Key Events Participating in the Pathogenesis of Alcoholic Liver Disease



Abstract
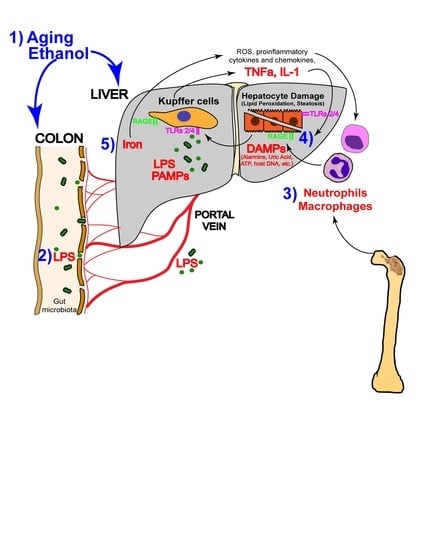
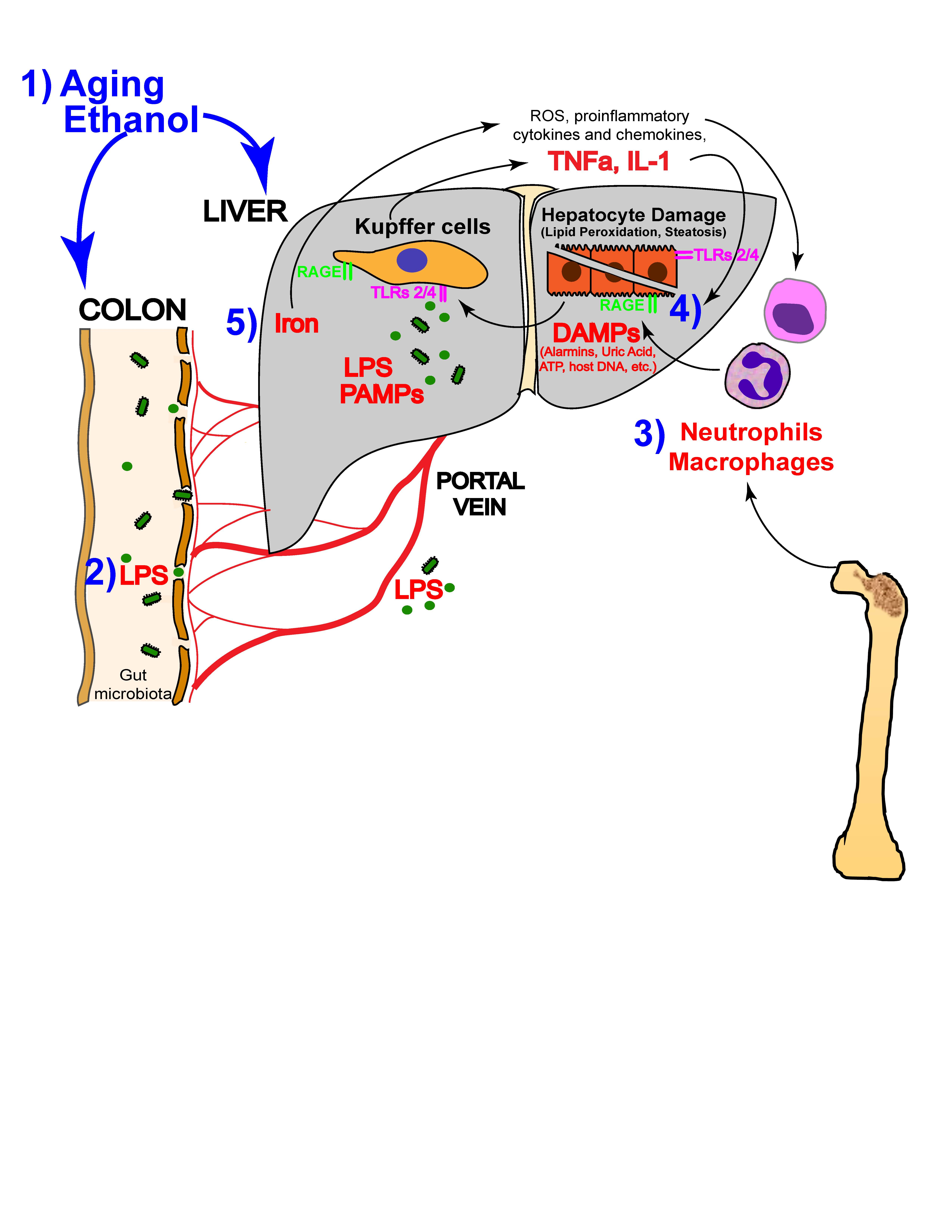
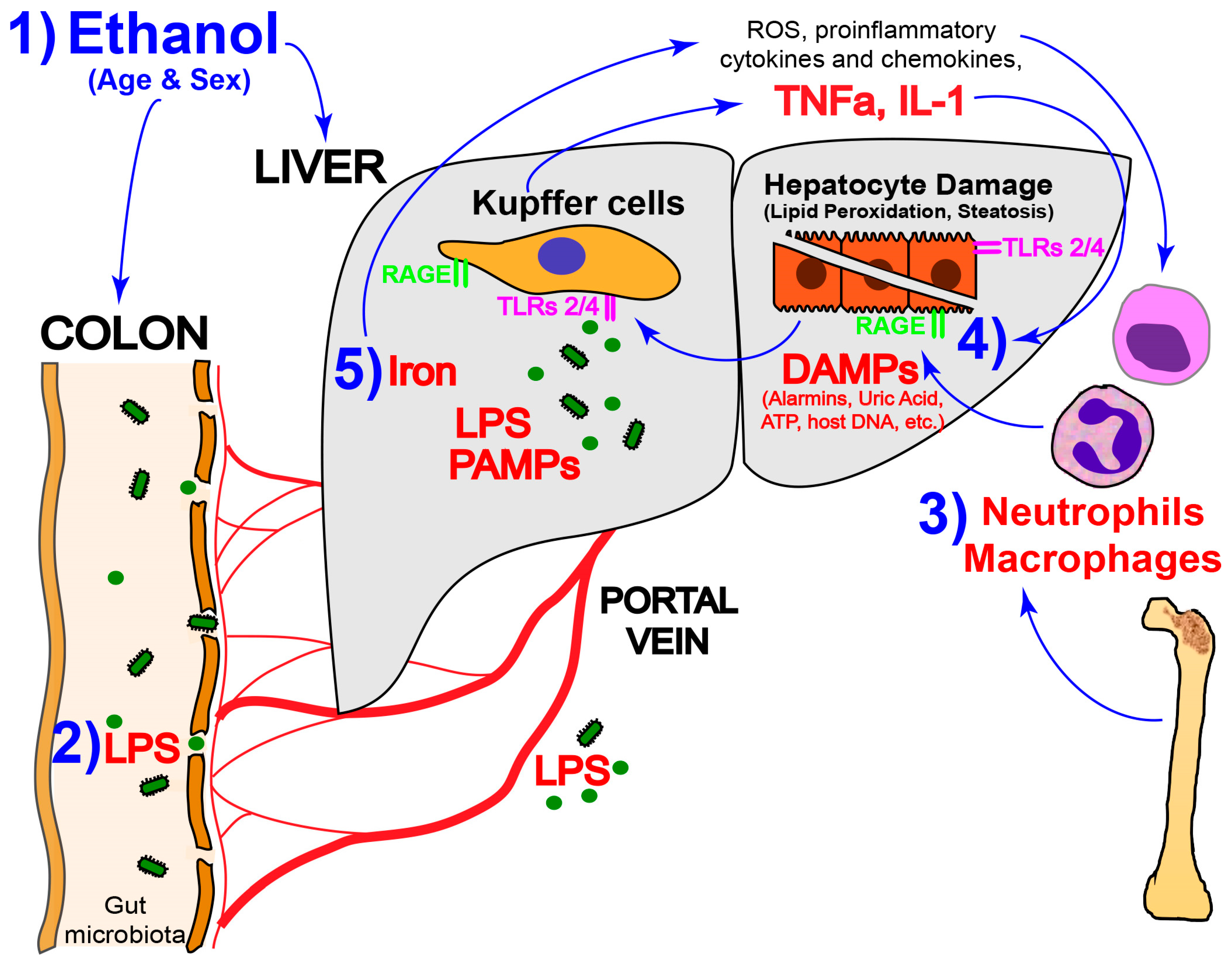
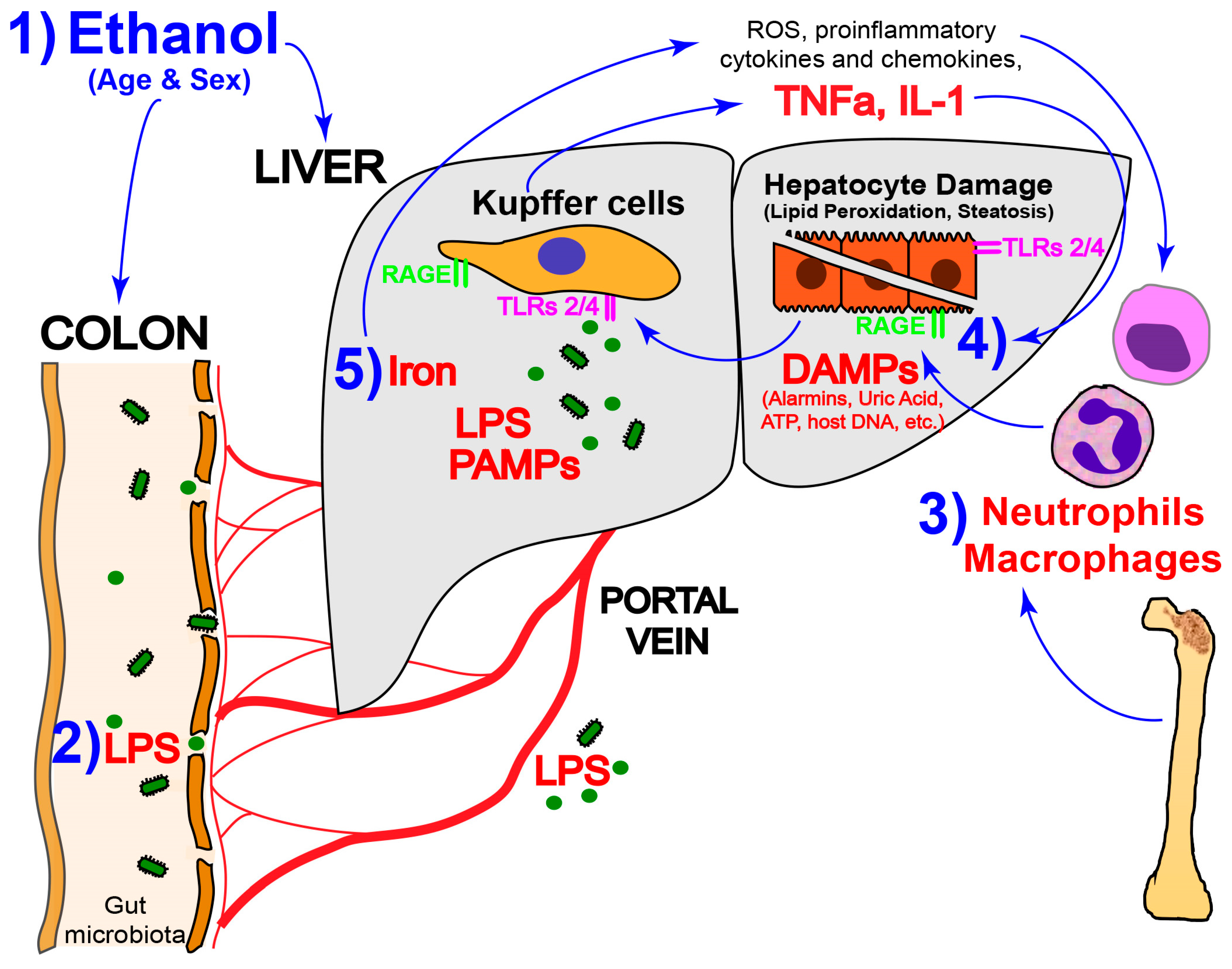
:
{kind=link}
{kind=link}
1. Introduction
2. Alcoholic Fatty Liver
3. Alcoholic Hepatitis
4. Alcoholic Cirrhosis
5. Key Players in ALD
5.1. ROS and Lipid Peroxidation
5.2. Sex Differences
5.3. Aging
5.4. Iron
5.5. Inflammation
5.5.1. Neutrophils
5.5.2. Macrophages
5.5.3. The Inflammasome
5.5.4. PAMPs and Damage-Associated Molecular Patterns
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Roh, Y.S.; Zhang, B.; Loomba, R.; Seki, E. TLR2 and TLR9 contribute to alcohol-mediated liver injury through induction of CXCL1 and neutrophil infiltration. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 309, G30–G41. [Google Scholar] [CrossRef] [PubMed]
- Joshi, K.; Kohli, A.; Manch, R.; Gish, R. Alcoholic liver disease: High risk or low risk for developing hepatocellular carcinoma? Clin. Liver Dis. 2016, 20, 563–580. [Google Scholar] [CrossRef] [PubMed]
- Stickel, F.; Moreno, C.; Hampe, J.; Morgan, M.Y. The genetics of alcohol dependence and alcohol-related liver disease. J. Hepatol. 2017, 66, 195–211. [Google Scholar] [CrossRef] [PubMed]
- Buch, S.; Stickel, F.; Trepo, E.; Way, M.; Herrmann, A.; Nischalke, H.D.; Brosch, M.; Rosendahl, J.; Berg, T.; Ridinger, M.; et al. A genome-wide association study confirms PNPLA3 and identifies TM6SF2 and MBOAT7 as risk loci for alcohol-related cirrhosis. Nat. Genet. 2015, 47, 1443–1448. [Google Scholar] [CrossRef] [PubMed]
- Viollet, B.; Guigas, B.; Leclerc, J.; Hebrard, S.; Lantier, L.; Mounier, R.; Andreelli, F.; Foretz, M. AMP-activated protein kinase in the regulation of hepatic energy metabolism: From physiology to therapeutic perspectives. Acta Physiol. 2009, 196, 81–98. [Google Scholar] [CrossRef] [PubMed]
- Bala, S.; Csak, T.; Saha, B.; Zatsiorsky, J.; Kodys, K.; Catalano, D.; Satishchandran, A.; Szabo, G. The pro-inflammatory effects of miR-155 promote liver fibrosis and alcohol-induced steatohepatitis. J. Hepatol. 2016, 64, 1378–1387. [Google Scholar] [CrossRef] [PubMed]
- Zeng, T.; Zhang, C.L.; Song, F.Y.; Zhao, X.L.; Xie, K.Q. Garlic oil alleviated ethanol-induced fat accumulation via modulation of SREBP-1, PPAR-α, and CYP2E1. Food Chem. Toxicol. 2012, 50, 485–491. [Google Scholar] [CrossRef] [PubMed]
- Altamirano, J.; Bataller, R. Alcoholic liver disease: Pathogenesis and new targets for therapy. Nat. Rev. Gastroenterol. Hepatol. 2011, 8, 491–501. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Wang, C.; Wang, C.; Zhao, H.; Zhao, C.; Chen, Y.; Wang, Y.; McClain, C.; Feng, W. Enhanced AMPK phosphorylation contributes to the beneficial effects of Lactobacillus rhamnosus GG supernatant on chronic-alcohol-induced fatty liver disease. J. Nutr. Biochem. 2015, 26, 337–344. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Villafranca, J.; Guillen, A.; Castro, J. Ethanol consumption impairs regulation of fatty acid metabolism by decreasing the activity of AMP-activated protein kinase in rat liver. Biochimie 2008, 90, 460–466. [Google Scholar] [CrossRef] [PubMed]
- Kirpich, I.A.; Miller, M.E.; Cave, M.C.; Joshi-Barve, S.; McClain, C.J. Alcoholic liver disease: Update on the role of dietary fat. Biomolecules 2016, 6, 1. [Google Scholar] [CrossRef] [PubMed]
- Teli, M.R.; Day, C.P.; Burt, A.D.; Bennett, M.K.; James, O.F. Determinants of progression to cirrhosis or fibrosis in pure alcoholic fatty liver. Lancet 1995, 346, 987–990. [Google Scholar] [CrossRef]
- Stickel, F. Alcoholic cirrhosis and hepatocellular carcinoma. Adv. Exp. Med. Biol. 2015, 815, 113–130. [Google Scholar] [PubMed]
