
Central Role of Glutamate Metabolism in the Maintenance of Nitrogen Homeostasis in Normal and Hyperammonemic Brain


Abstract
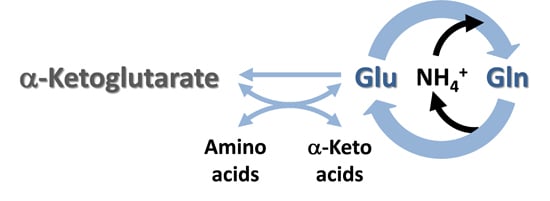
:
1. Introduction
2. Glutamate Formation in the Brain—Important Roles of Glutaminase and α-Ketoglutarate-Linked Aminotransferases
2.1. Glutaminase Versus Glutamate Dehydrogenase
2.2. α-Ketoglutarate/Glutamate-Linked Aminotransferases
2.3. Oxoprolinase
3. Major Routes for the Metabolism of Glutamate in the Brain
3.1. Glutamine Synthetase
3.2. α-Ketoglutarate/Glutamate-Linked Aminotransferases Coupled to the GDH Reaction
3.3. Ammonia Generated by Aminotransferase Reactions Coupled to the Purine Nucleotide Cycle
3.4. Conversion of L-Glutamate to GABA
3.5. GSH as a Glutamate Reservoir
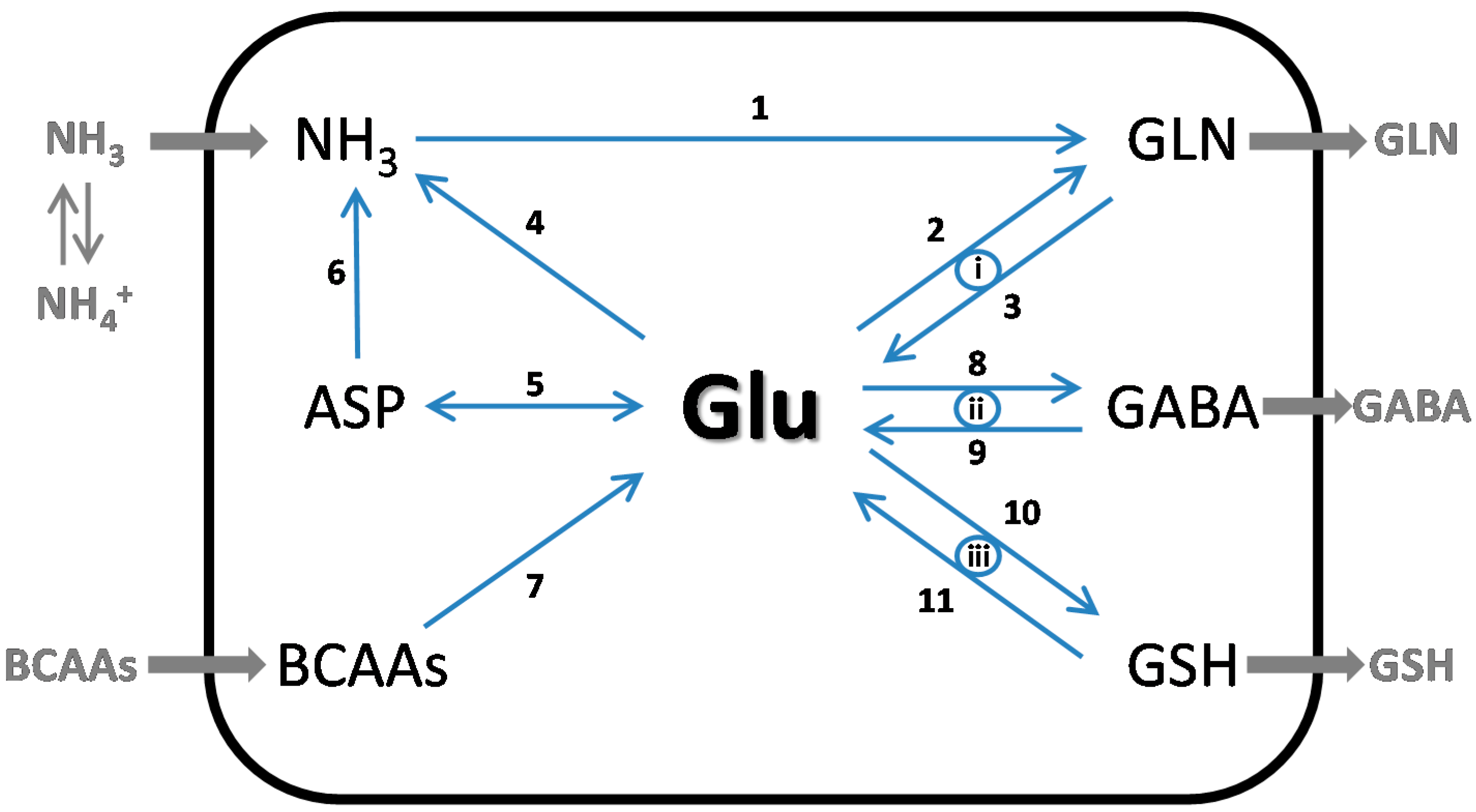
4. The Glutamate Buffer in the Brain
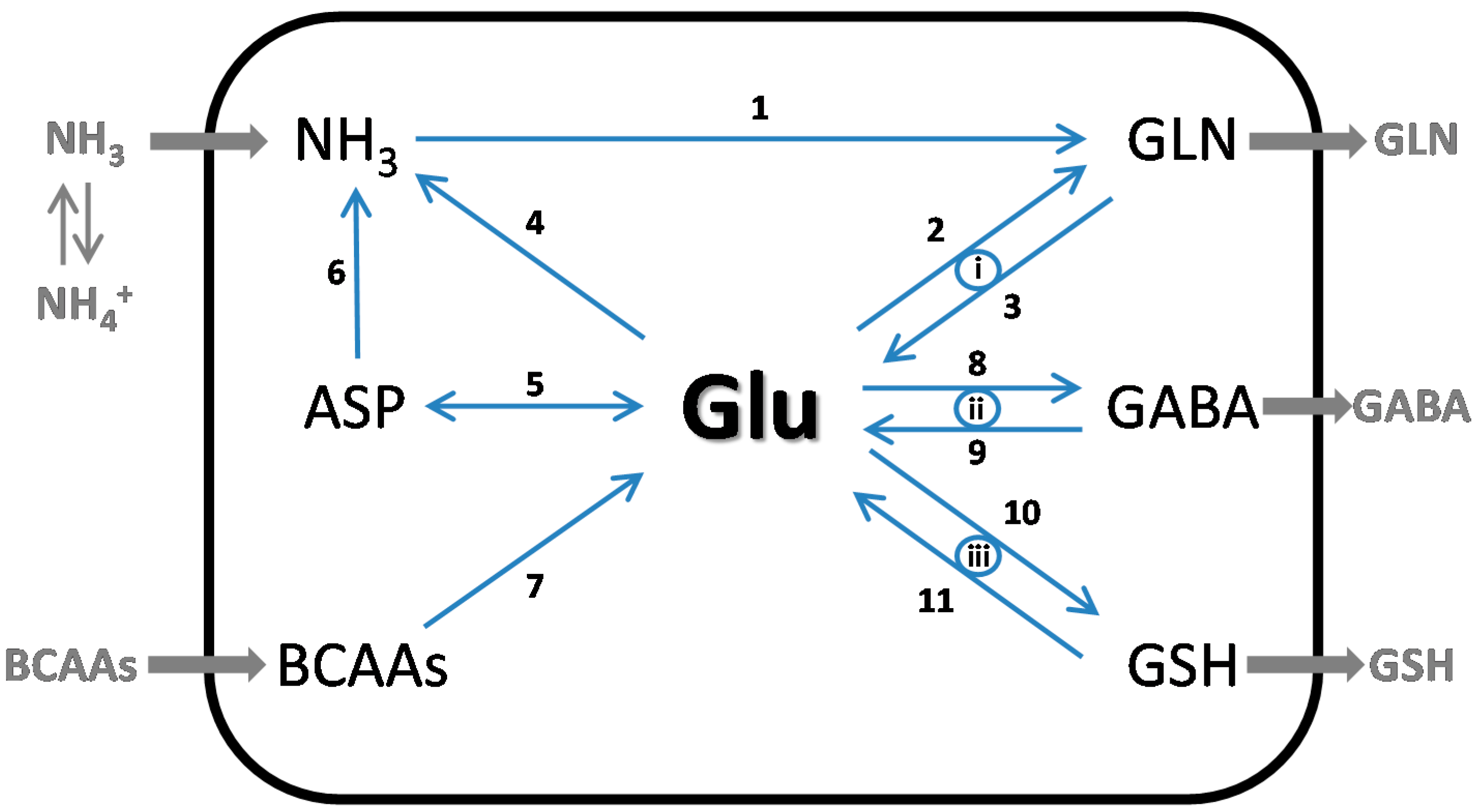
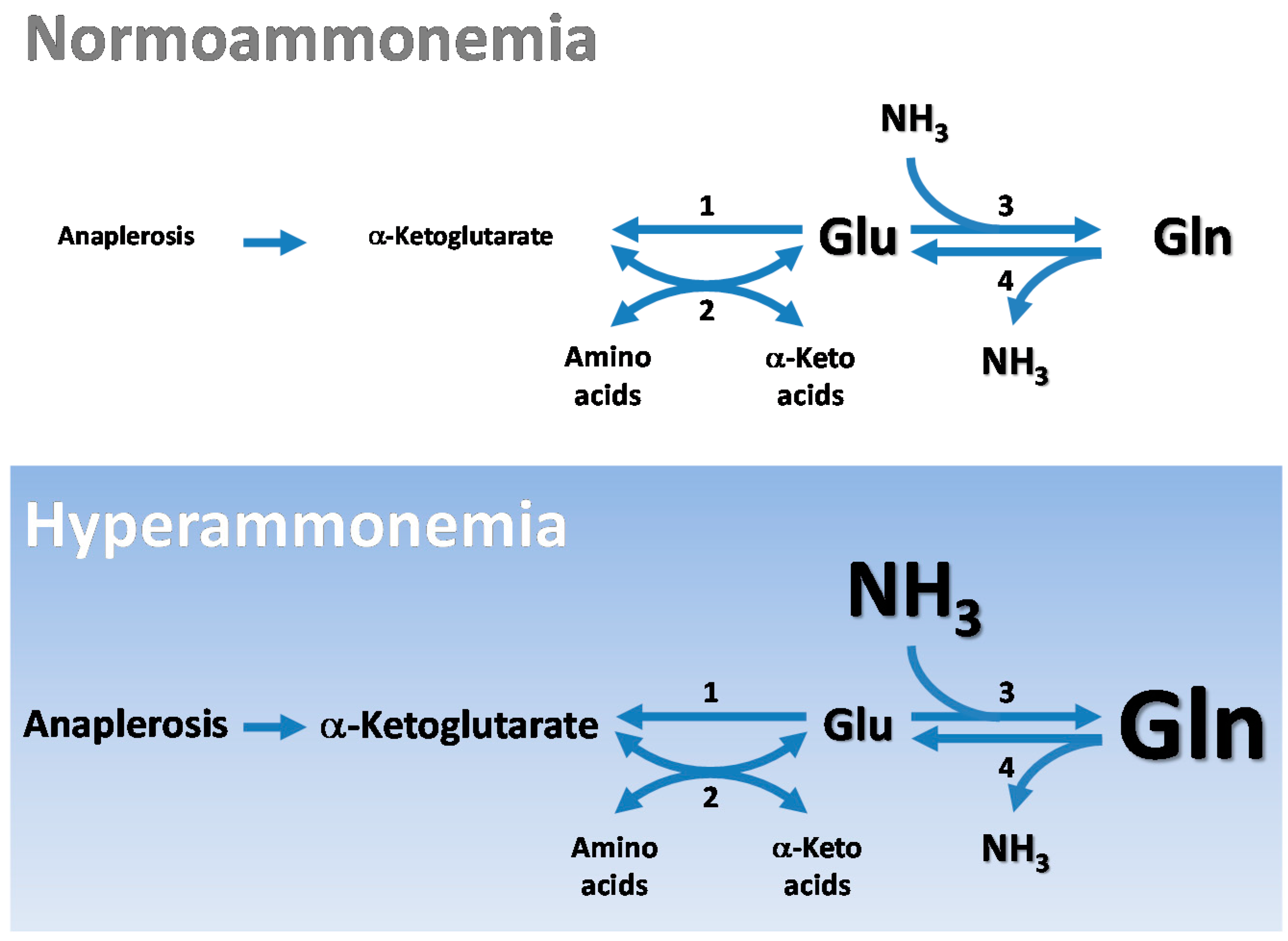
4.1. Glutamate as a Nitrogen Buffer in the Brain
4.2. Glutamate as an Energy Buffer in the Brain
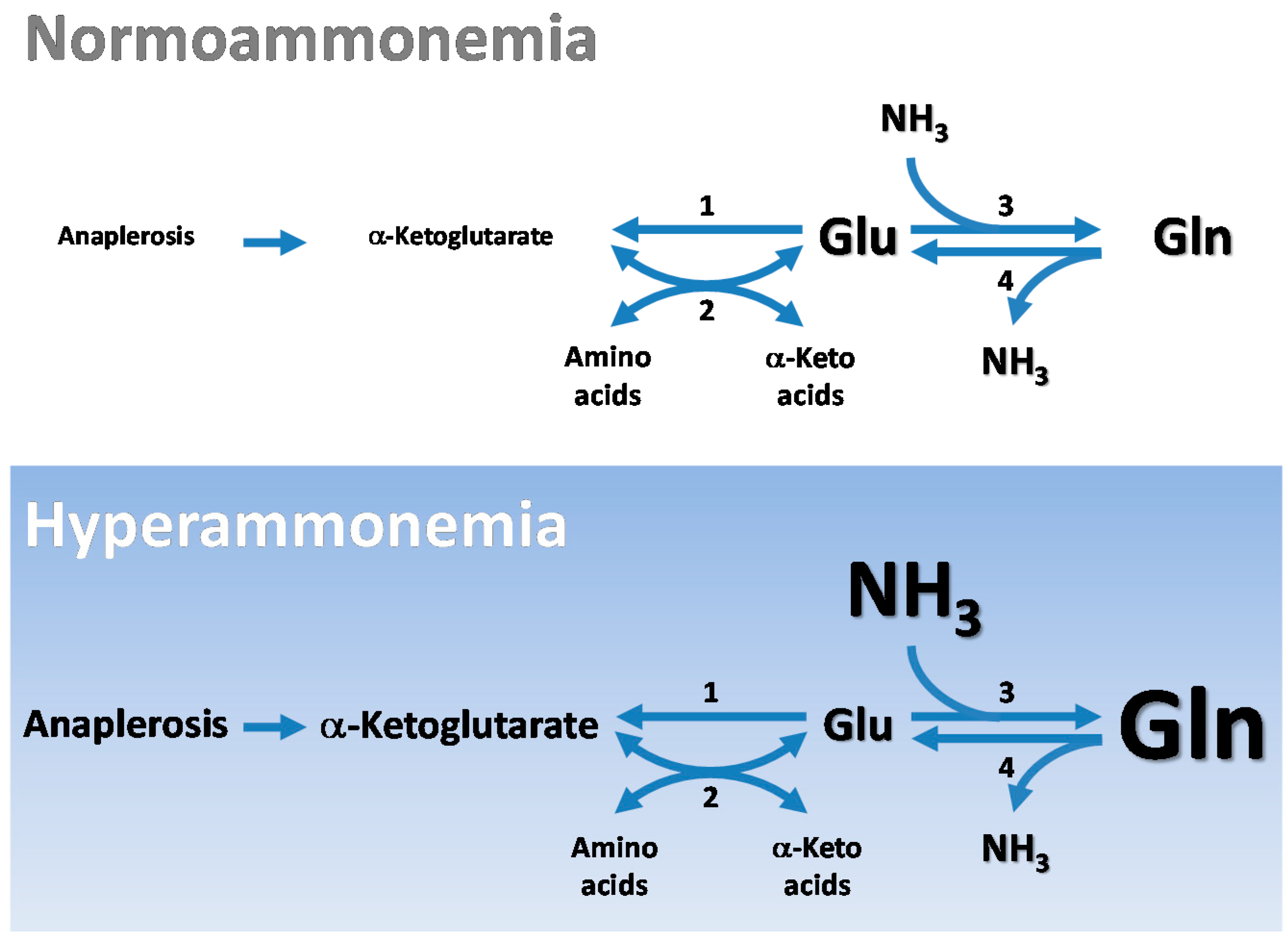
4.3. Cerebral Glutamate Buffering during Hyperammonemia
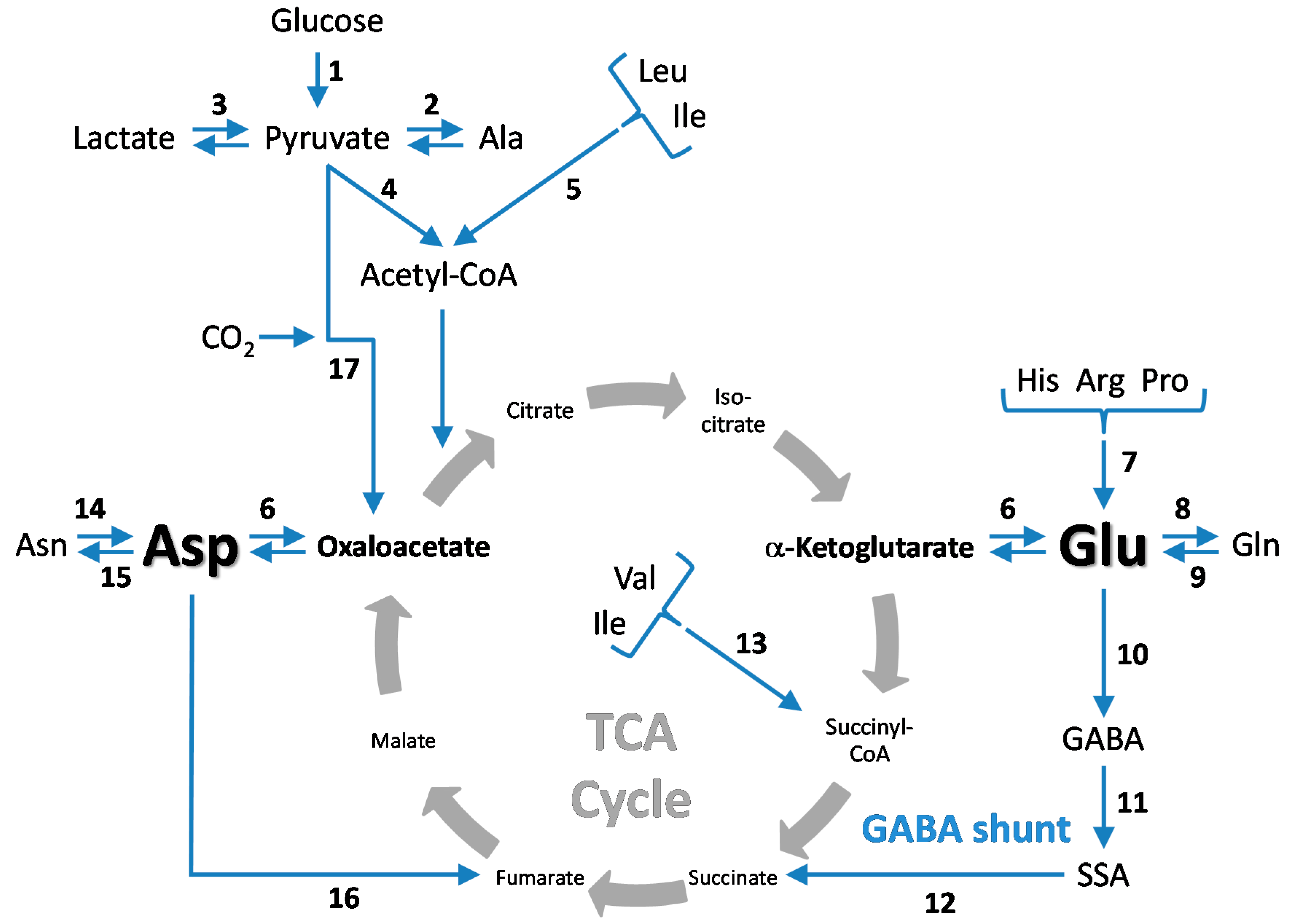
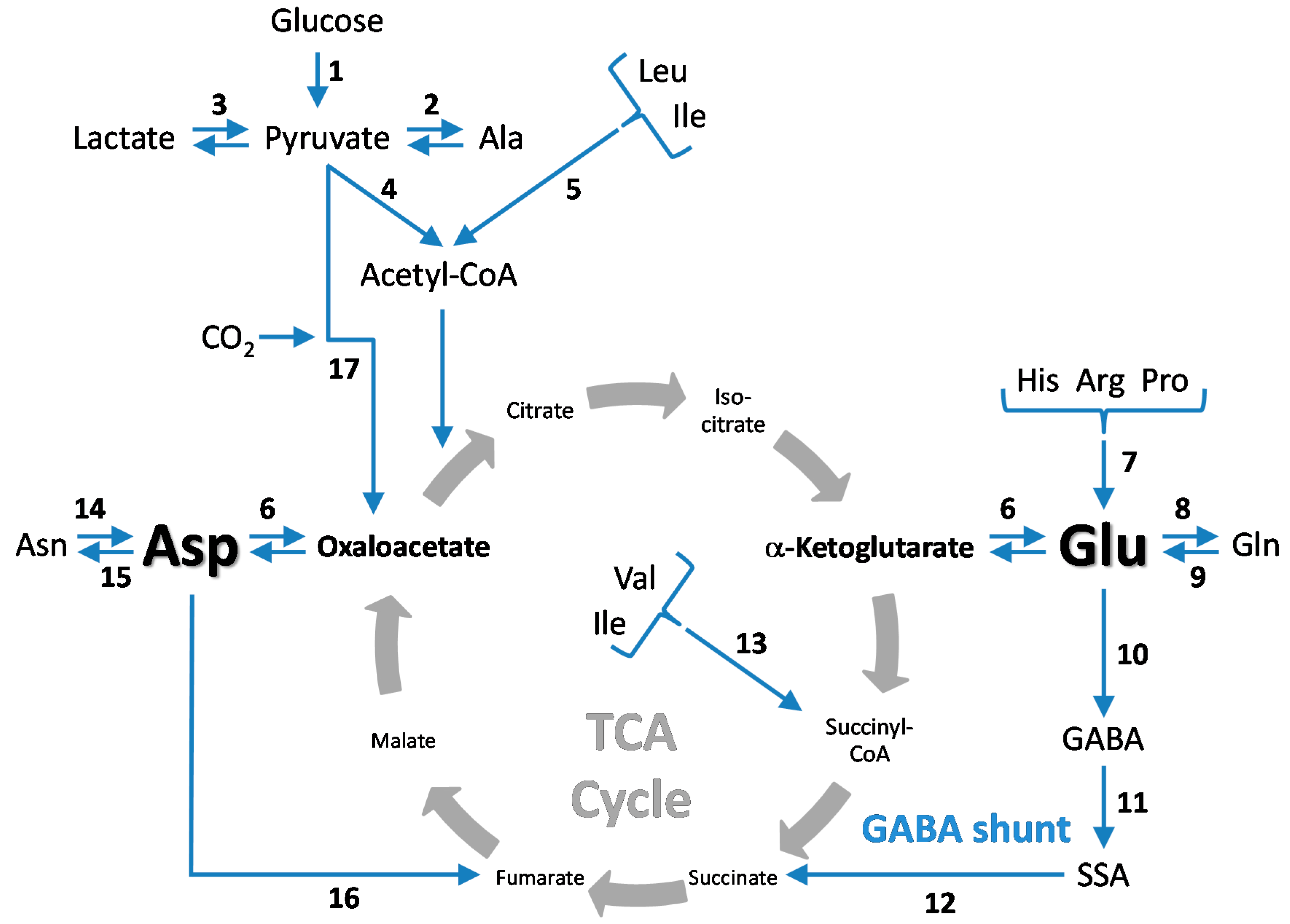
5. CO2 Fixation in the Brain—Stimulation by Hyperammonemia
5.1. Cerebral CO2 Fixation during Normoammonemia
5.2. Cerebral CO2 Fixation during Hyperammonemia
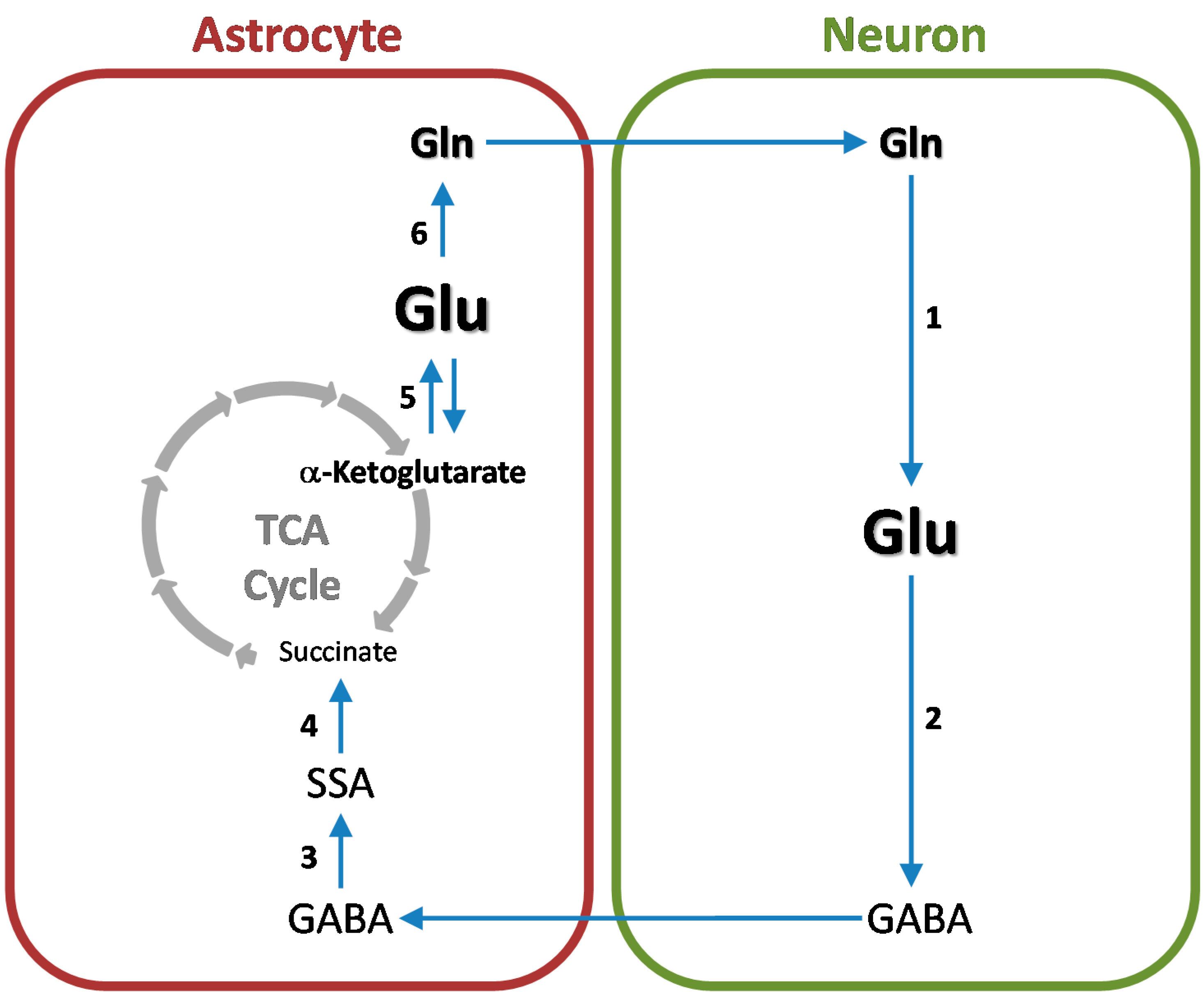
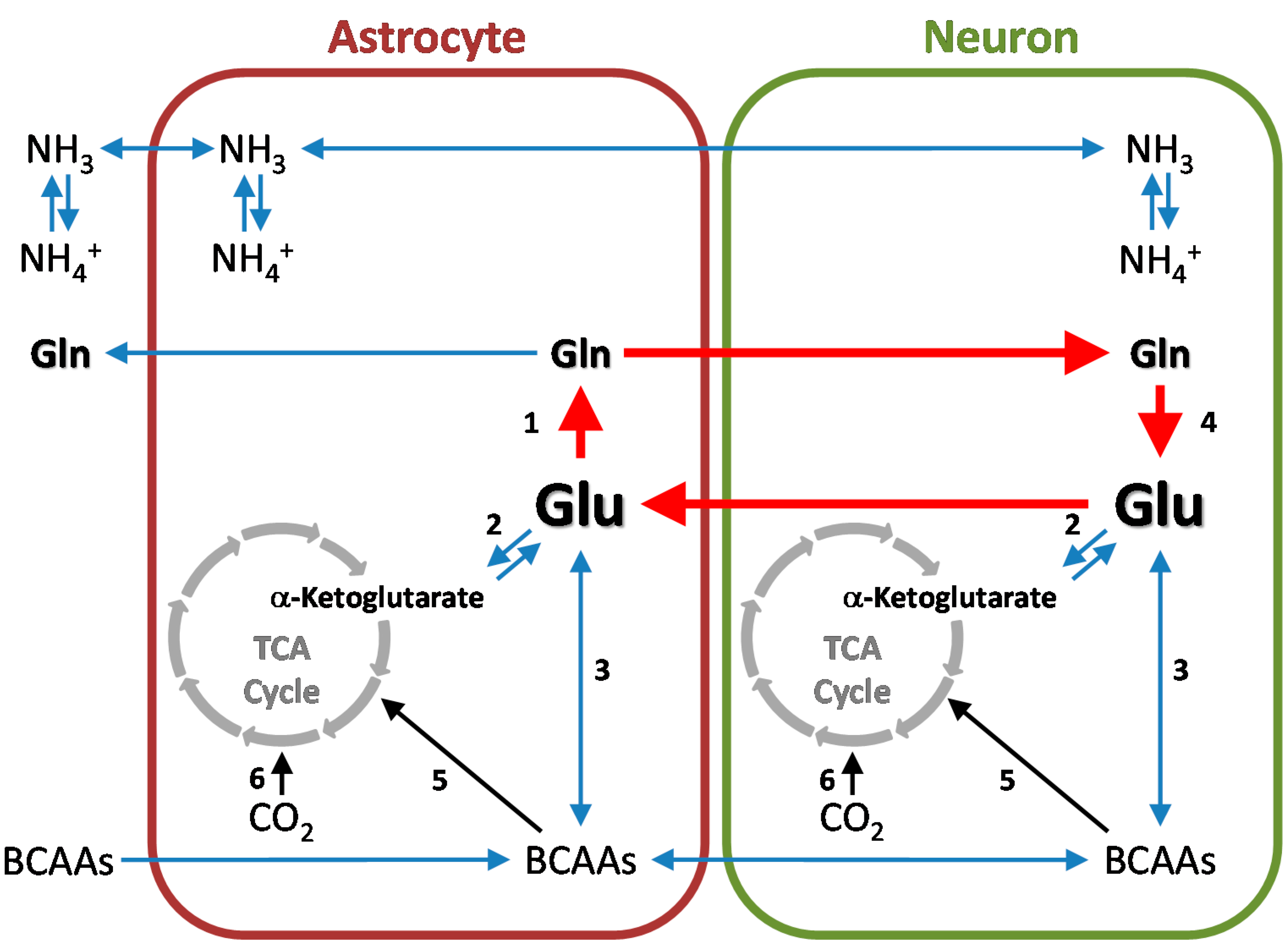
6. Cerebral Glutamine Cycle
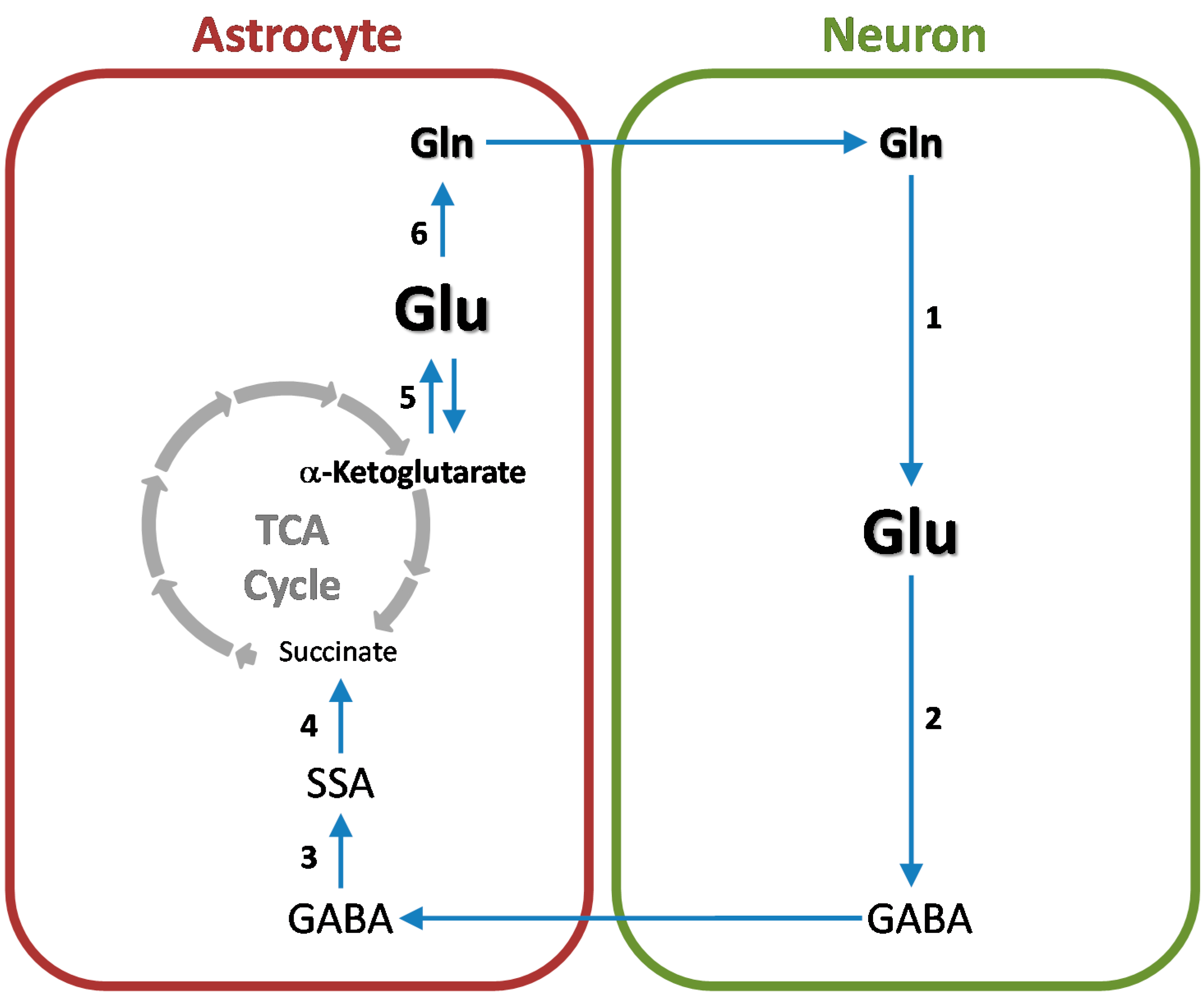
7. Cerebral Glutamine-GABA Cycle
8. Nitrogen Balance in the Glutamine and Glutamine-GABA Cycles
9. Disruption of Glutamate Homeostasis in Neurological Diseases
10. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| AD | Alzheimer disease |
| ALS | Amyotrophic lateral sclerosis |
| AspAT | Aspartate aminotransferase |
| BBB | Blood brain barrier |
| BCAT | Branched-chain aminotransferase |
| BCATc | Cytosolic isozyme of BCAT |
| BCATm | Mitochondrial isozyme of BCAT |
| CNS | Central nervous system |
| CSF | Cerebrospinal fluid |
| cyt | Cytosolic |
| GABA | γ-Aminobutyrate |
| GDH | Glutamate dehydrogenase |
| GLAST | Glutamate aspartate transporter |
| GSH | Glutathione |
| MR | Magnetic resonance (also nuclear magnetic resonance) |
| Mit | Mitochondrial |
| MSO | L-Methionine-S,R-sulfoximine |
| 5-OP | 5-Oxoproline |
| PNC | Purine nucleotide cycle |
| PrP | wild type prion protein |
| TCA | Tricarboxylic acid |
References
- Lajtha, A.B.; Maker, H.S.; Clarke, D.D. Chapter 17. Metabolism and transport of carbohydrates and amino acids. In Basic Neurochemistry, 3rd ed.; Siegel, D.J., Albers, R.W., Agranoff, B.W., Katzman, R., Eds.; Little, Brown and Company: Boston, MA, USA, 1981; pp. 329–353. [Google Scholar]
- Huxtable, R.J. Taurine in the central nervous system and the mammalian actions of taurine. Prog. Neurobiol. 1989, 32, 471–533. [Google Scholar] [CrossRef]
- Ripps, H.; Shen, W. Review: Taurine: A “very essential” amino acid. Mol. Vis. 2012, 18, 2673–2686. [Google Scholar] [PubMed]
- Albrecht, J.; Schousboe, A. Taurine interaction with neurotransmitter receptors in the CNS: An update. Neurochem. Res. 2005, 30, 1615–1621. [Google Scholar] [CrossRef] [PubMed]
- Dringen, R.; Brandmann, M.; Hohnholt, M.C.; Blumrich, E.M. Glutathione-Dependent Detoxification Processes in Astrocytes. Neurochem. Res. 2015, 40, 2570–2582. [Google Scholar] [CrossRef] [PubMed]
- Naini, A.B.; Vontzalidou, E.; Côté, L.J. Isocratic HPLC assay with electrochemical detection of free γ-aminobutyric acid in cerebrospinal fluid. Clin. Chem. 1993, 39, 247–250. [Google Scholar] [PubMed]
- Nicholls, D.G.; Sihra, T.S. Synaptosomes possess an exocytotic pool of glutamate. Nature 1986, 321, 772–773. [Google Scholar] [CrossRef] [PubMed]
- Featherstone, D.E.; Shippy, S.A. Regulation of synaptic transmission by ambient extracellular glutamate. Neuroscientist 2008, 14, 171–181. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Danbolt, N.C. Glutamate as a neurotransmitter in the healthy brain. J. Neural Transm. 2014, 121, 799–817. [Google Scholar] [CrossRef] [PubMed]
- Oldendorf, W.H.; Szabo, J. Amino acid assignment to one of three blood-brain barrier amino acid carriers. Am. J. Physiol. 1976, 230, 94–98. [Google Scholar] [PubMed]
- Márquez, J.; Cardona, C.; Campos-Sandoval, J.A.; Peñalver, A.; Tosina, M.; Matés, J.M.; Martín-Rufián, M. Mammalian glutaminase isozymes in brain. Metab. Brain Dis. 2013, 28, 133–137. [Google Scholar] [CrossRef] [PubMed]
- Spanaki, C.; Kotzamani, D.; Petraki, Z.; Drakos, E.; Plaitakis, A. Heterogeneous cellular distribution of glutamate dehydrogenase in brain and in non-neural tissues. Neurochem. Res. 2014, 39, 500–515. [Google Scholar] [CrossRef] [PubMed]
- Cooper, A.J.L.; Plum, F. Biochemistry and Physiology of brain ammonia. Physiol. Rev. 1997, 67, 440–519. [Google Scholar]
- Cooper, A.J.L.; McDonald, J.M.; Gelbard, A.S.; Gledhill, R.F.; Duffy, T.E. The metabolic fate of 13N-labeled ammonia in rat brain. J. Biol. Chem. 1979, 254, 4982–4992. [Google Scholar] [PubMed]
- Plaitakis, A.; Zaganas, I. Regulation of human glutamate dehydrogenases: Implications for glutamate, ammonia and energy metabolism in brain. J. Neurosci. Res. 2001, 66, 899–908. [Google Scholar] [CrossRef] [PubMed]
- Cooper, A.J.L. 13N as a tracer for studying glutamate metabolism. Neurochem. Int. 2011, 59, 456–464. [Google Scholar] [CrossRef] [PubMed]
- Felig, P.; Wahren, J.; Ahlborg, G. Uptake of individual amino acids by the human brain. Proc. Soc. Exp. Biol. Med. 1973, 142, 230–231. [Google Scholar] [CrossRef] [PubMed]
- Braunstein, A.E. Les voies principales de l’assimilation et dissimilation de l’azote chez les animaux. Adv. Enzymol. Relat. Subj. Biochem. 1957, 19, 335–389. [Google Scholar] [PubMed]
- Cooper, A.J.L.; Meister, A. An appreciation of Professor Alexander E. Braunstein. The discovery and scope of enzymatic transamination. Biochimie 1989, 71, 387–404. [Google Scholar] [CrossRef]
- Cooper, A.J.L. The role of glutamine synthetase and glutamate dehydrogenase in cerebral ammonia homeostasis. Neurochem. Res. 2012, 37, 2439–2455. [Google Scholar] [CrossRef] [PubMed]
