Alcohol, Adipose Tissue and Lipid Dysregulation

Abstract
:1. Introduction
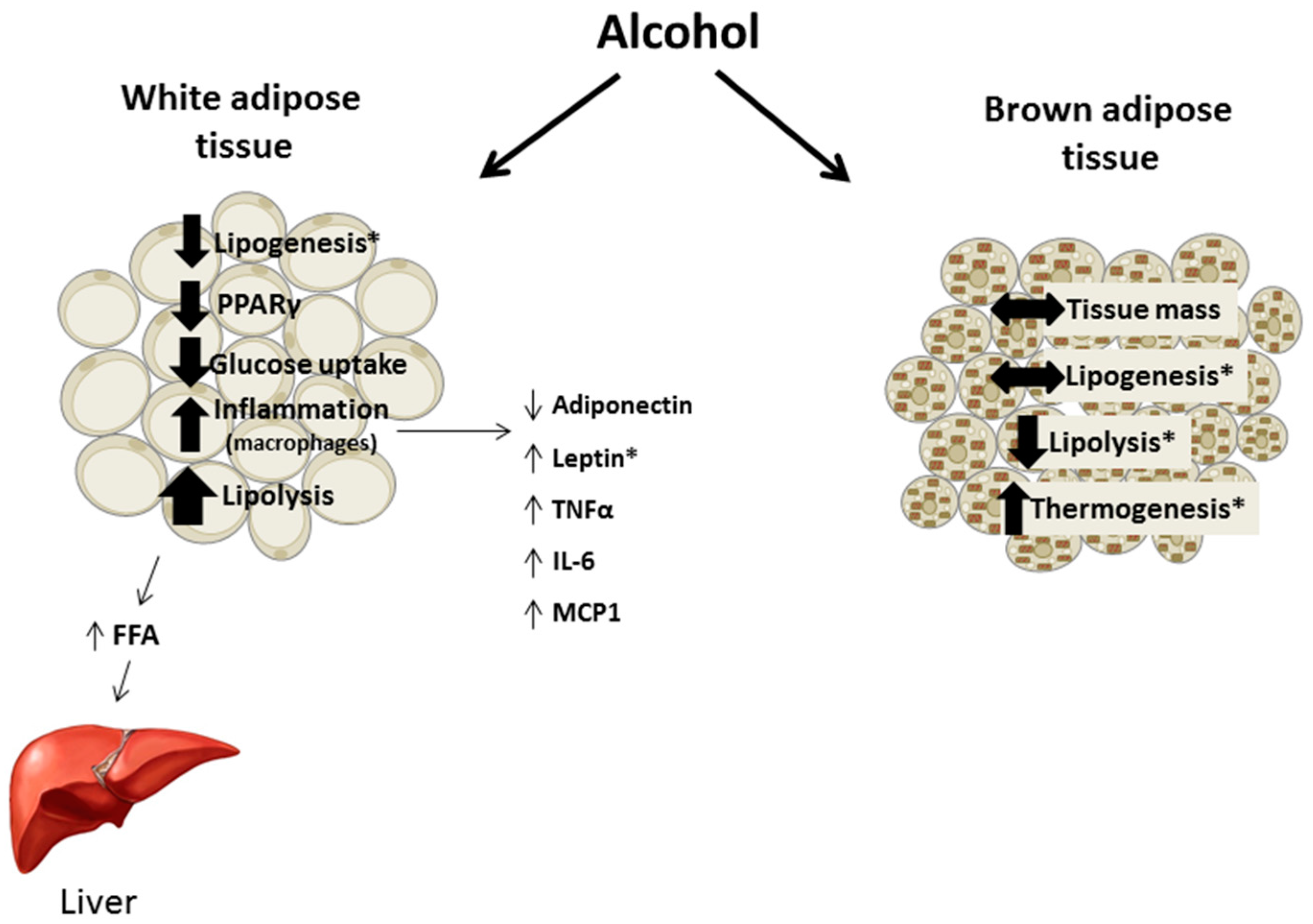
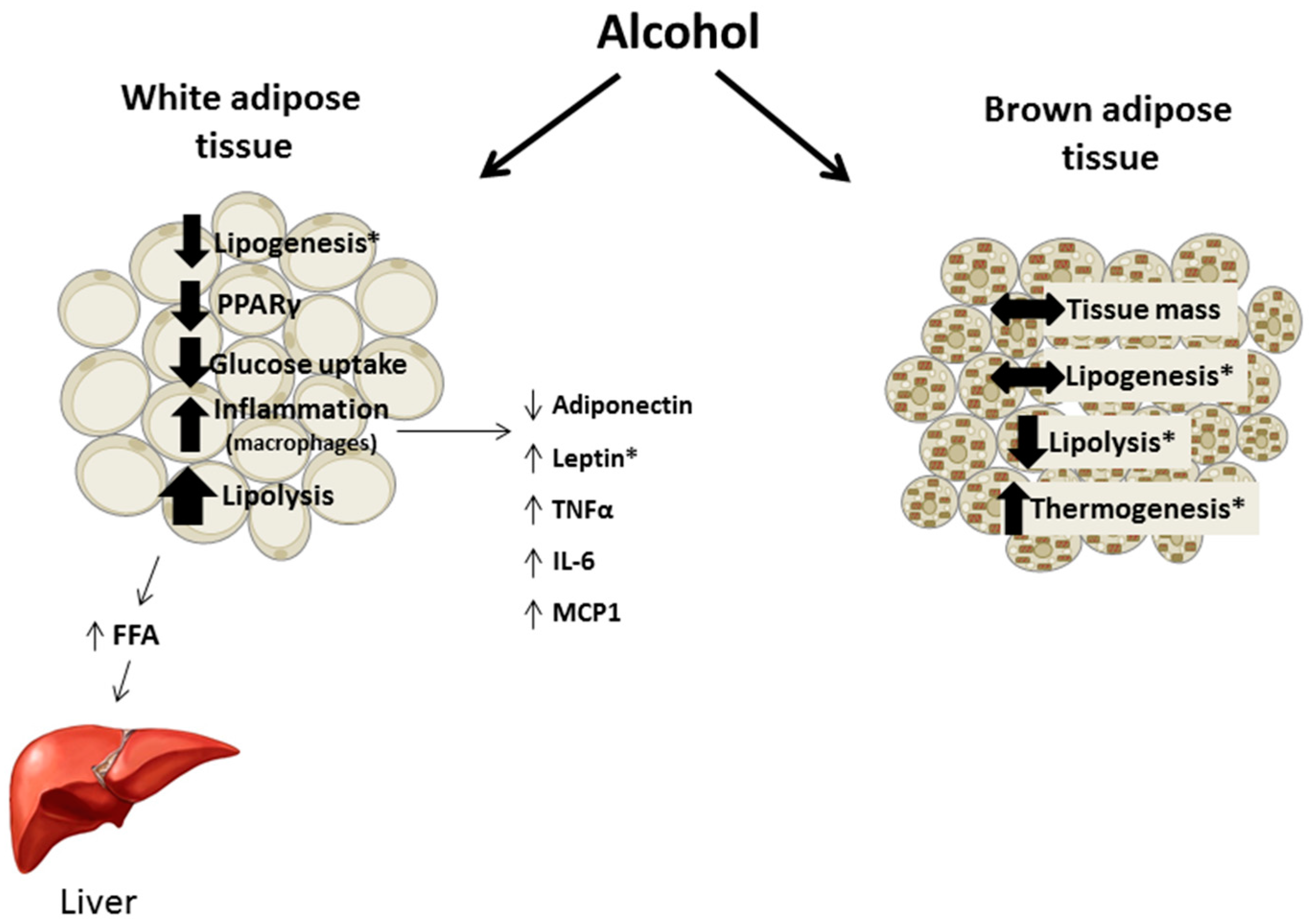
2. Chronic Alcohol and Adipose Tissue Mass
3. Regulation of Lipid Balance
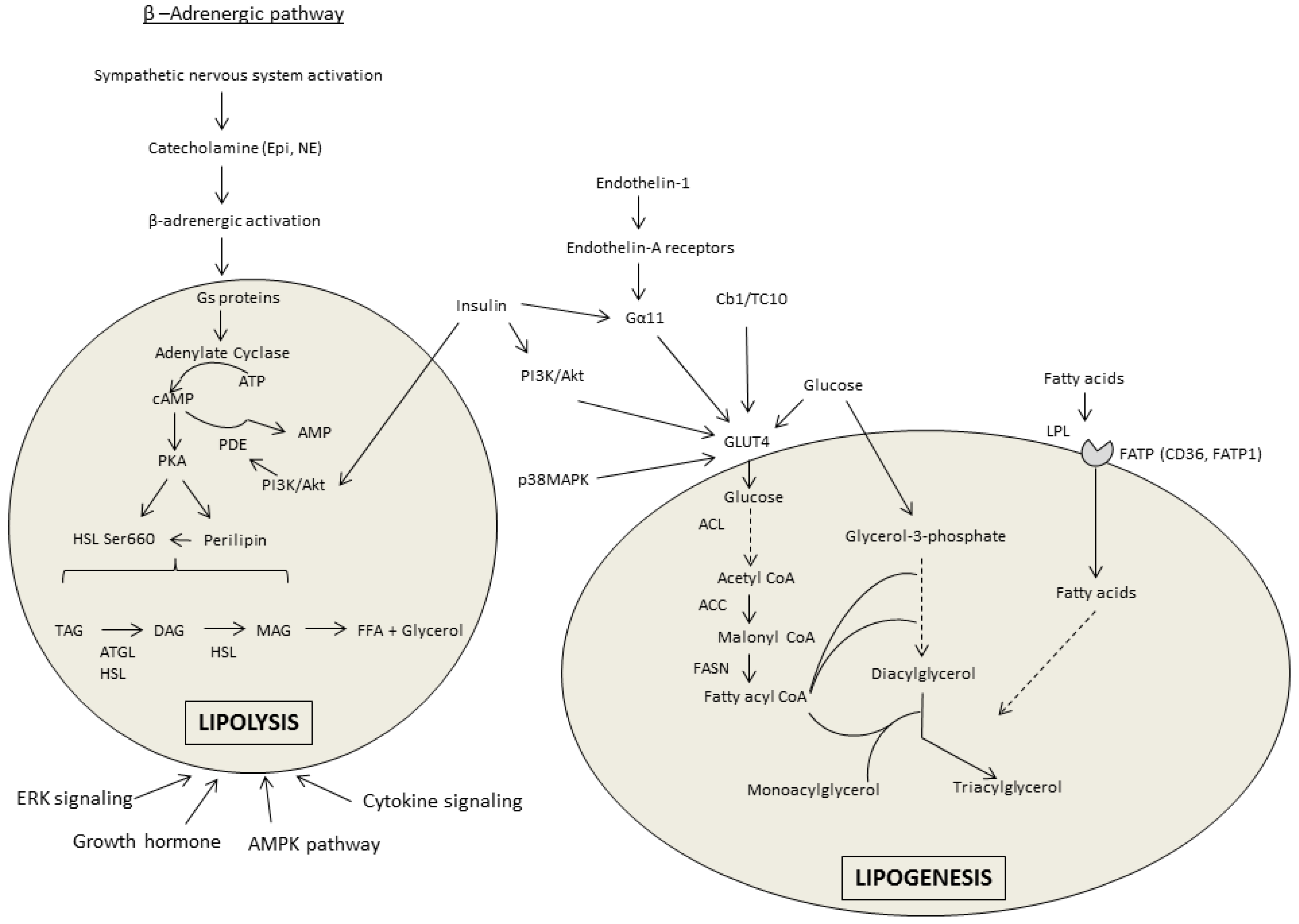
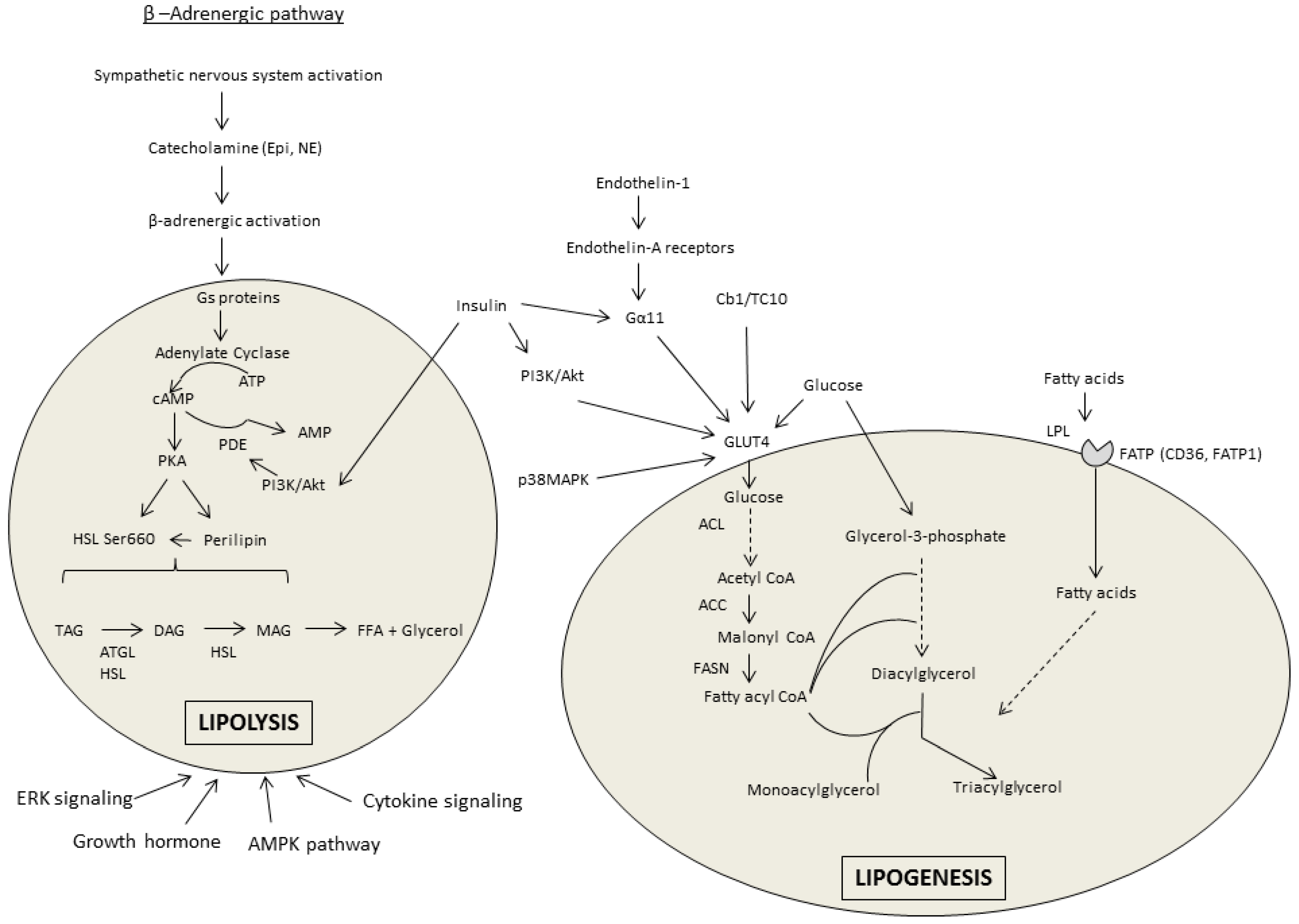
3.1. Lipolysis
3.2. Lipogenesis
3.3. Acute Alcohol and Lipid Balance
3.4. Adipose Tissue-Liver Crosstalk
3.5. Glucose Homeostasis in Adipose Tissue
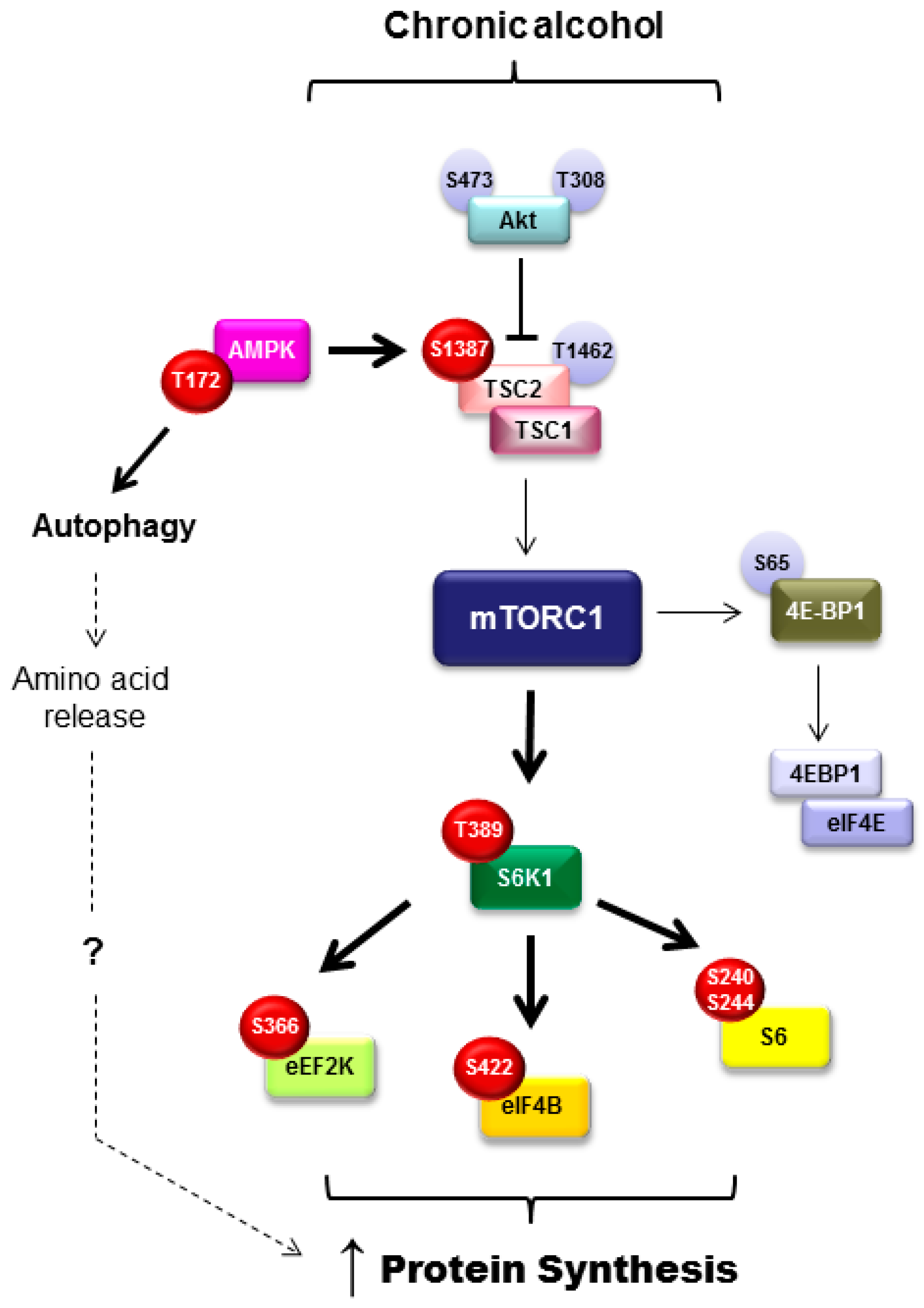
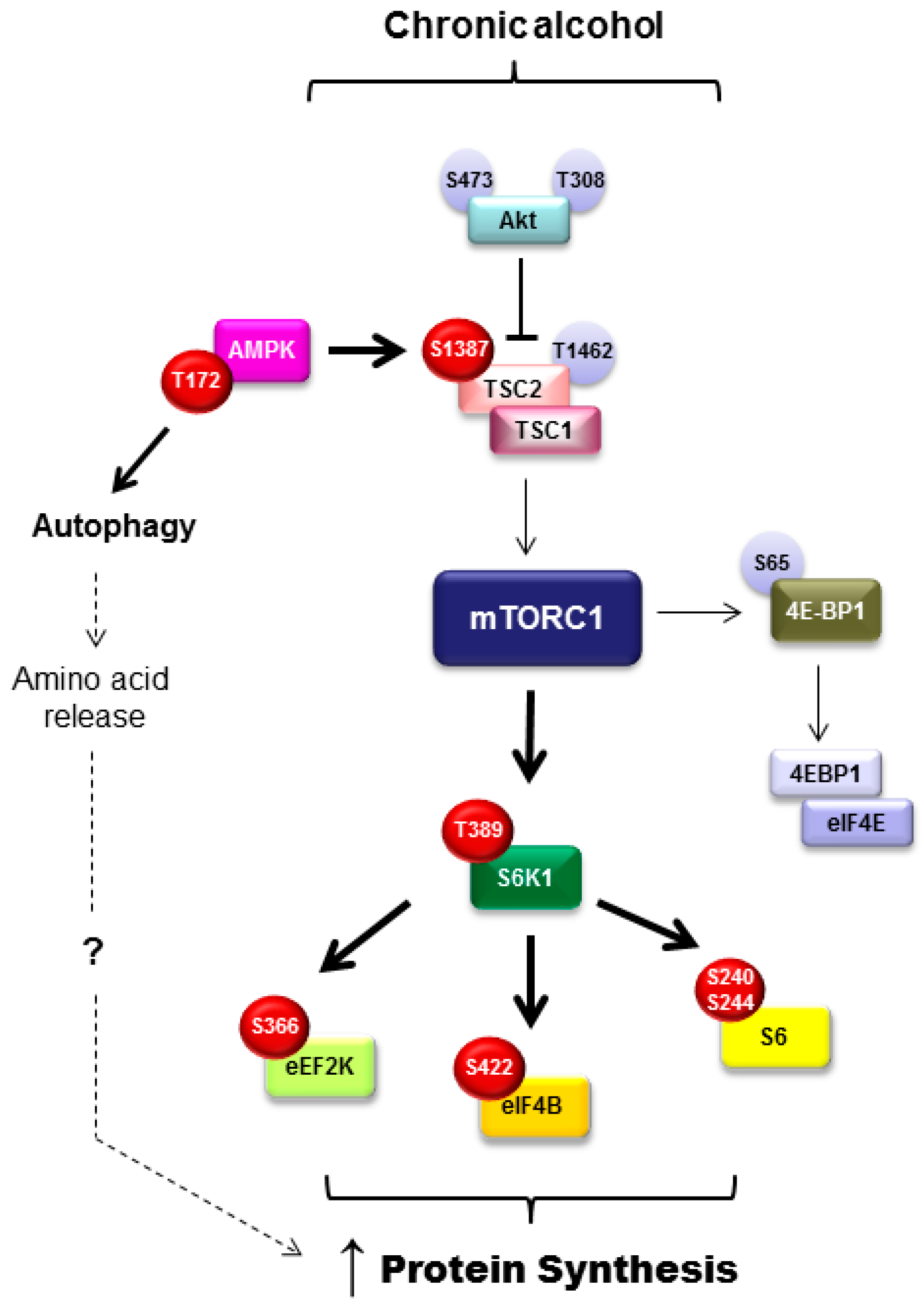
3.6. Mammalian Target of Rapamycin Activity in Adipose Tissue
4. Alcohol Metabolism
5. Adipokines
5.1. Adiponectin
5.2. Leptin
5.3. Resistin
5.4. Chemerin and Visfatin
6. Inflammatory Cytokines
7. Brown Adipose Tissue
8. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Luo, L.; Liu, M. Adipose tissue in control of metabolism. J. Endocrinol. 2016, 231, R77–R99. [Google Scholar] [CrossRef] [PubMed]
- Frühbeck, G.; Méndez-Giménez, L.; Fernández-Formoso, J.A.; Fernández, S.; Rodríguez, A. Regulation of adipocyte lipolysis. Nutr. Res. Rev. 2014, 27, 63–93. [Google Scholar] [CrossRef] [PubMed]
- Hosogai, N.; Fukuhara, A.; Oshima, K.; Miyata, Y.; Tanaka, S.; Segawa, K.; Furukawa, S.; Tochino, Y.; Komuro, R.; Matsuda, M.; et al. Adipose tissue hypoxia in obesity and its impact on adipocytokine dysregulation. Diabetes 2007, 56, 901–911. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Indias, I.; Tinahones, F.J. Impaired adipose tissue expandability and lipogenic capacities as ones of the main causes of metabolic disorders. J. Diabetes Res. 2015, 2015, 970375. [Google Scholar] [CrossRef] [PubMed]
- Pravdova, E.; Fickova, M. Alcohol intake modulates hormonal activity of adipose tissue. Endocr. Regul. 2006, 40, 91–104. [Google Scholar] [PubMed]
- Baraona, E.; Lieber, C.S. Effects of ethanol on lipid metabolism. J. Lipid Res. 1979, 20, 289–315. [Google Scholar] [PubMed]
- Zhong, W.; Zhao, Y.; Tang, Y.; Wei, X.; Shi, X.; Sun, W.; Sun, X.; Yin, X.; Kim, S.; McClain, C.J.; et al. Chronic alcohol exposure stimulates adipose tissue lipolysis in mice: Role of reverse triglyceride transport in the pathogenesis of alcoholic steatosis. Am. J. Pathol. 2012, 180, 998–1007. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Shi, X.; Zhong, W.; Zhao, Y.; Tang, Y.; Sun, W.; Yin, X.; Bogdanov, B.; Kim, S.; McClain, C.; et al. Chronic Alcohol Exposure Disturbs Lipid Homeostasis at the Adipose Tissue-Liver Axis in Mice: Analysis of Triacylglycerols Using High-Resolution Mass Spectrometry in Combination with In Vivo Metabolite Deuterium Labeling. PLoS ONE 2013, 8, e55382. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Zhang, X.J.; Feng, K.; He, C.; Li, P.; Hu, Y.J.; Su, H.; Wan, J.B. Dietary α-linolenic acid-rich flaxseed oil prevents against alcoholic hepatic steatosis via ameliorating lipid homeostasis at adipose tissue-liver axis in mice. Sci. Rep. 2016, 6, 26826. [Google Scholar] [CrossRef] [PubMed]
- Tan, X.; Sun, X.; Li, Q.; Zhao, Y.; Zhong, W.; Jia, W.; McClain, C.J.; Zhou, Z. Leptin deficiency contributes to the pathogenesis of alcoholic fatty liver disease in mice. Am. J. Pathol. 2012, 181, 1279–1286. [Google Scholar] [CrossRef] [PubMed]
- Dou, X.; Xia, Y.; Chen, J.; Qian, Y.; Li, S.; Zhang, X.; Song, Z. Rectification of impaired adipose tissue methylation status and lipolytic response contributes to hepatoprotective effect of betaine in a mouse model of alcoholic liver disease. Br. J. Pharmacol. 2014, 171, 4073–4086. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Tang, Y.; Tan, X.; Li, Q.; Zhong, W.; Jia, W.; McClain, C.J.; Zhou, Z. Activation of peroxisome proliferator-activated receptor-γ by rosiglitazone improves lipid homeostasis at the adipose tissue-liver axis in ethanol-fed mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 302, G548–G557. [Google Scholar] [CrossRef] [PubMed]
