMed]
- Mathew, R.; Karantza-Wadsworth, V.; White, E. Role of autophagy in cancer. Nat. Rev. Cancer 2007, 7, 961–967. [Google Scholar] [CrossRef] [PubMed]
- Duprez, L.; Wirawan, E.; Vanden Berghe, T.; Vandenabeele, P. Major cell death pathways at a glance. Microbes Infect. 2009, 11, 1050–1062. [Google Scholar] [CrossRef] [PubMed]
- Stankov, M.V.; El Khatib, M.; Kumar Thakur, B.; Heitmann, K.; Panayotova-Dimitrova, D.; Schoening, J.; Bourquin, J.P.; Schweitzer, N.; Leverkus, M.; Welte, K.; et al. Histone deacetylase inhibitors induce apoptosis in myeloid leukemia by suppressing autophagy. Leukemia 2014, 28, 577–588. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Ng, S.; Wang, J.; Zhou, J.; Tan, S.; Yang, N.; Lin, Q.; Xia, D.; Shen, H.; Zhang, J.; et al. Histone deacetylase inhibitors induce autophagy through FOXO1-dependent pathways. Autophagy 2015, 11, 629–642. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Klionsky, D.J. Regulation mechanisms and signaling pathways of autophagy. Annu. Rev. Genet. 2009, 43, 67–93. [Google Scholar] [CrossRef] [PubMed]
- Rosenfeldt, M.T.; Ryan, K.M. The role of autophagy in tumour development and cancer therapy. Expert Rev. Mol. Med. 2009, 11, e36. [Google Scholar] [CrossRef] [PubMed]
- Lopez, G.; Torres, K.; Lev, D. Autophagy blockade enhances HDAC inhibitors’ pro-apoptotic: Effects potential implications for the treatment of a therapeutic-resistant malignancy. Autophagy 2011, 7, 40–41. [Google Scholar] [CrossRef]
- Carew, J.S.; Medina, E.C.; Esquivel, J.A.; Mahalingam, D.; Swords, R.; Kelly, K.; Zhang, H.; Huang, P.; Mita, A.C.; Mita, M.M.; et al. Autophagy inhibition enhances vorinostat-induced apoptosis via ubiquitinated protein accumulation. J. Cell. Mol. Med. 2010, 14, 2448–2459. [Google Scholar] [CrossRef] [PubMed]
- Reggiori, F.; Ungermann, C. Autophagosome Maturation and Fusion. J. Mol. Biol. 2017, 429, 486–496. [Google Scholar] [CrossRef] [PubMed]
- Rubinsztein, D.C.; Shpilka, T.; Elazar, Z. Mechanisms of autophagosome biogenesis. Curr. Biol. 2012, 22, R29–R34. [Google Scholar] [CrossRef] [PubMed]
- Shibutani, S.; Yoshimori, T. A current perspective of autophagosome biogenesis. Cell Res. 2014, 24, 58–68. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Yoshimori, T.; Ohsumi, Y. The role of Atg proteins in autophagosome formation. Annu. Rev. Cell Dev. Biol. 2011, 27, 107–132. [Google Scholar] [CrossRef] [PubMed]
- Antonioli, M.; Di Rienzo, M.; Piacentini, M.; Fimia, G. Emerging mechanisms in initiating and terminating autophagy. Trends Biochem. Sci. 2017, 42, 28–41. [Google Scholar] [CrossRef] [PubMed]
- Lorin, S.; Hamaï, A.; Mehrpour, M.; Codogno, P. Autophagy regulation and its role in cancer. Semin. Cancer Biol. 2013, 23, 361–379. [Google Scholar] [CrossRef] [PubMed]
- Maiuri, M.C.; Tasdemir, E.; Criollo, A.; Morselli, E.; Vicencio, J.M.; Carnuccio, R.; Kroemer, G. Control of autophagy by oncogenes and tumor suppressor genes. Cell Death Differ. 2009, 16, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Pyo, J.O.; Nah, J.; Jung, Y.K. Molecules and their functions in autophagy. Exp. Mol. Med. 2012, 44, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Bento, C.F.; Renna, M.; Ghislat, G.; Puri, C.; Ashkenazi, A.; Vicinanza, M.; Menzies, F.M.; Rubinsztein, D.C. Mammalian Autophagy: How Does It Work? Annu. Rev. Biochem. 2016, 85, 685–713. [Google Scholar] [CrossRef] [PubMed]
- Noda, T. Regulation of autophagy through TORC1 and mTORC1. Biomolecules 2017, 7, 52. [Google Scholar] [CrossRef] [PubMed]
- Maiuri, M.; Criollo, A.; Tasdemir, E.; Vicencio, J.M.; Tajeddine, N.; Hickmann, J.; Geneste, O.; Kroemer, G. BH3-only proteins and BH3 mimetics induce autophagy by competively disrupting the interaction between Beclin 1 and Bcl-2/Bcl-X(L). Autophagy 2007, 3, 374–376. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.H.; Jackson, S.; Seaman, M.; Brown, K.; Kempkes, B.; Hibshoosh, H.; Levine, B. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 1999, 402, 672–676. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Sarbassov, D.; Ali, S.; King, J.; Latek, R.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell 2002, 110, 163–175. [Google Scholar] [CrossRef]