- Cooper, A.J.L.; Mora, S.N.; Cruz, N.F.; Gelbard, A.S. Cerebral ammonia metabolism in hyperammonemic rats. J. Neurochem. 1985, 44, 1716–1723. [Google Scholar] [CrossRef] [PubMed]
- Dadsetan, S.; Kukolj, E.; Bak, L.K.; Sørensen, M.; Ott, P.; Vilstrup, H.; Schousboe, A.; Keiding, S.; Waagepetersen, H.S. Brain alanine formation as an ammonia-scavenging pathway during hyperammonemia: Effects of glutamine synthetase inhibition in rats and astrocyte-neuron co-cultures. J. Cereb. Blood Flow Metab. 2013, 33, 1235–1241. [Google Scholar] [CrossRef] [PubMed]
- Sweatt, A.J.; Wood, M.; Suryawan, A.; Wallin, R.; Willingham, M.C.; Hutson, S.M. Branched-chain amino acid catabolism: Unique segregation of pathway enzymes in organ systems and peripheral nerves. Am. J. Physiol. Endocrinol. Metab. 2004, 286, E64–E76. [Google Scholar] [CrossRef] [PubMed]
- Hull, J.; Patel, V.B.; Hutson, S.M.; Conway, M.E. New insights into the role of the branched-chain aminotransferase proteins in the human brain. J. Neurosci. Res. 2015, 93, 987–998. [Google Scholar] [CrossRef] [PubMed]
- Van der Werf, P.; Meister, A. The metabolic formation and utilization of 5-oxo-L-proline (L-pyroglutamate, L-pyrrolidone carboxylate). Adv. Enzymol. Relat. Areas Mol. Biol. 1975, 43, 519–556. [Google Scholar] [PubMed]
- Li, L.Y.; Seddon, A.P.; Meister, A.; Inubushi, T. Study of the 5-oxoprolinase reaction by 13C NMR. J. Biol. Chem. 1989, 264, 3096–3101. [Google Scholar] [PubMed]
- Orlowski, M.; Wilk, S. Intermediates of the γ-glutamyl cycle in mouse tissues. Influence of administration of amino acids on pyrrolidone carboxylate and γ-glutamyl amino acids. Eur. J. Biochem. 1975, 53, 581–590. [Google Scholar] [CrossRef] [PubMed]
- Bowser, T.E.; Trawick, M.L. Probing the specificity of γ-glutamylamine cyclotransferase: An enzyme involved in the metabolism of transglutaminase-catalyzed protein crosslinks. Amino Acids 2013, 44, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, S.; Futagi, Y.; Kobayashi, M.; Ogura, J.; Iseki, K. Functional characterization of 5-oxoproline transport via SLC16A1/MCT1. J. Biol. Chem. 2015, 290, 2303–2311. [Google Scholar] [CrossRef] [PubMed]
- Wilk, S.; Orlowski, M. Determination of pyrrolidone carboxylate and γ-glutamyl amino acids by gas chromatography. Anal. Biochem. 1975, 69, 100–113. [Google Scholar] [CrossRef]
- Cooper, A.J.L.; Dhar, A.K.; Kutt, H.; Duffy, T.E. Determination of 2-pyrrolidone-5-carboxylic and α-ketoglutaramic acids in human cerebrospinal fluid by gas chromatography. Anal. Biochem. 1980, 103, 118–126. [Google Scholar] [CrossRef]
- Hook, G.; Yu, J.; Toneff, T.; Kindy, M.; Hook, V. Brain pyroglutamate amyloid-β is produced by cathepsin B and is reduced by the cysteine protease inhibitor E64d, representing a potential Alzheimer’s disease therapeutic. J. Alzheimers Dis. 2014, 41, 129–149. [Google Scholar] [PubMed]
- Pivtoraiko, V.N.; Abrahamson, E.E.; Leurgans, S.E.; DeKosky, S.T.; Mufson, E.J.; Ikonomovic, M.D. Cortical pyroglutamate amyloid-β levels and cognitive decline in Alzheimer’s disease. Neurobiol. Aging 2015, 36, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Levintow, L.; Meister, A. Reversibility of the enzymatic synthesis of glutamine. J. Biol. Chem. 1954, 209, 265–280. [Google Scholar] [PubMed]
- Wang, Y.1.; Kudoh, J.; Kubota, R.; Asakawa, S.; Minoshima, S.; Shimizu, N. Chromosomal mapping of a family of human glutamine synthetase genes: Functional gene (GLUL) on 1q25, pseudogene (GLULP) on 9p13, and three related genes (GLULL1, GLULL2, GLULL3) on 5q33, 11p15, and 11q24. Genomics 1996, 37, 195–199. [Google Scholar] [PubMed]
- Boksha, I.S.; Tereshkina, E.B.; Burbaeva, G.S. Glutamine synthetase and glutamine synthetase-like protein from human brain: Purification and comparative characterization. J. Neurochem. 2000, 75, 2574–2582. [Google Scholar] [CrossRef] [PubMed]
- Boksha, I.S.; Schönfeld, H.J.; Langen, H.; Müller, F.; Tereshkina, E.B.; Burbaeva, G.S. Glutamine synthetase isolated from human brain: Octameric structure and homology of partial primary structure with human liver glutamine synthetase. Biochemistry (Mosc.) 2002, 67, 1012–1020. [Google Scholar] [CrossRef] [PubMed]
- Meister, A. Glutamine synthetase from mammalian tissues. Adv. Enzymol. Relat. Areas Mol. Biol. 1985, 113, 185–199. [Google Scholar]
- Shin, D.; Park, C. N-terminal extension of canine glutamine synthetase created by splicing alters its enzymatic property. J. Biol. Chem. 2004, 279, 1184–1190. [Google Scholar] [CrossRef] [PubMed]
- Jeitner, T.M.; Battaile, K.; Cooper, A.J.L. Critical evaluation of the changes in glutamine synthetase activity in models of cerebral stroke. Neurochem. Res. 2015, 40, 2544–2556. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Hernandez, A.; Bell, K.P.; Norenberg, M.D. Glutamine synthetase: Glial localization in brain. Science 1977, 195, 1356–1358. [Google Scholar] [CrossRef] [PubMed]
- Norenberg, M.D.; Martinez-Hernandez, A. Fine structural localization of glutamine synthetase in astrocytes of rat brain. Brain Res. 1979, 161, 303–310. [Google Scholar] [CrossRef]
- Bernstein, H.G.; Bannier, J.; Meyer-Lotz, G.; Steiner, J.; Keilhoff, G.; Dobrowolny, H.; Walter, M.; Bogerts, B. Distribution of immunoreactive glutamine synthetase in the adult human and mouse brain. Qualitative and quantitative observations with special emphasis on extra-astroglial protein localization. J. Chem. Neuroanat. 2014, 61, 33–50. [Google Scholar] [CrossRef] [PubMed]
- Robinson, S.R. Neuronal expression of glutamine synthetase in Alzheimer’s disease indicates a profound impairment of metabolic interactions with astrocytes. Neurochem. Int. 2000, 36, 471–482. [Google Scholar] [CrossRef]
- Robinson, S.R. Changes in the cellular distribution of glutamine synthetase in Alzheimer’s disease. J. Neurosci. Res. 2001, 66, 972–980. [Google Scholar] [CrossRef] [PubMed]
- Parli, J.A.; Godfrey, D.A.; Ross, C.D. Separate enzymatic microassays for aspartate aminotransferase isoenzymes. Biochim. Biophys. Acta 1987, 925, 175–184. [Google Scholar] [CrossRef]
