
DNA Damage Signalling and Repair Inhibitors: The Long-Sought-After Achilles’ Heel of Cancer

 and
and
Abstract
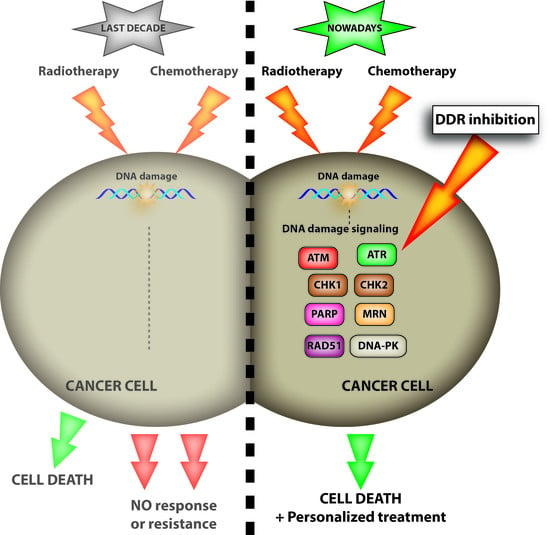
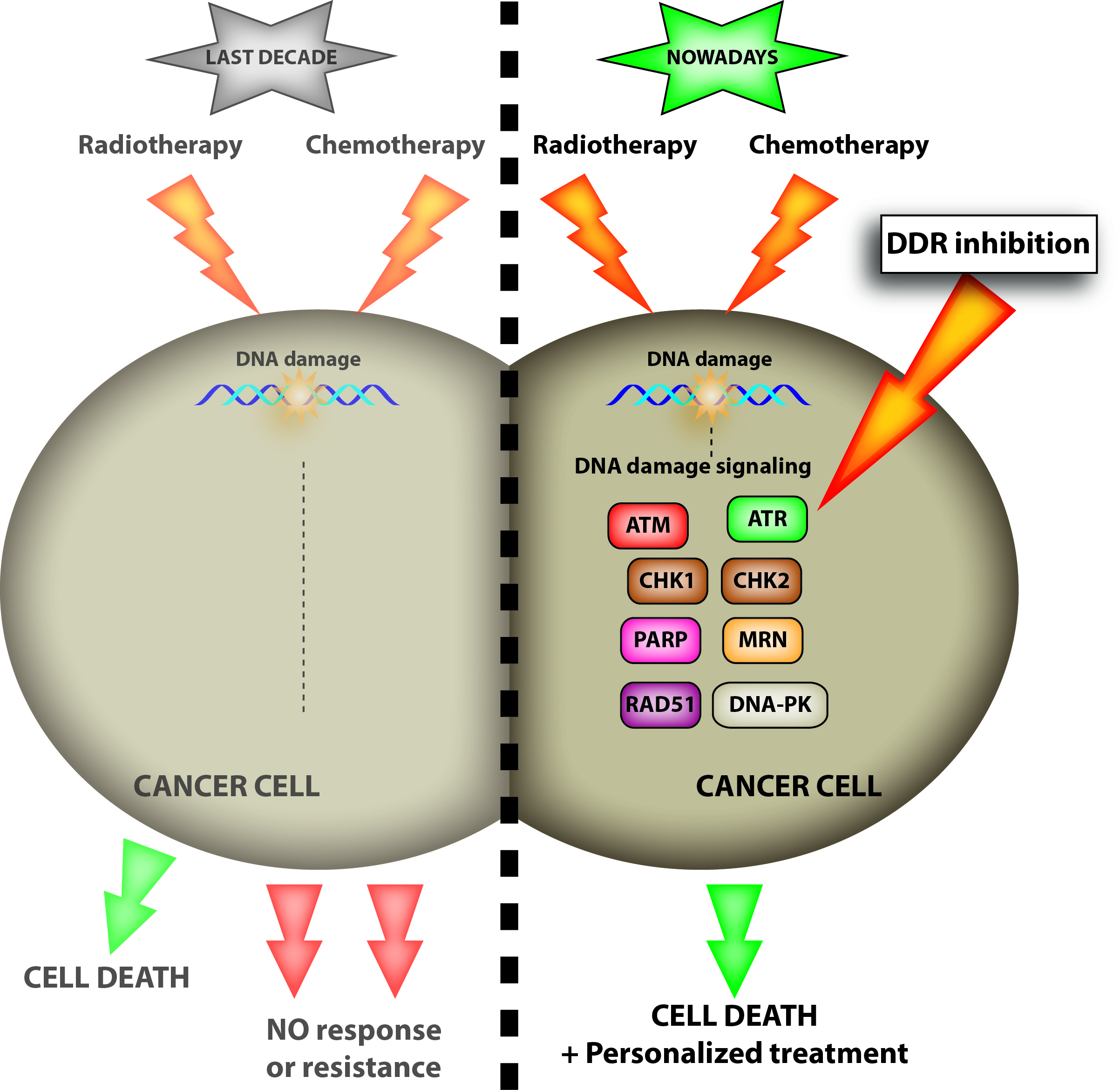{kind=link}
{kind=link}
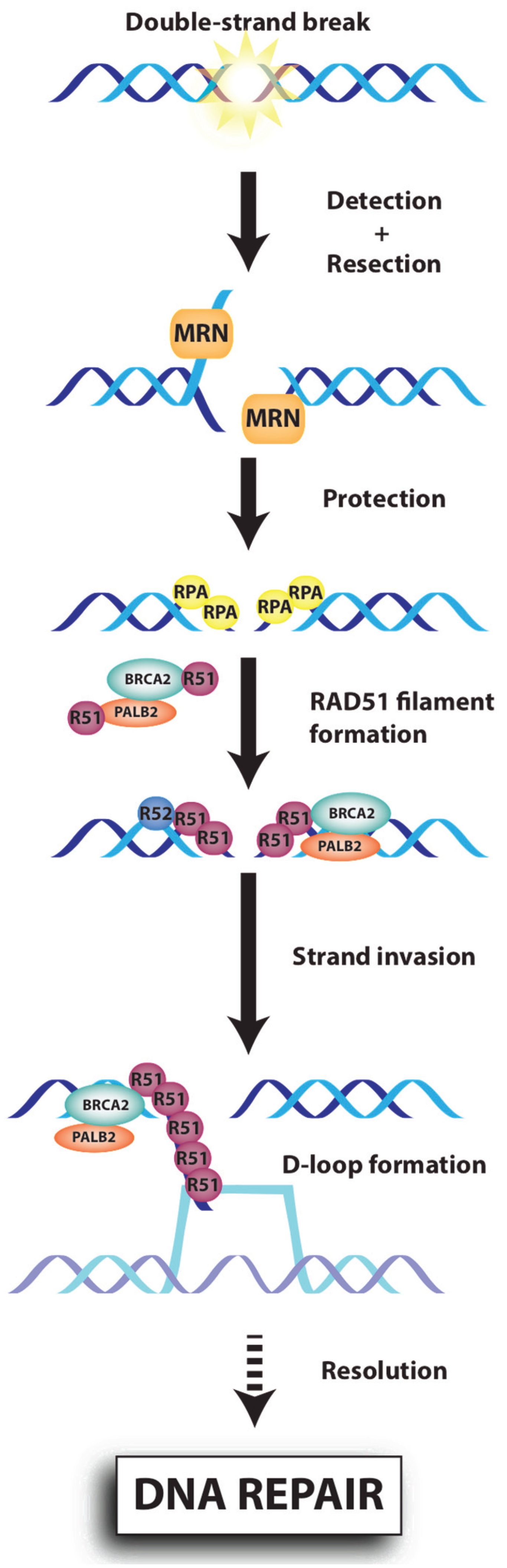{kind=link}
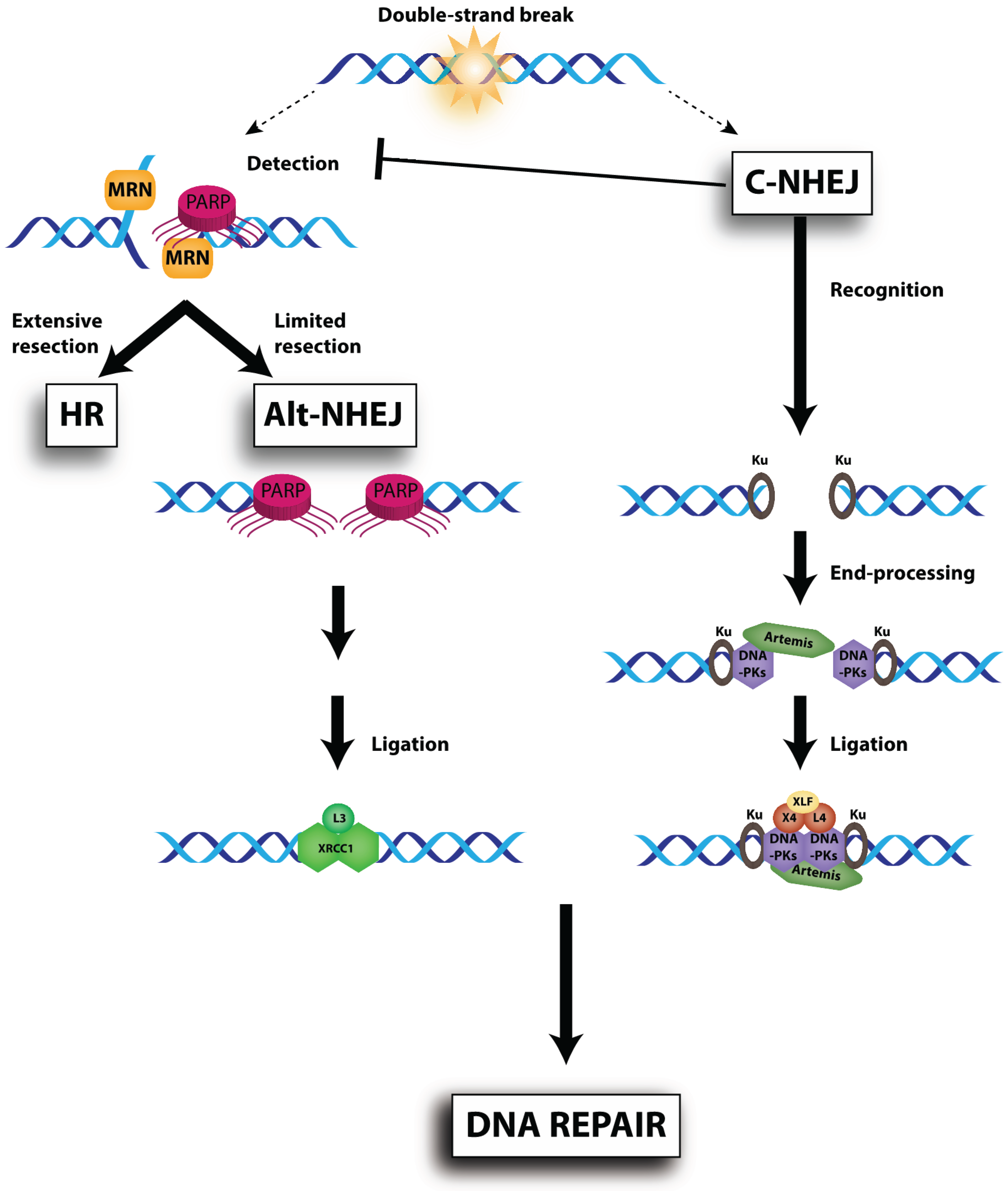{kind=link}
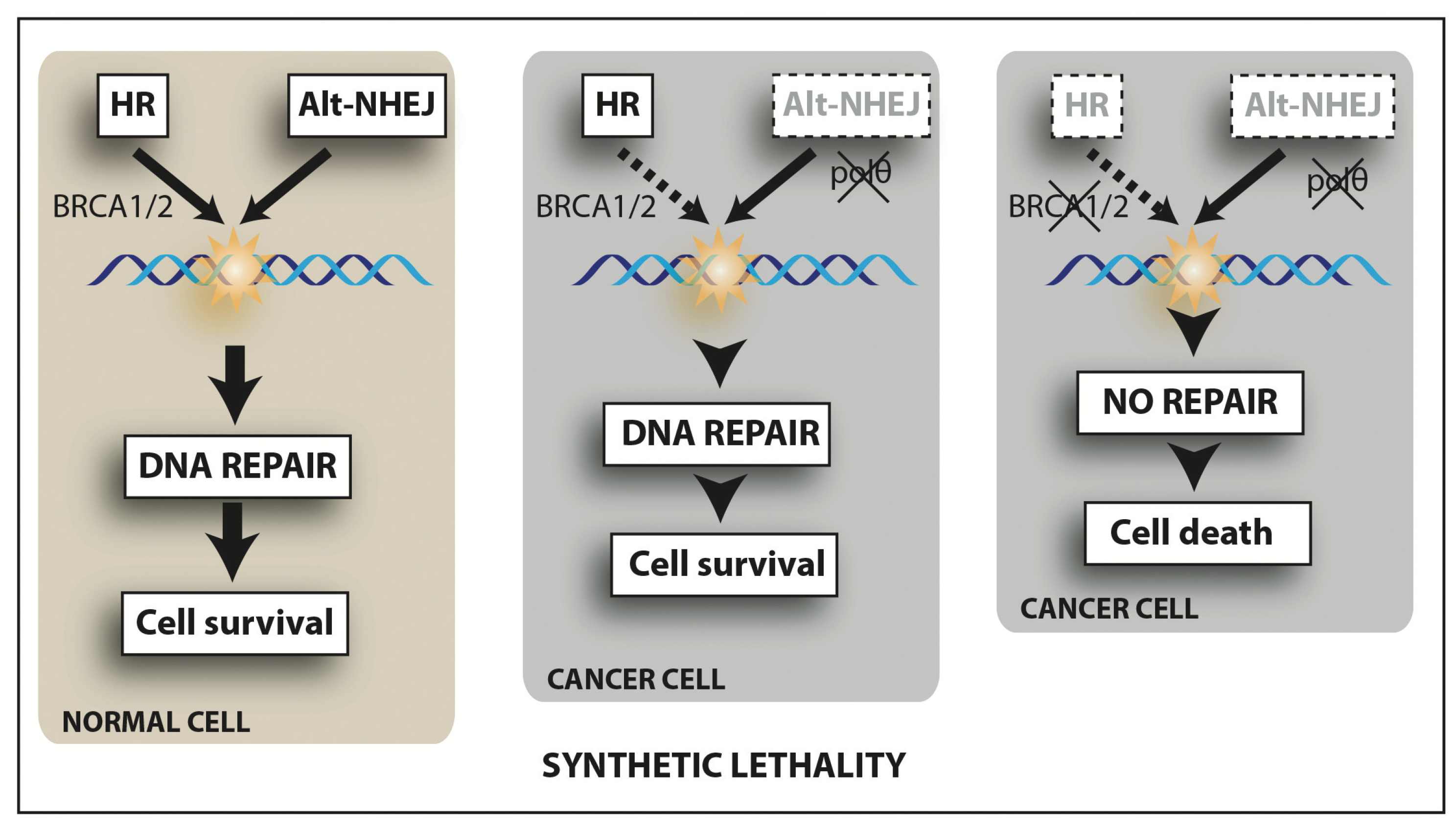{kind=link}
1. Introduction
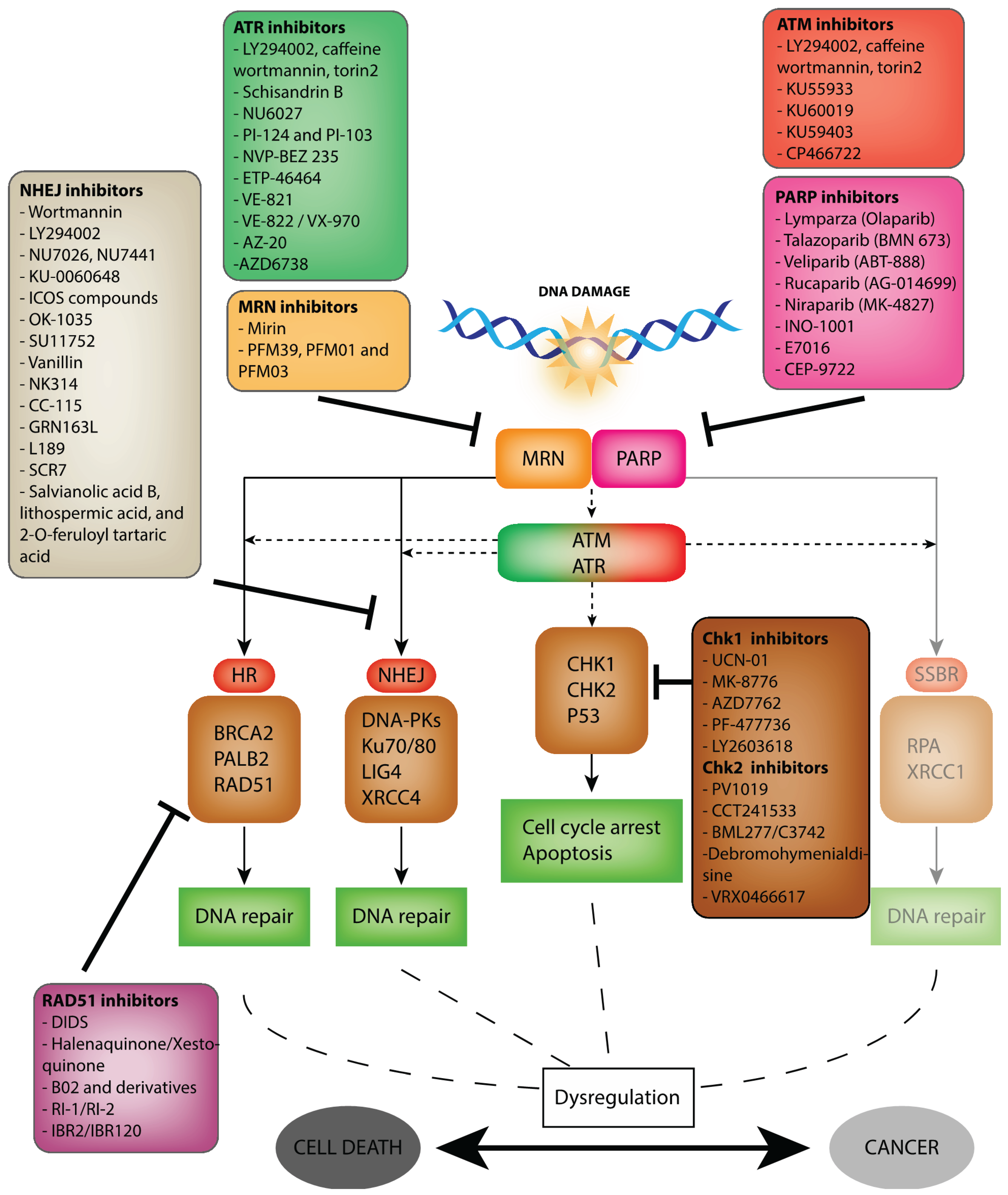
2. The ATM and ATR Kinases
2.1. Dual ATM/ATR Inhibitors
2.1.1. Caffeine, Wortmannin and LY294002
2.1.2. Torin2
2.2. Selective ATM Inhibitors
2.2.1. KU-55933
2.2.2. KU-60019
2.2.3. KU-59403
2.2.4. CP466722
2.3. Selective ATR Inhibitors
2.3.1. Schisandrin B
2.3.2. NU6027
2.3.3. PI-124 and PI-103
2.3.4. NVP-BEZ235
2.3.5. ETP-46464
2.3.6. VE-821
2.3.7. VE-822/VX-970
2.3.8. AZ-20
2.3.9. AZD6738
3. The Chk1 Kinase
3.1. Roles of Chk1 in the DNA Damage Response
3.2. Phosphorylation of Chk1
3.3. Targeting Chk1 as a Novel Strategy in Cancer Therapy
3.4. Inhibitors of Chk1
3.4.1. UCN-01 (7-Hydroxystaurosporine)
3.4.2. MK-8776 Inhibitor (Previously Known as SCH900776) (Pyrazolo [1,5-a] Pyrimidine Derivative)
3.4.3. AZD7762
3.4.4. PF-477736
3.4.5. LY2603618
3.4.6. Other Inhibitors
4. The Chk2 Kinase
4.1. PV1019 ([7-Nitro-1H-Indole-2-Carboxylic Acid {(4-[1-(Guanidinohydrazone)-Ethyl]-Phenyl}-Amide]
4.2. CCT241533
4.3. 2-(4-(4-Chlorophenoxy)Phenyl)-1H-Benzimidazole-5-Carboxamide Hydrate/BML277/C3742
4.4. Debromohymenialdisine and Analogues
4.5. VRX0466617
5. The Poly(ADP) Ribose Polymerase
5.1. Talazoparib (BMN 673; BioMarin Pharmaceutical Inc., Novato, CA, USA)
5.2. Veliparib (ABT-888; AbbVie Inc., North Chicago, IL, USA)
5.3. Rucaparib (AG-014699; Clovis Oncology, Boulder, CO, USA)
5.4. Niraparib (MK-4827; Tesaro Inc., Waltham, MA, USA)
5.5. INO-1001 (Selleck Chemicals, Euromedex, France)
5.6. E7016 (Eisai Co., Ltd; Tokyo, Japan)
5.7. CEP-9722 (Cephalon, Inc.; Frazer, PA, USA)
5.8. PARP Trapping: Mechanism of Action of PARP Inhibitors
6. The MRE11-RAD50-NBS1 Complex
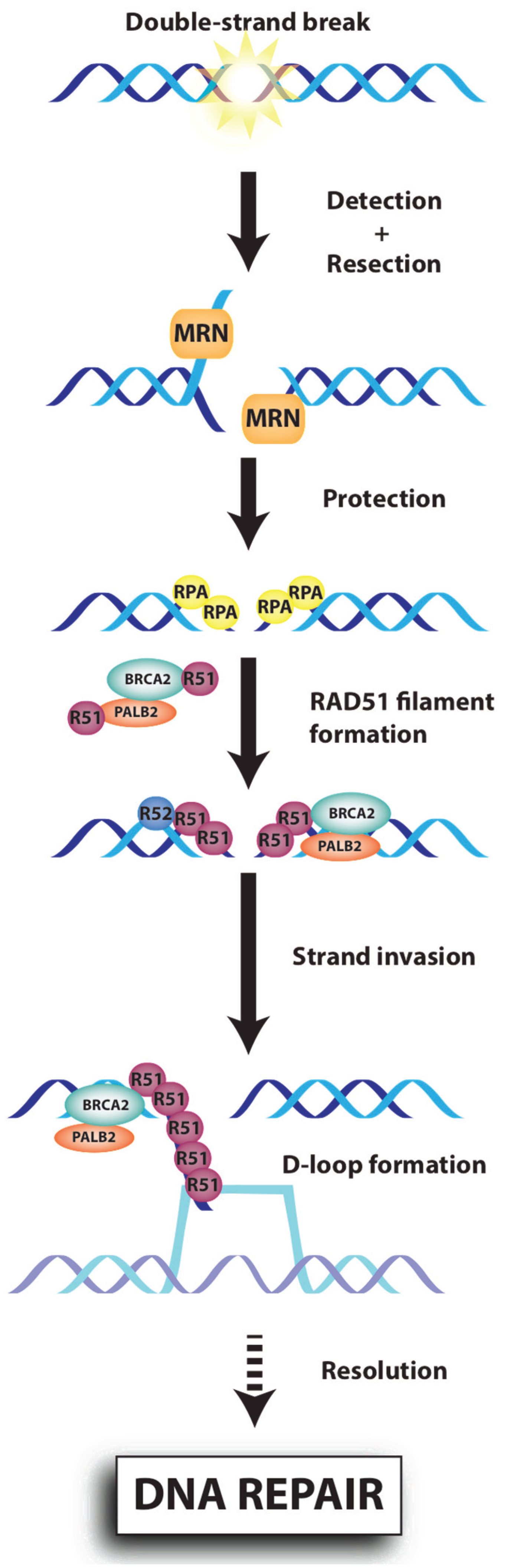
6.1. Mirin
6.2. PFM39, PFM01 and PFM03
7. The RAD51 Protein, a Central HR Protein
7.1. DIDS
7.2. Halenaquinone/Xestoquinone
7.3. B02 and Derivatives
7.4. RI-1 and RI-2
7.5. IBR2 and IBR120
8. The RAD52 Protein
9. The Non-Homologous End-Joining (NHEJ) Proteins
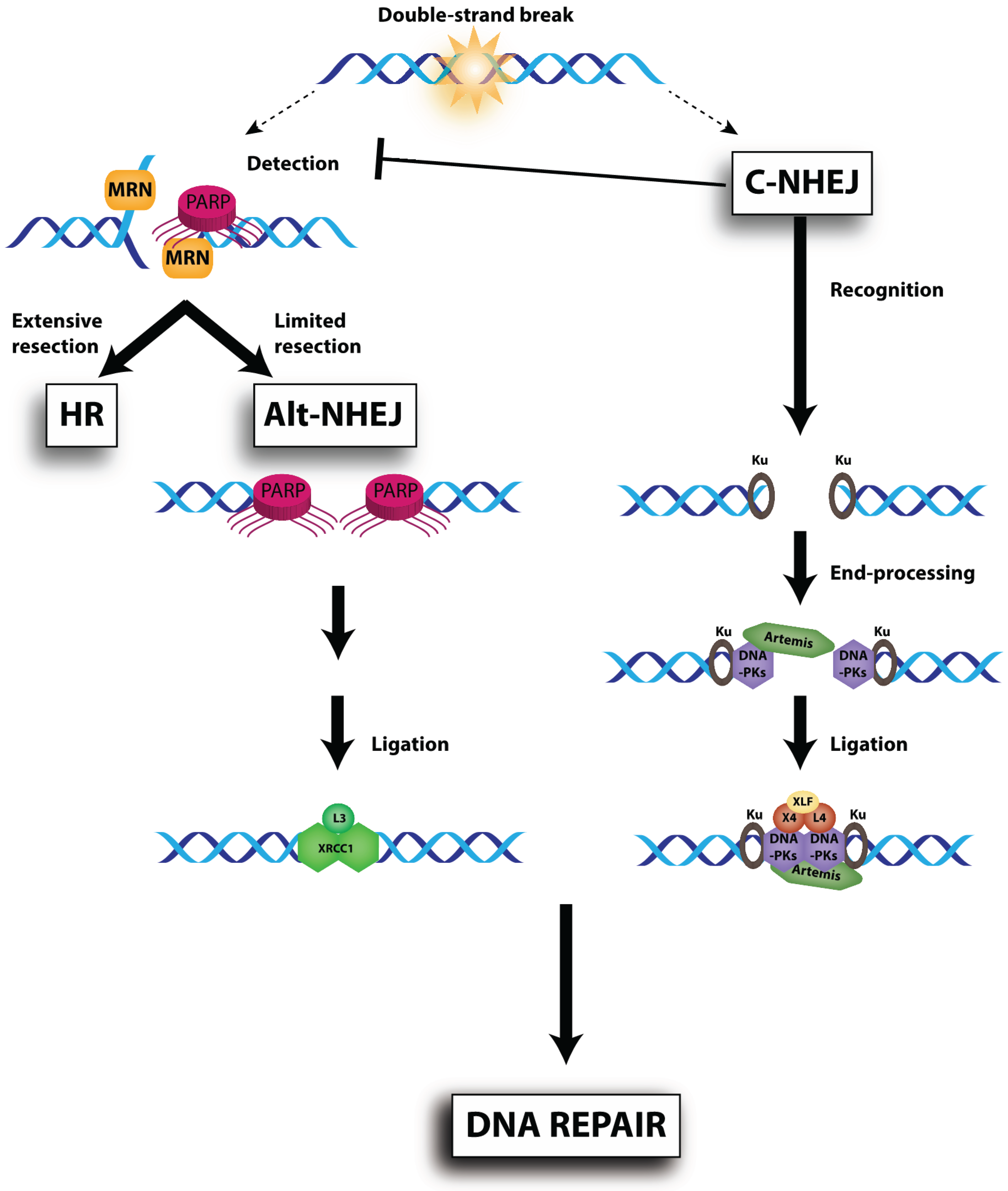
9.1. Selective DNA-PK Inhibitors
9.1.1. Wortmannin
9.1.2. LY294002
9.1.3. NU7026
9.1.4. NU7441
9.1.5. KU-0060648
9.1.6. ICOS Compounds
9.1.7. OK-1035
9.1.8. SU11752
9.1.9. Vanillin
9.1.10. NK314
9.1.11. CC-115
9.2. Nucleotide-Based Inhibitors of DNA-PK
GRN163L
9.3. Ku70/Ku80 Inhibitors
9.4. DNA Ligase IV Inhibitors
9.4.1. L189
9.4.2. SCR7
9.5. XRCC4 Inhibitors
Salvianolic Acid B, Lithospermic Acid, and 2-O-Feruloyl Tartaric Acid
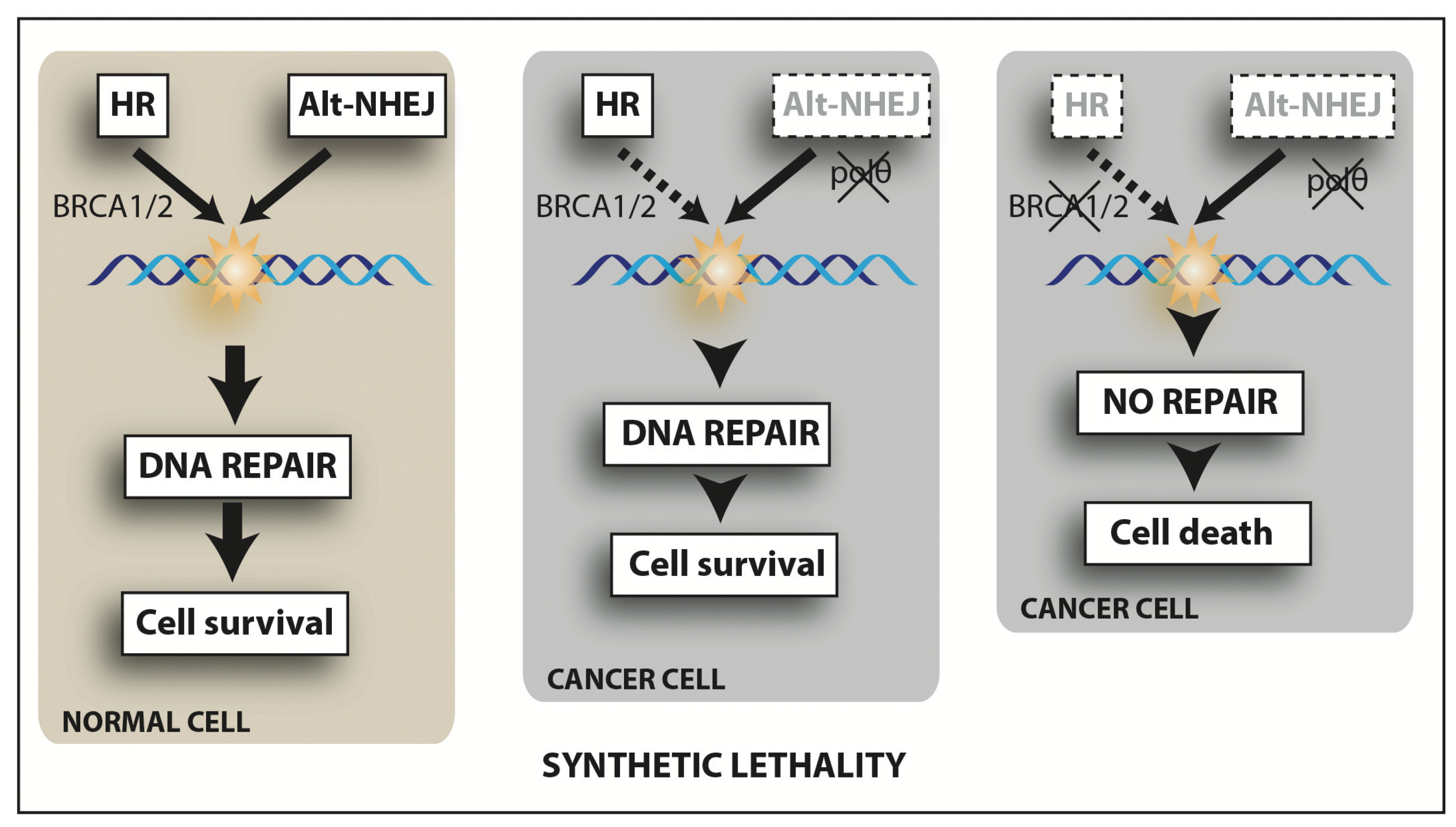
9.6. Alt-NHEJ Inhibitors
10. Conclusions
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ciccia, A.; Elledge, S.J. The DNA damage response: Making it safe to play with knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [PubMed]
- Savitsky, K.; Sfez, S.; Tagle, D.A.; Ziv, Y.; Sartiel, A.; Collins, F.S.; Shiloh, Y.; Rotman, G. The complete sequence of the coding region of the ATM gene reveals similarity to cell cycle regulators in different species. Hum. Mol. Genet. 1995, 4, 2025–2032. [Google Scholar] [CrossRef] [PubMed]
- Shiloh, Y. Ataxia telangiectasia and the nijmegen breakage syndrome: Related disorders but genes apart. Annu. Rev. Genet. 1997, 31, 635–662. [Google Scholar] [CrossRef] [PubMed]
- Bentley, N.J.; Holtzman, D.A.; Flaggs, G.; Keegan, K.S.; DeMaggio, A.; Ford, J.C.; Hoekstra, M.; Carr, A.M. The schizosaccharomyces pombe Rad3 checkpoint gene. EMBO J. 1996, 15, 6641–6651. [Google Scholar] [PubMed]
- Cortez, D.; Guntuku, S.; Qin, J.; Elledge, S.J. ATR and ATRIP: Partners in checkpoint signaling. Science 2001, 294, 1713–1716. [Google Scholar] [CrossRef] [PubMed]
- San Filippo, J.; Sung, P.; Klein, H. Mechanism of eukaryotic homologous recombination. Ann. Rev. Biochem. 2008, 77, 229–257. [Google Scholar] [CrossRef] [PubMed]
- de Klein, A.; Muijtjens, M.; van Os, R.; Verhoeven, Y.; Smit, B.; Carr, A.M.; Lehmann, A.R.; Hoeijmakers, J.H. Targeted disruption of the cell-cycle checkpoint gene ATR leads to early embryonic lethality in mice. Curr. Biol. 2000, 10, 479–482. [Google Scholar] [CrossRef]
- Brown, E.J.; Baltimore, D. ATR disruption leads to chromosomal fragmentation and early embryonic lethality. Genes Dev. 2000, 14, 397–402. [Google Scholar] [PubMed]
- O’Driscoll, M.; Ruiz-Perez, V.L.; Woods, C.G.; Jeggo, P.A.; Goodship, J.A. A splicing mutation affecting expression of ataxia-telangiectasia and Rad3-related protein (ATR) results in seckel syndrome. Nat. Genet. 2003, 33, 497–501. [Google Scholar] [CrossRef] [PubMed]
- Marechal, A.; Zou, L. DNA damage sensing by the ATM and ATR kinases. Cold Spring Harb. Symp. Quant. Biol. 2013. [Google Scholar] [CrossRef] [PubMed]
- Cortez, D. Unwind and slow down: Checkpoint activation by helicase and polymerase uncoupling. Genes Dev. 2005, 19, 1007–1012. [Google Scholar] [CrossRef] [PubMed]
- Cimprich, K.A.; Cortez, D. Atr: An essential regulator of genome integrity. Nat. Rev. Mol. Cell. Biol. 2008, 9, 616–627. [Google Scholar] [CrossRef] [PubMed]
- Sarkaria, J.N.; Busby, E.C.; Tibbetts, R.S.; Roos, P.; Taya, Y.; Karnitz, L.M.; Abraham, R.T. Inhibition of ATM and ATR kinase activities by the radiosensitizing agent, caffeine. Cancer Res. 1999, 59, 4375–4382. [Google Scholar] [PubMed]
- Cliby, W.A.; Roberts, C.J.; Cimprich, K.A.; Stringer, C.M.; Lamb, J.R.; Schreiber, S.L.; Friend, S.H. Overexpression of a kinase-inactive ATR protein causes sensitivity to DNA-damaging agents and defects in cell cycle checkpoints. EMBO J. 1998, 17, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, U.K.; Senderowicz, A.M.; Ferbeyre, G. Rna silencing of checkpoint regulators sensitizes p53-defective prostate cancer cells to chemotherapy while sparing normal cells. Cancer Res. 2005, 65, 2872–2881. [Google Scholar] [CrossRef] [PubMed]
- Cliby, W.A.; Lewis, K.A.; Lilly, K.K.; Kaufmann, S.H. S phase and G2 arrests induced by topoisomerase I poisons are dependent on ATR kinase function. J. Biol. Chem. 2002, 277, 1599–1606. [Google Scholar] [CrossRef] [PubMed]
- Won, J.; Kim, M.; Kim, N.; Ahn, J.H.; Lee, W.G.; Kim, S.S.; Chang, K.Y.; Yi, Y.W.; Kim, T.K. Retraction: Small molecule-based reversible reprogramming of cellular lifespan. Nat. Chem. Biol. 2008, 4, 431. [Google Scholar] [CrossRef] [PubMed]
- Abraham, R.T. Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes Dev. 2001, 15, 2177–2196. [Google Scholar] [CrossRef] [PubMed]
- Price, B.D.; Youmell, M.B. The phosphatidylinositol 3-kinase inhibitor wortmannin sensitizes murine fibroblasts and human tumor cells to radiation and blocks induction of p53 following DNA damage. Cancer Res. 1996, 56, 246–250. [Google Scholar] [PubMed]
- Sarkaria, J.N.; Tibbetts, R.S.; Busby, E.C.; Kennedy, A.P.; Hill, D.E.; Abraham, R.T. Inhibition of phosphoinositide 3-kinase related kinases by the radiosensitizing agent wortmannin. Cancer Res. 1998, 58, 4375–4382. [Google Scholar] [PubMed]
- Blasina, A.; Price, B.D.; Turenne, G.A.; McGowan, C.H. Caffeine inhibits the checkpoint kinase atm. Curr. Biol. 1999, 9, 1135–1138. [Google Scholar] [CrossRef]
- Tsabar, M.; Eapen, V.V.; Mason, J.M.; Memisoglu, G.; Waterman, D.P.; Long, M.J.; Bishop, D.K.; Haber, J.E. Caffeine impairs resection during DNA break repair by reducing the levels of nucleases Sae2 and Dna2. Nucleic Acids Res. 2015. [Google Scholar] [CrossRef] [PubMed]
- Newton, R.; Broughton, L.J.; Lind, M.J.; Morrison, P.J.; Rogers, H.J.; Bradbrook, I.D. Plasma and salivary pharmacokinetics of caffeine in man. Eur. J. Clin. Pharmacol. 1981, 21, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Karve, S.; Werner, M.E.; Sukumar, R.; Cummings, N.D.; Copp, J.A.; Wang, E.C.; Li, C.; Sethi, M.; Chen, R.C.; Pacold, M.E.; et al. Revival of the abandoned therapeutic wortmannin by nanoparticle drug delivery. Proc. Natl. Acad. Sci. USA 2012, 109, 8230–8235. [Google Scholar] [CrossRef] [PubMed]
- Vlahos, C.J.; Matter, W.F.; Hui, K.Y.; Brown, R.F. A specific inhibitor of phosphatidylinositol 3-kinase, 2-(4-morpholinyl)-8-phenyl-4h-1-benzopyran-4-one (ly294002). J. Biol. Chem. 1994, 269, 5241–5248. [Google Scholar] [PubMed]
- Hall-Jackson, C.A.; Cross, D.A.; Morrice, N.; Smythe, C. Atr is a caffeine-sensitive, DNA-activated protein kinase with a substrate specificity distinct from DNA-PK. Oncogene 1999, 18, 6707–6713. [Google Scholar] [PubMed]
- Hammond, E.M.; Denko, N.C.; Dorie, M.J.; Abraham, R.T.; Giaccia, A.J. Hypoxia links ATR and p53 through replication arrest. Mol. Cell. Biol. 2002, 22, 1834–1843. [Google Scholar] [CrossRef] [PubMed]
- Goodarzi, A.A.; Lees-Miller, S.P. Biochemical characterization of the ataxia-telangiectasia mutated (atm) protein from human cells. DNA repair 2004, 3, 753–767. [Google Scholar] [CrossRef] [PubMed]