- Sancho-Bru, P.; Altamirano, J.; Rodrigo-Torres, D.; Coll, M.; Millan, C.; Jose Lozano, J.; Miquel, R.; Arroyo, V.; Caballeria, J.; Gines, P.; et al. Liver progenitor cell markers correlate with liver damage and predict short-term mortality in patients with alcoholic hepatitis. Hepatology 2012, 55, 1931–1941. [Google Scholar] [CrossRef] [PubMed]
- Dubuquoy, L.; Louvet, A.; Lassailly, G.; Truant, S.; Boleslawski, E.; Artru, F.; Maggiotto, F.; Gantier, E.; Buob, D.; Leteurtre, E.; et al. Progenitor cell expansion and impaired hepatocyte regeneration in explanted livers from alcoholic hepatitis. Gut 2015, 64, 1949–1960. [Google Scholar] [CrossRef] [PubMed]
- Odena, G.; Chen, J.; Lozano, J.J.; Altamirano, J.; Rodrigo-Torres, D.; Affo, S.; Morales-Ibanez, O.; Matsushita, H.; Zou, J.; Dumitru, R.; et al. LPS-TLR4 pathway mediates ductular cell expansion in alcoholic hepatitis. Sci. Rep. 2016, 6, 35610. [Google Scholar] [CrossRef] [PubMed]
- Laursen, T.L.; Stoy, S.; Deleuran, B.; Vilstrup, H.; Gronbaek, H.; Sandahl, T.D. The damage-associated molecular pattern HMGB1 is elevated in human alcoholic hepatitis, but does not seem to be a primary driver of inflammation. APMIS 2016, 124, 741–747. [Google Scholar] [CrossRef] [PubMed]
- Lackner, C.; Spindelboeck, W.; Haybaeck, J.; Douschan, P.; Rainer, F.; Terracciano, L.; Haas, J.; Berghold, A.; Bataller, R.; Stauber, R.E. Histological parameters and alcohol abstinence determine long-term prognosis in patients with alcoholic liver disease. J. Hepatol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Saenz-de-Sicilia, M.; Duvoor, C.; Altamirano, J.; Chavez-Araujo, R.; Prado, V.; de Lourdes Candolo-Martinelli, A.; Holanda-Almeida, P.; Becerra-Martins-de-Oliveira, B.; Fernandez-de-Almeida, S.; Bataller, R.; et al. A Day-4 Lille Model Predicts Response to Corticosteroids and Mortality in Severe Alcoholic Hepatitis. Am. J. Gastroenterol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Rehm, J.; Samokhvalov, A.V.; Shield, K.D. Global burden of alcoholic liver diseases. J. Hepatol. 2013, 59, 160–168. [Google Scholar] [CrossRef] [PubMed]
- Scaglione, S.; Kliethermes, S.; Cao, G.; Shoham, D.; Durazo, R.; Luke, A.; Volk, M.L. The epidemiology of cirrhosis in the united states: A population-based study. J. Clin. Gastroenterol. 2015, 49, 690–696. [Google Scholar] [CrossRef] [PubMed]
- Lemoinne, S.; Cadoret, A.; Rautou, P.E.; El Mourabit, H.; Ratziu, V.; Corpechot, C.; Rey, C.; Bosselut, N.; Barbu, V.; Wendum, D.; et al. Portal myofibroblasts promote vascular remodeling underlying cirrhosis formation through the release of microparticles. Hepatology 2015, 61, 1041–1055. [Google Scholar] [CrossRef] [PubMed]
- Leung, T.M.; Nieto, N. CYP2E1 and oxidant stress in alcoholic and non-alcoholic fatty liver disease. J. Hepatol. 2013, 58, 395–398. [Google Scholar] [CrossRef] [PubMed]
- Bankoglu, E.E.; Tschopp, O.; Schmitt, J.; Burkard, P.; Jahn, D.; Geier, A.; Stopper, H. Role of PTEN in Oxidative Stress and DNA Damage in the Liver of Whole-Body Pten Haplodeficient Mice. PLoS ONE 2016, 11, e0166956. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Han, D.; Xu, R.; Wu, H.; Qu, C.; Wang, F.; Wang, X.; Zhao, Y. Glycine protects against high sucrose and high fat-induced non-alcoholic steatohepatitis in rats. Oncotarget 2016, 7, 80223–80237. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Lee, D.H.; Park, M.S.; Jung, Y.S.; Hong, J.T. CCR5 deficiency exacerbates alcoholic fatty liver disease through pro-inflammatory cytokines and chemokines-induced hepatic inflammation. J. Gastroenterol. Hepatol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Lee, D.E.; Kang, J.H.; Nam, M.J.; Park, J.W.; Kang, B.S.; Lee, D.S.; Lee, H.S.; Kwon, O.S. New potential biomarker proteins for alcoholic liver disease identified by a comparative proteomics approach. J. Cell. Biochem. 2016. [Google Scholar] [CrossRef] [PubMed]