- Laplante, M.; Sabatini, D. mTOR signaling. Cold Spring Harb Perspect Biol. 2012, 4, a011593. [Google Scholar] [CrossRef] [PubMed]
- Zoncu, R.; Efeyan, A.; Sabatini, D. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat. Rev. Mol. Cell Biol. 2011, 12, 21–35. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kundu, M.; Viollet, B.; Kun-Liang, G. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Jung, C.H.; Jun, C.B.; Ro, S.; Kim, Y.; Otto, N.M.; Cao, J.; Kundu, M.; Kim, D. ULK-Atg13-FIP200 Complexes Mediate mTOR Signaling to the Autophagy Machinery. Mol. Biol. Cell. 2009, 20, 1992–2003. [Google Scholar] [CrossRef] [PubMed]
- Ganley, I.G.; Lam, D.H.; Wang, J.; Ding, X.; Chen, S.; Jiang, X. ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. Autophagy. J. Biol. Chem. 2009, 284, 12297–12305. [Google Scholar] [CrossRef] [PubMed]
- Rosenbluth, J.M.; Mays, D.J.; Pino, M.F.; Tang, L.J.; Pietenpol, J.A. A Gene Signature-Based Approach Identifies mTOR as a Regulator of p73. Mol. Cell. Biol. 2008, 28, 5951–5964. [Google Scholar] [CrossRef] [PubMed]
- Rosenbluth, J.M.; Pietenpol, J.A. mTOR regulates autophagy-associated genes downstream of p73. Autophagy 2009, 2, 1–7. [Google Scholar] [CrossRef]
- He, C.; Levine, B. The Beclin 1 interactome. Curr. Opin. Cell Biol. 2010, 22, 140–149. [Google Scholar] [CrossRef] [PubMed]
- Maiuri, M.; Criollo, A.; Kroemer, G. Crosstalk between apoptosis and autophagy within the Beclin 1 interactome. EMBO J. 2010, 29, 515–516. [Google Scholar] [CrossRef] [PubMed]
- Zalckvar, E.; Berissi, H.; Mizrachy, L.; Idelchuk, Y.; Koren, I.; Eisenstein, M.; Sabanay, H.; Pinkas-Kramarski, R.; Kimchi, A. DAP-kinase mediated phosphorylation on the BH3 domain of beclin 1 promotes dissociation of beclin 1 from Bcl-XL and induction of autophagy. EMBO Rep. 2009, 10, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Geng, J.; Klionsky, D.J. The Atg8 and ATG12 ubiquitin-like conjugation systems in macroautophagy. ´Protein modifications: Beyond the usual suspects´ review series. EMBO Rep. 2008, 9, 859–864. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Sugita, H.; Yoshimori, T.; Ohsumi, Y. A new protein conjugation system in human. The counterpart of the yeast Apg12p conjugation system essential for autophagy. J. Biol. Chem. 1998, 273, 33889–33892. [Google Scholar] [CrossRef] [PubMed]
- Polson, H.E.; de Lartigue, J.; Ridgen, D.J.; Reedijk, M.; Urbe, S.; Clague, M.J.; Tooze, S.A. Mammalian Atg18 (WIPI2) localizes to omegasome-anchored phagophores and positively regulates LC3 lipidation. Autophagy 2010, 6, 506–522. [Google Scholar] [CrossRef] [PubMed]
- Seibenhener, M.L.; Geetha, T.; Wooten, M.W. Sequestosome 1/p62—More than just a scaffold. FEBS Lett. 2007, 581, 175–179. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Seibenhener, M.L.; Calderilla-Barbosa, L.; Diaz-Meco, M.-T.; Moscat, J.; Jiang, J.; Wooten, M.W.; Wooten, M.C. SQSTM1/p62 interacts with HDAC6 and regulates deacetylase activity. PLoS ONE 2013, 8, e76016. [Google Scholar] [CrossRef] [PubMed]
- Mathew, R.; Karp, C.M.; Beaudoin, B.; Vuong, N.; Chen, G.; Chen, H.Y.; Bray, K.; Reddy, A.; Bhanot, G.; Gelinas, C.; et al. Autophagy Suppresses Tumorigenesis through Elimination of p62. Cell 2009, 137, 1062–1075. [Google Scholar] [CrossRef] [PubMed]
- Bjørkøy, G.; Lamark, T.; Pankiv, S.; Øvervatn, A.; Brech, A.; Johansen, T. Monitoring autophagic degradation of p62/SQSTM1. Meth. Enzym. 2009, 452, 181–197. [Google Scholar] [PubMed]
- Xie, Z.; Klionsky, D.J. Autophagosome formation: core machinery and adaptations. Nat. Cell Biol. 2007, 9, 1102–1109. [Google Scholar] [CrossRef] [PubMed]