- Stiff, T.; O’Driscoll, M.; Rief, N.; Iwabuchi, K.; Löbrich, M.; Jeggo, P.A. Atm and DNA-PK function redundantly to phosphorylate H2AX after exposure to ionizing radiation. Cancer Res. 2004, 64, 2390–2396. [Google Scholar] [CrossRef] [PubMed]
- Cleary, J.M.; Shapiro, G.I. Development of phosphoinositide-3 kinase pathway inhibitors for advanced cancer. Curr. Oncol. Rep. 2010, 12, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Gharbi, S.I.; Zvelebil, M.J.; Shuttleworth, S.J.; Hancox, T.; Saghir, N.; Timms, J.F.; Waterfield, M.D. Exploring the specificity of the PI3K family inhibitor LY294002. Biochem. J. 2007, 404, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Wang, J.; Kang, S.A.; Thoreen, C.C.; Hur, W.; Ahmed, T.; Sabatini, D.M.; Gray, N.S. Discovery of 9-(6-aminopyridin-3-yl)-1-(3-(trifluoromethyl)phenyl)benzo[h][1,6]naphthyridin-2(1h)-one (torin2) as a potent, selective and orally available mtor inhibitor for treatment of cancer. J. Med. Chem. 2011, 54, 1473–1480. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Xu, C.; Kirubakaran, S.; Zhang, X.; Hur, W.; Liu, Y.; Kwiatkowski, N.P.; Wang, J.; Westover, K.D.; Gao, P.; et al. Characterization of Torin2, an ATP-competitive inhibitor of mTOR, ATM and ATR. Cancer Res. 2013, 73, 2574–2586. [Google Scholar] [CrossRef] [PubMed]
- Hickson, I.; Zhao, Y.; Richardson, C.J.; Green, S.J.; Martin, N.M.B.; Orr, A.I.; Reaper, P.M.; Jackson, S.P.; Curtin, N.J.; Smith, G.C.M. Identification and characterization of a novel and specific inhibitor of the ataxia-telangiectasia mutated kinase ATM. Cancer Res. 2004, 64, 9152–9159. [Google Scholar] [CrossRef] [PubMed]
- Weber, A.M.; Ryan, A.J. ATM and ATR as therapeutic targets in cancer. Pharmacol. Ther. 2015, 149, 124–138. [Google Scholar] [CrossRef] [PubMed]
- Golding, S.E.; Rosenberg, E.; Valerie, N.; Hussaini, I.; Frigerio, M.; Cockcroft, X.F.; Chong, W.Y.; Hummersone, M.; Rigoreau, L.; Menear, K.A.; et al. Improved ATM kinase inhibitor KU-60019 radiosensitizes glioma cells, compromises insulin, AKT and ERK prosurvival signaling, and inhibits migration and invasion. Mol. Cancer Ther. 2009, 8, 2894–2902. [Google Scholar] [CrossRef] [PubMed]
- Crescenzi, E.; Palumbo, G.; de Boer, J.; Brady, H.J.M. Ataxia telangiectasia mutated and P21CIP1 modulate cell survival of drug-induced senescent tumor cells: Implications for chemotherapy. Clin. Cancer Res. 2008, 14, 1877–1887. [Google Scholar] [CrossRef] [PubMed]
- Eaton, J.S.; Lin, Z.P.; Sartorelli, A.C.; Bonawitz, N.D.; Shadel, G.S. Ataxia-telangiectasia mutated kinase regulates ribonucleotide reductase and mitochondrial homeostasis. J. Clin. Invest. 2007, 117, 2723–2734. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Shen, Y.; Chen, Y.; Hsieh, J.T.; Kong, Z. The atm inhibitor KU55933 sensitizes radioresistant bladder cancer cells with dab2ip gene defect. Int. J. Radiat. Biol. 2015, 91, 368–378. [Google Scholar] [CrossRef] [PubMed]
- Toulany, M.; Mihatsch, J.; Holler, M.; Chaachouay, H.; Rodemann, H.P. Cisplatin-mediated radiosensitization of non-small cell lung cancer cells is stimulated by ATM inhibition. Radiother. Oncol. 2014, 111, 228–236. [Google Scholar] [CrossRef] [PubMed]
- Vecchio, D.; Daga, A.; Carra, E.; Marubbi, D.; Raso, A.; Mascelli, S.; Nozza, P.; Garrè, M.L.; Pitto, F.; Ravetti, J.L.; et al. Pharmacokinetics, pharmacodynamics and efficacy on pediatric tumors of the glioma radiosensitizer ku60019. Int. J. Cancer 2015, 136, 1445–1457. [Google Scholar] [CrossRef] [PubMed]
- Biddlestone-Thorpe, L.; Sajjad, M.; Rosenberg, E.; Beckta, J.M.; Valerie, N.C.K.; Tokarz, M.; Adams, B.R.; Wagner, A.F.; Khalil, A.; Gilfor, D.; et al. ATM kinase inhibition preferentially sensitizes p53-mutant glioma to ionizing radiation. Clin. Cancer Res. 2013, 19, 3189–3200. [Google Scholar] [CrossRef] [PubMed]
- McCabe, N.; Hanna, C.; Walker, S.M.; Gonda, D.; Li, J.; Wikstrom, K.; Savage, K.I.; Butterworth, K.T.; Chen, C.; Harkin, D.P.; et al. Mechanistic rationale to target PTEN-deficient tumor cells with inhibitors of the DNA damage response kinase ATM. Cancer Res. 2015. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Mao, C.; Wu, J.; Li, S.; Ma, R.; Cao, H.; Ji, M.; Jing, C.; Tang, J. Improved ataxia telangiectasia mutated kinase inhibitor KU60019 provides a promising treatment strategy for non-invasive breast cancer. Oncology Lett. 2014, 8, 2043–2048. [Google Scholar] [CrossRef] [PubMed]
- Batey, M.A.; Zhao, Y.; Kyle, S.; Richardson, C.; Slade, A.; Martin, N.M.; Lau, A.; Newell, D.R.; Curtin, N.J. Preclinical evaluation of a novel ATM inhibitor, KU59403, in vitro and in vivo in p53 functional and dysfunctional models of human cancer. Mol. Cancer Ther. 2013, 12, 959–967. [Google Scholar] [CrossRef] [PubMed]
- Rainey, M.D.; Charlton, M.E.; Stanton, R.V.; Kastan, M.B. Transient inhibition of ATM kinase is sufficient to enhance cellular sensitivity to ionizing radiation. Cancer Res. 2008, 68, 7466–7474. [Google Scholar] [CrossRef] [PubMed]
- Węsierska-Gądek, J.; Heinzl, S. Interactions between ataxia telangiectasia mutated kinase inhibition, poly(adp-ribose) polymerase-1 inhibition and brca1 status in breast cancer cells. J. Cancer Prev. 2014, 19, 125–136. [Google Scholar] [CrossRef] [PubMed]
- Nishida, H.; Tatewaki, N.; Nakajima, Y.; Magara, T.; Ko, K.M.; Hamamori, Y.; Konishi, T. Inhibition of ATR protein kinase activity by schisandrin b in DNA damage response. Nucleic Acids Res. 2009, 37, 5678–5689. [Google Scholar] [CrossRef] [PubMed]
- Tatewaki, N.; Nishida, H.; Yoshida, M.; Ando, H.; Kondo, S.; Sakamaki, T.; Konishi, T. Differential effect of schisandrin b stereoisomers on ATR-mediated DNA damage checkpoint signaling. J. Pharmacol. Sci. 2013, 122, 138–148. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Lu, Q.; Shen, Y.; Hu, X. Schisandrin b enhances doxorubicin-induced apoptosis of cancer cells but not normal cells. Biochem. Pharmacol. 2006, 71, 584–595. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Liu, Z.; Sun, J.; Pan, Q.; Sun, F.; Yan, Z.; Hu, X. Schisandrin B prevents doxorubicin-induced chronic cardiotoxicity and enhances its anticancer activity in vivo. PLoS ONE 2011, 6, e28335. [Google Scholar] [CrossRef] [PubMed]
- Hou, W.; Gao, W.; Wang, D.; Liu, Q.; Zheng, S.; Wang, Y. The protecting effect of deoxyschisandrin and schisandrin b on HaCat cells against UVB-induced damage. PLoS ONE 2015, 10, e0127177. [Google Scholar] [CrossRef] [PubMed]
- Peasland, A.; Wang, L.Z.; Rowling, E.; Kyle, S.; Chen, T.; Hopkins, A.; Cliby, W.A.; Sarkaria, J.; Beale, G.; Edmondson, R.J.; et al. Identification and evaluation of a potent novel ATR inhibitor, NU6027, in breast and ovarian cancer cell lines. Br. J. Cancer 2011, 105, 372–381. [Google Scholar] [CrossRef] [PubMed]
- Sultana, R.; Abdel-Fatah, T.; Perry, C.; Moseley, P.; Albarakti, N.; Mohan, V.; Seedhouse, C.; Chan, S.; Madhusudan, S. Ataxia telangiectasia mutated and Rad3 related (ATR) protein kinase inhibition is synthetically lethal in XRCC1 deficient ovarian cancer cells. PLoS ONE 2013, 8, e57098. [Google Scholar] [CrossRef] [PubMed]
- Knight, Z.A.; Gonzalez, B.; Feldman, M.E.; Zunder, E.R.; Goldenberg, D.D.; Williams, O.; Loewith, R.; Stokoe, D.; Balla, A.; Toth, B.; et al. A pharmacological map of the PI3-K family defines a role for p110α in insulin signaling. Cell 2006, 125, 733–747. [Google Scholar] [CrossRef] [PubMed]
- Jang, N.Y.; Kim, D.H.; Cho, B.J.; Choi, E.J.; Lee, J.S.; Wu, H.G.; Chie, E.K.; Kim, I.A. Radiosensitization with combined use of olaparib and PI-103 in triple-negative breast cancer. BMC Cancer 2015, 15, 89. [Google Scholar] [CrossRef] [PubMed]
- Toledo, L.I.; Murga, M.; Zur, R.; Soria, R.; Rodriguez, A.; Martinez, S.; Oyarzabal, J.; Pastor, J.; Bischoff, J.R.; Fernandez-Capetillo, O. A cell-based screen identifies Atr inhibitors with synthetic lethal properties for cancer-associated mutations. Nat. Struct. Mol. Biol. 2011, 18, 721–727. [Google Scholar] [CrossRef] [PubMed]
- Maira, S.M.; Stauffer, F.; Brueggen, J.; Furet, P.; Schnell, C.; Fritsch, C.; Brachmann, S.; Chene, P.; de Pover, A.; Schoemaker, K.; et al. Identification and characterization of NVP-BEZ235, a new orally available dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor with potent in vivo antitumor activity. Mol. Cancer Ther. 2008, 7, 1851–1863. [Google Scholar] [CrossRef] [PubMed]
- Matsushima, M.; Kikuchi, E.; Matsumoto, K.; Hattori, S.; Takeda, T.; Kosaka, T.; Miyajima, A.; Oya, M. Intravesical dual PI3K/mTOR complex 1/2 inhibitor NVP-BEZ235 therapy in an orthotopic bladder cancer model. Int. J. Oncol. 2015, 47, 377–383. [Google Scholar] [CrossRef] [PubMed]
- Echeverry, N.; Ziltener, G.; Barbone, D.; Weder, W.; Stahel, R.A.; Broaddus, V.C.; Felley-Bosco, E. Inhibition of autophagy sensitizes malignant pleural mesothelioma cells to dual PI3K/mTOR inhibitors. Cell Death Dis. 2015. [Google Scholar] [CrossRef] [PubMed]