- Zhong, W.; Zhao, Y.; Sun, X.; Song, Z.; McClain, C.J.; Zhou, Z. Dietary zinc deficiency exaggerates ethanol-induced liver injury in mice: Involvement of intrahepatic and extrahepatic factors. PLoS ONE 2013, 8, e76522. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Liu, Y.; Xiao, J.; Liu, L.; Chen, S.; Mohammadi, M.; McClain, C.J.; Li, X.; Feng, W. FGF21 mediates alcohol-induced adipose tissue lipolysis by activation of systemic release of catecholamine in mice. J. Lipid Res. 2015, 56, 1481–1491. [Google Scholar] [CrossRef] [PubMed]
- Crowell, K.T.; Steiner, J.L.; Coleman, C.S.; Lang, C.H. Decreased whole-body fat mass produced by chronic alcohol consumption is associated with activation of S6K1-mediated protein synthesis and increased autophagy in epididymal white adipose tissue. Alcohol. Clin. Exp. Res. 2016, 40, 1832–1845. [Google Scholar] [CrossRef] [PubMed]
- Pravdova, E.; Macho, L.; Fickova, M. Alcohol intake modifies leptin, adiponectin and resistin serum levels and their mRNA expressions in adipose tissue of rats. Endocr. Regul. 2009, 43, 117–125. [Google Scholar] [PubMed]
- Pravdová, E.; Macho, L.; Hlavácová, N.; Ficková, M. Long-time alcohol intake modifies resistin secretion and expression of resistin gene in adipose tissue. Gen. Physiol. Biophys. 2007, 26, 221–229. [Google Scholar] [PubMed]
- Lee, H.I.; Lee, M.K. Coordinated regulation of scopoletin at adipose tissue-liver axis improved alcohol-induced lipid dysmetabolism and inflammation in rats. Toxicol. Lett. 2015, 237, 210–218. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Zhong, W.; Sun, X.; Sun, Q.; Tan, X.; Li, Q.; Zhou, Z. Visceral white adipose tissue is susceptible to alcohol-induced lipodystrophy in rats: Role of acetaldehyde. Alcohol. Clin. Exp. Res. 2015, 39, 416–423. [Google Scholar] [CrossRef] [PubMed]
- Wilkes, J.J.; DeForrest, L.L.; Nagy, L.E. Chronic ethanol feeding in a high-fat diet decreases insulin-stimulated glucose transport in rat adipocytes. Am. J. Physiol. 1996, 271, E477–E484. [Google Scholar] [PubMed]
- Tang, H.; Sebastian, B.M.; Axhemi, A.; Chen, X.; Hillian, A.D.; Jacobsen, D.W.; Nagy, L.E. Ethanol-induced oxidative stress via the CYP2E1 pathway disrupts adiponectin secretion from adipocytes. Alcohol. Clin. Exp. Res. 2012, 36, 214–222. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Li, M.; Zheng, D.; Chen, Q.; Liu, W.; Feng, L. Adipose tissue hypoxia and low-grade inflammation: A possible mechanism for ethanol-related glucose intolerance? Br. J. Nutr. 2015, 113, 1355–1364. [Google Scholar] [CrossRef] [PubMed]
- Feng, L.; Song, Y.F.; Guan, Q.B.; Liu, H.J.; Ban, B.; Dong, H.X.; Hou, X.L.; Lee, K.O.; Gao, L.; Zhao, J.J. Long-term ethanol exposure inhibits glucose transporter 4 expression via an AMPK-dependent pathway in adipocytes. Acta Pharmacol. Sin. 2010, 31, 329–340. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.C.; Li, S.Y.; Cao, M.F.; Jiang, X.Y.; Feng, L.; Zhao, J.J.; Gao, L. Effects of chronic ethanol consumption on levels of adipokines in visceral adipose tissues and sera of rats. Acta Pharmacol. Sin. 2010, 31, 461–469. [Google Scholar] [CrossRef] [PubMed]
- Feng, L.; Gao, L.; Guan, Q.; Hou, X.; Wan, Q.; Wang, X.; Zhao, J. Long-term moderate ethanol consumption restores insulin sensitivity in high-fat-fed rats by increasing SLC2A4 (GLUT4) in the adipose tissue by AMP-activated protein kinase activation. J. Endocrinol. 2008, 199, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Addolorato, G.; Capristo, E.; Marini, M.; Santini, P.; Scognamiglio, U.; Attilia, M.L.; Messineo, D.; Sasso, G.F.; Gasbarrini, G.; Ceccanti, M. Body composition changes induced by chronic ethanol abuse: Evaluation by dual energy X-ray absorptiometry. Am. J. Gastroenterol. 2000, 95, 2323–2327. [Google Scholar] [CrossRef] [PubMed]
- Santolaria, F.; Pérez-Cejas, A.; Alemán, M.R.; González-Reimers, E.; Milena, A.; de la Vega, M.J.; Martínez-Riera, A.; Gómez-Rodríguez, M.A. Low serum leptin levels and malnutrition in chronic alcohol misusers hospitalized by somatic complications. Alcohol Alcohol. 2003, 38, 60–66. [Google Scholar] [CrossRef] [PubMed]
- Addolorato, G.; Capristo, E.; Greco, A.V.; Stefanini, G.F.; Gasbarrini, G. Energy expenditure, substrate oxidation, and body composition in subjects with chronic alcoholism: New findings from metabolic assessment. Alcohol. Clin. Exp. Res. 1997, 21, 962–967. [Google Scholar] [CrossRef] [PubMed]