- Fitzpatrick, S.M.; Cooper, A.J.L.; Duffy, T.E. Use of β-methylene-D,L-aspartate to assess the role of aspartate aminotransferase in cerebral oxidative metabolism. J. Neurochem. 1983, 41, 1370–1383. [Google Scholar] [CrossRef] [PubMed]
- Cheeseman, A.J.; Clark, J.B. Influence of the malate-aspartate shuttle on oxidative metabolism in synaptosomes. J. Neurochem. 1988, 50, 1559–1565. [Google Scholar] [CrossRef] [PubMed]
- Williamson, D.H.; Lund, P.; Krebs, H.A. The redox state of free nicotinamide-adenine dinucleotide in the cytoplasm and mitochondria of rat liver. Biochem. J. 1967, 103, 514–527. [Google Scholar] [CrossRef] [PubMed]
- Treberg, J.R.; Brosnan, M.E.; Watford, M.; Brosnan, J.T. On the reversibility of glutamate dehydrogenase and the source of hyperammonemia in the hyperinsulinism/hyperammonemia syndrome. Adv. Enzyme Regul. 2010, 50, 34–43. [Google Scholar] [CrossRef] [PubMed]
- Howse, D.C.; Duffy, T.E. Control of the redox state of the pyridine nucleotides in the rat cerebral cortex. Effect of electroshock-induced seizures. J. Neurochem. 1975, 24, 935–940. [Google Scholar] [CrossRef] [PubMed]
- Erecińska, M.; Pleasure, D.; Nelson, D.; Nissim, I.; Yudkoff, M. Cerebral aspartate utilization: Near-equilibrium relationships in aspartate aminotransferase reaction. J. Neurochem. 1993, 60, 1696–1706. [Google Scholar] [CrossRef] [PubMed]
- Cooper, A.J.L.; Nieves, E.; Coleman, A.E.; Filc-DeRicco, S.; Gelbard, A.S. Short-term metabolic fate of [13N]ammonia in rat liver in vivo. J. Biol. Chem. 1987, 262, 1073–1080. [Google Scholar] [PubMed]
- Fell, D.A. Enzymes, metabolites and fluxes. J. Exp. Bot. 2005, 56, 267–272. [Google Scholar] [CrossRef] [PubMed]
- Duffy, T.E.; Plum, F.; Cooper, A.J.L. Cerebral ammonia metabolism in vivo. In Glutamine, Glutamate and GABA in the Central Nervous System; Hertz, L., Kvamme, E., McGeer, E.G., Schousboe, A., Eds.; Alan R. Liss, Inc.: New York, NY, USA, 1983; pp. 371–388. [Google Scholar]
- Spanaki, C.; Kotzamani, D.; Kleopa, K.; Plaitakis, A. Evolution of GLUD2 glutamate dehydrogenase allows expression in human cortical neurons. Mol. Neurobiol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Spanaki, C.; Kotzamani, D.; Petraki, Z.; Drakos, E.; Plaitakis, A. Expression of human GLUD1 and GLUD2 glutamate dehydrogenases in steroid producing tissues. Mol. Cell. Endocrinol. 2015, 415, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Arevalo, M.A.; Azcoitia, I.; Garcia-Segura, L.M. The neuroprotective actions of oestradiol and oestrogen receptors. Nat. Rev. Neurosci. 2015, 16, 17–29. [Google Scholar] [CrossRef] [PubMed]
- Lowenstein, J.M. Ammonia production in muscle and other tissues: The purine nucleotide cycle. Physiol. Rev. 1972, 52, 382–414. [Google Scholar] [PubMed]
- Schultz, V.; Lowenstein, J.M. Purine nucleotide cycle. Evidence for the occurrence of the cycle in brain. J. Biol. Chem. 1976, 251, 485–492. [Google Scholar] [PubMed]
- Schultz, V.; Lowenstein, J.M. The purine nucleotide cycle. Studies of ammonia production and interconversions of adenine and hypoxanthine nucleotides and nucleosides by rat brain in situ. J. Biol. Chem. 1978, 253, 1938–1943. [Google Scholar] [PubMed]
- Zulfiqar, M.; Lin, D.D.; Van der Graaf, M.; Barker, P.B.; Fahrner, J.A.; Marie, S.; Morava, E.; De Boer, L.; Willemsen, M.A.; Vining, E.; et al. Novel proton MR spectroscopy findings in adenylosuccinate lyase deficiency. J. Magn. Reson. Imag. 2013, 37, 974–980. [Google Scholar] [CrossRef] [PubMed]
- Knecht, K.; Wiesmüller, K.H.; Gnau, V.; Jung, G.; Meyermann, R.; Todd, K.G.; Hamprecht, B. AMP deaminase in rat brain: Localization in neurons and ependymal cells. J. Neurosci. Res. 2001, 66, 941–950. [Google Scholar] [CrossRef] [PubMed]
- Lowenstein, J.M.; Goodman, M.N. The purine nucleotide cycle in skeletal muscle. Fed. Proc. 1978, 37, 2308–2312. [Google Scholar] [PubMed]
- Müller, A.F.; Leuthardt, F. Die Umwandlung der Glutaminsaure in Asparaginsaure in den Mitochondrien der Leber. (mit Bemerkung über das Vorkommen einer Transaminase in Clostridium Welchii). Helv. Chim. Acta 1950, 33, 268–273. [Google Scholar] [CrossRef]
- Kishore, N.; Tewari, Y.B.; Goldberg, R.N. An investigation of the equilibrium of the reaction {L-aspartate(aq)+2-oxoglutarate(aq)=oxaloacetate(aq)+L-glutamate(aq)}. J. Chem. Thermodyn. 1978, 30, 1373–1384. [Google Scholar] [CrossRef]
- Lowenstein, J.M. The tricarboxylic acid cycle. In Metabolic Pathways, 3rd ed.; Greenberg, D.M., Ed.; Academic Press: New York, NY, USA, 1967; Volume 1, pp. 146–270. [Google Scholar]
- Cooper, A.J.L. L-Glutamate (2-oxoglutarate) aminotransferases. In Glutamine and Glutamate in Mammals; Kwamme, E., Ed.; CRC Press, Inc.: Boca Raton, FL, USA, 1981; Volume I, pp. 445–461. [Google Scholar]
- Balázs, R.; Machiyama, Y.; Hammond, B.J.; Julian, T.; Richter, D. The operation of the γ-aminobutyrate bypath of the tricarboxylic acid cycle in brain tissue in vitro. Biochem. J. 1970, 116, 445–461. [Google Scholar] [PubMed]
- Salminen, A.; Jouhten, P.; Sarajärvi, T.; Haapasalo, A.; Hiltunen, M. Hypoxia and GABA shunt activation in the pathogenesis of Alzheimer’s disease. Neurochem. Int. 2016, 92, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Douglas, G.W.; Mortensen, R.A. The rate of metabolism of brain and liver glutathione in the rat studied with C14-glycine. J. Biol. Chem. 1956, 222, 581–585. [Google Scholar] [PubMed]
- Rehncrona, S.; Folbergrová, J.; Smith, D.S.; Siesjö, B.K. Influence of complete and pronounced incomplete cerebral ischemia and subsequent recirculation on cortical concentrations of oxidized and reduced glutathione in the rat. J. Neurochem. 1980, 34, 477–486. [Google Scholar] [CrossRef] [PubMed]
- Cooper, A.J.L.; Pulsinelli, W.A.; Duffy, T.E. Glutathione and ascorbate during ischemia and postischemic reperfusion in rat brain. J. Neurochem. 1980, 35, 1242–1245. [Google Scholar] [CrossRef] [PubMed]
- Guitart, K.; Loers, G.; Schachner, M.; Kleene, R. Prion protein regulates glutathione metabolism and neural glutamate and cysteine uptake via excitatory amino acid transporter 3. J. Neurochem. 2015, 133, 558–571. [Google Scholar] [CrossRef] [PubMed]