- Li, H.H.; Tyburski, J.B.; Wang, Y.W.; Strawn, S.; Moon, B.H.; Kallakury, B.V.; Gonzalez, F.J.; Fornace, A.J., Jr. Modulation of fatty acid and bile acid metabolism by peroxisome proliferator-activated receptor α protects against alcoholic liver disease. Alcohol. Clin. Exp. Res. 2014, 38, 1520–1531. [Google Scholar] [CrossRef] [PubMed]
- Kwon, H.J.; Won, Y.S.; Park, O.; Chang, B.; Duryee, M.J.; Thiele, G.E.; Matsumoto, A.; Singh, S.; Abdelmegeed, M.A.; Song, B.J.; et al. Aldehyde dehydrogenase 2 deficiency ameliorates alcoholic fatty liver but worsens liver inflammation and fibrosis in mice. Hepatology 2014, 60, 146–157. [Google Scholar] [CrossRef] [PubMed]
- Holmuhamedov, E.L.; Czerny, C.; Beeson, C.C.; Lemasters, J.J. Ethanol suppresses ureagenesis in rat hepatocytes: Role of acetaldehyde. J. Biol. Chem. 2012, 287, 7692–7700. [Google Scholar] [CrossRef] [PubMed]
- Mani, V.; Arivalagan, S.; Siddique, A.I.; Namasivayam, N. Antioxidant and anti-inflammatory role of zingerone in ethanol-induced hepatotoxicity. Mol. Cell. Biochem. 2016, 421, 169–181. [Google Scholar] [CrossRef] [PubMed]
- Comporti, M.; Signorini, C.; Leoncini, S.; Gardi, C.; Ciccoli, L.; Giardini, A.; Vecchio, D.; Arezzini, B. Ethanol-induced oxidative stress: Basic knowledge. Genes Nutr. 2010, 5, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Portari, G.V.; Ovidio, P.P.; Deminice, R.; Jordao, A.A., Jr. Protective effect of treatment with thiamine or benfotiamine on liver oxidative damage in rat model of acute ethanol intoxication. Life Sci. 2016, 162, 21–24. [Google Scholar] [CrossRef] [PubMed]
- Bradford, B.U.; Rusyn, I. Swift increase in alcohol metabolism (SIAM): Understanding the phenomenon of hypermetabolism in liver. Alcohol 2005, 35, 13–17. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Jia, Z.; Misra, H.; Li, Y.R. Oxidative stress and redox signaling mechanisms of alcoholic liver disease: Updated experimental and clinical evidence. J. Dig. Dis. 2012, 13, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Lieber, C.S. Metabolism of alcohol. Clin. Liver Dis. 2005, 9, 1–35. [Google Scholar] [CrossRef] [PubMed]
- Baraona, E.; Lieber, C.S. Effects of ethanol on lipid metabolism. J. Lipid Res. 1979, 20, 289–315. [Google Scholar] [PubMed]
- Howarth, D.L.; Vacaru, A.M.; Tsedensodnom, O.; Mormone, E.; Nieto, N.; Costantini, L.M.; Snapp, E.L.; Sadler, K.C. Alcohol disrupts endoplasmic reticulum function and protein secretion in hepatocytes. Alcohol. Clin. Exp. Res. 2012, 36, 14–23. [Google Scholar] [CrossRef] [PubMed]
- Cederbaum, A.I. Alcohol metabolism. Clin. Liver Dis. 2012, 16, 667–685. [Google Scholar] [CrossRef] [PubMed]
- Baraona, E.; Abittan, C.S.; Dohmen, K.; Moretti, M.; Pozzato, G.; Chayes, Z.W.; Schaefer, C.; Lieber, C.S. Gender differences in pharmacokinetics of alcohol. Alcohol. Clin. Exp. Res. 2001, 25, 502–507. [Google Scholar] [CrossRef] [PubMed]
- Mansouri, A.; Fromenty, B.; Berson, A.; Robin, M.A.; Grimbert, S.; Beaugrand, M.; Erlinger, S.; Pessayre, D. Multiple hepatic mitochondrial DNA deletions suggest premature oxidative aging in alcoholic patients. J. Hepatol. 1997, 27, 96–102. [Google Scholar] [CrossRef]
- Fromenty, B.; Grimbert, S.; Mansouri, A.; Beaugrand, M.; Erlinger, S.; Rotig, A.; Pessayre, D. Hepatic mitochondrial DNA deletion in alcoholics: Association with microvesicular steatosis. Gastroenterology 1995, 108, 193–200. [Google Scholar] [CrossRef]