- Addolorato, G.; Capristo, E.; Greco, A.V.; Stefanini, G.F.; Gasbarrini, G. Influence of chronic alcohol abuse on body weight and energy metabolism: Is excess ethanol consumption a risk factor for obesity or malnutrition? J. Intern. Med. 1998, 244, 387–395. [Google Scholar] [CrossRef] [PubMed]
- Naveau, S.; Cassard-Doulcier, A.M.; Njiké-Nakseu, M.; Bouchet-Delbos, L.; Barri-Ova, N.; Boujedidi, H.; Dauvois, B.; Balian, A.; Maitre, S.; Prévot, S.; et al. Harmful effect of adipose tissue on liver lesions in patients with alcoholic liver disease. J. Hepatol. 2010, 52, 895–902. [Google Scholar] [CrossRef] [PubMed]
- Ren, R.Z.; Zhang, X.; Xu, J.; Zhang, H.Q.; Yu, C.X.; Cao, M.F.; Gao, L.; Guan, Q.B.; Zhao, J.J. Chronic ethanol consumption increases the levels of chemerin in the serum and adipose tissue of humans and rats. Acta Pharmacol. Sin. 2012, 33, 652–659. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Davidoff, A.J.; Brown, R.A. Acetaldehyde depresses shortening and intracellular Ca2+ transients in adult rat ventricular myocytes. Cell. Mol. Biol. 1997, 43, 825–834. [Google Scholar] [PubMed]
- Englund Ogge, L.; Brohall, G.; Behre, C.J.; Schmidt, C.; Fagerberg, B. Alcohol consumption in relation to metabolic regulation, inflammation, and adiponectin in 64-year-old caucasian women: A population-based study with a focus on impaired glucose regulation. Diabetes Care 2006, 29, 908–913. [Google Scholar] [CrossRef] [PubMed]
- Makita, S.; Abiko, A.; Nagai, M.; Yonezawa, S.; Koshiyama, M.; Ohta, M.; Nakamura, M. Influence of daily alcohol consumption on serum adiponectin levels in men. Metabolism 2013, 62, 411–416. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.K.; Kim, M.K.; Shin, J.; Choi, B.Y. A cross-sectional analysis of the relationship between daily alcohol consumption and serum adiponectin levels among adults aged 40 years or more in a rural area of Korea. Eur. J. Clin. Nutr. 2013, 67, 841–847. [Google Scholar] [CrossRef] [PubMed]
- Sierksma, A.; Patel, H.; Ouchi, N.; Kihara, S.; Funahashi, T.; Heine, R.J.; Grobbee, D.E.; Kluft, C.; Hendriks, H.F. Effect of moderate alcohol consumption on adiponectin, tumor necrosis factor-α, and insulin sensitivity. Diabetes Care 2004, 27, 184–189. [Google Scholar] [CrossRef] [PubMed]
- Joosten, M.M.; Beulens, J.W.; Kersten, S.; Hendriks, H.F. Moderate alcohol consumption increases insulin sensitivity and ADIPOQ expression in postmenopausal women: A randomised, crossover trial. Diabetologia 2008, 51, 1375–1381. [Google Scholar] [CrossRef] [PubMed]
- Brandhagen, M.; Forslund, H.B.; Lissner, L.; Winkvist, A.; Lindroos, A.K.; Carlsson, L.M.; Sjöström, L.; Larsson, I. Alcohol and macronutrient intake patterns are related to general and central adiposity. Eur. J. Clin. Nutr. 2012, 66, 305–313. [Google Scholar] [CrossRef] [PubMed]
- Kang, L.; Nagy, L.E. Chronic Ethanol Feeding Suppresses β-Adrenergic Receptor-Stimulated Lipolysis in Adipocytes Isolated from Epididymal Fat. Endocrinology 2006, 147, 4330–4338. [Google Scholar] [CrossRef] [PubMed]
- Kang, L.; Chen, X.; Sebastian, B.M.; Pratt, B.T.; Bederman, I.R.; Alexander, J.C.; Previs, S.F.; Nagy, L.E. Chronic ethanol and triglyceride turnover in white adipose tissue in rats: Inhibition of the anti-lipolytic action of insulin after chronic ethanol contributes to increased triglyceride degradation. J. Biol. Chem. 2007, 282, 28465–28473. [Google Scholar] [CrossRef] [PubMed]
- Lang, C.H.; Derdak, Z.; Wands, J.R. Strain-dependent differences for suppression of insulin-stimulated glucose uptake in skeletal and cardiac muscle by ethanol. Alcohol. Clin. Exp. Res. 2014, 38, 897–910. [Google Scholar] [CrossRef] [PubMed]
- Yki-Järvinen, H.; Koivisto, V.A.; Ylikahri, R.; Taskinen, M.R. Acute effects of ethanol and acetate on glucose kinetics in normal subjects. Am. J. Physiol. 1988, 254, E175–E180. [Google Scholar] [PubMed]
- Itaya, K. Effect of ethanol on adrenaline-stimulated glucose uptake in rat white adipose tissue. J. Pharm. Pharmacol. 1979, 31, 536–541. [Google Scholar] [CrossRef] [PubMed]