- Simonsen, A.; Tooze, S. Coordination of membrane events during autophagy by multiple class III PI3-kinase complexes. J. Cell Biol. 2009, 186, 773–782. [Google Scholar] [CrossRef] [PubMed]
- Vousden, K.H.; Lane, D.P. p53 in health and disease. Nat. Rev. Mol. Cell Biol. 2007, 8, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Toledo, F.; Wahl, G.M. Regulating the p53 pathway: In vitro hypotheses, in vivo veritas. Nat. Rev. Cancer 2006, 6, 909–923. [Google Scholar] [CrossRef] [PubMed]
- Brooks, C.L.; Gu, W. Ubiquitination, phosphorylation and acetylation: The molecular basis for p53 regulation. Curr. Opin. Cell Biol. 2003, 15, 164–171. [Google Scholar] [CrossRef]
- Brooks, C.L.; Gu, W. The impact of acetylation and deacetylation on the p53 pathway. Protein Cell 2011, 2, 456–462. [Google Scholar] [CrossRef] [PubMed]
- Vousden, K.; Ryan, K. p53 and metabolism. Nat. Rev. Cancer 2009, 9, 691–700. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Morselli, E.; Kepp, O.; Maiuri, M.C.; Kroemer, G. Defective autophagy control by the p53 rheostat in cancer. Cell Cycle 2010, 9, 250–255. [Google Scholar] [CrossRef] [PubMed]
- Maiuri, M.; Galluzi, L.; Morselli, E.; Kepp, O.; Malik, S.; Kroemer, G. Autophagy regulation by p53. Curr. Opin. Cell Biol. 2010, 22, 181185. [Google Scholar] [CrossRef] [PubMed]
- Brady, C.A.; Attardi, L.D. P53 At a Glance. J. Cell Sci. 2010, 123, 2527–2532. [Google Scholar] [CrossRef] [PubMed]
- Yonish-Rouach, E.; Resnitzky, D.; Lotem, J.; Sachs, L.; Kimchi, A.; Oren, M. Wild-type p53 induces apoptosis of myeloid leukaemic cells that is inhibited by interleukin-6. Nature 1991, 352, 345–347. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z. p53 regulation of the IGF-1/AKT/mTOR pathways and the endosomal compartment. Cold Spring Harb. Perspect. Biol. 2010, 2, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Arico, S.; Petiot, A.; Bauvy, C.; Dubbelhuis, P.F.; Meijer, A.J.; Codogno, P.; Ogier-Denis, E. The Tumor Suppressor PTEN Positively Regulates Macroautophagy by Inhibiting the Phosphatidylinositol 3-Kinase/Protein Kinase B Pathway. J. Biol. Chem. 2001, 276, 35243–35246. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Zhang, H.; Levine, A.J.; Jin, S. The coordinate regulation of the p53 and mTOR pathways in cells. Proc. Natl. Acad. Sci. USA 2005, 102, 8204–8209. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Budanov, A.V.; Park, E.J.; Birse, R.; Kim, T.E.; Guy, A.; Ocorr, K.; Ellisman, M.H.; Bodmer, R.; Bier, E. Sestrin as a feedback inhibitor of TOR that prevents age-related pathologies. Science 2010, 327, 1223–1228. [Google Scholar] [CrossRef] [PubMed]
- Budanov, A.V.; Karin, M. p53 target genes sestrin1 and sestrin2 connect genotoxic stress and mTOR signaling. Cell 2008, 134, 451–460. [Google Scholar] [CrossRef] [PubMed]
- Crighton, D.; Wilkinson, S.; O’Prey, J.; Syed, N.; Smith, P.; Harrison, P.R.; Gasco, M.; Garrone, O.; Crook, T.; Ryan, K.M. DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell 2006. [Google Scholar] [CrossRef] [PubMed]
- Crighton, D.; Wilkinson, S.; Ryan, K. DRAM links autophagy to p53 and programmed cell death. Autophagy 2007, 3, 72–74. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.Y.; Gang, H.; Aviv, Y.; Dhingra, R.; Margulets, V.; Kirshenbaum, L.A. p53 mediates autophagy and cell death by a mechanism contingent on Bnip3. Hypertension 2013, 62, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Yee, K.S.; Wilkinson, S.; James, J.; Ryan, K.M.; Vousden, K.H. PUMA and Bax-induced autophagy contributes to apoptosis. Cell Death Differ. 2010, 16, 1135–1145. [Google Scholar] [CrossRef] [PubMed]
- Pattingre, S.; Tassa, A.; Qu, X.; Garuti, R.; Liang, X.; Mizushima, N.; Packer, M.; Schneider, M.; Levine, B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 2005, 122, 927–939. [Google Scholar] [CrossRef] [PubMed]
- Pimkina, J.; Humbey, O.; Zilfou, J.; Jarnik, M.; Murphy, M. ARF induces autophagy by virtue of interaction with Bcl-xl. J. Biol. Chem. 2009, 284, 2803–2810. [Google Scholar] [CrossRef] [PubMed]