- Ramirez, T.; Li, Y.M.; Yin, S.; Xu, M.J.; Feng, D.; Zhou, Z.; Zang, M.; Mukhopadhyay, P.; Varga, Z.V.; Pacher, P.; et al. Aging aggravates alcoholic liver injury and fibrosis in mice by downregulating sirtuin 1 expression. J. Hepatol. 2016, in press. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.H.; Kisseleva, T.; Brenner, D.A. Aging and liver disease. Curr. Opin. Gastroenterol. 2015, 31, 184–191. [Google Scholar] [CrossRef] [PubMed]
- Tajiri, K.; Shimizu, Y. Liver physiology and liver diseases in the elderly. World J. Gastroenterol. 2013, 19, 8459–8467. [Google Scholar] [CrossRef] [PubMed]
- Ma, T.Y.; Hollander, D.; Dadufalza, V.; Krugliak, P. Effect of aging and caloric restriction on intestinal permeability. Exp. Gerontol. 1992, 27, 321–333. [Google Scholar] [CrossRef]
- Farhadi, A.; Banan, A.; Fields, J.; Keshavarzian, A. Intestinal barrier: An interface between health and disease. J. Gastroenterol. Hepatol. 2003, 18, 479–497. [Google Scholar] [CrossRef] [PubMed]
- Datz, C.; Felder, T.K.; Niederseer, D.; Aigner, E. Iron homeostasis in the metabolic syndrome. Eur. J. Clin. Investig. 2013, 43, 215–224. [Google Scholar] [CrossRef] [PubMed]
- Winterbourn, C.C. Toxicity of iron and hydrogen peroxide: The fenton reaction. Toxicol. Lett. 1995, 82–83, 969–974. [Google Scholar] [CrossRef]
- Houglum, K.; Filip, M.; Witztum, J.L.; Chojkier, M. Malondialdehyde and 4-hydroxynonenal protein adducts in plasma and liver of rats with iron overload. J. Clin. Investig. 1990, 86, 1991–1998. [Google Scholar] [CrossRef] [PubMed]
- Pietrangelo, A. Mechanisms of iron hepatotoxicity. J. Hepatol. 2016, 65, 226–227. [Google Scholar] [CrossRef] [PubMed]
- Varghese, J.; James, J.V.; Sagi, S.; Chakraborty, S.; Sukumaran, A.; Ramakrishna, B.; Jacob, M. Decreased hepatic iron in response to alcohol may contribute to alcohol-induced suppression of hepcidin. Br. J. Nutr. 2016, 115, 1978–1986. [Google Scholar] [CrossRef] [PubMed]
- Pietrangelo, A. Iron and the liver. Liver Int. 2016, 36, 116–123. [Google Scholar] [CrossRef] [PubMed]
- Ambade, A.; Satishchandran, A.; Gyongyosi, B.; Lowe, P.; Szabo, G. Adult mouse model of early hepatocellular carcinoma promoted by alcoholic liver disease. World J. Gastroenterol. 2016, 22, 4091–4108. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; He, Y.; Zhou, Z.; Ramirez, T.; Gao, Y.; Gao, Y.; Ross, R.A.; Cao, H.; Cai, Y.; Xu, M.; et al. MicroRNA-223 ameliorates alcoholic liver injury by inhibiting the IL-6-p47phox-oxidative stress pathway in neutrophils. Gut 2016. [Google Scholar] [CrossRef] [PubMed]
- Bukong, T.N.; Iracheta-Vellve, A.; Saha, B.; Ambade, A.; Satishchandran, A.; Gyongyosi, B.; Lowe, P.; Catalano, D.; Kodys, K.; Szabo, G. Inhibition of spleen tyrosine kinase activation ameliorates inflammation, cell death, and steatosis in alcoholic liver disease. Hepatology 2016, 64, 1057–1071. [Google Scholar] [CrossRef] [PubMed]
- Stoy, S.; Dige, A.; Sandahl, T.D.; Laursen, T.L.; Buus, C.; Hokland, M.; Vilstrup, H. Cytotoxic t lymphocytes and natural killer cells display impaired cytotoxic functions and reduced activation in patients with alcoholic hepatitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 308, G269–G276. [Google Scholar] [CrossRef] [PubMed]
- Stadlbauer, V.; Mookerjee, R.P.; Wright, G.A.; Davies, N.A.; Jurgens, G.; Hallstrom, S.; Jalan, R. Role of Toll-like receptors 2, 4, and 9 in mediating neutrophil dysfunction in alcoholic hepatitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2009, 296, G15–G22. [Google Scholar] [CrossRef] [PubMed]
- Andrews, K.; Abdelsamed, H.; Yi, A.K.; Miller, M.A.; Fitzpatrick, E.A. TLR2 regulates neutrophil recruitment and cytokine production with minor contributions from TLR9 during hypersensitivity pneumonitis. PLoS ONE 2013, 8, e73143. [Google Scholar] [CrossRef] [PubMed]