- Schrauwen, S.; Depreeuw, J.; Coenegrachts, L.; Hermans, E.; Lambrechts, D.; Amant, F. Dual blockade of PI3K/AKT/mTOR (NVP-BEZ235) and Ras/Raf/MEK (AZD6244) pathways synergistically inhibit growth of primary endometrioid endometrial carcinoma cultures, whereas NVP-BEZ235 reduces tumor growth in the corresponding xenograft models. Gynecol. Oncol. 2015, 138, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Konstantinidou, G.; Bey, E.A.; Rabellino, A.; Schuster, K.; Maira, M.S.; Gazdar, A.F.; Amici, A.; Boothman, D.A.; Scaglioni, P.P. Dual phosphoinositide 3-kinase/mammalian target of rapamycin blockade is an effective radiosensitizing strategy for the treatment of non-small cell lung cancer harboring K-RAS mutations. Cancer Res. 2009, 69, 7644–7652. [Google Scholar] [CrossRef] [PubMed]
- Charrier, J.D.; Durrant, S.J.; Golec, J.M.; Kay, D.P.; Knegtel, R.M.; MacCormick, S.; Mortimore, M.; O’Donnell, M.E.; Pinder, J.L.; Reaper, P.M.; et al. Discovery of potent and selective inhibitors of ataxia telangiectasia mutated and Rad3 related (ATR) protein kinase as potential anticancer agents. J. Med. Chem. 2011, 54, 2320–2330. [Google Scholar] [CrossRef] [PubMed]
- Reaper, P.M.; Griffiths, M.R.; Long, J.M.; Charrier, J.-D.; MacCormick, S.; Charlton, P.A.; Golec, J.M.C.; Pollard, J.R. Selective killing of ATM- or p53-deficient cancer cells through inhibition of ATR. Nat. Chem. Biol. 2011, 7, 428–430. [Google Scholar] [CrossRef] [PubMed]
- Pires, I.M.; Olcina, M.M.; Anbalagan, S.; Pollard, J.R.; Reaper, P.M.; Charlton, P.A.; McKenna, W.G.; Hammond, E.M. Targeting radiation-resistant hypoxic tumour cells through ATR inhibition. Br. J. Cancer 2012, 107, 291–299. [Google Scholar] [CrossRef] [PubMed]
- Prevo, R.; Fokas, E.; Reaper, P.M.; Charlton, P.A.; Pollard, J.R.; McKenna, W.G.; Muschel, R.J.; Brunner, T.B. The novel ATR inhibitor VE-821 increases sensitivity of pancreatic cancer cells to radiation and chemotherapy. Cancer Biol. Ther. 2012, 13, 1072–1081. [Google Scholar] [CrossRef] [PubMed]
- Huntoon, C.J.; Flatten, K.S.; Wahner Hendrickson, A.E.; Huehls, A.M.; Sutor, S.L.; Kaufmann, S.H.; Karnitz, L.M. ATR inhibition broadly sensitizes ovarian cancer cells to chemotherapy independent of BRCA status. Cancer Res. 2013, 73, 3683–3691. [Google Scholar] [CrossRef] [PubMed]
- Fokas, E.; Prevo, R.; Pollard, J.R.; Reaper, P.M.; Charlton, P.A.; Cornelissen, B.; Vallis, K.A.; Hammond, E.M.; Olcina, M.M.; Gillies McKenna, W.; et al. Targeting ATR in vivo using the novel inhibitor VE-822 results in selective sensitization of pancreatic tumors to radiation. Cell Death Dis. 2012. [Google Scholar] [CrossRef] [PubMed]
- Hall, A.B.; Newsome, D.; Wang, Y.; Boucher, D.M.; Eustace, B.; Gu, Y.; Hare, B.; Johnson, M.A.; Li, H.; Milton, S.; et al. Potentiation of tumor responses to DNA damaging therapy by the selective ATR inhibitor VX-970. Oncotarget 2014, 5, 5674–5685. [Google Scholar] [CrossRef] [PubMed]
- Nakahara, T.; Tanaka, K.; Ohno, S.I.; Egawa, N.; Yugawa, T.; Kiyono, T. Activation of NF-κB by human papillomavirus 16 E1 limits E1-dependent viral replication through degradation of E1. J. Vir. 2015, 89, 5040–5059. [Google Scholar] [CrossRef] [PubMed]
- Foote, K.M.; Blades, K.; Cronin, A.; Fillery, S.; Guichard, S.S.; Hassall, L.; Hickson, I.; Jacq, X.; Jewsbury, P.J.; McGuire, T.M.; et al. Discovery of 4-{4-[(3r)-3-methylmorpholin-4-yl]-6-[1-(methylsulfonyl)cyclopropyl]pyrimidin-2-yl}-1h-indole (az20): A potent and selective inhibitor of ATR protein kinase with monotherapy in vivo antitumor activity. J. Med. Chem. 2013, 56, 2125–2138. [Google Scholar] [CrossRef] [PubMed]
- Clack, G.; Lau, A.; Pierce, A.; Smith, S.; Stephens, C. O6.3 ATR inhibitor AZD6738. Ann. Oncol. 2015. [Google Scholar] [CrossRef]
- Rhind, N.; Russell, P. Chk1 and cds1: Linchpins of the DNA damage and replication checkpoint pathways. J. Cell Sci. 2000, 113, 3889–3896. [Google Scholar] [PubMed]
- Xu, N.; Libertini, S.; Black, E.J.; Lao, Y.; Hegarat, N.; Walker, M.; Gillespie, D.A. Cdk-mediated phosphorylation of Chk1 is required for efficient activation and full checkpoint proficiency in response to DNA damage. Oncogene 2012, 31, 1086–1094. [Google Scholar] [CrossRef] [PubMed]
- Goto, H.; Kasahara, K.; Inagaki, M. Novel insights into Chk1 regulation by phosphorylation. Cell Struct. Funct. 2015, 40, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Goto, H.; Izawa, I.; Li, P.; Inagaki, M. Novel regulation of checkpoint kinase 1: Is checkpoint kinase 1 a good candidate for anti-cancer therapy? Cancer Sci. 2012, 103, 1195–1200. [Google Scholar] [CrossRef] [PubMed]
- Merry, C.; Fu, K.; Wang, J.; Yeh, I.J.; Zhang, Y. Targeting the checkpoint kinase Chk1 in cancer therapy. Cell Cycle 2010, 9, 279–283. [Google Scholar] [CrossRef] [PubMed]
- Walker, M.; Black, E.J.; Oehler, V.; Gillespie, D.A.; Scott, M.T. Chk1 C-terminal regulatory phosphorylation mediates checkpoint activation by de-repression of Chk1 catalytic activity. Oncogene 2009, 28, 2314–2323. [Google Scholar] [CrossRef] [PubMed]
- Smits, V.A.; Reaper, P.M.; Jackson, S.P. Rapid pikk-dependent release of Chk1 from chromatin promotes the DNA-damage checkpoint response. Curr. Biol. 2006, 16, 150–159. [Google Scholar] [CrossRef] [PubMed]
- Bryant, C.; Rawlinson, R.; Massey, A.J. Chk1 inhibition as a novel therapeutic strategy for treating triple-negative breast and ovarian cancers. BMC Cancer 2014. [Google Scholar] [CrossRef] [PubMed]
- Sorensen, C.S.; Hansen, L.T.; Dziegielewski, J.; Syljuasen, R.G.; Lundin, C.; Bartek, J.; Helleday, T. The cell-cycle checkpoint kinase Chk1 is required for mammalian homologous recombination repair. Nat. Cell Biol. 2005, 7, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Morgan, M.A.; Parsels, L.A.; Zhao, L.; Parsels, J.D.; Davis, M.A.; Hassan, M.C.; Arumugarajah, S.; Hylander-Gans, L.; Morosini, D.; Simeone, D.M.; et al. Mechanism of radiosensitization by the chk1/2 inhibitor azd7762 involves abrogation of the G2 checkpoint and inhibition of homologous recombinational DNA repair. Cancer Res. 2010, 70, 4972–4981. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Yu, L.; Bowen, J.; Gorovsky, M.A.; Allis, C.D. Phosphorylation of histone H3 is required for proper chromosome condensation and segregation. Cell 1999, 97, 99–109. [Google Scholar] [CrossRef]
- Chen, Z.; Xiao, Z.; Gu, W.Z.; Xue, J.; Bui, M.H.; Kovar, P.; Li, G.; Wang, G.; Tao, Z.F.; Tong, Y.; et al. Selective Chk1 inhibitors differentially sensitize p53-deficient cancer cells to cancer therapeutics. Int. J. Cancer 2006, 119, 2784–2794. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Xiao, Z.; Chen, J.; Ng, S.C.; Sowin, T.; Sham, H.; Rosenberg, S.; Fesik, S.; Zhang, H. Human Chk1 expression is dispensable for somatic cell death and critical for sustaining G2 DNA damage checkpoint. Mol. Cancer Ther. 2003, 2, 543–548. [Google Scholar] [PubMed]
- Benada, J.; Macurek, L. Targeting the checkpoint to kill cancer cells. Biomolecules 2015, 5, 1912–1937. [Google Scholar] [CrossRef] [PubMed]
- Sarmento, L.M.; Povoa, V.; Nascimento, R.; Real, G.; Antunes, I.; Martins, L.R.; Moita, C.; Alves, P.M.; Abecasis, M.; Moita, L.F.; et al. Chk1 overexpression in T-cell acute lymphoblastic leukemia is essential for proliferation and survival by preventing excessive replication stress. Oncogene 2015, 34, 2978–2990. [Google Scholar] [CrossRef] [PubMed]
- Grabauskiene, S.; Bergeron, E.J.; Chen, G.; Thomas, D.G.; Giordano, T.J.; Beer, D.G.; Morgan, M.A.; Reddy, R.M. Checkpoint kinase 1 protein expression indicates sensitization to therapy by checkpoint kinase 1 inhibition in non-small cell lung cancer. J. Surg. Res. 2014, 187, 6–13. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Hunter, T. Roles of Chk1 in cell biology and cancer therapy. Int. J. Cancer 2014, 134, 1013–1023. [Google Scholar] [CrossRef] [PubMed]
- Curtin, N.J. DNA repair dysregulation from cancer driver to therapeutic target. Nat. Rev. Cancer 2012, 12, 801–817. [Google Scholar] [CrossRef] [PubMed]
- Daud, A.I.; Ashworth, M.T.; Strosberg, J.; Goldman, J.W.; Mendelson, D.; Springett, G.; Venook, A.P.; Loechner, S.; Rosen, L.S.; Shanahan, F.; et al. Phase I dose-escalation trial of checkpoint kinase 1 inhibitor MK-8776 as Monotherapy and in combination with gemcitabine in patients with advanced solid tumors. J. Clin. Oncol. 2015, 33, 1060–1066. [Google Scholar] [CrossRef] [PubMed]
- Garrett, M.D.; Collins, I. Anticancer therapy with checkpoint inhibitors: What, where and when? Trends Pharmacol. Sci. 2011, 32, 308–316. [Google Scholar] [CrossRef] [PubMed]
- Prudhomme, M. Novel checkpoint 1 inhibitors. Recent Pat. Anticancer Drug Discov. 2006, 1, 55–68. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Fan, S.; Eastman, A.; Worland, P.J.; Sausville, E.A.; O’Connor, P.M. Ucn-01: A potent abrogator of G2 checkpoint function in cancer cells with disrupted p53. J. Natl. Cancer Inst. 1996, 88, 956–965. [Google Scholar] [CrossRef] [PubMed]