- Zalckvar, E.; Berissi, H.; Eisenstein, M.; A, K. Phosphorylation of Beclin 1 by DAP-kinase promotes autophagy by weakening its interactions with Bcl-2 and Bcl-XL. Autophagy 2009, 5, 720–722. [Google Scholar] [CrossRef] [PubMed]
- Harrison, B.; Kraus, M.; Burch, L.; Stevens, C.; Craig, A.; Gordon-Weeks, P.; Hupp, T. DAPK-1 binding to a linear peptide motif in MAP1B stimulates autophagy and membrane blebbing. J. Biol. Chem. 2008, 283, 9999–10014. [Google Scholar] [CrossRef] [PubMed]
- Kenzelmann Broz, D.; Mello, S.S.; Bieging, K.T.; Jiang, D.; Dusek, R.L.; Brady, C.A.; Sidow, A.; Attardi, L.D. Global genomic profiling reveals an extensive p53-regulated autophagy program contributing to key p53 responses. Genes Dev. 2013, 27, 1016–1031. [Google Scholar] [CrossRef] [PubMed]
- Tasdemir, E.; Maiuri, M.C.; Galluzzi, L.; Vitale, I.; Djavaheri-Mergny, M.; D’Amelio, M.; Criollo, A.; Morselli, E.; Zhu, C.; Harper, F.; et al. Regulation of autophagy by cytoplasmic p53. Nat. Cell Biol. 2008, 10, 676–687. [Google Scholar] [CrossRef] [PubMed]
- Morselli, E.; Shen, S.; Ruckenstuhl, C.; Bauer, M.; Mariño, G.; Galluzzi, L.; Criollo, A.; Michaud, M.; Maiuri, M.; Chano, T.; et al. p53 inhibits autophagy by interacting with the human ortholog of yeast Atg17, RB1CC1/FIP200. Cell Cycle 2011, 10, 2763–2769. [Google Scholar] [CrossRef] [PubMed]
- Bensaad, K.; Tsuruta, A.; Selak, M.A.; Nieves Calvo Vidal, M.; Katsunori, N.; Bartrons, R.; Gottlieb, E.; Vousen, K.H. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell 2006, 126, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, R.; Ash, D.; Shaha, C. Beclin-1—p53 interaction is crucial for cell fate determination in embryonal carcinoma cells. J. Cell Mol. Med. 2014, 18, 2275–2286. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Xia, H.; Kim, M.; Xu, L.; Li, Y.; Zhang, L.; Cai, Y.; Norberg, H.V.; Zhang, T.; Furuya, T.; et al. Beclin1 controls the levels of p53 by regulating the deubiquitination activity of USP10 and USP13. Cell 2011, 147, 223–234. [Google Scholar] [CrossRef] [PubMed]
- Balaburski, G.M.; Hontz, R.D.; Murphy, M.E. P53 and ARF: Unexpected players in autophagy. Trends Cell Biol. 2010, 20, 363–369. [Google Scholar] [CrossRef] [PubMed]
- Reef, S.; Zalckvar, E.; Shifman, O.; Bialik, S.; Sabanay, H.; Oren, M.; Kimchi, A. A Short Mitochondrial Form of p19ARF Induces Autophagy and Caspase-Independent Cell Death. Mol. Cell 2006, 22, 463–475. [Google Scholar] [CrossRef] [PubMed]
- Abida, W.M.; Gu, W. p53-dependent and p53-independent activation of autophagy by ARF. Cancer Res. 2008, 68, 352–357. [Google Scholar] [CrossRef] [PubMed]
- Budina-Kolomets, A.; Hontz, R.D.; Pimkina, J.; Murphy, M.E. A conserved domain in exon 2 coding for the human and murine ARF tumor suppressor protein is required for autophagy induction. Autophagy 2013, 9, 1553–1565. [Google Scholar] [CrossRef] [PubMed]
- Ueda, Y.; Koya, T.; Yoneda-Kato, N.; Kato, J.Y. Small mitochondrial ARF (smARF) is located in both the nucleus and cytoplasm, induces cell death, and activates p53 in mouse fibroblasts. FEBS Lett. 2008, 582, 1459–1464. [Google Scholar] [CrossRef] [PubMed]
- Green, D.R.; Kroemer, G. Cytoplasmic functions of the tumour suppressor p53. Nature 2009, 458, 1127–1130. [Google Scholar] [CrossRef] [PubMed]
- Comel, A.; Sorrentino, G.; Capaci, V.; Del Sal, G. The cytoplasmic side of p53´s oncosuppressive activities. FEBS Lett. 2014, 588, 2600–2609. [Google Scholar] [CrossRef] [PubMed]
- Morselli, E.; Tasdemir, E.; Maiuri, M.C.; Galluzzi, L.; Kepp, O.; Criollo, A.; Vicencio, J.M.; Soussi, T.; Kroemer, G. Mutant p53 protein localized in the cytoplasm inhibits autophagy. Cell Cycle 2008, 7, 3056–3061. [Google Scholar] [CrossRef] [PubMed]
- Bensaad, K.; Cheung, E.C.; Vousden, K.H. Modulation of intracellular ROS levels by TIGAR controls autophagy. EMBO J. 2009, 28, 3015–3026. [Google Scholar] [CrossRef] [PubMed]