- Petrasek, J.; Iracheta-Vellve, A.; Csak, T.; Satishchandran, A.; Kodys, K.; Kurt-Jones, E.A.; Fitzgerald, K.A.; Szabo, G. Sting-IRF3 pathway links endoplasmic reticulum stress with hepatocyte apoptosis in early alcoholic liver disease. Proc. Natl. Acad. Sci. USA 2013, 110, 16544–16549. [Google Scholar] [CrossRef] [PubMed]
- Krysko, D.V.; Kaczmarek, A.; Krysko, O.; Heyndrickx, L.; Woznicki, J.; Bogaert, P.; Cauwels, A.; Takahashi, N.; Magez, S.; Bachert, C.; et al. TLR-2 and TLR-9 are sensors of apoptosis in a mouse model of doxorubicin-induced acute inflammation. Cell Death Differ. 2011, 18, 1316–1325. [Google Scholar] [CrossRef] [PubMed]
- Huebener, P.; Pradere, J.P.; Hernandez, C.; Gwak, G.Y.; Caviglia, J.M.; Mu, X.; Loike, J.D.; Jenkins, R.E.; Antoine, D.J.; Schwabe, R.F. The HMGB1/rage axis triggers neutrophil-mediated injury amplification following necrosis. J. Clin. Investig. 2015, 125, 539–550. [Google Scholar] [CrossRef] [PubMed]
- Bangert, A.; Andrassy, M.; Muller, A.M.; Bockstahler, M.; Fischer, A.; Volz, C.H.; Leib, C.; Goser, S.; Korkmaz-Icoz, S.; Zittrich, S.; et al. Critical role of RAGE and HMGB1 in inflammatory heart disease. Proc. Natl. Acad. Sci. USA 2016, 113, E155–E164. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Yu, M.; Zhao, J.; Martin, B.N.; Roychowdhury, S.; McMullen, M.R.; Wang, E.; Fox, P.L.; Yamasaki, S.; Nagy, L.E.; et al. IRAKM-Mincle axis links cell death to inflammation: Pathophysiological implications for chronic alcoholic liver disease. Hepatology 2016, 64, 1978–1993. [Google Scholar] [CrossRef] [PubMed]
- Hirsiger, S.; Simmen, H.P.; Werner, C.M.; Wanner, G.A.; Rittirsch, D. Danger signals activating the immune response after trauma. Mediat. Inflamm. 2012, 2012, 315941. [Google Scholar] [CrossRef] [PubMed]
- Gross, S.R.; Sin, C.G.; Barraclough, R.; Rudland, P.S. Joining S100 proteins and migration: For better or for worse, in sickness and in health. Cell Mol. Life Sci. 2014, 71, 1551–1579. [Google Scholar] [CrossRef] [PubMed]
- Bertheloot, D.; Latz, E. HMGB1, IL-1α, IL-33 and S100 proteins: Dual-function alarmins. Cell. Mol. Immunol. 2017, 14, 43–64. [Google Scholar] [CrossRef] [PubMed]
- Rider, P.; Carmi, Y.; Guttman, O.; Braiman, A.; Cohen, I.; Voronov, E.; White, M.R.; Dinarello, C.A.; Apte, R.N. IL-1α and IL-1β recruit different myeloid cells and promote different stages of sterile inflammation. J. Immunol. 2011, 187, 4835–4843. [Google Scholar] [CrossRef] [PubMed]
- Dinarello, C.A.; Simon, A.; van der Meer, J.W. Treating inflammation by blocking interleukin-1 in a broad spectrum of diseases. Nat. Rev. Drug Discov. 2012, 11, 633–652. [Google Scholar] [CrossRef] [PubMed]
- Lodder, J.; Denaes, T.; Chobert, M.N.; Wan, J.; El-Benna, J.; Pawlotsky, J.M.; Lotersztajn, S.; Teixeira-Clerc, F. Macrophage autophagy protects against liver fibrosis in mice. Autophagy 2015, 11, 1280–1292. [Google Scholar] [CrossRef] [PubMed]
- Varga, T.; Mounier, R.; Horvath, A.; Cuvellier, S.; Dumont, F.; Poliska, S.; Ardjoune, H.; Juban, G.; Nagy, L.; Chazaud, B. Highly dynamic transcriptional signature of distinct macrophage subsets during sterile inflammation, resolution, and tissue repair. J. Immunol. 2016, 196, 4771–4782. [Google Scholar] [CrossRef] [PubMed]
- Pradere, J.P.; Kluwe, J.; De Minicis, S.; Jiao, J.J.; Gwak, G.Y.; Dapito, D.H.; Jang, M.K.; Guenther, N.D.; Mederacke, I.; Friedman, R.; et al. Hepatic macrophages but not dendritic cells contribute to liver fibrosis by promoting the survival of activated hepatic stellate cells in mice. Hepatology 2013, 58, 1461–1473. [Google Scholar] [CrossRef] [PubMed]
- Gao, B.; Bataller, R. Alcoholic liver disease: Pathogenesis and new therapeutic targets. Gastroenterology 2011, 141, 1572–1585. [Google Scholar] [CrossRef] [PubMed]