- Welch, S.; Hirte, H.W.; Carey, M.S.; Hotte, S.J.; Tsao, M.S.; Brown, S.; Pond, G.R.; Dancey, J.E.; Oza, A.M. Ucn-01 in combination with topotecan in patients with advanced recurrent ovarian cancer: A study of the princess margaret hospital phase II consortium. Gynecol. Oncol. 2007, 106, 305–310. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Christensen, S.D.; Frankel, P.H.; Margolin, K.A.; Agarwala, S.S.; Luu, T.; Mack, P.C.; Lara, P.N., Jr.; Gandara, D.R. A phase II study of cell cycle inhibitor UCN-01 in patients with metastatic melanoma: A california cancer consortium trial. Invest. New Drugs 2012, 30, 741–748. [Google Scholar] [CrossRef] [PubMed]
- Guzi, T.J.; Paruch, K.; Dwyer, M.P.; Labroli, M.; Shanahan, F.; Davis, N.; Taricani, L.; Wiswell, D.; Seghezzi, W.; Penaflor, E.; et al. Targeting the replication checkpoint using sch 900776, a potent and functionally selective Chk1 inhibitor identified via high content screening. Mol. Cancer Ther. 2011, 10, 591–602. [Google Scholar] [CrossRef] [PubMed]
- Engelke, C.G.; Parsels, L.A.; Qian, Y.; Zhang, Q.; Karnak, D.; Robertson, J.R.; Tanska, D.M.; Wei, D.; Davis, M.A.; Parsels, J.D.; et al. Sensitization of pancreatic cancer to chemoradiation by the Chk1 inhibitor MK8776. Clin. Cancer Res. 2013, 19, 4412–4421. [Google Scholar] [CrossRef] [PubMed]
- Grabauskiene, S.; Bergeron, E.J.; Chen, G.; Chang, A.C.; Lin, J.; Thomas, D.G.; Giordano, T.J.; Beer, D.G.; Morgan, M.A.; Reddy, R.M. Chk1 levels correlate with sensitization to pemetrexed by Chk1 inhibitors in non-small cell lung cancer cells. Lung Cancer 2013, 82, 477–484. [Google Scholar] [CrossRef] [PubMed]
- Chernikova, S.B.; Game, J.C.; Brown, J.M. Inhibiting homologous recombination for cancer therapy. Cancer Biol. Ther. 2012, 13, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Venkatesha, V.A.; Parsels, L.A.; Parsels, J.D.; Zhao, L.; Zabludoff, S.D.; Simeone, D.M.; Maybaum, J.; Lawrence, T.S.; Morgan, M.A. Sensitization of pancreatic cancer stem cells to gemcitabine by Chk1 inhibition. Neoplasia 2012, 14, 519–525. [Google Scholar] [CrossRef] [PubMed]
- Hermann, P.C.; Huber, S.L.; Herrler, T.; Aicher, A.; Ellwart, J.W.; Guba, M.; Bruns, C.J.; Heeschen, C. Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem Cell 2007, 1, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Landau, H.J.; McNeely, S.C.; Nair, J.S.; Comenzo, R.L.; Asai, T.; Friedman, H.; Jhanwar, S.C.; Nimer, S.D.; Schwartz, G.K. The checkpoint kinase inhibitor AZD7762 potentiates chemotherapy-induced apoptosis of p53-mutated multiple myeloma cells. Mol. Cancer Ther. 2012, 11, 1781–1788. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.K.; James, J.; Annunziata, C.M. Topotecan synergizes with chek1 (Chk1) inhibitor to induce apoptosis in ovarian cancer cells. BMC Cancer 2015. [Google Scholar] [CrossRef] [PubMed]
- Network., C.G.A.R. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [Google Scholar] [CrossRef]
- King, C.; Diaz, H.; Barnard, D.; Barda, D.; Clawson, D.; Blosser, W.; Cox, K.; Guo, S.; Marshall, M. Characterization and preclinical development of LY2603618: A selective and potent Chk1 inhibitor. Invest. New Drugs 2014, 32, 213–226. [Google Scholar] [CrossRef] [PubMed]
- Calvo, E.; Chen, V.J.; Marshall, M.; Ohnmacht, U.; Hynes, S.M.; Kumm, E.; Diaz, H.B.; Barnard, D.; Merzoug, F.F.; Huber, L.; et al. Preclinical analyses and phase I evaluation of lY2603618 administered in combination with pemetrexed and cisplatin in patients with advanced cancer. Invest. New Drugs 2014, 32, 955–968. [Google Scholar] [CrossRef] [PubMed]
- Carrassa, L.; Damia, G. Unleashing Chk1 in cancer therapy. Cell Cycle 2011, 10, 2121–2128. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, S.; Huang, M.; Elledge, S.J. Linkage of atm to cell cycle regulation by the Chk2 protein kinase. Science 1998, 282, 1893–1897. [Google Scholar] [CrossRef] [PubMed]
- Ahn, J.; Urist, M.; Prives, C. The Chk2 protein kinase. DNA repair 2004, 3, 1039–1047. [Google Scholar] [CrossRef] [PubMed]
- Kass, E.M.; Ahn, J.; Tanaka, T.; Freed-Pastor, W.A.; Keezer, S.; Prives, C. Stability of checkpoint kinase 2 is regulated via phosphorylation at serine 456. J. Biol. Chem. 2007, 282, 30311–30321. [Google Scholar] [CrossRef] [PubMed]
- Lovly, C.M.; Yan, L.; Ryan, C.E.; Takada, S.; Piwnica-Worms, H. Regulation of Chk2 ubiquitination and signaling through autophosphorylation of serine 379. Mol. Cell. Biol. 2008, 28, 5874–5885. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Matsuoka, S.; Carpenter, P.B.; Elledge, S.J. 53BP1, a mediator of the DNA damage checkpoint. Science 2002, 298, 1435–1438. [Google Scholar] [CrossRef] [PubMed]
- Lou, Z.; Minter-Dykhouse, K.; Wu, X.; Chen, J. Mdc1 is coupled to activated Chk2 in mammalian DNA damage response pathways. Nature 2003, 421, 957–961. [Google Scholar] [CrossRef] [PubMed]
- Mochan, T.A.; Venere, M.; DiTullio, R.A.; Halazonetis, T.D. 53BP1 and NFBD1/MDC1-Nbs1 function in parallel interacting pathways activating ataxia-telangiectasia mutated (ATM) in response to DNA damage. Cancer Res. 2003, 63, 8586–8591. [Google Scholar] [PubMed]
- Lou, Z.; Minter-Dykhouse, K.; Franco, S.; Gostissa, M.; Rivera, M.A.; Celeste, A.; Manis, J.P.; van Deursen, J.; Nussenzweig, A.; Paull, T.T.; et al. MDC1 maintains genomic stability by participating in the amplification of ATM-dependent DNA damage signals. Mol. Cell 2006, 21, 187–200. [Google Scholar] [CrossRef] [PubMed]
- Minter-Dykhouse, K.; Ward, I.; Huen, M.S.Y.; Chen, J.; Lou, Z. Distinct versus overlapping functions of MDC1 and 53BP1 in DNA damage response and tumorigenesis. J. Cell Biol. 2008, 181, 727–735. [Google Scholar] [CrossRef] [PubMed]
- Blasina, A.; de Weyer, I.V.; Laus, M.C.; Luyten, W.H.; Parker, A.E.; McGowan, C.H. A human homologue of the checkpoint kinase Cds1 directly inhibits Cdc25 phosphatase. Curr. Biol. 1999, 9, 1–10. [Google Scholar] [CrossRef]
- Bahassi, E.M.; Ovesen, J.L.; Riesenberg, A.L.; Bernstein, W.Z.; Hasty, P.E.; Stambrook, P.J. The checkpoint kinases Chk1 and Chk2 regulate the functional associations between hBRCA2 and Rad51 in response to DNA damage. Oncogene 2008, 27, 3977–3985. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Willers, H.; Feng, Z.; Ghosh, J.C.; Kim, S.; Weaver, D.T.; Chung, J.H.; Powell, S.N.; Xia, F. Chk2 phosphorylation of BRCA1 regulates DNA double-strand break repair. Mol. Cell Biol. 2004, 24, 708–718. [Google Scholar] [CrossRef] [PubMed]
- Lountos, G.T.; Jobson, A.G.; Tropea, J.E.; Self, C.R.; Zhang, G.; Pommier, Y.; Shoemaker, R.H.; Waugh, D.S. Structural characterization of inhibitor complexes with checkpoint kinase 2 (Chk2), a drug target for cancer therapy. J. Struct. Biol. 2011, 176, 292–301. [Google Scholar] [CrossRef] [PubMed]
- Pommier, Y.; Weinstein, J.N.; Aladjem, M.I.; Kohn, K.W. Chk2 molecular interaction map and rationale for Chk2 inhibitors. Clin. Cancer Res. 2006, 12, 2657–2661. [Google Scholar] [CrossRef] [PubMed]
- Hirao, A.; Kong, Y.Y.; Matsuoka, S.; Wakeham, A.; Ruland, J.; Yoshida, H.; Liu, D.; Elledge, S.J.; Mak, T.W. DNA damage-induced activation of p53 by the checkpoint kinase Chk2. Science 2000, 287, 1824–1827. [Google Scholar] [CrossRef] [PubMed]
- Jobson, A.G.; Cardellina, J.H.; Scudiero, D.; Kondapaka, S.; Zhang, H.; Kim, H.; Shoemaker, R.; Pommier, Y. Identification of a bis-guanylhydrazone [4,4'-diacetyldiphenylurea-bis(guanylhydrazone); NSC 109555] as a novel chemotype for inhibition of Chk2 kinase. Mol. Pharmacol. 2007, 72, 876–884. [Google Scholar] [CrossRef] [PubMed]
- Lountos, G.T.; Tropea, J.E.; Zhang, D.; Jobson, A.G.; Pommier, Y.; Shoemaker, R.H.; Waugh, D.S. Crystal structure of checkpoint kinase 2 in complex with NSC 109555, a potent and selective inhibitor. Protein Sci. 2009, 18, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Jobson, A.G.; Lountos, G.T.; Lorenzi, P.L.; Llamas, J.; Connelly, J.; Cerna, D.; Tropea, J.E.; Onda, A.; Zoppoli, G.; Kondapaka, S.; et al. Cellular inhibition of checkpoint kinase 2 (Chk2) and potentiation of camptothecins and radiation by the novel Chk2 inhibitor PV1019 [7-nitro-1h-indole-2-carboxylic acid {4-[1-(guanidinohydrazone)-ethyl]-phenyl}-amide]. J. Pharm. Exp. Ther. 2009, 331, 816–826. [Google Scholar] [CrossRef] [PubMed]
- Caldwell, J.J.; Welsh, E.J.; Matijssen, C.; Anderson, V.E.; Antoni, L.; Boxall, K.; Urban, F.; Hayes, A.; Raynaud, F.I.; Rigoreau, L.J.M.; et al. Structure-based design of potent and selective 2-(quinazolin-2-yl)phenol inhibitors of checkpoint kinase 2. J. Med. Chem. 2011, 54, 580–590. [Google Scholar] [CrossRef] [PubMed]
- Anderson, V.E.; Walton, M.I.; Eve, P.D.; Boxall, K.J.; Antoni, L.; Caldwell, J.J.; Aherne, W.; Pearl, L.H.; Oliver, A.W.; Collins, I.; et al. CCT241533 is a potent and selective inhibitor of Chk2 that potentiates the cytotoxicity of PARP inhibitors. Cancer Res. 2011, 71, 463–472. [Google Scholar] [CrossRef] [PubMed]