- Frayn, K.N.; Coppack, S.W.; Walsh, P.E.; Butterworth, H.C.; Humphreys, S.M.; Pedrosa, H.C. Metabolic responses of forearm and adipose tissues to acute ethanol ingestion. Metabolism 1990, 39, 958–966. [Google Scholar] [CrossRef]
- Shih, M.F.; Taberner, P.V. Changes in adipose tissue hormone-sensitive lipase activity and camp during ethanol withdrawal. Eur. J. Pharmacol. 2000, 409, 223–231. [Google Scholar] [CrossRef]
- Shih, M.F.; Taberner, P.V. Effects of acute and chronic ethanol administration on the response of mouse adipose tissue hormone-sensitive lipase to alpha(2)-adrenoceptor activation bu UK 14304. Alcohol Alcohol. 2001, 36, 381–387. [Google Scholar] [CrossRef] [PubMed]
- Shih, M.F.; Taberner, P.V. Dose-dependent effects of chronic ethanol on mouse adipose tissue lipase activity and cyclic AMPp accumulation. Br. J. Pharmacol. 1997, 120, 721–727. [Google Scholar] [CrossRef] [PubMed]
- Inagaki, T.; Dutchak, P.; Zhao, G.; Ding, X.; Gautron, L.; Parameswara, V.; Li, Y.; Goetz, R.; Mohammadi, M.; Esser, V.; et al. Endocrine regulation of the fasting response by PPARα-mediated induction of fibroblast growth factor 21. Cell Metab. 2007, 5, 415–425. [Google Scholar] [CrossRef] [PubMed]
- Tian, C.; Jin, X.; Ye, X.; Wu, H.; Ren, W.; Zhang, R.; Long, J.; Ying, C. Long term intake of 0.1% ethanol decreases serum adiponectin by suppressing PPARγ expression via p38 MAPK pathway. Food Chem. Toxicol. 2014, 65, 329–334. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Sebastian, B.M.; Tang, H.; McMullen, M.M.; Axhemi, A.; Jacobsen, D.W.; Nagy, L.E. Taurine supplementation prevents ethanol-induced decrease in serum adiponectin and reduces hepatic steatosis in rats. Hepatology 2009, 49, 1554–1562. [Google Scholar] [CrossRef] [PubMed]
- Shen, Z.; Liang, X.; Rogers, C.Q.; Rideout, D.; You, M. Involvement of adiponectin-SIRT1-AMPK signaling in the protective action of rosiglitazone against alcoholic fatty liver in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2010, 298, G364–G374. [Google Scholar] [CrossRef] [PubMed]
- Woollett, L.A.; Baldner-Shank, G.L.; Aprahamian, S.; Engen, R.L.; Beitz, D.C. Adaptation of lipogenesis and lipolysis to dietary ethanol. Alcohol. Clin. Exp. Res. 1987, 11, 336–339. [Google Scholar] [CrossRef] [PubMed]
- Lomeo, F.; Khokher, M.A.; Dandona, P. Ethanol and its novel metabolites inhibit insulin action on adipocytes. Diabetes 1988, 37, 912–915. [Google Scholar] [CrossRef] [PubMed]
- Siler, S.Q.; Neese, R.A.; Hellerstein, M.K. De novo lipogenesis, lipid kinetics, and whole-body lipid balances in humans after acute alcohol consumption. Am. J. Clin. Nutr. 1999, 70, 928–936. [Google Scholar] [PubMed]
- Suter, P.M.; Schutz, Y.; Jequier, E. The effect of ethanol on fat storage in healthy subjects. N. Engl. J. Med. 1992, 326, 983–987. [Google Scholar] [CrossRef] [PubMed]
- Sonko, B.J.; Prentice, A.M.; Murgatroyd, P.R.; Goldberg, G.R.; van de Ven, M.L.; Coward, W.A. Effect of alcohol on postmeal fat storage. Am. J. Clin. Nutr. 1994, 59, 619–625. [Google Scholar] [PubMed]
- Clugston, R.D.; Yuen, J.J.; Hu, Y.; Abumrad, N.A.; Berk, P.D.; Goldberg, I.J.; Blaner, W.S.; Huang, L.S. CD36-deficient mice are resistant to alcohol- and high-carbohydrate-induced hepatic steatosis. J. Lipid Res. 2014, 55, 239–246. [Google Scholar] [CrossRef] [PubMed]
- Clugston, R.D.; Jiang, H.; Lee, M.X.; Piantedosi, R.; Yuen, J.J.; Ramakrishnan, R.; Lewis, M.J.; Gottesman, M.E.; Huang, L.S.; Goldberg, I.J.; et al. Altered hepatic lipid metabolism in C57BL/6 mice fed alcohol: A targeted lipidomic and gene expression study. J. Lipid Res. 2011, 52, 2021–2031. [Google Scholar] [CrossRef] [PubMed]
- Ge, F.; Zhou, S.; Hu, C.; Lobdell, H.; Berk, P.D. Insulin- and leptin-regulated fatty acid uptake plays a key causal role in hepatic steatosis in mice with intact leptin signaling but not in ob/ob or db/db mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2010, 299, G855–G866. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Dou, X.; Li, S.; Zhang, X.; Sun, X.; Zhou, Z.; Song, Z. Nuclear factor (erythroid-derived 2)-like 2 activation-induced hepatic very-low-density lipoprotein receptor overexpression in response to oxidative stress contributes to alcoholic liver disease in mice. Hepatology 2014, 59, 1381–1392. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Sun, Q.; Zhong, W.; Sun, X.; Zhou, Z. Hepatic Peroxisome Proliferator-Activated Receptor Gamma Signaling Contributes to Alcohol-Induced Hepatic Steatosis and Inflammation in Mice. Alcohol. Clin. Exp. Res. 2016, 40, 988–999. [Google Scholar] [CrossRef] [PubMed]