- Rice, M.E.; Russo-Menna, I. Differential compartmentalization of brain ascorbate and glutathione between neurons and glia. Neuroscience 1998, 82, 1213–1223. [Google Scholar] [CrossRef]
- Sun, X.; Shih, A.Y.; Johannssen, H.C.; Erb, H.; Li, P.; Murphy, T.H. Two-photon imaging of glutathione levels in intact brain indicates enhanced redox buffering in developing neurons and cells at the cerebrospinal fluid and blood-brain interface. J. Biol. Chem. 2006, 281, 17420–17431. [Google Scholar] [CrossRef] [PubMed]
- Dringen, R.; Pfeiffer, B.; Hamprecht, B. Synthesis of the antioxidant glutathione in neurons: Supply by astrocytes of CysGly as precursor for neuronal glutathione. J. Neurosci. 1999, 19, 562–569. [Google Scholar] [PubMed]
- Dringen, R. Hirrlinger J Glutathione pathways in the brain. Biol. Chem. 2003, 384, 505–516. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Swanson, R.A. The glutamate transporters EAAT2 and EAAT3 mediate cysteine uptake in cortical neuron cultures. J. Neurochem. 2003, 84, 1332–1339. [Google Scholar] [CrossRef] [PubMed]
- Ye, Z.C.; Rothstein, J.D.; Sontheimer, H. Compromised glutamate transport in human glioma cells: Reduction-mislocalization of sodium-dependent glutamate transporters and enhanced activity of cystine-glutamate exchange. J. Neurosci. 1999, 19, 10767–10777. [Google Scholar] [PubMed]
- Glowatzki, E.; Cheng, N.; Hiel, H.; Yi, E.; Tanaka, K.; Ellis-Davies, G.C.; Rothstein, J.D.; Bergles, D.E. The glutamate-aspartate transporter GLAST mediates glutamate uptake at inner hair cell afferent synapses in the mammalian cochlea. J. Neurosci. 2006, 26, 7659–7564. [Google Scholar] [CrossRef] [PubMed]
- Benediktsson, A.M.; Marrs, G.S.; Tu, J.C.; Worley, P.F.; Rothstein, J.D.; Bergles, D.E.; Dailey, M.E. Neuronal activity regulates glutamate transporter dynamics in developing astrocytes. Glia 2012, 60, 175–188. [Google Scholar] [CrossRef] [PubMed]
- Fuhrmann, M.; Bittner, T.; Mitteregger, G.; Haider, N.; Moosmang, S.; Kretzschmar, H.; Herms, J. Loss of the cellular prion protein affects the Ca2+ homeostasis in hippocampal CA1 neurons. J. Neurochem. 2006, 98, 1876–1885. [Google Scholar] [CrossRef] [PubMed]
- Krebs, B.; Wiebelitz, A.; Balitzki-Korte, B.; Vassallo, N.; Paluch, S.; Mitteregger, G.; Onodera, T.; Kretzschmar, H.A.; Herms, J. Cellular prion protein modulates the intracellular calcium response to hydrogen peroxide. J. Neurochem. 2007, 100, 358–367. [Google Scholar] [CrossRef] [PubMed]
- Harris, J.L.; Choi, I.Y.; Brooks, W.M. Probing astrocyte metabolism in vivo: Proton magnetic resonance spectroscopy in the injured and aging brain. Front. Aging Neurosci. 2015. [Google Scholar] [CrossRef] [PubMed]
- Mason, G.F.; Rothman, D.L.; Behar, K.L.; Shulman, R.G. NMR determination of the TCA cycle rate and α-ketoglutarate/glutamate exchange rate in rat brain. J. Cereb. Blood Flow Metab. 1992, 12, 434–447. [Google Scholar] [CrossRef] [PubMed]
- Mason, G.F.; Gruetter, R.; Rothman, D.L.; Behar, K.L.; Shulman, R.G.; Novotny, E.J. Simultaneous determination of the rates of the TCA cycle, glucose utilization, α-ketoglutarate/glutamate exchange, and glutamine synthesis in human brain by NMR. J. Cereb. Blood Flow Metab. 1995, 15, 12–25. [Google Scholar] [CrossRef] [PubMed]
- Xin, L.; Lanz, B.; Lei, H.; Gruetter, R. Assessment of metabolic fluxes in the mouse brain in vivo using 1H-[13C] NMR spectroscopy at 14.1 Tesla. J. Cereb. Blood Flow Metab. 2015, 35, 759–765. [Google Scholar] [CrossRef] [PubMed]
- Pamiljans, V.; Krishnaswamy, P.R.; Dumville, G.; Meister, A. Studies on the mechanism of glutamine synthesis; isolation and properties of the enzyme from sheep brain. Biochemistry 1962, 1, 153–158. [Google Scholar] [CrossRef] [PubMed]
- Raichle, M.E.; Larson, K.B. The significance of the NH3-NH4+ equilibrium on the passage of 13N-ammonia from blood to brain. A new regional residue detection model. Circ. Res. 1981, 48, 913–937. [Google Scholar] [CrossRef] [PubMed]
- Girard, G.; Giguère, J.F.; Butterworth, R.F. Region-selective reductions in activities of glutamine synthetase in rat brain following portacaval anastomosis. Metab. Brain Dis. 1993, 8, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Butterworth, R.F.; Lavoie, J.; Layrargues, G.P.; Giguère, J.F. Cerebral amino acid changes in hepatic encephalopathy: Biochemical-neuropathological correlations. In Biochemical Pathology of the Astrocytes; Norenberg, M.D., Hertz, L., Schousboe, A., Eds.; Alan R. Liss, Inc.: New York, NY, USA, 1983; pp. 481–482. [Google Scholar]
- Singh, S.; Mondal, P.; Trigun, S.K. Acute liver failure in rats activates glutamine-glutamate cycle but declines antioxidant enzymes to induce oxidative stress in cerebral cortex and cerebellum. PLoS ONE 2014, 9, e95855. [Google Scholar] [CrossRef] [PubMed]
- Gjedde, A.; Lockwood, A.H.; Duffy, T.E.; Plum, F. Cerebral blood flow and metabolism in chronically hyperammonemic rats: Effect of an acute ammonia challenge. Ann. Neurol. 1978, 3, 325–330. [Google Scholar] [CrossRef] [PubMed]
- Hindfelt, B.; Plum, F.; Duffy, T.E. Effect of acute ammonia intoxication on cerebral metabolism in rats with portacaval shunts. J. Clin. Investig. 1977, 59, 386–396. [Google Scholar] [CrossRef] [PubMed]
- Butterworth, R.F.; Giguère, J.F. Cerebral aminoacids in portal-systemic encephalopathy: Lack of evidence for altered γ-aminobutyric acid (GABA) function. Metab. Brain Dis. 1986, 1, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Cudalbu, C.; Lanz, B.; Duarte, J.M.; Morgenthaler, F.D.; Pilloud, Y.; Mlynárik, V.; Gruetter, R. Cerebral glutamine metabolism under hyperammonemia determined in vivo by localized 1H and 15N NMR spectroscopy. J. Cereb. Blood Flow Metab. 2012, 32, 696–708. [Google Scholar] [CrossRef] [PubMed]
- Mardini, H.; Smith, F.E.; Record, C.O.; Blamire, A.M. Magnetic resonance quantification of water and metabolites in the brain of cirrhotics following induced hyperammonaemia. J. Hepatol. 2011, 54, 1154–1160. [Google Scholar] [CrossRef] [PubMed]
- Chavarria, L.; Alonso, J.; García-Martínez, R.; Simón-Talero, M.; Ventura-Cots, M.; Ramírez, C.; Torrens, M.; Vargas, V.; Rovira, A.; Córdoba, J. Brain magnetic resonance spectroscopy in episodic hepatic encephalopathy. J. Cereb. Blood Flow Metab. 2013, 33, 272–277. [Google Scholar] [CrossRef] [PubMed]