- Arienti, K.L.; Brunmark, A.; Axe, F.U.; McClure, K.; Lee, A.; Blevitt, J.; Neff, D.K.; Huang, L.; Crawford, S.; Pandit, C.R.; et al. Checkpoint kinase inhibitors: Sar and radioprotective properties of a series of 2-arylbenzimidazoles. J. Med. Chem. 2005, 48, 1873–1885. [Google Scholar] [CrossRef] [PubMed]
- Matijssen, C.; Silva-Santisteban, M.C.; Westwood, I.M.; Siddique, S.; Choi, V.; Sheldrake, P.; van Montfort, R.L.M.; Blagg, J. Benzimidazole inhibitors of the protein kinase Chk2: Clarification of the binding mode by flexible side chain docking and protein-ligand crystallography. Bioorg. Med. Chem. 2012, 20, 6630–6639. [Google Scholar] [CrossRef] [PubMed]
- Curman, D.; Cinel, B.; Williams, D.E.; Rundle, N.; Block, W.D.; Goodarzi, A.A.; Hutchins, J.R.; Clarke, P.R.; Zhou, B.B.; Lees-Miller, S.P.; et al. Inhibition of the g2 DNA damage checkpoint and of protein kinases Chk1 and Chk2 by the marine sponge alkaloid debromohymenialdisine. J. Biol. Chem. 2001, 276, 17914–17919. [Google Scholar] [CrossRef] [PubMed]
- Oliver, A.W.; Paul, A.; Boxall, K.J.; Barrie, S.E.; Aherne, G.W.; Garrett, M.D.; Mittnacht, S.; Pearl, L.H. Trans-activation of the DNA-damage signalling protein kinase Chk2 by t-loop exchange. EMBO J. 2006, 25, 3179–3190. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.; Tepe, J.J. Potent inhibition of checkpoint kinase activity by a hymenialdisine-derived indoloazepine. Bioorg. Med. Chem. Lett. 2004, 14, 4319–4321. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.N.T.; Saleem, R.S.Z.; Luderer, M.J.; Hovde, S.; Henry, R.W.; Tepe, J.J. Radioprotection by hymenialdisine-derived checkpoint kinase 2 inhibitors. ACS Chem. Biol. 2012, 7, 172–184. [Google Scholar] [CrossRef] [PubMed]
- Saleem, R.S.Z.; Lansdell, T.A.; Tepe, J.J. Synthesis and evaluation of debromohymenialdisine-derived Chk2 inhibitors. Bioorg. Med. Chem. 2012, 20, 1475–1481. [Google Scholar] [CrossRef] [PubMed]
- Larson, G.; Yan, S.; Chen, H.; Rong, F.; Hong, Z.; Wu, J.Z. Identification of novel, selective and potent Chk2 inhibitors. Bioorg. Med. Chem. Lett. 2007, 17, 172–175. [Google Scholar] [CrossRef] [PubMed]
- Carlessi, L.; Buscemi, G.; Larson, G.; Hong, Z.; Wu, J.Z.; Delia, D. Biochemical and cellular characterization of VRX0466617, a novel and selective inhibitor for the checkpoint kinase Chk2. Mol. Cancer Ther. 2007, 6, 935–944. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; La Rose, J.; Zhang, H.; Takemura, H.; Kohn, K.W.; Pommier, Y. Ucn-01 inhibits p53 up-regulation and abrogates gamma-radiation-induced g(2)-m checkpoint independently of p53 by targeting both of the checkpoint kinases, Chk2 and Chk1. Cancer Res. 2002, 62, 5743–5748. [Google Scholar] [PubMed]
- Zabludoff, S.D.; Deng, C.; Grondine, M.R.; Sheehy, A.M.; Ashwell, S.; Caleb, B.L.; Green, S.; Haye, H.R.; Horn, C.L.; Janetka, J.W.; et al. AZD7762, a novel checkpoint kinase inhibitor, drives checkpoint abrogation and potentiates DNA-targeted therapies. Mol. Cancer Ther. 2008, 7, 2955–2966. [Google Scholar] [CrossRef] [PubMed]
- Matthews, D.J.; Yakes, F.M.; Chen, J.; Tadano, M.; Bornheim, L.; Clary, D.O.; Tai, A.; Wagner, J.M.; Miller, N.; Kim, Y.D.; et al. Pharmacological abrogation of s-phase checkpoint enhances the anti-tumor activity of gemcitabine in vivo. Cell cycle 2007, 6, 104–110. [Google Scholar] [CrossRef] [PubMed]
- Ame, J.C.; Spenlehauer, C.; de Murcia, G. The parp superfamily. BioEssays 2004, 26, 882–893. [Google Scholar] [CrossRef] [PubMed]
- Otto, H.; Reche, P.A.; Bazan, F.; Dittmar, K.; Haag, F.; Koch-Nolte, F. In silico characterization of the family of PARP-like poly(ADP-ribosyl)transferases (pARTs). BMC Genomics 2005. [Google Scholar] [CrossRef] [PubMed]
- Tartier, L.; Spenlehauer, C.; Newman, H.C.; Folkard, M.; Prise, K.M.; Michael, B.D.; Menissier-de Murcia, J.; de Murcia, G. Local DNA damage by proton microbeam irradiation induces poly(adp-ribose) synthesis in mammalian cells. Mutagenesis 2003, 18, 411–416. [Google Scholar] [CrossRef] [PubMed]
- Malanga, M.; Althaus, F.R. The role of poly(adp-ribose) in the DNA damage signaling network. Biochem. Cell Biol. 2005, 83, 354–364. [Google Scholar] [CrossRef] [PubMed]
- Curtin, N.J.; Szabo, C. Therapeutic applications of PARP inhibitors: Anticancer therapy and beyond. JMAM 2013, 34, 1217–1256. [Google Scholar] [CrossRef] [PubMed]
- Ekblad, T.; Camaioni, E.; Schuler, H.; Macchiarulo, A. Parp inhibitors: Polypharmacology versus selective inhibition. FEBS J. 2013, 280, 3563–3575. [Google Scholar] [CrossRef] [PubMed]
- Lupo, B.; Trusolino, L. Inhibition of poly(adp-ribosyl)ation in cancer: Old and new paradigms revisited. Biochim. Biophys. Acta 2014, 1846, 201–215. [Google Scholar] [CrossRef] [PubMed]
- Kummar, S.; Chen, A.; Parchment, R.E.; Kinders, R.J.; Ji, J.; Tomaszewski, J.E.; Doroshow, J.H. Advances in using parp inhibitors to treat cancer. BMC Medicine 2012. [Google Scholar] [CrossRef] [PubMed]
- Rouleau, M.; Patel, A.; Hendzel, M.J.; Kaufmann, S.H.; Poirier, G.G. PARP inhibition: PARP1 and beyond. Nat. Rev. Cancer 2010, 10, 293–301. [Google Scholar] [CrossRef] [PubMed]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.J.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef] [PubMed]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(adp-ribose) polymerase. Nature 2005, 434, 913–917. [Google Scholar] [CrossRef] [PubMed]
- Tutt, A.N.; Lord, C.J.; McCabe, N.; Farmer, H.; Turner, N.; Martin, N.M.; Jackson, S.P.; Smith, G.C.; Ashworth, A. Exploiting the DNA repair defect in BRCA mutant cells in the design of new therapeutic strategies for cancer. Cold Spring Harb. Symp. Quant. Biol. 2005, 70, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Petermann, E.; Orta, M.L.; Issaeva, N.; Schultz, N.; Helleday, T. Hydroxyurea-stalled replication forks become progressively inactivated and require two different Rad51-mediated pathways for restart and repair. Mol. Cell 2010, 37, 492–502. [Google Scholar] [CrossRef] [PubMed]
- Schlacher, K.; Christ, N.; Siaud, N.; Egashira, A.; Wu, H.; Jasin, M. Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by Mre11. Cell 2011, 145, 529–542. [Google Scholar] [CrossRef] [PubMed]
- Fong, P.C.; Boss, D.S.; Yap, T.A.; Tutt, A.; Wu, P.; Mergui-Roelvink, M.; Mortimer, P.; Swaisland, H.; Lau, A.; O’Connor, M.J.; et al. Inhibition of poly(adp-ribose) polymerase in tumors from BRCA mutation carriers. N. Engl. J. Med. 2009, 361, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Tutt, A.; Robson, M.; Garber, J.E.; Domchek, S.M.; Audeh, M.W.; Weitzel, J.N.; Friedlander, M.; Arun, B.; Loman, N.; Schmutzler, R.K.; et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: A proof-of-concept trial. Lancet 2010, 376, 235–244. [Google Scholar] [CrossRef]
- Audeh, M.W.; Carmichael, J.; Penson, R.T.; Friedlander, M.; Powell, B.; Bell-McGuinn, K.M.; Scott, C.; Weitzel, J.N.; Oaknin, A.; Loman, N.; et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer: A proof-of-concept trial. Lancet 2010, 376, 245–251. [Google Scholar] [CrossRef]
- Turner, N.; Tutt, A.; Ashworth, A. Hallmarks of “brcaness” in sporadic cancers. Nat. Struct. Mol. Biol. 2004, 4, 814–819. [Google Scholar]
- McCabe, N.; Turner, N.C.; Lord, C.J.; Kluzek, K.; Bialkowska, A.; Swift, S.; Giavara, S.; O’Connor, M.J.; Tutt, A.N.; Zdzienicka, M.Z.; et al. Deficiency in the repair of DNA damage by homologous recombination and sensitivity to poly(ADP-Ribose) polymerase inhibition. Cancer Res. 2006, 66, 8109–8115. [Google Scholar] [CrossRef] [PubMed]
- Peng, G.; Lin, S.Y. Exploiting the homologous recombination DNA repair network for targeted cancer therapy. World J. Clin. Oncol. 2011, 2, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.C.; Ashworth, A. Biomarkers of PARP inhibitor sensitivity. Breast Cancer Res.. Treat. 2011, 127, 283–286. [Google Scholar] [CrossRef] [PubMed]
- Yap, T.A.; Sandhu, S.K.; Carden, C.P.; de Bono, J.S. Poly(Adp-Ribose) polymerase (PARP) inhibitors: Exploiting a synthetic lethal strategy in the clinic. CA Cancer J. Clin. 2011, 61, 31–49. [Google Scholar] [CrossRef] [PubMed]
- Ferraris, D.V. Evolution of poly(ADP-Ribose) polymerase-1 (PARP-1) inhibitors. From concept to clinic. J. Med. Chem. 2010, 53, 4561–4584. [Google Scholar] [CrossRef] [PubMed]
- Davar, D.; Beumer, J.H.; Hamieh, L.; Tawbi, H. Role of PARP inhibitors in cancer biology and therapy. Curr. Med. Chem. 2012, 19, 3907–3921. [Google Scholar] [CrossRef] [PubMed]
- Kopetz, S.; Mita, M.M.; Mok, I.; Sankhala, K.K.; Moseley, J.; Sherman, B.M.; Bradley, C.R.; Tolcher, A.W. First in human phase I study of BSI-201, a small molecule inhibitor of poly ADP-Ribose polymerase (PARP) in subjects with advanced solid tumors. J. Clin. Oncol. 2008, 26, 3577. [Google Scholar]