- Xu, A.; Wang, Y.; Keshaw, H.; Xu, L.Y.; Lam, K.S.; Cooper, G.J. The fat-derived hormone adiponectin alleviates alcoholic and nonalcoholic fatty liver diseases in mice. J. Clin. Investig. 2003, 112, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Steiner, J.L.; Crowell, K.T.; Lang, C.H. Impact of alcohol on glycemic control and insulin action. Biomolecules 2015, 5, 2223–2246. [Google Scholar] [CrossRef] [PubMed]
- Kang, L.; Sebastian, B.M.; Pritchard, M.T.; Pratt, B.T.; Previs, S.F.; Nagy, L.E. Chronic ethanol-induced insulin resistance is associated with macrophage infiltration into adipose tissue and altered expression of adipocytokines. Alcohol. Clin. Exp. Res. 2007, 31, 1581–1588. [Google Scholar] [CrossRef] [PubMed]
- Poirier, L.A.; Rachdaoui, N.; Nagy, L.E. GLUT4 vesicle trafficking in rat adipocytes after ethanol feeding: Regulation by heterotrimeric G-proteins. Biochem. J. 2001, 354, 323–330. [Google Scholar] [CrossRef] [PubMed]
- Souza-Smith, F.M.; Ford, S.M.; Simon, L.; Molina, P.E. Repeated binge-like alcohol intoxication; depot specific adipose tissue immuno-metabolic dysregulation. Shock 2017. [Google Scholar] [CrossRef] [PubMed]
- Rachdaoui, N.; Sebastian, B.M.; Nagy, L.E. Chronic ethanol feeding impairs endothelin-1-stimulated glucose uptake via decreased Gα11 expression in rat adipocytes. Am. J. Physiol. Endocrinol. Metab. 2003, 285, E303–E310. [Google Scholar] [CrossRef] [PubMed]
- Sebastian, B.M.; Nagy, L.E. Decreased insulin-dependent glucose transport by chronic ethanol feeding is associated with dysregulation of the cbl/TC10 pathway in rat adipocytes. Am. J. Physiol. Endocrinol. Metab. 2005, 289, E1077–E1084. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; Dong, L.Q.; Liu, F. Recent Advances in Adipose mTOR Signaling and Function: Therapeutic prospects. Trends Pharmacol. Sci. 2016, 37, 303–317. [Google Scholar] [CrossRef] [PubMed]
- Steiner, J.L.; Lang, C.H. Dysregulation of skeletal muscle protein metabolism by alcohol. Am. J. Physiol. Endocrinol. Metab. 2015, 308, E699–E712. [Google Scholar] [CrossRef] [PubMed]
- Sebastian, B.M.; Roychowdhury, S.; Tang, H.; Hillian, A.D.; Feldstein, A.E.; Stahl, G.L.; Takahashi, K.; Nagy, L.E. Identification of a cytochrome P4502E1/Bid/C1q-dependent axis mediating inflammation in adipose tissue after chronic ethanol feeding to mice. J. Biol. Chem. 2011, 286, 35989–35997. [Google Scholar] [CrossRef] [PubMed]
- Muralidhara, D.V.; Desautels, M. Effects of ethanol consumption on brown adipose tissue thermogenic capacity in mice. Physiol. Behav. 1996, 60, 639–644. [Google Scholar] [CrossRef]
- Stern, J.H.; Rutkowski, J.M.; Scherer, P.E. Adiponectin, leptin, and fatty acids in the maintenance of metabolic homeostasis through adipose tissue crosstalk. Cell Metab. 2016, 23, 770–784. [Google Scholar] [CrossRef] [PubMed]
- Song, Z.; Zhou, Z.; Deaciuc, I.; Chen, T.; McClain, C.J. Inhibition of adiponectin production by homocysteine: A potential mechanism for alcoholic liver disease. Hepatology 2008, 47, 867–879. [Google Scholar] [CrossRef] [PubMed]
- Ford, S.M.; Simon, L.; Vande Stouwe, C.; Allerton, T.; Mercante, D.E.; Byerley, L.O.; Dufour, J.P.; Bagby, G.J.; Nelson, S.; Molina, P.E. Chronic binge alcohol administration impairs glucose-insulin dynamics and decreases adiponectin in asymptomatic simian immunodeficiency virus-infected macaques. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2016, 311, R888–R897. [Google Scholar] [CrossRef] [PubMed]
- Thakur, V.; Pritchard, M.T.; McMullen, M.R.; Nagy, L.E. Adiponectin normalizes LPS-stimulated TNF-α production by rat Kupffer cells after chronic ethanol feeding. Am. J. Physiol. Gastrointest. Liver Physiol. 2006, 290, G998–G1007. [Google Scholar] [CrossRef] [PubMed]