- Yousefi, S.; Perozzo, R.; Schmid, I.; Ziemiecki, A.; Schaffner, T.; Scapozza, L.; Brunner, T.; Simon, H.-U. Calpain-mediated cleavage of Atg5 switches autophagy to apoptosis. Nat. Cell Biol. 2006, 8, 1124–1132. [Google Scholar] [CrossRef] [PubMed]
- Muller, P.A.J.; Vousden, K.H. P53 Mutations in Cancer. Nat. Cell Biol. 2013, 15, 2–8. [Google Scholar] [CrossRef] [PubMed]
- Vousden, K.H.; Lu, X. Live or let die: the cell´s response to p53. Nat. Rev. Cancer 2002, 2, 594–604. [Google Scholar] [CrossRef] [PubMed]
- Dittmer, D.; Pati, S.; Zambetti, G.; Chu, S.; Teresky, A.K.; Moore, M.; Finlay, C.; Levine, A.J. Gain of function mutations in p53. Nat. Genet. 1993, 4, 42–46. [Google Scholar] [CrossRef] [PubMed]
- Santoro, R.; Strano, S.; Blandino, G. Transcriptional regulation by mutant p53 and oncogenesis. Subcell. Biochem. 2014, 85, 91–103. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Ma, Q.; Zhang, M.; Wang, X.; Zhang, D.; Li, W.; Wang, F.; Wu, E. Alterations of TP53 are associated with a poor outcome for patients with hepatocellular carcinoma: evidence from a systematic review and metaanalysis. Eur. J. Cancer 2012, 48, 2328–2338. [Google Scholar] [CrossRef] [PubMed]
- Cordani, M.; Butera, G.; Pacchiana, R.; Donadelli, M. Molecular interplay between mutant p53 proteins and autophagy in cancer cells. Biochim. Biophys. Acta Rev. Cancer 2017, 1867, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Fröhlich, L.F.; Mrakovcic, M.; Smole, C.; Zatloukal, K. Molecular mechanism leading to SAHA-induced autophagy in tumor cells: evidence for a p53-dependent pathway. Cancer Cell Int. 2016, 16. [Google Scholar] [CrossRef] [PubMed]
- Cordani, M.; Oppici, E.; Dando, I.; Butturini, E.; Dalla Pozza, E.; Nadal-Serrano, M.; Oliver, J.; Roca, P.; Mariotto, S.; Cellini, B.; et al. Mutant p53 proteins counteract autophagic mechanism sensitizing cancer cells to mTOR inhibition. Mol. Oncol. 2016, 10, 1008–1029. [Google Scholar] [CrossRef] [PubMed]
- Qu, X.; Yu, J.; Bhagat, G.; Furuya, N.; Hibshoosh, H.; Troxel, A.; Rosen, J.; Eskelinen, E.; Mizushima, N.; Ohsumi, Y.; et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J. Clin. Investig. 2003, 112, 1809–1820. [Google Scholar] [CrossRef] [PubMed]
- Yue, Z.; Jin, S.; Yang, C.; Levine, A.J.; Heintz, N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc. Natl. Acad. Sci. USA 2003, 100, 15077–15082. [Google Scholar] [CrossRef] [PubMed]
- Napoli, M.; Flores, E.R. The family that eats together stays together: New p53 family transcriptional targets in autophagy. Genes Dev. 2013, 27, 971–974. [Google Scholar] [CrossRef] [PubMed]
- Dokmanovic, M.; Clarke, C.; Marks, P.A. Histone Deacetylase Inhibitors: Overview and Perspectives. Mol. Cancer Res. 2007, 5, 981–989. [Google Scholar] [CrossRef] [PubMed]
- Bolden, J.; Peart, M.; Johnstone, R. Anticancer activities of histone deacetylase inhibitors. Nat. Rev. Drug Discov. 2006, 5, 769–784. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhu, W.-G. Targeting Histone Deacetylases for Cancer Therapy: From Molecular Mechanisms to Clinical Implications. Int. J. Biol. Sci. 2014, 10, 757–770. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Li, M.; Tang, Y.; Laszkowska, M.; Roeder, R.G.; Gu, W. Acetylation of p53 augments its site-specific DNA binding both in vitro and in vivo. Proc. Natl. Acad. Sci. USA 2003, 101, 2259–2264. [Google Scholar] [CrossRef]
- Bali, P.; Pranpat, M.; Bradner, J.; Balasis, M.; Fiskus, W.; Guo, F.; Rocha, K.; Kumaraswamy, S.; Boyapalle, S.; Atadja, P.; et al. Inhibition of histone deacetylase 6 acetylates and disrupts the chaperone function of heat shock protein 90: A novel basis for antileukemia activity of histone deacetylase inhibitors. J. Biol. Chem. 2005, 280, 26729–26734. [Google Scholar] [CrossRef] [PubMed]
- Newbold, A.; Falkenberg, K.J.; Prince, H.M.; Johnstone, R.W. How do tumor cells respond to HDAC inhibition? FEBS J. 2016, 283, 4032–4046. [Google Scholar] [CrossRef] [PubMed]