- Ratnakumari, L.; Qureshi, I.A.; Butterworth, R.F. Effects of congenital hyperammonemia on the cerebral and hepatic levels of the intermediates of energy metabolism in spf mice. Biochem. Biophys. Res. Commun. 1992, 184, 746–751. [Google Scholar] [CrossRef]
- Lai, J.C.K.; Cooper, A.J.L. Brain α-ketoglutarate dehydrogenase complex: Kinetic properties, regional distribution, and effects of inhibitors. J. Neurochem. 1986, 47, 1376–1386. [Google Scholar] [CrossRef] [PubMed]
- Cooper, A.J.L.; Vergara, F.; Duffy, T.E. Cerebral glutamine synthetase. In Glutamine, Glutamate and GABA in the Central Nervous System; Hertz, L., Kvamme, E., McGeer, E.G., Schousboe, A., Eds.; Alan R. Liss, Inc.: New York, NY, USA, 1983; pp. 77–93. [Google Scholar]
- Bak, L.K.; Waagepetersen, H.S.; Sørensen, M.; Ott, P.; Vilstrup, H.; Keiding, S.; Schousboe, A. Role of branched chain amino acids in cerebral ammonia homeostasis related to hepatic encephalopathy. Metab. Brain Dis. 2013, 28, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Hassel, B. Carboxylation and anaplerosis in neurons and glia. Mol. Neurobiol. 2000, 22, 21–40. [Google Scholar] [CrossRef]
- Hassel, B.; Bråthe, A. Neuronal pyruvate carboxylation supports formation of transmitter glutamate. J. Neurosci. 2000, 20, 1342–1347. [Google Scholar] [PubMed]
- Hassel, B.; Bråthe, A. Cerebral metabolism of lactate in vivo: Evidence for neuronal pyruvate carboxylation. J. Cereb. Blood Flow Metab. 2000, 20, 327–336. [Google Scholar] [CrossRef] [PubMed]
- McKenna, M.C.; Stevenson, J.H.; Huang, X.; Tildon, J.T.; Zielke, C.L.; Hopkins, I.B. Mitochondrial malic enzyme activity is much higher in mitochondria from cortical synaptic terminals compared with mitochondria from primary cultures of cortical neurons or cerebellar granule cells. Neurochem. Int. 2000, 36, 451–459. [Google Scholar] [CrossRef]
- Dienel, G.A.; McKenna, M.C. A dogma-breaking concept: Glutamate oxidation in astrocytes is the source of lactate during aerobic glycolysis in resting subjects. J. Neurochem. 2014, 131, 395–398. [Google Scholar] [CrossRef] [PubMed]
- Sonnewald, U. Glutamate synthesis has to be matched by its degradation—Where do all the carbons go? J. Neurochem. 2014, 131, 399–406. [Google Scholar] [CrossRef] [PubMed]
- Yu, A.C.; Drejer, J.; Hertz, L.; Schousboe, A. Pyruvate carboxylase activity in primary cultures of astrocytes and neurons. J. Neurochem. 1983, 41, 1484–1487. [Google Scholar] [CrossRef] [PubMed]
- Shank, R.P.; Bennett, G.S.; Freytag, S.O.; Campbell, G.L. Pyruvate carboxylase: An astrocyte-specific enzyme implicated in the replenishment of amino acid neurotransmitter pools. Brain Res. 1985, 329, 364–367. [Google Scholar] [CrossRef]
- Schousboe, A.; Scafidi, S.; Bak, L.K.; Waagepetersen, H.S.; McKenna, M.C. Glutamate metabolism in the brain focusing on astrocytes. Adv. Neurobiol. 2014, 11, 13–30. [Google Scholar] [PubMed]
- Öz, G.; Berkich, D.A.; Henry, P.G.; Xu, Y.; LaNoue, K.; Hutson, S.M.; Gruetter, R. Neuroglial metabolism in the awake rat brain: CO2 fixation increases with brain activity. J. Neurosci. 2004, 24, 11273–11279. [Google Scholar] [CrossRef] [PubMed]
- Lanz, B.; Xin, L.; Millet, P.; Gruetter, R. In vivo quantification of neuro-glial metabolism and glial glutamate concentration using 1H-[13C] MRS at 14.1T. J. Neurochem. 2014, 128, 125–139. [Google Scholar] [CrossRef] [PubMed]
- Gruetter, R.; Seaquist, E.R.; Kim, S.; Ugurbil, K. Localized in vivo 13C-NMR of glutamate metabolism in the human brain: Initial results at 4Tesla. Dev. Neurosci. 1998, 20, 380–388. [Google Scholar] [CrossRef] [PubMed]
- Gruetter, R.; Seaquist, E.R.; Ugurbil, K. A mathematical model of compartmentalized neurotransmitter metabolism in the human brain. Am. J. Physiol. Endocrinol. Metab. 2001, 281, E100–E112. [Google Scholar] [PubMed]
- Aureli, T.; Di Cocco, M.E.; Calvani, M.; Conti, F. The entry of [1–13C]glucose into biochemical pathways reveals a complex compartmentation and metabolite trafficking between glia and neurons: A study by 13C-NMR spectroscopy. Brain Res. 1997, 765, 218–227. [Google Scholar] [CrossRef]
- Sibson, N.R.; Mason, G.F.; Shen, J.; Cline, G.W.; Herskovits, A.Z.; Wall, J.E.; Behar, K.L.; Rothman, D.L.; Shulman, R.G. In vivo 13C NMR measurement of neurotransmitter glutamate cycling, anaplerosis and TCA cycle flux in rat brain during [2–13C]glucose infusion. J. Neurochem. 2001, 76, 975–989. [Google Scholar] [CrossRef] [PubMed]
- Lapidot, A.; Gopher, A. Cerebral metabolic compartmentation. Estimation of glucose flux via pyruvate carboxylase/pyruvate dehydrogenase by 13C NMR isotopomer analysis of D-[U-13C]glucose metabolites. J. Biol. Chem. 1994, 269, 27198–27208. [Google Scholar] [PubMed]
- Clark, G.M.; Eiseman, B. Studies in ammonia metabolism. IV. Biochemical changes in brain tissue of dogs during ammonia induced coma. New Engl. J. Med. 1958, 259, 178–180. [Google Scholar] [CrossRef] [PubMed]
- Berl, S.; Takagaki, G.; Clarke, D.D.; Waelsch, H. Carbon dioxide fixation in the brain. J. Biol. Chem. 1962, 237, 2570–2573. [Google Scholar] [PubMed]
- Sarma, M.K.; Huda, A.; Nagarajan, R.; Hinkin, C.H.; Wilson, N.; Gupta, R.K.; Frias-Martinez, E.; Sayre, J.; Guze, B.; Han, S.H.; et al. Multi-dimensional MR spectroscopy: Towards a better understanding of hepatic encephalopathy. Metab. Brain Dis. 2011, 26, 173–184. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.C.; Chou, K.H.; Chen, C.L.; Chen, H.L.; Lu, C.H.; Li, S.H.; Huang, C.C.; Lin, C.P.; Cheng, Y.F. Longitudinal brain white matter alterations in minimal hepatic encephalopathy before and after liver transplantation. PLoS ONE 2014, 9, e105887. [Google Scholar] [CrossRef] [PubMed]
- Schousboe, A.; Waagepetersen, H.S.; Leke, R.; Bak, L.K. Effects of hyperammonemia on brain energy metabolism: Controversial findings in vivo and in vitro. Metab. Brain Dis. 2014, 29, 913–917. [Google Scholar] [CrossRef] [PubMed]
- Ott, P.; Vilstrup, H. Cerebral effects of ammonia in liver disease: Current hypotheses. Metab. Brain Dis. 2014, 29, 901–911. [Google Scholar] [CrossRef] [PubMed]