- Kaufman, B.; Shapira-Frommer, R.; Schmutzler, R.K.; Audeh, M.W.; Friedlander, M.; Balmaña, J.; Mitchell, G.; Fried, G.; Stemmer, S.M.; Hubert, A.; et al. Olaparib monotherapy in patients with advanced cancer and a germ-line BRCA1/2 mutation: An open-label phase II study. J. Clin. Oncol. 2013. [Google Scholar] [CrossRef]
- Mendes-Pereira, A.M.; Martin, S.A.; Brough, R.; McCarthy, A.; Taylor, J.R.; Kim, J.S.; Waldman, T.; Lord, C.J.; Ashworth, A. Synthetic lethal targeting of pten mutant cells with PARP inhibitors. EMBO Mol. Med. 2009, 1, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Forster, M.D.; Dedes, K.J.; Sandhu, S.; Frentzas, S.; Kristeleit, R.; Ashworth, A.; Poole, C.J.; Weigelt, B.; Kaye, S.B.; Molife, L.R. Treatment with olaparib in a patient with pten-deficient endometrioid endometrial cancer. Nat. Rev. Clin. Oncol. 2011, 8, 302–306. [Google Scholar] [CrossRef] [PubMed]
- Dedes, K.J.; Wetterskog, D.; Mendes-Pereira, A.M.; Natrajan, R.; Lambros, M.B.; Geyer, F.C.; Vatcheva, R.; Savage, K.; Mackay, A.; Lord, C.J.; et al. PTEN deficiency in endometrioid endometrial adenocarcinomas predicts sensitivity to PARP inhibitors. Sci. Transl. Med. 2010. [Google Scholar] [CrossRef] [PubMed]
- Hunt, C.R.; Gupta, A.; Horikoshi, N.; Pandita, T.K. Does pten loss impair DNA double-strand break repair by homologous recombination? Clin. Cancer Res. 2012, 18, 920–922. [Google Scholar] [CrossRef] [PubMed]
- Fraser, M.; Zhao, H.; Luoto, K.R.; Lundin, C.; Coackley, C.; Chan, N.; Joshua, A.M.; Bismar, T.A.; Evans, A.; Helleday, T.; et al. PTEN deletion in prostate cancer cells does not associate with loss of Rad51 function: Implications for radiotherapy and chemotherapy. Clin. Cancer Res. 2012, 18, 1015–1027. [Google Scholar] [CrossRef] [PubMed]
- Minami, D.; Takigawa, N.; Takeda, H.; Takata, M.; Ochi, N.; Ichihara, E.; Hisamoto, A.; Hotta, K.; Tanimoto, M.; Kiura, K. Synergistic effect of olaparib with combination of cisplatin on PTEN-deficient lung cancer cells. Mol. Cancer Res. 2013, 11, 140–148. [Google Scholar] [CrossRef] [PubMed]
- Faraoni, I.; Compagnone, M.; Lavorgna, S.; Angelini, D.F.; Cencioni, M.T.; Piras, E.; Panetta, P.; Ottone, T.; Dolci, S.; Venditti, A.; et al. BRCA1, PARP1 and γH2AX in acute myeloid leukemia: Role as biomarkers of response to the PARP inhibitor olaparib. Biochim. Biophys. Acta 2015, 1852, 462–472. [Google Scholar] [CrossRef] [PubMed]
- Ajimura, M.; Leem, S.H.; Ogawa, H. Identification of new genes required for meiotic recombination in saccharomyces cerevisiae. Genetics 1993, 133, 51–66. [Google Scholar] [PubMed]
- Parry, J.M.; Davies, P.J.; Evans, W.E. The effects of “cell age” upon the lethal effects of physical and chemical mutagens in the yeast, saccharomyces cerevisiae. Mol. Gen. Genet. 1976, 146, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Carney, J.P.; Maser, R.S.; Olivares, H.; Davis, E.M.; Le Beau, M.; Yates, J.R.; Hays, L.; Morgan, W.F.; Petrini, J.H. The hMre11/hRad50 protein complex and nijmegen breakage syndrome: Linkage of double-strand break repair to the cellular DNA damage response. Cell 1998, 93, 477–486. [Google Scholar] [CrossRef]
- Stewart, G.S.; Maser, R.S.; Stankovic, T.; Bressan, D.A.; Kaplan, M.I.; Jaspers, N.G.; Raams, A.; Byrd, P.J.; Petrini, J.H.; Taylor, A.M. The DNA double-strand break repair gene hMRE11 is mutated in individuals with an ataxia-telangiectasia-like disorder. Cell 1999, 99, 577–587. [Google Scholar] [CrossRef]
- Waltes, R.; Kalb, R.; Gatei, M.; Kijas, A.W.; Stumm, M.; Sobeck, A.; Wieland, B.; Varon, R.; Lerenthal, Y.; Lavin, M.F.; et al. Human Rad50 deficiency in a nijmegen breakage syndrome-like disorder. Am. J. Hum. Genet. 2009, 84, 605–616. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Weaver, D.T. Conditional gene targeted deletion by cre recombinase demonstrates the requirement for the double-strand break repair MRE11 protein in murine embryonic stem cells. Nucleic Acids Res. 1997, 25, 2985–2991. [Google Scholar] [CrossRef] [PubMed]
- Luo, G.; Yao, M.S.; Bender, C.F.; Mills, M.; Bladl, A.R.; Bradley, A.; Petrini, J.H. Disruption of mRad50 causes embryonic stem cell lethality, abnormal embryonic development, and sensitivity to ionizing radiation. Proc. Natl. Acad. Sci. USA 1999, 96, 7376–7381. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Petersen, S.; Tessarollo, L.; Nussenzweig, A. Targeted disruption of the nijmegen breakage syndrome gene NBS1 leads to early embryonic lethality in mice. Curr. Biol. 2001, 11, 105–109. [Google Scholar] [CrossRef]
- Williams, R.S.; Moncalian, G.; Williams, J.S.; Yamada, Y.; Limbo, O.; Shin, D.S.; Groocock, L.M.; Cahill, D.; Hitomi, C.; Guenther, G.; et al. Mre11 dimers coordinate DNA end bridging and nuclease processing in double-strand-break repair. Cell 2008, 135, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Dery, U.; Coulombe, Y.; Rodrigue, A.; Stasiak, A.; Richard, S.; Masson, J.Y. A glycine-arginine domain in control of the human MRE11 DNA repair protein. Mol. Cell Biol. 2008, 28, 3058–3069. [Google Scholar] [CrossRef] [PubMed]
- De Jager, M.; Dronkert, M.L.; Modesti, M.; Beerens, C.E.; Kanaar, R.; van Gent, D.C. DNA-binding and strand-annealing activities of human Mre11: Implications for its roles in DNA double-strand break repair pathways. Nucleic Acids Res. 2001, 29, 1317–1325. [Google Scholar] [CrossRef] [PubMed]
- Paull, T.T.; Gellert, M. The 3' to 5' exonuclease activity of Mre 11 facilitates repair of DNA double-strand breaks. Mol. Cell 1998, 1, 969–979. [Google Scholar] [CrossRef]
- Paull, T.T.; Gellert, M. NBS1 potentiates ATP-driven DNA unwinding and endonuclease cleavage by the Mre11/Rad50 complex. Genes Dev. 1999, 13, 1276–1288. [Google Scholar] [CrossRef] [PubMed]
- De Jager, M.; van Noort, J.; van Gent, D.C.; Dekker, C.; Kanaar, R.; Wyman, C. Human Rad50/Mre11 is a flexible complex that can tether DNA ends. Mol. Cell 2001, 8, 1129–1135. [Google Scholar] [CrossRef]
- Williams, R.S.; Dodson, G.E.; Limbo, O.; Yamada, Y.; Williams, J.S.; Guenther, G.; Classen, S.; Glover, J.N.M.; Iwasaki, H.; Russell, P.; et al. Nbs1 flexibly tethers Ctp1 and Mre11-Rad50 to coordinate DNA double-strand break processing and repair. Cell 2009, 139, 87–99. [Google Scholar] [CrossRef] [PubMed]
- Chapman, J.R.; Jackson, S.P. Phospho-dependent interactions between NBS1 and MDC1 mediate chromatin retention of the Mrn complex at sites of DNA damage. EMBO Rep. 2008, 9, 795–801. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Shi, L.Z.; Wong, C.C.; Han, X.; Hwang, P.Y.; Truong, L.N.; Zhu, Q.; Shao, Z.; Chen, D.J.; Berns, M.W.; et al. The interaction of CtIP and Nbs1 connects CDK and ATM to regulate HR-mediated double-strand break repair. PLoS Genet. 2013, 9, e1003277. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, J.; Tauchi, H.; Sakamoto, S.; Nakamura, A.; Morishima, K.; Matsuura, S.; Kobayashi, T.; Tamai, K.; Tanimoto, K.; Komatsu, K. NBS1 localizes to γ-H2AX foci through interaction with the FHA/BRCT domain. Curr. Biol. 2002, 12, 1846–1851. [Google Scholar] [CrossRef]
- Paull, T.T. Making the best of the loose ends: Mre11/Rad50 complexes and Sae2 promote DNA double-strand break resection. DNA repair 2010, 9, 1283–1291. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Paull, T.T. ATM activation by DNA double-strand breaks through the Mre11-Rad50-Nbs1 complex. Science 2005, 308, 551–554. [Google Scholar] [CrossRef] [PubMed]
- Ferretti, L.P.; Lafranchi, L.; Sartori, A.A. Controlling DNA-end resection: A new task for CDKs. Front. Genet. 2013. [Google Scholar] [CrossRef] [PubMed]
- Shibata, A.; Moiani, D.; Arvai, A.S.; Perry, J.; Harding, S.M.; Genois, M.M.; Maity, R.; van Rossum-Fikkert, S.; Kertokalio, A.; Romoli, F.; et al. DNA double-strand break repair pathway choice is directed by distinct Mre11 nuclease activities. Mol. Cell 2014, 53, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Dupré, A.; Boyer-Chatenet, L.; Sattler, R.M.; Modi, A.P.; Lee, J.-H.; Nicolette, M.L.; Kopelovich, L.; Jasin, M.; Baer, R.; Paull, T.T.; et al. A forward chemical genetic screen reveals an inhibitor of the Mre11-Rad50-Nbs1 complex. Nat. Chem. Biol. 2008, 4, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Rass, E.; Grabarz, A.; Plo, I.; Gautier, J.; Bertrand, P.; Lopez, B.S. Role of Mre11 in chromosomal nonhomologous end joining in mammalian cells. Nat. Struct. Mol. Biol. 2009, 16, 819–824. [Google Scholar] [CrossRef] [PubMed]
- Tsuzuki, T.; Fujii, Y.; Sakumi, K.; Tominaga, Y.; Nakao, K.; Sekiguchi, M.; Matsushiro, A.; Yoshimura, Y.; Morita, T. Targeted disruption of the Rad51 gene leads to lethality in embryonic mice. Proc. Natl. Acad. Sci. USA 1996, 93, 6236–6240. [Google Scholar] [CrossRef] [PubMed]
- Baumann, P.; Benson, F.E.; West, S.C. Human rad51 protein promotes ATP-dependent homologous pairing and strand transfer reactions in vitro. Cell 1996, 87, 757–766. [Google Scholar] [CrossRef]