- Esfandiari, F.; You, M.; Villanueva, J.A.; Wong, D.H.; French, S.W.; Halsted, C.H. S-Adenosylmethionine Attenuates Hepatic Lipid Synthesis in Micropigs Fed Ethanol With a Folate-Deficient Diet. Alcohol. Clin. Exp. Res. 2007, 31, 1231–1239. [Google Scholar] [CrossRef] [PubMed]
- Strbák, V.; Benický, J.; Macho, L.; Jezová, D.; Nikodémová, M. Four-week ethanol intake decreases food intake and body weight but does not affect plasma leptin, corticosterone, and insulin levels in pubertal rats. Metabolism 1998, 47, 1269–1273. [Google Scholar] [CrossRef]
- Bell, S.; Britton, A. The role of alcohol consumption in regulating circulating levels of adiponectin: A prospective cohort study. J. Clin. Endocrinol. Metab. 2015, 100, 2763–2768. [Google Scholar] [CrossRef] [PubMed]
- Beulens, J.W.; de Zoete, E.C.; Kok, F.J.; Schaafsma, G.; Hendriks, H.F. Effect of moderate alcohol consumption on adipokines and insulin sensitivity in lean and overweight men: A diet intervention study. Eur. J. Clin. Nutr. 2008, 62, 1098–1105. [Google Scholar] [CrossRef] [PubMed]
- Hillemacher, T.; Weinland, C.; Heberlein, A.; Gröschl, M.; Schanze, A.; Frieling, H.; Wilhelm, J.; Kornhuber, J.; Bleich, S. Increased levels of adiponectin and resistin in alcohol dependence—Possible link to craving. Drug Alcohol Depend. 2009, 99, 333–337. [Google Scholar] [CrossRef] [PubMed]
- Thamer, C.; Haap, M.; Fritsche, A.; Haering, H.; Stumvoll, M. Relationship between moderate alcohol consumption and adiponectin and insulin sensitivity in a large heterogeneous population. Diabetes Care 2004, 27, 1240. [Google Scholar] [CrossRef] [PubMed]
- Stejskal, D.; Růzicka, V.; Fanfrdlová, G.; Kolár, V.; Bartek, J. High adiponectin and TNF-α levels in moderate drinkers suffering from liver steatosis: Comparison with non drinkers suffering from similar hepatopathy. Biomed. Papers 2005, 149, 93–99. [Google Scholar] [CrossRef]
- Beulens, J.W.; van Loon, L.J.; Kok, F.J.; Pelsers, M.; Bobbert, T.; Spranger, J.; Helander, A.; Hendriks, H.F. The effect of moderate alcohol consumption on adiponectin oligomers and muscle oxidative capacity: A human intervention study. Diabetologia 2007, 50, 1388–1392. [Google Scholar] [CrossRef] [PubMed]
- Joosten, M.M.; Witkamp, R.F.; Hendriks, H.F. Alterations in total and high-molecular-weight adiponectin after 3 weeks of moderate alcohol consumption in premenopausal women. Metabolism 2011, 60, 1058–1063. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Sebastian, B.M.; Nagy, L.E. Chronic ethanol feeding to rats decreases adiponectin secretion by subcutaneous adipocytes. Am. J. Physiol. Endocrinol. Metab. 2007, 292, E621–E628. [Google Scholar] [CrossRef] [PubMed]
- Voican, C.S.; Njiké-Nakseu, M.; Boujedidi, H.; Barri-Ova, N.; Bouchet-Delbos, L.; Agostini, H.; Maitre, S.; Prévot, S.; Cassard-Doulcier, A.M.; Naveau, S.; et al. Alcohol withdrawal alleviates adipose tissue inflammation in patients with alcoholic liver disease. Liver Int. 2015, 35, 967–978. [Google Scholar] [CrossRef] [PubMed]
- Wandler, A.; Bruun, J.M.; Nielsen, M.P.; Richelsen, B. Ethanol exerts anti-inflammatory effects in human adipose tissue in vitro. Mol. Cell. Endocrinol. 2008, 296, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Souza-Smith, F.M.; Siggins, R.W.; Molina, P.E. Mesenteric lymphatic-perilymphatic adipose crosstalk: Role in alcohol-induced perilymphatic adipose tissue inflammation. Alcohol. Clin. Exp. Res. 2015, 39, 1380–1387. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Hamilton, J.L.; Bird, M.D.; Chen, M.M.; Ramirez, L.; Zahs, A.; Kovacs, E.J.; Makowski, L. Adipose inflammation and macrophage infiltration after binge ethanol and burn injury. Alcohol. Clin. Exp. Res. 2014, 38, 204–213. [Google Scholar] [CrossRef] [PubMed]
- Obradovic, T.; Meadows, G.G. Chronic ethanol consumption increases plasma leptin levels and alters leptin receptors in the hypothalamus and the perigonadal fat of C57BL/6 mice. Alcohol. Clin. Exp. Res. 2002, 26, 255–262. [Google Scholar] [CrossRef] [PubMed]