- Fitzpatrick, S.M.; Hetherington, H.P.; Behar, K.L.; Shulman, R.G. Effects of acute hyperammonemia on cerebral amino acid metabolism and pHi in vivo, measured by 1H and 31P nuclear magnetic resonance. J. Neurochem. 1989, 52, 741–749. [Google Scholar] [CrossRef] [PubMed]
- Rackayova, V.; Braissant, O.; McLin, V.A.; Berset, C.; Lanz, B.; Cudalbu, C. 1H and 31P magnetic resonance spectroscopy in a rat model of chronic hepatic encephalopathy: In vivo longitudinal measurements of brain energy metabolism. Metab. Brain Dis. 2015. [Google Scholar] [CrossRef] [PubMed]
- Brusilow, S.W.; Koehler, R.C.; Traystman, R.J.; Cooper, A.J.L. Astrocyte glutamine synthetase: Importance in hyperammonemic syndromes and potential target for therapy. Neurotherapeutics 2010, 7, 452–470. [Google Scholar] [CrossRef] [PubMed]
- Butterworth, R.F. Pathophysiology of brain dysfunction in hyperammonemic syndromes: The many faces of glutamine. Mol. Genet. Metab. 2014, 113, 113–117. [Google Scholar] [CrossRef] [PubMed]
- Bosoi, C.R.; Zwingmann, C.; Marin, H.; Parent-Robitaille, C.; Huynh, J.; Tremblay, M.; Rose, C.F. Increased brain lactate is central to the development of brain edema in rats with chronic liver disease. J. Hepatol. 2014, 60, 554–560. [Google Scholar] [CrossRef] [PubMed]
- Berl, S.; Takagaki, G.; Clarke, D.D.; Waelsch, H. Metabolic compartments in vivo. Ammonia and glutamic acid metabolism in brain and liver. J. Biol. Chem. 1962, 237, 2562–2569. [Google Scholar] [PubMed]
- Benjamin, A.M.; Quastel, J.H. Metabolism of amino acids and ammonia in rat brain cortex slices in vitro: A possible role of ammonia in brain function. J. Neurochem. 1975, 25, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Nagaraja, T.N.; Brookes, N. Intracellular acidification induced by passive and active transport of ammonium ions in astrocytes. Am. J. Physiol. 1998, 274, C883–C891. [Google Scholar] [PubMed]
- Rothman, D.L.; De Feyter, H.M.; de Graaf, R.A.; Mason, G.F.; Behar, K.L. 13C MRS studies of neuroenergetics and neurotransmitter cycling in humans. NMR Biomed. 2011, 24, 943–957. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.B.; de Graaf, R.A.; Mason, G.F.; Rothman, D.L.; Shulman, R.G.; Behar, K.L. The contribution of GABA to glutamate/glutamine cycling and energy metabolism in the rat cortex in vivo. Proc. Natl. Acad. Sci. USA 2005, 102, 5588–5593. [Google Scholar] [CrossRef] [PubMed]
- Erecińska, M.; Zaleska, M.M.; Nissim, I.; Nelson, D.; Dagani, F.; Yudkoff, M. Glucose and synaptosomal glutamate metabolism: Studies with [15N]glutamate. J. Neurochem. 1988, 51, 892–902. [Google Scholar] [CrossRef] [PubMed]
- Ward, H.K.; Bradford, H.F. Relative activities of glutamine synthetase and glutaminase in mammalian synaptosomes. J. Neurochem. 1979, 33, 339–342. [Google Scholar] [CrossRef] [PubMed]
- Dennis, S.C.; Lai, J.C.K.; Clark, J.B. The distribution of glutamine synthetase in subcellular fractions of rat brain. Brain Res. 1980, 197, 469–475. [Google Scholar] [CrossRef]
- Tranah, T.H.; Vijay, G.K.; Ryan, J.M.; Shawcross, D.L. Systemic inflammation and ammonia in hepatic encephalopathy. Metab. Brain Dis. 2013, 28, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Jayakumar, A.R.; Rama Rao, K.V.; Norenberg, M.D. Neuroinflammation in hepatic encephalopathy: Mechanistic aspects. J. Clin. Exp. Hepatol. 2015, 5, S21–S28. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, K.; Kanamatsu, T.; Takezawa, Y.; Kohsaka, S. Up-regulation of glutamine synthesis in microglia activated with endotoxin. Neurosci. Lett. 2015, 591, 99–104. [Google Scholar] [CrossRef] [PubMed]
- Morioka, N.; Harano, S.; Tokuhara, M.; Idenoshita, Y.; Zhang, F.F.; Hisaoka-Nakashima, K.; Nakata, Y. Stimulation of α7 nicotinic acetylcholine receptor regulates glutamate transporter GLAST via basic fibroblast growth factor production in cultured cortical microglia. Brain Res. 2015, 1625, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Beaulieu, C.; Somogyi, P. Targets and quantitative distribution of gabaergic synapses in the visual cortex of the cat. Eur. J. Neurosci. 1990, 2, 296–303. [Google Scholar] [CrossRef] [PubMed]
- Walls, A.B.; Waagepetersen, H.S.; Bak, L.K.; Schousboe, A.; Sonnewald, U. The glutamine-glutamate/GABA cycle: Function, regional differences in glutamate and GABA production and effects of interference with GABA metabolism. Neurochem. Res. 2015, 40, 402–409. [Google Scholar] [CrossRef] [PubMed]
- Berl, S.; Frigyesi, T.L. The turnover of glutamate, glutamine, aspartate and GABA labeled with [1–14C]acetate in caudate nucleus, thalamus and motor cortex (cat). Brain Res. 1969, 12, 444–455. [Google Scholar] [CrossRef]
- Van den Berg, C.J.; Krzalic, L.; Mela, P.; Waelsch, H. Compartmentation of glutamate metabolism in brain. Evidence for the existence of two different tricarboxylic acid cycles in brain. Biochem. J. 1969, 113, 281–290. [Google Scholar] [CrossRef] [PubMed]
- Van den Berg, C.J.; Ronda, G. The incorporation of double-labelled acetate into glutamate ad related amino acids from adult mouse brain: Compartmentation of amino acid metabolism in brain. J. Neurochem. 1976, 27, 1443–1448. [Google Scholar] [CrossRef] [PubMed]
- Wyss, M.T.; Magistretti, P.J.; Buck, A.; Weber, B. Labeled acetate as a marker of astrocytic metabolism. J. Cereb. Blood Flow Metab. 2011, 31, 1668–1674. [Google Scholar] [CrossRef] [PubMed]
- Badar-Goffer, R.S.; Bachelard, H.S.; Morris, P.G. Cerebral metabolism of acetate and glucose studied by 13C-n.m.r. spectroscopy. A technique for investigating metabolic compartmentation in the brain. Biochem. J. 1990, 266, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Sonnewald, U.; Westergaard, N.; Hassel, B.; Muller, T.B.; Unsgard, G.; Fonnum, F.; Hertz, L.; Schousboe, A.; Petersen, S.B. NMR spectroscopic studies of 13C acetate and 13C glucose metabolism in neocortical astrocytes: Evidence for mitochondrial heterogeneity. Dev. Neurosci. 1993, 15, 351–358. [Google Scholar] [CrossRef] [PubMed]