- Sykes, S.M.; Mellert, H.S.; Holbert, M.A.; Li, K.; Lane, W.S.; Mcmahon, S.B. Acetylation of the p53 DNA binding domain regulates apoptosis induction. Mol. Cell 2007, 24, 841–851. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Luo, J.; Zhang, W. Tip60-Dependent Acetylation of p53 Modulates the Decision between Cell-Cycle Arrest and Apoptosis. Mol. Cell 2006, 24, 827–839. [Google Scholar] [CrossRef] [PubMed]
- Barlev, N.A.; Liu, L.; Chehab, N.H.; Mansfield, K.; Harris, K.G.; Halazonetis, T.D.; Berger, S.L. Acetylation of p53 Activates Transcription through Recruitment of Coactivators/Histone Acetyltransferases. Mol. Cell 2001, 8, 1243–1254. [Google Scholar] [CrossRef]
- Buchwald, M.; Krämer, O.H.; Heinzel, T. HDACi - Targets beyond chromatin. Cancer Lett. 2009, 280, 160–167. [Google Scholar] [CrossRef] [PubMed]
- Li, A.G.; Piluso, L.G.; Cai, X.; Gadd, B.J.; Ladurner, A.G.; Liu, X. An Acetylation Switch in p53 Mediates Holo-TFIID Recruitment. Mol. Cell 2007, 28, 408–421. [Google Scholar] [CrossRef] [PubMed]
- Gu, W.; Roeder, R.G. Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell 1997, 90, 595–606. [Google Scholar] [CrossRef]
- Zhao, Y.; Lu, S.; Wu, L.; Chai, G.; Wang, H.; Chen, Y.; Sun, J.; Yu, Y.; Zhou, W.; Zheng, Q.; et al. Acetylation of p53 at Lysine 373/382 by the histone deacetylase inhibitor depsipeptide induces expression of p21 Waf1/Cip1. Mol. Cell Biol. 2006, 26, 2782–2790. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhong, Q. Histone deacetylase inhibitors and cell death. Cell. Mol. Life Sci. 2014, 71, 3885–3901. [Google Scholar] [CrossRef] [PubMed]
- Rosato, R.; Almenara, J.; Dai, Y.; Grant, S. Simultaneous activation of the intrinsic and extrinsic pathways by histone deacetylase (HDAC) inhibitors and tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) synergistically induces mitochondrial damage and apoptosis in human leukemia cells. Mol. Cancer Ther. 2003, 2, 1273–1284. [Google Scholar] [PubMed]
- Frew, A.J.; Johnstone, R.W.; Bolden, J.E. Enhancing the apoptotic and therapeutic effects of HDAC inhibitors. Cancer Lett. 2009, 280, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Shao, Y.; Gao, Z.; Marks, P.A.; Jiang, X. Apoptotic and autophagic cell death induced by histone deacetylase inhibitors. Proc. Natl. Acad. Sci. USA 2004, 101, 18030–18035. [Google Scholar] [CrossRef] [PubMed]
- Hrzenjak, A.; Kremser, M.; Strohmeier, B.; Moinfar, F.; Zatloukal, K.; Denk, H. SAHA induces caspase-independent, autophagic cell death of endometrial stromal sarcoma cells by influencing the mTOR pathway. J. Pathol. 2008, 216, 495–504. [Google Scholar] [CrossRef] [PubMed]
- Robert, T.; Vanoli, F.; Chiolo, I.; Shubassi, G.; Bernstein, K.; Rothstein, R.; Botrugno, O.; Parazzoli, D.; Oldani, A.; Minucci, S.; et al. HDACs link the DNA damage response, processing of double-strand breaks and autophagy. Nature 2011, 471, 74–79. [Google Scholar] [CrossRef] [PubMed]
- Gammoh, N.; Lam, D.; Puente, C.; Ganley, I.; Marks, P.A.; Jiang, X. Role of autophagy in histone deacetylase inhibitor-induced apoptotic and nonapoptotic cell death. Proc. Natl. Acad. Sci. USA 2012, 109, 6561–6565. [Google Scholar] [CrossRef] [PubMed]
- Koeneke, E.; Witt, O.; Oehme, I. HDAC family members intertwined in the regulation of autophagy: A druggable vulnerability in aggressive tumor entities. Cells 2015, 4, 135–168. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, S.; Tanaka, K.; Sakimura, R.; Okada, T.; Nakamura, T.; Li, Y.; Takasaki, M.; Nakabeppu, Y.; Iwamoto, Y. Suberoylanilide hydroxamic acid (SAHA) induces apoptosis or autophagy-associated cell death in chondrosarcoma cell lines. Anticancer Res. 2008, 28, 1585–1591. [Google Scholar] [PubMed]
- Gandesiri, M.; Chakilam, S.; Ivanovska, J.; Benderska, N.; Ocker, M.; Di Fazio, P.; Feoktistova, M.; Gali-Muhtasib, H.; Rave-Fränk, M.; Prante, O.; et al. DAPK plays an important role in panobinostat-induced autophagy and commits cells to apoptosis under autophagy deficient conditions. Apoptosis 2012, 17, 1300–1315. [Google Scholar] [CrossRef] [PubMed]