- Szkudelski, T.; Bialik, I.; Szkudelska, K. Adipocyte lipolysis, hormonal and metabolic changes in ethanol-drinking rats. J. Anim. Physiol. Anim. Nutr. 2004, 88, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Hiney, J.K.; Dearth, R.K.; Lara, F.; Wood, S.; Srivastava, V.; Les Dees, W. Effects of ethanol on leptin secretion and the leptin-induced luteinizing hormone (LH) release from late juvenile female rats. Alcohol. Clin. Exp. Res. 1999, 23, 1785–1792. [Google Scholar] [CrossRef] [PubMed]
- Maddalozzo, G.F.; Turner, R.T.; Edwards, C.H.; Howe, K.S.; Widrick, J.J.; Rosen, C.J.; Iwaniec, U.T. Alcohol alters whole body composition, inhibits bone formation, and increases bone marrow adiposity in rats. Osteoporos. Int. 2009, 20, 1529–1538. [Google Scholar] [CrossRef] [PubMed]
- Greco, A.V.; Mingrone, G.; Favuzzi, A.; Capristo, E.; Gniuli, D.; Addolorato, G.; Brunani, A.; Cavagnin, F.; Gasbarrini, G. Serum leptin levels in post-hepatitis liver cirrhosis. J. Hepatol. 2000, 33, 38–42. [Google Scholar] [CrossRef]
- Kalousová, M.; Zima, T.; Popov, P.; Spacek, P.; Braun, M.; Soukupová, J.; Pelinkova, K.; Kientsch-Engel, R. Advanced glycation end-products in patients with chronic alcohol misuse. Alcohol Alcohol. 2004, 39, 316–320. [Google Scholar] [PubMed]
- Wannamethee, S.G.; Tchernova, J.; Whincup, P.; Lowe, G.D.; Kelly, A.; Rumley, A.; Wallace, A.M.; Sattar, N. Plasma leptin: Associations with metabolic, inflammatory and haemostatic risk factors for cardiovascular disease. Atherosclerosis 2007, 191, 418–426. [Google Scholar] [CrossRef] [PubMed]
- Lagiou, P.; Signorello, L.B.; Mantzoros, C.S.; Trichopoulos, D.; Hsieh, C.C.; Trichopoulou, A. Hormonal, lifestyle, and dietary factors in relation to leptin among elderly men. Ann. Nutr. Metab. 1999, 43, 23–29. [Google Scholar] [CrossRef] [PubMed]
- De Silva, A.; De Courten, M.; Zimmet, P.; Nicholson, G.; Kotowicz, M.; Pasco, J.; Collier, G.R. Lifestyle factors fail to explain the variation in plasma leptin concentrations in women. Nutrition 1998, 14, 653–657. [Google Scholar] [CrossRef]
- Nicolás, J.M.; Fernández-Solà, J.; Fatjó, F.; Casamitjana, R.; Bataller, R.; Sacanella, E.; Tobías, E.; Badía, E.; Estruch, R. Increased circulating leptin levels in chronic alcoholism. Alcohol. Clin. Exp. Res. 2001, 25, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Donahue, R.P.; Zimmet, P.; Bean, J.A.; Decourten, M.; DeCarlo Donahue, R.A.; Collier, G.; Goldberg, R.B.; Prineas, R.J.; Skyler, J.; Schneiderman, N. Cigarette smoking, alcohol use, and physical activity in relation to serum leptin levels in a multiethnic population: The Miami Community Health Study. Ann. Epidemiol. 1999, 9, 108–113. [Google Scholar] [CrossRef]
- Henriksen, J.H.; Holst, J.J.; Møller, S.; Brinch, K.; Bendtsen, F. Increased circulating leptin in alcoholic cirrhosis: Relation to release and disposal. Hepatology 1999, 29, 1818–1824. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.Z.; Yang, S.Q.; Zeldin, G.; Diehl, A.M. Chronic Ethanol Consumption Induces the Production of Tumor Necrosis Factor-α and Related Cytokines in Liver and Adipose Tissue. Alcohol. Clin. Exp. Res. 1998, 22, 231S–237S. [Google Scholar] [CrossRef] [PubMed]
- Otaka, M.; Konishi, N.; Odashima, M.; Jin, M.; Wada, I.; Matsuhashi, T.; Ohba, R.; Watanabe, S. Effect of alcohol consumption on leptin level in serum, adipose tissue, and gastric mucosa. Dig. Dis. Sci. 2007, 52, 3066–3069. [Google Scholar] [CrossRef] [PubMed]
- Röjdmark, S.; Calissendorff, J.; Brismar, K. Alcohol ingestion decreases both diurnal and nocturnal secretion of leptin in healthy individuals. Clin. Endocrinol. 2001, 55, 639–647. [Google Scholar] [CrossRef]
- Chen, N.; Zhou, L.; Zhang, Z.; Xu, J.; Wan, Z.; Qin, L. Resistin induces lipolysis and suppresses adiponectin secretion in cultured human visceral adipose tissue. Regul. Pept. 2014, 194–195, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Goralski, K.B.; McCarthy, T.C.; Hanniman, E.A.; Zabel, B.A.; Butcher, E.C.; Parlee, S.D.; Muruganandan, S.; Sinal, C.J. Chemerin, a novel adipokine that regulates adipogenesis and adipocyte metabolism. J. Biol. Chem. 2007, 282, 28175–28188. [Google Scholar] [CrossRef] [PubMed]