- Sonnewald, U.; Westergaard, N.; Schousboe, A.; Svendsen, J.S.; Unsgard, G.; Petersen, S.B. Direct demonstration by [13C]NMR spectroscopy that glutamine from astrocytes is a precursor for GABA synthesis in neurons. Neurochem. Int. 1993, 22, 19–29. [Google Scholar] [CrossRef]
- Waniewski, R.A.; Martin, D.L. Preferential utilization of acetate by astrocytes is attributable to transport. J. Neurosci. 1998, 18, 5225–5233. [Google Scholar] [PubMed]
- Waniewski, R.A.; Martin, D.L. Astrocytes and synaptosomes transport and metabolize lactate and acetate differently. Neurochem. Res. 2004, 29, 209–217. [Google Scholar] [CrossRef] [PubMed]
- Calvetti, D.; Somersalo, E. Quantitative in silico analysis of neurotransmitter pathways under steady state conditions. Front. Endocrinol. (Lausanne) 2013. [Google Scholar] [CrossRef] [PubMed]
- Cooper, A.J.L. Quantitative analysis of neurotransmitter pathways under steady state conditions—A perspective. Front. Endocrinol. (Lausanne) 2013. [Google Scholar] [CrossRef] [PubMed]
- Benuck, M.; Lajtha, A. Aminotransferase activity in brain. Int. Rev. Neurobiol. 1975, 17, 85–129. [Google Scholar] [PubMed]
- Segal, H.L.; Beattie, D.S.; Hopper, S. Purification and properties of liver glutamic-alanine transaminase from normal and corticoid-treated rats. J. Biol. Chem. 1962, 237, 1914–1920. [Google Scholar]
- Fries, A.W.; Dadsetan, S.; Keiding, S.; Bak, L.K.; Schousboe, A.; Waagepetersen, H.S.; Simonsen, M.; Ott, P.; Vilstrup, H.; Sørensen, M. Effect of glutamine synthetase inhibition on brain and interorgan ammonia metabolism in bile duct ligated rats. J. Cereb. Blood Flow Metab. 2014, 34, 460–466. [Google Scholar] [CrossRef] [PubMed]
- Hutson, S.M.; Berkich, D.; Drown, P.; Xu, B.; Aschner, M.; LaNoue, K.F. Role of branched-chain aminotransferase isoenzymes and gabapentin in neurotransmitter metabolism. J. Neurochem. 1998, 71, 863–874. [Google Scholar] [CrossRef] [PubMed]
- Hutson, S.M.; Lieth, E.; LaNoue, K.F. Function of leucine in excitatory neurotransmitter metabolism in the central nervous system. J. Nutr. 2001, 131, 846S–850S. [Google Scholar] [PubMed]
- Berl, S.; Frigyesi, T.L. Metabolism of [14C]leucine and [14C]acetate in sensorimotor cortex, thalamus, caudate nucleus and cerebellum of the cat. J. Neurochem. 1968, 15, 965–970. [Google Scholar] [CrossRef] [PubMed]
- Berl, S.; Frigyesi, T.L. Comparison of cerebral regional metabolism of [14C]leucine following third ventricle and intravenous administration in the cat. J. Neurochem. 1969, 16, 405–415. [Google Scholar] [CrossRef] [PubMed]
- Yudkoff, M.; Daikhin, Y.; Grunstein, L.; Nissim, I.; Stern, J.; Pleasure, D.; Nissim, I. Astrocyte leucine metabolism: Significance of branched-chain amino acid transamination. J. Neurochem. 1996, 66, 378–385. [Google Scholar] [CrossRef] [PubMed]
- Yudkoff, M.; Daikhin, Y.; Nissim, I.; Horyn, O.; Luhovyy, B.; Lazarow, A.; Nissim, I. Brain amino acid requirements and toxicity: The example of leucine. J. Nutr. 2005, 135, 1531S–1538S. [Google Scholar] [PubMed]
- Rothman, D.L.; De Feyter, H.M.; Maciejewski, P.K.; Behar, K.L. Is there in vivo evidence for amino acid shuttles carrying ammonia from neurons to astrocytes? Neurochem. Res. 2012, 37, 2597–2612. [Google Scholar] [CrossRef] [PubMed]
- Pardo, B.; Rodrigues, T.B.; Contreras, L.; Garzón, M.; Llorente-Folch, I.; Kobayashi, K.; Saheki, T.; Cerdan, S.; Satrústegui, J. Brain glutamine synthesis requires neuronal-born aspartate as amino donor for glial glutamate formation. J. Cereb. Blood Flow Metab. 2011, 31, 90–101. [Google Scholar] [CrossRef] [PubMed]
- Hertz, L. Brain glutamine synthesis requires neuronal aspartate: A commentary. J. Cereb. Blood Flow Metab. 2011, 31, 384–387. [Google Scholar] [CrossRef] [PubMed]
- Dingledine, R.; Borges, K.; Bowie, D.; Traynelis, S.F. The glutamate receptor ion channels. Pharmacol. Rev. 1999, 51, 7–61. [Google Scholar] [PubMed]
- Greenamyre, J.T.; Porter, R.H. Anatomy and physiology of glutamate in the CNS. Neurology 1994, 44, S7–S13. [Google Scholar] [PubMed]
- Olney, J.W.; Sharpe, L.G. Brain lesions in an infant rhesus monkey treated with monosodium glutamate. Science 1969, 166, 386–388. [Google Scholar] [CrossRef] [PubMed]
- Olney, J.W. Brain lesions, obesity, and other disturbances in mice treated with monosodium glutamate. Science 1969, 164, 719–721. [Google Scholar] [CrossRef] [PubMed]
- Olney, J.W. Inciting excitotoxic cytocide among central neurons. Adv. Exp. Med. Biol. 1986, 203, 631–645. [Google Scholar] [PubMed]
- Choi, D.W. Glutamate neurotoxicity and diseases of the nervous system. Neuron 1988, 1, 623–634. [Google Scholar] [CrossRef]
- Nicholls, D.; Attwell, D. The release and uptake of excitatory amino acids. Trends Pharmacol. Sci. 1990, 11, 462–468. [Google Scholar] [CrossRef]
- Meldrum, B.; Garthwaite, J. Excitatory amino acid neurotoxicity and neurodegenerative disease. Trends Pharmacol. Sci. 1990, 11, 379–387. [Google Scholar] [CrossRef]
- Lipton, S.A.; Rosenberg, P.A. Excitatory amino acids as a final common pathway for neurologic disorders. New Engl. J. Med. 1994, 330, 613–622. [Google Scholar] [PubMed]
- Lewerenz, J.; Maher, P. Chronic glutamate toxicity in neurodegenerative diseases-What is the evidence? Front. Neurosci. 2015. [Google Scholar] [CrossRef] [PubMed]
- Krishnamurthy, K.; Laskowitz, D.T. Cellular and molecular mechanisms of secondary neuronal injury. In Translational Research in Traumatic Brain Injury; Laskowitz, D., Grant, G., Eds.; CRC Press/Taylor and Francis Group: Boca Raton, FL, USA, 2016. [Google Scholar]
- Prasad, K.N.; Bondy, S.C. Common biochemical defects linkage between post-traumatic stress disorders, mild traumatic brain injury (TBI) and penetrating TBI. Brain Res. 2015, 1599, 103–114. [Google Scholar] [CrossRef] [PubMed]
- Camacho, A.; Massieu, L. Role of glutamate transporters in the clearance and release of glutamate during ischemia and its relation to neuronal death. Arch. Med. Res. 2006, 37, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Kostandy, B.B. The role of glutamate in neuronal ischemic injury: The role of spark in fire. Neurol. Sci. 2012, 33, 223–237. [Google Scholar] [CrossRef] [PubMed]
- Coulter, D.A.; Eid, T. Astrocytic regulation of glutamate homeostasis in epilepsy. Glia 2012, 60, 1215–1226. [Google Scholar] [CrossRef] [PubMed]