- Buisson, R.; Dion-Cote, A.M.; Coulombe, Y.; Launay, H.; Cai, H.; Stasiak, A.Z.; Stasiak, A.; Xia, B.; Masson, J.Y. Cooperation of breast cancer proteins PALB2 and piccolo BRCA2 in stimulating homologous recombination. Nat. Struct. Mol. Biol. 2010, 17, 1247–1254. [Google Scholar] [CrossRef] [PubMed]
- Davies, A.A.; Masson, J.Y.; McIlwraith, M.J.; Stasiak, A.Z.; Stasiak, A.; Venkitaraman, A.R.; West, S.C. Role of BRCA2 in control of the Rad51 recombination and DNA repair protein. Mol. Cell 2001, 7, 273–282. [Google Scholar] [CrossRef]
- Takata, M.; Sasaki, M.S.; Tachiiri, S.; Fukushima, T.; Sonoda, E.; Schild, D.; Thompson, L.H.; Takeda, S. Chromosome instability and defective recombinational repair in knockout mutants of the five Rad51 paralogs. Mol. Cell Biol. 2001, 21, 2858–2866. [Google Scholar] [CrossRef] [PubMed]
- Pellegrini, L.; Yu, D.S.; Lo, T.; Anand, S.; Lee, M.; Blundell, T.L.; Venkitaraman, A.R. Insights into DNA recombination from the structure of a RAD51-BRCA2 complex. Nature 2002, 420, 287–293. [Google Scholar] [CrossRef] [PubMed]
- Aihara, H.; Ito, Y.; Kurumizaka, H.; Yokoyama, S.; Shibata, T. The N-terminal domain of the human Rad51 protein binds DNA: Structure and a DNA binding surface as revealed by NMR. J. Mol. Biol. 1999, 290, 495–504. [Google Scholar] [CrossRef] [PubMed]
- Haaf, T.; Golub, E.I.; Reddy, G.; Radding, C.M.; Ward, D.C. Nuclear foci of mammalian Rad51 recombination protein in somatic cells after DNA damage and its localization in synaptonemal complexes. Proc. Natl. Acad. Sci. USA 1995, 92, 2298–2302. [Google Scholar] [CrossRef] [PubMed]
- Raderschall, E.; Stout, K.; Freier, S.; Suckow, V.; Schweiger, S.; Haaf, T. Elevated levels of Rad51 recombination protein in tumor cells. Cancer Res. 2002, 62, 219–225. [Google Scholar] [PubMed]
- Maacke, H.; Jost, K.; Opitz, S.; Miska, S.; Yuan, Y.; Hasselbach, L.; Lüttges, J.; Kalthoff, H.; Stürzbecher, H.W. DNA repair and recombination factor Rad51 is over-expressed in human pancreatic adenocarcinoma. Oncogene 2000, 19, 2791–2795. [Google Scholar] [CrossRef] [PubMed]
- Maacke, H.; Opitz, S.; Jost, K.; Hamdorf, W.; Henning, W.; Krüger, S.; Feller, A.C.; Lopens, A.; Diedrich, K.; Schwinger, E.; et al. Over-expression of wild-type Rad51 correlates with histological grading of invasive ductal breast cancer. Int. J. Cancer 2000, 88, 907–913. [Google Scholar] [CrossRef]
- Mitra, A.; Jameson, C.; Barbachano, Y.; Sanchez, L.; Kote-Jarai, Z.; Peock, S.; Sodha, N.; Bancroft, E.; Fletcher, A.; Cooper, C.; et al. Overexpression of Rad51 occurs in aggressive prostatic cancer. Histopathology 2009, 55, 696–704. [Google Scholar] [CrossRef] [PubMed]
- Takenaka, T.; Yoshino, I.; Kouso, H.; Ohba, T.; Yohena, T.; Osoegawa, A.; Shoji, F.; Maehara, Y. Combined evaluation of Rad51 and ERCC1 expressions for sensitivity to platinum agents in non-small cell lung cancer. Int. J. Cancer 2007, 121, 895–900. [Google Scholar] [CrossRef] [PubMed]
- Henning, W.; Stürzbecher, H.W. Homologous recombination and cell cycle checkpoints: Rad51 in tumour progression and therapy resistance. Toxicology 2003, 193, 91–109. [Google Scholar] [CrossRef]
- Vispe, S.; Cazaux, C.; Lesca, C.; Defais, M. Overexpression of Rad51 protein stimulates homologous recombination and increases resistance of mammalian cells to ionizing radiation. Nucleic Acids Res. 1998, 26, 2859–2864. [Google Scholar] [CrossRef] [PubMed]
- Ito, M.; Yamamoto, S.; Nimura, K.; Hiraoka, K.; Tamai, K.; Kaneda, Y. Rad51 siRNA delivered by HVJ envelope vector enhances the anti-cancer effect of cisplatin. J. Gene Med. 2005, 7, 1044–1052. [Google Scholar] [CrossRef] [PubMed]
- Kiyohara, E.; Tamai, K.; Katayama, I.; Kaneda, Y. The combination of chemotherapy with HVJ-E containing Rad51 siRNA elicited diverse anti-tumor effects and synergistically suppressed melanoma. Gene Ther. 2012, 19, 734–741. [Google Scholar] [CrossRef] [PubMed]
- Macara, I.G.; Cantley, L.C. Mechanism of anion exchange across the red cell membrane by band 3: Interactions between stilbenedisulfonate and NAP-taurine binding sites. Biochemistry 1981, 20, 5695–5701. [Google Scholar] [CrossRef] [PubMed]
- Ishida, T.; Takizawa, Y.; Kainuma, T.; Inoue, J.; Mikawa, T.; Shibata, T.; Suzuki, H.; Tashiro, S.; Kurumizaka, H. Dids, a chemical compound that inhibits Rad51-mediated homologous pairing and strand exchange. Nucleic Acids Res. 2009, 37, 3367–3376. [Google Scholar] [CrossRef] [PubMed]
- Lamont, K.R.; Hasham, M.G.; Donghia, N.M.; Branca, J.; Chavaree, M.; Chase, B.; Breggia, A.; Hedlund, J.; Emery, I.; Cavallo, F.; et al. Attenuating homologous recombination stimulates an aid-induced antileukemic effect. J. Exp. Med. 2013, 210, 1021–1033. [Google Scholar] [CrossRef] [PubMed]
- Hasham, M.G.; Donghia, N.M.; Coffey, E.; Maynard, J.; Snow, K.J.; Ames, J.; Wilpan, R.Y.; He, Y.; King, B.L.; Mills, K.D. Widespread genomic breaks generated by activation-induced cytidine deaminase are prevented by homologous recombination. Nat. Immunol. 2010, 11, 820–826. [Google Scholar] [CrossRef] [PubMed]
- Takaku, M.; Kainuma, T.; Ishida-Takaku, T.; Ishigami, S.; Suzuki, H.; Tashiro, S.; van Soest, R.W.M.; Nakao, Y.; Kurumizaka, H. Halenaquinone, a chemical compound that specifically inhibits the secondary DNA binding of Rad51. Genes cells 2011, 16, 427–436. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.; Motlekar, N.A.; Burgwin, C.M.; Napper, A.D.; Diamond, S.L.; Mazin, A.V. Identification of specific inhibitors of human Rad51 recombinase using high-throughput screening. ACS Chem. Biol. 2011, 6, 628–635. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.; Mazina, O.M.; Zentner, I.J.; Cocklin, S.; Mazin, A.V. Inhibition of homologous recombination in human cells by targeting Rad51 recombinase. J. Med. Chem. 2012, 55, 3011–3020. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.; Mazin, A.V. A small molecule inhibitor of human Rad51 potentiates breast cancer cell killing by therapeutic agents in mouse xenografts. PLoS ONE 2014, 9, e100993. [Google Scholar] [CrossRef] [PubMed]
- Alagpulinsa, D.A.; Ayyadevara, S.; Shmookler Reis, R.J. A small-molecule inhibitor of Rad51 reduces homologous recombination and sensitizes multiple myeloma cells to doxorubicin. Front. Oncol. 2014. [Google Scholar] [CrossRef] [PubMed]
- Budke, B.; Logan, H.L.; Kalin, J.H.; Zelivianskaia, A.S.; Cameron McGuire, W.; Miller, L.L.; Stark, J.M.; Kozikowski, A.P.; Bishop, D.K.; Connell, P.P. RI-1: A chemical inhibitor of RAD51 that disrupts homologous recombination in human cells. Nucleic Acids Res. 2012, 40, 7347–7357. [Google Scholar] [CrossRef] [PubMed]
- Bee, L.; Fabris, S.; Cherubini, R.; Mognato, M.; Celotti, L. The efficiency of homologous recombination and non-homologous end joining systems in repairing double-strand breaks during cell cycle progression. PLoS ONE 2013, 8, e69061. [Google Scholar] [CrossRef] [PubMed]
- Budke, B.; Kalin, J.H.; Pawlowski, M.; Zelivianskaia, A.S.; Wu, M.; Kozikowski, A.P.; Connell, P.P. An optimized Rad51 inhibitor that disrupts homologous recombination without requiring michael acceptor reactivity. J. Med. Chem. 2013, 56, 254–263. [Google Scholar] [CrossRef] [PubMed]
- Jensen, R.B.; Carreira, A.; Kowalczykowski, S.C. Purified human BRCA2 stimulates Rad51-mediated recombination. Nature 2010, 467, 678–683. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Zhou, L.; Wu, G.; Konig, H.; Lin, X.; Li, G.; Qiu, X.L.; Chen, C.F.; Hu, C.M.; Goldblatt, E.; et al. A novel small molecule Rad51 inactivator overcomes imatinib-resistance in chronic myeloid leukaemia. EMBO Mol. Med. 2013, 5, 353–365. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Chen, H.; Guo, X.E.; Qiu, X.L.; Hu, C.M.; Chamberlin, A.R.; Lee, W.H. Synthesis, molecular modeling, and biological evaluation of novel Rad51 inhibitors. Eur. J. Med. Chem. 2015, 96, 196–208. [Google Scholar] [CrossRef] [PubMed]
- Game, J.C.; Mortimer, R.K. A genetic study of X-ray sensitive mutants in yeast. Mutat. Res. 1974, 24, 281–292. [Google Scholar] [CrossRef]
- Rijkers, T.; van Den Ouweland, J.; Morolli, B.; Rolink, A.G.; Baarends, W.M.; Van Sloun, P.P.; Lohman, P.H.; Pastink, A. Targeted inactivation of mouse Rad52 reduces homologous recombination but not resistance to ionizing radiation. Mol. Cell. Biol. 1998, 18, 6423–6429. [Google Scholar] [CrossRef] [PubMed]
- Van Dyck, E.; Stasiak, A.Z.; Stasiak, A.; West, S.C. Binding of double-strand breaks in DNA by human Rad52 protein. Nature 1999, 398, 728–731. [Google Scholar] [CrossRef] [PubMed]
- Baumann, P.; West, S.C. Heteroduplex formation by human Rad51 protein: Effects of DNA end-structure, hRP-A and hRad52. J. Mol. Biol. 1999, 291, 363–374. [Google Scholar] [CrossRef] [PubMed]
- Singleton, M.R.; Wentzell, L.M.; Liu, Y.; West, S.C.; Wigley, D.B. Structure of the single-strand annealing domain of human Rad52 protein. Proc. Natl. Acad. Sci. USA 2002, 99, 13492–13497. [Google Scholar] [CrossRef] [PubMed]