- Lang, C.H.; Dobrescu, C.; Bagby, G.J. Tumor necrosis factor impairs insulin action on peripheral glucose disposal and hepatic glucose output. Endocrinology 1992, 130, 43–52. [Google Scholar] [PubMed]
- Choe, S.S.; Huh, J.Y.; Hwang, I.J.; Kim, J.I.; Kim, J.B. Adipose tissue remodeling: Its role in energy metabolism and metabolic disorders. Front. Endocrinol. 2016, 7, 30. [Google Scholar] [CrossRef] [PubMed]
- Fulham, M.A.; Mandrekar, P. Sexual dimorphism in alcohol induced adipose inflammation relates to liver injury. PLoS ONE 2016, 11, e0164225. [Google Scholar] [CrossRef] [PubMed]
- Kortelainen, M.L.; Huttunen, P.; Hirvonen, J. Histochemical and biochemical detection of alcohol dehydrogenase in rat brown adipose tissue. Alcohol 1991, 8, 151–154. [Google Scholar] [CrossRef]
- Rothwell, N.J.; Stock, M.J. Influence of alcohol and sucrose consumption on energy balance and brown fat activity in the rat. Metabolism 1984, 33, 768–771. [Google Scholar] [CrossRef]
- Huttunen, P.; Kortelainen, M.L. Chronic alcohol intake induces the oxidative capacity of brown adipose tissue in the rat. Pharmacol. Biochem. Behav. 1988, 29, 53–57. [Google Scholar] [CrossRef]
- Huttunen, P.; Kortelainen, M.L.; Hirvonen, J. Foetal and Lactational Exposure to Alcohol Increases Oxidative Capacity of Brown Adipose Tissue in the Rat. A Possible Relationship to Cot Death. Br. J. Exp. Pathol. 1989, 70, 691–695. [Google Scholar] [PubMed]
- Larue-Achagiotis, C.; Poussard, A.M.; Louis-Sylvestre, J. Effect of interscapular brown adipose tissue denervation on body weight and feed efficiency in alcohol drinking rats. Physiol. Behav. 1989, 46, 195–197. [Google Scholar] [CrossRef]
- Huttunen, P.; Penttinen, J.; Hirvonen, J. The effect of ethanol and cold-adaptation on the survival of guinea pigs in severe cold. Z. Rechtsmed. 1980, 85, 289–294. [Google Scholar] [CrossRef] [PubMed]
- Huttunen, P.; Sämpi, M.; Myllylä, R. Ethanol-induced hypothermia and thermogenesis of brown adipose tissue in the rat. Alcohol 1998, 15, 315–318. [Google Scholar] [CrossRef]
- Yoshimoto, K.; Yasuhara, M.; Komura, S.; Misumi, Y.; Uchiyama, Y.; Kogure, A.; Hioki, C.; Wakabayashi, Y.; Satomi, Y.; Nishimura, A.; et al. Effects of ethanol on the induction of uncoupling protein-1 (UCP1) mRNA in the mouse brown adipose tissue. Tohoku J. Exp. Med. 2004, 204, 45–51. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Adipokine | Action | Plasma Acute Alcohol Chronic Alcohol | Adipose Tissue Acute Alcohol Chronic alcohol | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Rodent | Human | Rodent | Human | Rodent | Human | Rodent | Human | ||
| Adiponectin | Adipogenic; insulin sensitizer | ↓ [89] | ↓ [9,18,49,50,51,62,74,75,76,77] | ↑ [36,37,79,80,81,82,83,84] | ↔ [90] | ↓ [15,18,24,25,49,51,74,86]. | ↑ [37] | ||
| ↔ [66] | ↔ [10,12,25,78] | ↓ [34,35] | ↑ [16] | ||||||
| Leptin | Lipolysis, fatty acid oxidation, lipogenesis, insulin sensitivity; responsive to changes in fat mass | ↓ [104] | ↓ [105] | ↓ [10,93,94] | ↓ [27,101] | ↑ [104] | ↑ [16,22,24,103] | ↔ [30] | |
| ↔ [24] | ↔ [80,87,95,96,97,98,99] | ||||||||
| ↑ [24,91,92] | ↑ [100,102] | ||||||||
| Resistin | Stimulate lipolysis and fatty acid release, suppress adiponectin | ↑ [16,17,24] | ↑ [81] | ↔ [16,17,24] | |||||
| ↔ [80,81] | ↑ [16] | ||||||||
| Chemerin | Adipogenesis and adipocyte differentiation | ↑ [31] | ↑ [31] | ↑ [31] | ↑ [31] | ||||
| Visfatin | Glucose metabolism | ↔ [89] | ↑ [24] | ||||||
| ↔ [24] | |||||||||
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Steiner, J.L.; Lang, C.H. Alcohol, Adipose Tissue and Lipid Dysregulation. Biomolecules 2017, 7, 16. https://doi.org/10.3390/biom7010016
Steiner JL, Lang CH. Alcohol, Adipose Tissue and Lipid Dysregulation. Biomolecules. 2017; 7(1):16. https://doi.org/10.3390/biom7010016
Chicago/Turabian StyleSteiner, Jennifer L., and Charles H. Lang. 2017. "Alcohol, Adipose Tissue and Lipid Dysregulation" Biomolecules 7, no. 1: 16. https://doi.org/10.3390/biom7010016
APA StyleSteiner, J. L., & Lang, C. H. (2017). Alcohol, Adipose Tissue and Lipid Dysregulation. Biomolecules, 7(1), 16. https://doi.org/10.3390/biom7010016