- Cao, Q.; Yu, C.; Xue, R.; Hsueh, W.; Pan, P.; Chen, Z.; Wang, S.; McNutt, M.; Gu, J. Autophagy induced by suberoylanilide hydroxamic acid in Hela S3 cells involves inhibition of protein kinase B and up-regulation of Beclin 1. Int. J. Biochem. Cell Biol. 2008, 40, 272–283. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.-L.; Yang, P.-M.; Shun, C.-T.; Wu, M.-S.; Weng, J.-R.; Chen, C.-C. Autophagy potentiates the anti-cancer effects of the histone deacetylase inhibitors in hepatocellular carcinoma. Autophagy 2010, 6, 1057–1065. [Google Scholar] [CrossRef] [PubMed]
- Mrakovcic, M.; Fröhlich, L.F. Regulation of HDAC inhibitor-triggered autophagy. Sci. Fed. J. Oncol. Cancer Res. 2017, 1, 2–4. [Google Scholar]
- Li, J.; Liu, R.; Lei, Y.; Wang, K.; Lau, Q.C.; Xie, N.; Zhou, S.; Nie, C.; Chen, L.; Wei, Y.; et al. Proteomic analysis revealed association of aberrant ROS signaling with suberoylanilide hydroxamic acid-induced autophagy in Jurkat T-leukemia cells. Autophagy 2010, 6, 711–724. [Google Scholar] [CrossRef] [PubMed]
- Ungerstedt, J.S.; Sowa, Y.; Xu, W.; Shao, Y.; Dokmanovic, M.; Perez, G.; Ngo, L.; Holmgren, A.; Jiang, X.; Marks, P.A. Role of thioredoxin in the response of normal and transformed cells to histone deacetylase inhibitors. Proc. Natl. Acad. Sci. USA 2005, 102, 673–678. [Google Scholar] [CrossRef] [PubMed]
- Chiao, M.; Cheng, W.; Yang, Y.; Shen, C.; Chiao, M.; Cheng, W.; Yang, Y.; Shen, C.; Ko, J. Suberoylanilide hydroxamic acid (SAHA) causes tumor growth slowdown and triggers autophagy in glioblastoma stem cells. Autophagy 2013, 9, 1509–1526. [Google Scholar] [CrossRef] [PubMed]
- Carew, J.S.; Nawrocki, S.T.; Kahue, C.N.; Zhang, H.; Yang, C.; Chung, L.; Houghton, J.A.; Huang, P.; Giles, F.J.; Cleveland, J.L. Targeting autophagy augments the anticancer activity of the histone deacetylase inhibitor SAHA to overcome Bcr-Abl—Mediated drug resistance. Blood 2007, 110, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Hrzenjak, A.; Moinfar, F.; Kremser, M.; Strohmeier, B.; Staber, P.; Zatloukal, K.; Denk, H. Valproate inhibiton of histone deacetylase 2 affects differentiation and decreases proliferation of endometrial stromal sarcoma cells. Mol. Cancer Ther. 2006, 5, 2203–2210. [Google Scholar] [CrossRef] [PubMed]
- Hrzenjak, A.; Moinfar, F.; Kremser, M.-L.; Strohmeier, B.; Petru, E.; Zatloukal, K.; Denk, H. Histone deacetylase inhibitor vorinostat suppresses the growth of uterine sarcomas in vitro and in vivo. Mol. Cancer 2010, 9. [Google Scholar] [CrossRef] [PubMed]
- Fröhlich, L.F.; Mrakovcic, M.; Smole, C.; Lahiri, P.; Zatloukal, K. Epigenetic silencing of apoptosis-inducing gene expression can be efficiently overcome by combined SAHA and TRAIL treatment in uterine sarcoma cells. PLoS ONE 2014, 9, e91558. [Google Scholar] [CrossRef] [PubMed]
- Scherz-Shouval, R.; Weidberg, H.; Gonen, C.; Wilder, S.; Elazar, Z.; Oren, M. p53-dependent regulation of autophagy protein LC3 supports cancer cell survival under prolonged starvation. Proc. Natl. Acad. Sci. USA 2010, 107, 18511–18516. [Google Scholar] [CrossRef] [PubMed]
- Mrakovcic, M.; Kleinheinz, J.; Fröhlich, L.F. Histone deacetylase inhibitor-induced autophagy in tumor cells: Implications for p53. Int. J. Mol. Sci. 2017, 18, 1883. [Google Scholar] [CrossRef]
- Mrakovcic, M.; Fröhlich, L.F. Mutational and Epimutational Analysis of Cell Death- Resistant Tumor Cells: Clues to Molecular Carcinogenesis and Cancer Therapy. Ann. Mutagen. 2017, 1, 1–2. [Google Scholar]