- Arriazu, E.; Ge, X.; Leung, T.M.; Magdaleno, F.; Lopategi, A.; Lu, Y.; Kitamura, N.; Urtasun, R.; Theise, N.; Antoine, D.J.; et al. Signalling via the osteopontin and high mobility group box-1 axis drives the fibrogenic response to liver injury. Gut 2016. [Google Scholar] [CrossRef] [PubMed]
- Ge, X.; Leung, T.M.; Arriazu, E.; Lu, Y.; Urtasun, R.; Christensen, B.; Fiel, M.I.; Mochida, S.; Sørensen, E.S.; Nieto, N. Osteopontin binding to lipopolysaccharide lowers tumor necrosis factor-α and prevents early alcohol-induced liver injury in mice. Hepatology 2014, 59, 1600–1616. [Google Scholar] [CrossRef] [PubMed]
- Ge, X.; Lu, Y.; Leung, T.M.; Sorensen, E.S.; Nieto, N. Milk osteopontin, a nutritional approach to prevent alcohol-induced liver injury. Am. J. Physiol. Gastrointest. Liver Physiol. 2013, 304, G929–G939. [Google Scholar] [CrossRef] [PubMed]
- Kolaczkowska, E.; Kubes, P. Neutrophil recruitment and function in health and inflammation. Nat. Rev. Immunol. 2013, 13, 159–175. [Google Scholar] [CrossRef] [PubMed]
- Amanzada, A.; Moriconi, F.; Mansuroglu, T.; Cameron, S.; Ramadori, G.; Malik, I.A. Induction of chemokines and cytokines before neutrophils and macrophage recruitment in different regions of rat liver after TAA administration. Lab. Investig. 2014, 94, 235–247. [Google Scholar] [CrossRef] [PubMed]
- De Filippo, K.; Henderson, R.B.; Laschinger, M.; Hogg, N. Neutrophil chemokines KC and macrophage-inflammatory protein-2 are newly synthesized by tissue macrophages using distinct TLR signaling pathways. J. Immunol. 2008, 180, 4308–4315. [Google Scholar] [CrossRef] [PubMed]
- Fraczek, J.; Kim, T.W.; Xiao, H.; Yao, J.; Wen, Q.; Li, Y.; Casanova, J.L.; Pryjma, J.; Li, X. The Kinase Activity of IL-1 Receptor-associated Kinase 4 is Required for Interleukin-1 Receptor/Toll-like Receptor-induced TAK1-dependent NFκB Activation. J. Biol. Chem. 2008, 283, 31697–31705. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.W.; Staschke, K.; Bulek, K.; Yao, J.; Peters, K.; Oh, K.H.; Vandenburg, Y.; Xiao, H.; Qian, W.; Hamilton, T.; et al. A critical role for IRAK4 kinase activity in Toll-like receptor-mediated innate immunity. J. Exp. Med. 2007, 204, 1025–1036. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.; Kim, T.W.; Qin, J.; Jiang, Z.; Qian, Y.; Xiao, H.; Lu, Y.; Qian, W.; Gulen, M.F.; Sizemore, N.; et al. Interleukin-1 (IL-1)-induced TAK1-dependent versus MEKK3-dependent NFκB activation pathways bifurcate at IL-1 receptor-associated kinase modification. J. Biol. Chem. 2007, 282, 6075–6089. [Google Scholar] [CrossRef] [PubMed]
- Franchi, L.; Eigenbrod, T.; Munoz-Planillo, R.; Nunez, G. The inflammasome: A caspase-1-activation platform that regulates immune responses and disease pathogenesis. Nat. Immunol. 2009, 10, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Szabo, G.; Petrasek, J. Inflammasome activation and function in liver disease. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 387–400. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F.; Burns, K.; Tschopp, J. The Inflammasome: A Molecular Platform Triggering Activation of Inflammatory Caspases and Processing of proIL-β. Mol. Cell 2002, 10, 417–426. [Google Scholar] [CrossRef]
- Schroder, K.; Tschopp, J. The inflammasomes. Cell 2010, 140, 821–832. [Google Scholar] [CrossRef] [PubMed]
- Hoque, R.; Farooq, A.; Ghani, A.; Gorelick, F.; Mehal, W.Z. Lactate reduces liver and pancreatic injury in Toll-like receptor- and inflammasome-mediated inflammation via GPR81-mediated suppression of innate immunity. Gastroenterology 2014, 146, 1763–1774. [Google Scholar] [CrossRef] [PubMed]
- Prochnicki, T.; Mangan, M.S.; Latz, E. Recent insights into the molecular mechanisms of the NLRP3 inflammasome activation. F1000Research 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Cui, K.; Yan, G.; Xu, C.; Chen, Y.; Wang, J.; Zhou, R.; Bai, L.; Lian, Z.; Wei, H.; Sun, R.; et al. Invariant NKT cells promote alcohol-induced steatohepatitis through interleukin-1β in mice. J. Hepatol. 2015, 62, 1311–1318. [Google Scholar] [CrossRef] [PubMed]