- Johnston, M.V. Excitotoxicity in perinatal brain injury. Brain Pathol. 2005, 15, 234–240. [Google Scholar] [CrossRef] [PubMed]
- Zádori, D.; Veres, G.; Szalárdy, L.; Klivényi, P.; Toldi, J.; Vécsei, L. Glutamatergic dysfunctioning in Alzheimer’s disease and related therapeutic targets. J. Alzheimers Dis. 2014, 42, S177–S187. [Google Scholar] [PubMed]
- Ong, W.Y.; Tanaka, K.; Dawe, G.S.; Ittner, L.M.; Farooqui, A.A. Slow excitotoxicity in Alzheimer’s disease. J. Alzheimers Dis. 2013, 35, 643–668. [Google Scholar] [PubMed]
- Rothstein, J.D.; Tsai, G.; Kuncl, R.W.; Clawson, L.; Cornblath, D.R.; Drachman, D.B.; Pestronk, A.; Stauch, B.L.; Coyle, J.T. Abnormal excitatory amino acid metabolism in amyotrophic lateral sclerosis. Ann. Neurol. 1990, 28, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Rothstein, J.D. Excitotoxicity and neurodegeneration in amyotrophic lateral sclerosis. Clin. Neurosci. 1995, 3, 348–59. [Google Scholar] [PubMed]
- King, A.E.; Woodhouse, A.; Kirkcaldie, M.T.; Vickers, J.C. Excitotoxicity in ALS: Overstimulation, or overreaction? Exp. Neurol. 2016, 275, 162–171. [Google Scholar] [CrossRef] [PubMed]
- Blasco, H.; Mavel, S.; Corcia, P.; Gordon, P.H. The glutamate hypothesis in ALS: Pathophysiology and drug development. Curr. Med. Chem. 2014, 21, 3551–3575. [Google Scholar] [CrossRef] [PubMed]
- André, V.M.; Cepeda, C.; Levine, M.S. Dopamine and glutamate in Huntington’s disease: A balancing act. CNS Neurosci. Ther. 2010, 16, 163–178. [Google Scholar] [CrossRef] [PubMed]
- Meredith, G.E.; Totterdell, S.; Beales, M.; Meshul, C.K. Impaired glutamate homeostasis and programmed cell death in a chronic MPTP mouse model of Parkinson’s disease. Exp. Neurol. 2009, 219, 334–340. [Google Scholar] [CrossRef] [PubMed]
- Ambrosi, G.; Cerri, S.; Blandini, F. A further update on the role of excitotoxicity in the pathogenesis of Parkinson’s disease. J. Neural Transm. 2014, 121, 849–859. [Google Scholar] [CrossRef] [PubMed]
- Hoque, A.; Hossain, M.I.; Ameen, S.S.; Ang, C.S.; Williamson, N.; Ng, D.C.; Chueh, A.; Roulston, C.; Cheng, H.C. A beacon of hope in stroke therapy-Blockade of pathologically activated cellular events in excitotoxic neuronal death as potential neuroprotective strategies. Pharmacol. Ther. 2016, in press. [Google Scholar] [CrossRef] [PubMed]
- Fontana, A.C. Current approaches to enhance glutamate transporter function and expression. J. Neurochem. 2015, 134, 982–1007. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Foster, J.B.; Lin, C.L. Glutamate transporter EAAT2: Regulation, function, and potential as a therapeutic target for neurological and psychiatric disease. Cell. Mol. Life Sci. 2015, 72, 3489–3506. [Google Scholar] [CrossRef] [PubMed]
- Soni, N.; Reddy, B.V.; Kumar, P. GLT-1 transporter: An effective pharmacological target for various neurological disorders. Pharmacol. Biochem. Behav. 2014, 127, 70–81. [Google Scholar] [CrossRef] [PubMed]
- Khanna, S.; Briggs, Z.; Rink, C. Inducible glutamate oxaloacetate transaminase as a therapeutic target against ischemic stroke. Antioxid. Redox Signal. 2015, 22, 175–186. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Mato, M.; Ramos-Cabrer, P.; Sobrino, T.; Blanco, M.; Ruban, A.; Mirelman, D.; Menendez, P.; Castillo, J.; Campos, F. Human recombinant glutamate oxaloacetate transaminase 1 (GOT1) supplemented with oxaloacetate induces a protective effect after cerebral ischemia. Cell Death Dis. 2014. [Google Scholar] [CrossRef] [PubMed]
- Ghoddoussi, F.; Galloway, M.P.; Jambekar, A.; Bame, M.; Needleman, R.; Brusilow, W.S. Methionine sulfoximine, an inhibitor of glutamine synthetase, lowers brain glutamine and glutamate in a mouse model of ALS. J. Neurol. Sci. 2010, 290, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Bame, M.; Pentiak, P.A.; Needleman, R.; Brusilow, W.S. Effect of sex on lifespan, disease progression, and the response to methionine sulfoximine in the SOD1 G93A mouse model for ALS. Gend. Med. 2012, 9, 524–535. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cat | Rat | Human | |
|---|---|---|---|
| Glutamate | 7.90 (9.88) | 11.6 (14.5) | 6.00 (7.50) |
| Taurine | 2.30 (2.88) | 6.60 (8.25) | 0.93 (1.16) |
| Glutamine | 2.80 (3.50) | 4.50 (5.63) | 5.80 (7.25) |
| Aspartate | 1.70 (2.13) | 2.60 (3.25) | 0.96 (1.20) |
| γ-Aminobutyrate | 1.40 (1.75) | 2.30 (2.88) | 0.42 (0.53) |
| Glycine | 0.78 (0.98) | 0.68 (0.85) | 0.40 (0.50) |
| Alanine | 0.48 (0.60) | 0.65 (0.81) | 0.25 (0.31) |
| Serine | 0.48 (0.60) | 0.98 (1.23) | 0.44 (0.55) |
| Glutathione | 0.49 (0.61) | 2.60 (3.25) | 0.20 (0.25) |
| Metabolite | Control (NaAc-Infused) | NH4Ac-Infused | MSO-Treated + NaAc-Infused | MSO-Treated + NH4Ac-Infused |
|---|---|---|---|---|
| Ammonia (blood) | 0.191 ± 0.063 | 0.710 ± 0.150 * | 0.432 ± 0.089 * | 1.02 ± 0.07 * |
| Ammonia | 0.326 ± 0.063 | 0.985 ± 0.084 | 0.855 ± 0.031 | 2.48 ± 0.06 |
| α-Ketoglutarate | 0.096 ± 0.011 | 0.112 ± 0.011 | 0.157 ± 0.006 * | 0.141 ± 0.018 * |
| L-Glutamate | 12.9 ± 0.4 | 11.1 ± 0.3 * | 10.1 ± 0.3 * | 11.6 ± 0.4 *,† |
| L-Glutamine | 8.04 ± 0.019 | 18.5 ± 0.8 * | 3.79 ± 0.54 * | 6.97 ± 1.00 * |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cooper, A.J.L.; Jeitner, T.M. Central Role of Glutamate Metabolism in the Maintenance of Nitrogen Homeostasis in Normal and Hyperammonemic Brain. Biomolecules 2016, 6, 16. https://doi.org/10.3390/biom6020016
Cooper AJL, Jeitner TM. Central Role of Glutamate Metabolism in the Maintenance of Nitrogen Homeostasis in Normal and Hyperammonemic Brain. Biomolecules. 2016; 6(2):16. https://doi.org/10.3390/biom6020016
Chicago/Turabian StyleCooper, Arthur J. L., and Thomas M. Jeitner. 2016. "Central Role of Glutamate Metabolism in the Maintenance of Nitrogen Homeostasis in Normal and Hyperammonemic Brain" Biomolecules 6, no. 2: 16. https://doi.org/10.3390/biom6020016
APA StyleCooper, A. J. L., & Jeitner, T. M. (2016). Central Role of Glutamate Metabolism in the Maintenance of Nitrogen Homeostasis in Normal and Hyperammonemic Brain. Biomolecules, 6(2), 16. https://doi.org/10.3390/biom6020016