- Carlisi, D.; Vassallo, B.; Lauricella, M.; Emanuele, S.; D’Anneo, A.; Di Leonardo, E.; Di Fazio, P.; Vento, R.; Tesoriere, G. Histone deacetylase inhibitors induce in human hepatoma HepG2 cells acetylation of p53 and histones in correlation with apoptotic effects. Int. J. Oncol. 2008, 32, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Park, E.Y.; Woo, Y.; Kim, S.J.; Kim, D.H.; Lee, E.K.; De, U.; Kim, K.S.; Lee, J.; Jung, J.H.; Ha, K.T.; et al. Anticancer effects of a new SIRT inhibitor, MHY2256, against human breast cancer MCF-7 cells via regulation of MDM2-p53 binding. Int. J. Biol. Sci. 2016, 12, 1555–1567. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.H.; Cao, L.; Mostoslavsky, R.; Lombard, D.B.; Liu, J.; Bruns, N.E.; Tsokos, M.; Alt, F.W.; Finkel, T. A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy. Proc. Natl. Acad. Sci. USA 2008, 105, 3374–3379. [Google Scholar] [CrossRef] [PubMed]
- Morselli, E.; Galluzzi, L.; Kepp, O.; Vicencio, J.M.; Criollo, A.; Maiuri, M.C.; Kroemer, G. Anti- and pro-tumor functions of autophagy. Biochim. Biophys. Acta 2009, 1793, 1524–1532. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.; Karantza-Wadsworth, V. Role and regulation of autophagy in cancer. Biochim. Biophys. Acta Mol. Cell Res. 2009, 1793, 1516–1523. [Google Scholar] [CrossRef] [PubMed]
- Amaravadi, R.; Kimmelman, A.C.; White, E. Recent insights into the function of autophagy in cancer. Genes Dev. 2016, 30, 1913–1930. [Google Scholar] [CrossRef] [PubMed]
- Moreau, K.; Luo, S.; Rubinsztein, D.C. Cytoprotective roles for autophagy. Curr. Opin. Cell Biol. 2010, 22, 206–211. [Google Scholar] [CrossRef] [PubMed]
- Degenhardt, K.; Mathew, R.; Beaudoin, B.; Bray, K.; Anderson, D.; Chen, G.; Mukherjee, C.; Shi, Y.; Gélinas, C.; Fan, Y.; et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell 2006, 10, 51–64. [Google Scholar] [CrossRef] [PubMed]
- White, E. The role for autophagy in cancer. J. Clin. Investig. 2015, 125, 42–46. [Google Scholar] [CrossRef] [PubMed]
- Yanxiang Guo, J.; White, E. Autophagy, metabolism, and cancer. Cold Spring Harb. Symp. Quant. Biol. 2016, LXXXI, 1–6. [Google Scholar] [CrossRef]
- Galluzzi, L.; Bravo-San Pedro, J.M.; Levine, B.; Green, D.R.; Kroemer, G. Pharmacological modulation of autophagy: Therapeutic potential and persisting obstacles. Nat. Rev. Drug Discov. 2017, 16, 487–511. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| p53-Induced Autophagy | Molecular Mechanism | Additional Mechanism | Ref. |
|---|---|---|---|
| Activation | TSC2 upregulation | mTOR inhibition | [68] |
| AMPK/PTEN upregulation | mTOR inhibition | [69] | |
| Sestrin 1 and 2 upregulation | mTOR inhibition | [70] | |
| DRAM upregulation | Direct autophagosome formation | [73] | |
| Downregulation of BCL-2, BCL-xL, MCL-1 | Release of BECN1 | [76] | |
| Upregulation of BAX, BAD, BNIP3, PUMA | Release of BECN1 | [76] | |
| p14ARF upregulation | BCL-xL mediated BECN1 release | [77] | |
| DAPK-1 upregulation | Release of BECN1 by phosphorylation | [78] | |
| DAPK-1 upregulation | Inhibition of MAP1B-LC3 | [79] | |
| p63 and p73 upregulation | ATG5, ATG7, UVRAG upregulation | [45,80] | |
| Inhibition | AMPK inhibition | mTOR activation | [81] |
| FIP200 interaction | ? | [82] | |
| TIGAR upregulation | Downregulation of glycolysis, suppression of ROS | [83] | |
| BECN1 interaction | BECN1 ubiquitination and degradation | [84] | |
| BECN1 interaction | De-ubiquitination of p53 via USP10 and USP13 | [85] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mrakovcic, M.; Fröhlich, L.F. p53-Mediated Molecular Control of Autophagy in Tumor Cells. Biomolecules 2018, 8, 14. https://doi.org/10.3390/biom8020014
Mrakovcic M, Fröhlich LF. p53-Mediated Molecular Control of Autophagy in Tumor Cells. Biomolecules. 2018; 8(2):14. https://doi.org/10.3390/biom8020014
Chicago/Turabian StyleMrakovcic, Maria, and Leopold F. Fröhlich. 2018. "p53-Mediated Molecular Control of Autophagy in Tumor Cells" Biomolecules 8, no. 2: 14. https://doi.org/10.3390/biom8020014
APA StyleMrakovcic, M., & Fröhlich, L. F. (2018). p53-Mediated Molecular Control of Autophagy in Tumor Cells. Biomolecules, 8(2), 14. https://doi.org/10.3390/biom8020014