- Dinarello, C.A. Interleukin-1β, interleukin-18, and the interleukin-1β converting enzyme. Ann. N. Y. Acad. Sci. 1998, 856, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Mehta, R.; Neupane, A.; Wang, L.; Goodman, Z.; Baranova, A.; Younossi, Z.M. Expression of NALPs in adipose and the fibrotic progression of non-alcoholic fatty liver disease in obese subjects. BMC Gastroenterol. 2014, 14, 208. [Google Scholar] [CrossRef] [PubMed]
- Bracey, N.A.; Gershkovich, B.; Chun, J.; Vilaysane, A.; Meijndert, H.C.; Wright, J.R., Jr.; Fedak, P.W.; Beck, P.L.; Muruve, D.A.; Duff, H.J. Mitochondrial NLRP3 protein induces reactive oxygen species to promote Smad protein signaling and fibrosis independent from the inflammasome. J. Biol. Chem. 2014, 289, 19571–19584. [Google Scholar] [CrossRef] [PubMed]
- Lozano-Ruiz, B.; Bachiller, V.; Garcia-Martinez, I.; Zapater, P.; Gomez-Hurtado, I.; Moratalla, A.; Gimenez, P.; Bellot, P.; Frances, R.; Such, J.; et al. Absent in melanoma 2 triggers a heightened inflammasome response in ascitic fluid macrophages of patients with cirrhosis. J. Hepatol. 2015, 62, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Richardson, M.B.; Williams, S.J. Mcl and mincle: C-type lectin receptors that sense damaged self and pathogen-associated molecular patterns. Front. Immunol. 2014, 5, 288. [Google Scholar] [CrossRef] [PubMed]
- Wree, A.; Marra, F. The inflammasome in liver disease. J. Hepatol. 2016, 65, 1055–1056. [Google Scholar] [CrossRef] [PubMed]
- Iracheta-Vellve, A.; Petrasek, J.; Satishchandran, A.; Gyongyosi, B.; Saha, B.; Kodys, K.; Fitzgerald, K.A.; Kurt-Jones, E.A.; Szabo, G. Inhibition of sterile danger signals, uric acid and ATP, prevents inflammasome activation and protects from alcoholic steatohepatitis in mice. J. Hepatol. 2015, 63, 1147–1155. [Google Scholar] [CrossRef] [PubMed]
- Ioannou, G.N.; Van Rooyen, D.M.; Savard, C.; Haigh, W.G.; Yeh, M.M.; Teoh, N.C.; Farrell, G.C. Cholesterol-lowering drugs cause dissolution of cholesterol crystals and disperse Kupffer cell crown-like structures during resolution of NASH. J. Lipid Res. 2015, 56, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Hendrikx, T.; Bieghs, V.; Walenbergh, S.M.; van Gorp, P.J.; Verheyen, F.; Jeurissen, M.L.; Steinbusch, M.M.; Vaes, N.; Binder, C.J.; Koek, G.H.; et al. Macrophage specific caspase-1/11 deficiency protects against cholesterol crystallization and hepatic inflammation in hyperlipidemic mice. PLoS ONE 2013, 8, e78792. [Google Scholar] [CrossRef] [PubMed]
- Lundback, P.; Lea, J.D.; Sowinska, A.; Ottosson, L.; Furst, C.M.; Steen, J.; Aulin, C.; Clarke, J.I.; Kipar, A.; Klevenvall, L.; et al. A novel high mobility group box 1 neutralizing chimeric antibody attenuates drug-induced liver injury and postinjury inflammation in mice. Hepatology 2016, 64, 1699–1710. [Google Scholar] [CrossRef] [PubMed]
- Antoine, D.J.; Jenkins, R.E.; Dear, J.W.; Williams, D.P.; McGill, M.R.; Sharpe, M.R.; Craig, D.G.; Simpson, K.J.; Jaeschke, H.; Park, B.K. Molecular forms of HMGB1 and keratin-18 as mechanistic biomarkers for mode of cell death and prognosis during clinical acetaminophen hepatotoxicity. J. Hepatol. 2012, 56, 1070–1079. [Google Scholar] [CrossRef] [PubMed]
- Antoine, D.J.; Williams, D.P.; Kipar, A.; Laverty, H.; Park, B.K. Diet restriction inhibits apoptosis and HMGB1 oxidation and promotes inflammatory cell recruitment during acetaminophen hepatotoxicity. Mol. Med. 2010, 16, 479–490. [Google Scholar] [CrossRef] [PubMed]
- Kang, R.; Zhang, Q.; Hou, W.; Yan, Z.; Chen, R.; Bonaroti, J.; Bansal, P.; Billiar, T.R.; Tsung, A.; Wang, Q.; et al. Intracellular HMGB1 inhibits inflammatory nucleosome release and limits acute pancreatitis in mice. Gastroenterology 2014, 146, 1097–1107. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Fu, S.; Fan, X.G.; Lotze, M.T.; Zeh, H.J., 3rd; Tang, D.; Kang, R. Nuclear DAMP Complex-mediatedRAGE-Dependent Macrophage Cell Death. Biochem. Biophys. Res. Commun. 2015, 458, 650–655. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Hou, W.; Zhang, Q.; Kang, R.; Fan, X.G.; Tang, D. Emerging role of high-mobility group box 1 (HMGB1) in liver diseases. Mol. Med. 2013, 19, 357–366. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.X.; Sun, H.; Liu, Q.; Guo, H.; Gong, J.P. LPS induces HMGB1 relocation and release by activating the NF-κB-CBP signal transduction pathway in the murine macrophage-like cell line RAW264.7. J. Surg. Res. 2012, 175, 88–100. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.M.; Park, E.J.; Kim, J.H.; Park, S.W.; Kim, H.J.; Chang, K.C. Ethyl pyruvate inhibits the acetylation and release of HMGB1 via effects on SIRT1/STAT signaling in LPS-activated RAW264.7 cells and peritoneal macrophages. Int. Immunopharmacol. 2016, 41, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Ge, X.; Antoine, D.J.; Lu, Y.; Arriazu, E.; Leung, T.M.; Klepper, A.L.; Branch, A.D.; Fiel, M.I.; Nieto, N. High mobility group box-1 (HMGB1) participates in the pathogenesis of alcoholic liver disease (ALD). J. Biol. Chem. 2014, 289, 22672–22691. [Google Scholar] [CrossRef] [PubMed]
- Farooq, A.; Hoque, R.; Ouyang, X.; Farooq, A.; Ghani, A.; Ahsan, K.; Guerra, M.; Mehal, W.Z. Activation of N-methyl-d-aspartate receptor downregulates inflammasome activity and liver inflammation via a β-arrestin-2 pathway. Am. J. Physiol. Gastrointest. Liver Physiol. 2014, 307, G732–G740. [Google Scholar] [CrossRef] [PubMed]
- O’Shea, R.S.; Dasarathy, S.; McCullough, A.J.; Practice Guideline Committee of the American Association for the Study of Liver Diseases; Practice Parameters Committee of the American College of Gastroenterology. Alcoholic liver disease. Hepatology 2010, 51, 307–328. [Google Scholar] [PubMed]
- Mir, H.; Meena, A.S.; Chaudhry, K.K.; Shukla, P.K.; Gangwar, R.; Manda, B.; Padala, M.K.; Shen, L.; Turner, J.R.; Dietrich, P.; et al. Occludin deficiency promotes ethanol-induced disruption of colonic epithelial junctions, gut barrier dysfunction and liver damage in mice. Biochim. Biophys. Acta 2016, 1860, 765–774. [Google Scholar] [CrossRef] [PubMed]
- Ferrere, G.; Wrzosek, L.; Cailleux, F.; Turpin, W.; Puchois, V.; Spatz, M.; Ciocan, D.; Rainteau, D.; Humbert, L.; Hugot, C.; et al. Fecal microbiota manipulation prevents dysbiosis and alcohol-induced liver injury in mice. J. Hepatol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Rao, R. Endotoxemia and gut barrier dysfunction in alcoholic liver disease. Hepatology 2009, 50, 638–644. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Magdaleno, F.; Blajszczak, C.C.; Nieto, N. Key Events Participating in the Pathogenesis of Alcoholic Liver Disease. Biomolecules 2017, 7, 9. https://doi.org/10.3390/biom7010009
Magdaleno F, Blajszczak CC, Nieto N. Key Events Participating in the Pathogenesis of Alcoholic Liver Disease. Biomolecules. 2017; 7(1):9. https://doi.org/10.3390/biom7010009
Chicago/Turabian StyleMagdaleno, Fernando, Chuck C. Blajszczak, and Natalia Nieto. 2017. "Key Events Participating in the Pathogenesis of Alcoholic Liver Disease" Biomolecules 7, no. 1: 9. https://doi.org/10.3390/biom7010009
APA StyleMagdaleno, F., Blajszczak, C. C., & Nieto, N. (2017). Key Events Participating in the Pathogenesis of Alcoholic Liver Disease. Biomolecules, 7(1), 9. https://doi.org/10.3390/biom7010009