- Kagawa, W.; Kurumizaka, H.; Ishitani, R.; Fukai, S.; Nureki, O.; Shibata, T.; Yokoyama, S. Crystal structure of the homologous-pairing domain from the human Rad52 recombinase in the undecameric form. Mol. Cell 2002, 10, 359–371. [Google Scholar] [CrossRef]
- Kagawa, W.; Kagawa, A.; Saito, K.; Ikawa, S.; Shibata, T.; Kurumizaka, H.; Yokoyama, S. Identification of a second DNA binding site in the human Rad52 protein. J. Biol. Chem. 2008, 283, 24264–24273. [Google Scholar] [CrossRef] [PubMed]
- Lieberman, R.; Xiong, D.; James, M.; Han, Y.; Amos, C.I.; Wang, L.; You, M. Functional characterization of RAD52 as a lung cancer susceptibility gene in the 12p13.33 locus. Mol. Carcinogen. 2015. [Google Scholar] [CrossRef] [PubMed]
- Lok, B.H.; Carley, A.C.; Tchang, B.; Powell, S.N. RAD52 inactivation is synthetically lethal with deficiencies in BRCA1 and PALB2 in addition to BRCA2 through RAD51-mediated homologous recombination. Oncogene 2013, 32, 3552–3558. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Scott, S.P.; Bussen, W.; Sharma, G.G.; Guo, G.; Pandita, T.K.; Powell, S.N. Rad52 inactivation is synthetically lethal with BRCA2 deficiency. Proc. Natl. Acad. Sci. USA 2011, 108, 686–691. [Google Scholar] [CrossRef] [PubMed]
- Grimme, J.M.; Spies, M. Fret-based assays to monitor DNA binding and annealing by Rad52 recombination mediator protein. Methods Mol. Biol. 2011, 745, 463–483. [Google Scholar] [PubMed]
- Grimme, J.M.; Honda, M.; Wright, R.; Okuno, Y.; Rothenberg, E.; Mazin, A.V.; Ha, T.; Spies, M. Human Rad52 binds and wraps single-stranded DNA and mediates annealing via two hRad52-ssDNA complexes. Nucleic Acids Res. 2010, 38, 2917–2930. [Google Scholar] [CrossRef] [PubMed]
- Mao, Z.; Bozzella, M.; Seluanov, A.; Gorbunova, V. Comparison of nonhomologous end joining and homologous recombination in human cells. DNA Repair 2008, 7, 1765–1771. [Google Scholar] [CrossRef] [PubMed]
- Mao, Z.; Bozzella, M.; Seluanov, A.; Gorbunova, V. DNA repair by nonhomologous end joining and homologous recombination during cell cycle in human cells. Cell cycle 2008, 7, 2902–2906. [Google Scholar] [CrossRef] [PubMed]
- Lieber, M.R.; Gu, J.; Lu, H.; Shimazaki, N.; Tsai, A.G. Nonhomologous DNA end joining (NHEJ) and chromosomal translocations in humans. Subcell. Biochem. 2010, 50, 279–296. [Google Scholar] [PubMed]
- Davidson, D.; Amrein, L.; Panasci, L.; Aloyz, R. Small molecules, inhibitors of DNA-PK, targeting DNA repair, and beyond. Front. Pharmacol. 2013. [Google Scholar] [CrossRef] [PubMed]
- Wymann, M.P.; Bulgarelli-Leva, G.; Zvelebil, M.J.; Pirola, L.; Vanhaesebroeck, B.; Waterfield, M.D.; Panayotou, G. Wortmannin inactivates phosphoinositide 3-kinase by covalent modification of LYS-802, a residue involved in the phosphate transfer reaction. Mol. Cell. Biol. 1996, 16, 1722–1733. [Google Scholar] [CrossRef] [PubMed]
- Collis, S.J.; DeWeese, T.L.; Jeggo, P.A.; Parker, A.R. The life and death of DNA-PK. Oncogene 2004, 24, 949–961. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Zeng, X.; Gao, C.; Ming, P.; Zhang, J.; Guo, C.; Zhou, L.; Lu, Y.; Wang, L.; Huang, L.; et al. A new arylbenzofuran derivative functions as an anti-tumour agent by inducing DNA damage and inhibiting PARP activity. Sci. Reports 2015. [Google Scholar] [CrossRef] [PubMed]
- Wennström, S.; Downward, J. Role of phosphoinositide 3-kinase in activation of ras and mitogen-activated protein kinase by epidermal growth factor. Mol. Cell. Biol. 1999, 19, 4279–4288. [Google Scholar] [CrossRef] [PubMed]
- Veuger, S.J.; Curtin, N.J.; Richardson, C.J.; Smith, G.C.M.; Durkacz, B.W. Radiosensitization and DNA repair inhibition by the combined use of novel inhibitors of DNA-dependent protein kinase and poly(ADP-Ribose) polymerase-1. Cancer Res. 2003, 63, 6008–6015. [Google Scholar] [PubMed]
- Willmore, E.; de Caux, S.; Sunter, N.J.; Tilby, M.J.; Jackson, G.H.; Austin, C.A.; Durkacz, B.W. A novel DNA-dependent protein kinase inhibitor, NU7026, potentiates the cytotoxicity of topoisomerase ii poisons used in the treatment of leukemia. Blood 2004, 103, 4659–4665. [Google Scholar] [CrossRef] [PubMed]
- Nutley, B.P.; Smith, N.F.; Hayes, A.; Kelland, L.R.; Brunton, L.; Golding, B.T.; Smith, G.C.M.; Martin, N.M.B.; Workman, P.; Raynaud, F.I. Preclinical pharmacokinetics and metabolism of a novel prototype DNA-pk inhibitor nu7026. Br. J. Cancer 2005, 93, 1011–1018. [Google Scholar] [CrossRef] [PubMed]
- Niazi, M.T.; Mok, G.; Heravi, M.; Lee, L.; Vuong, T.; Aloyz, R.; Panasci, L.; Muanza, T. Effects of DNA-dependent protein kinase inhibition by NU7026 on dna repair and cell survival in irradiated gastric cancer cell line n87. Curr. Oncol. 2014, 21, 91–96. [Google Scholar] [CrossRef] [PubMed]
- Tavecchio, M.; Munck, J.; Cano, C.; Newell, D.; Curtin, N. Further characterisation of the cellular activity of the DNA-PK inhibitor, NU7441, reveals potential cross-talk with homologous recombination. Cancer Chemother. Pharmacol. 2012, 69, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Thomas, H.D.; Batey, M.A.; Cowell, I.G.; Richardson, C.J.; Griffin, R.J.; Calvert, A.H.; Newell, D.R.; Smith, G.C.M.; Curtin, N.J. Preclinical evaluation of a potent novel DNA-dependent protein kinase inhibitor NU7441. Cancer Res. 2006, 66, 5354–5362. [Google Scholar] [CrossRef] [PubMed]
- Tichy, A.; Durisova, K.; Salovska, B.; Pejchal, J.; Zarybnicka, L.; Vavrova, J.; Dye, N.; Sinkorova, Z. Radio-sensitization of human leukaemic molt-4 cells by DNA-dependent protein kinase inhibitor, NU7441. Radiat. Environ. Biophys. 2014, 53, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhang, F.; Zhang, W.; Chen, L.; Gao, N.; Men, Y.; Xu, X.; Jiang, Y. Harmine suppresses homologous recombination repair and inhibits proliferation of hepatoma cells. Cancer Biol. Ther. 2015. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Zhang, J.; Wu, H.; Han, E. DNA-PKcs interference sensitizes colorectal cancer cells to a mTOR kinase inhibitor WAY-600. Biochem. Biophys. Res. Commun. 2015, 466, 547–553. [Google Scholar] [CrossRef] [PubMed]
- Munck, J.M.; Batey, M.A.; Zhao, Y.; Jenkins, H.; Richardson, C.J.; Cano, C.; Tavecchio, M.; Barbeau, J.; Bardos, J.; Cornell, L.; et al. Chemosensitisation of cancer cells by KU-0060648; a dual inhibitor of DNA-PK and PI-3K. Mol. Cancer Ther. 2012, 11, 1789–1798. [Google Scholar] [CrossRef] [PubMed]
- Robert, F.; Barbeau, M.; Éthier, S.; Dostie, J.; Pelletier, J. Pharmacological inhibition of DNA-PK stimulates Cas9-mediated genome editing. Genome Med. 2015, 7, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Kashishian, A.; Douangpanya, H.; Clark, D.; Schlachter, S.T.; Eary, C.T.; Schiro, J.G.; Huang, H.; Burgess, L.E.; Kesicki, E.A.; Halbrook, J. DNA-dependent protein kinase inhibitors as drug candidates for the treatment of cancer. Mol. Cancer Ther. 2003, 2, 1257–1264. [Google Scholar] [PubMed]
- Allen, C.; Halbrook, J.; Nickoloff, J.A. Interactive competition between homologous recombination and non-homologous end Joining11NIH grant CA77693 to JAN. Mol. Cancer Res. 2003, 1, 913–920. [Google Scholar] [PubMed]
- Bailey, S.M.; Brenneman, M.A.; Halbrook, J.; Nickoloff, J.A.; Ullrich, R.L.; Goodwin, E.H. The kinase activity of DNA-PK is required to protect mammalian telomeres. DNA Repair 2004, 3, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Yasaei, H.; Gozaly-Chianea, Y.; Slijepcevic, P. Analysis of telomere length and function in radiosensitive mouse and human cells in response to DNA-PKcs inhibition. Genome Integr. 2013. [Google Scholar] [CrossRef] [PubMed]
- Shinohara, E.T.; Geng, L.; Tan, J.; Chen, H.; Shir, Y.; Edwards, E.; Halbrook, J.; Kesicki, E.A.; Kashishian, A.; Hallahan, D.E. DNA-dependent protein kinase is a molecular target for the development of noncytotoxic radiation-sensitizing drugs. Cancer Res. 2005, 65, 4987–4992. [Google Scholar] [CrossRef] [PubMed]
- Davidson, D.; Grenier, J.; Martinez-Marignac, V.; Amrein, L.; Shawi, M.; Tokars, M.; Aloyz, R.; Panasci, L. Effects of the novel DNA dependent protein kinase inhibitor, IC486241, on the DNA damage response to doxorubicin and cisplatin in breast cancer cells. Invest. New Drugs 2012, 30, 1736–1742. [Google Scholar] [CrossRef] [PubMed]
- Davidson, D.; Coulombe, Y.; Martinez-Marignac, V.; Amrein, L.; Grenier, J.; Hodkinson, K.; Masson, J.Y.; Aloyz, R.; Panasci, L. Irinotecan and DNA-PKcs inhibitors synergize in killing of colon cancer cells. Invest. New Drugs 2012, 30, 1248–1256. [Google Scholar] [CrossRef] [PubMed]
- Take, Y.; Kumano, M.; Hamano, Y.; Fukatsu, H.; Teraoka, H.; Nishimura, S.; Okuyama, A. OK-1035, a selective inhibitor of DNA-dependent protein kinase. Biochem. Biophys. Res. Commun. 1995, 215, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Take, Y.; Kumano, M.; Teraoka, H.; Nishimura, S.; Okuyama, A. DNA-dependent protein kinase inhibitor (OK-1035) suppresses p21 expression in HCT116 cells containing wild-type p53 induced by adriamycin. Biochem. Biophys. Res. Commun. 1996, 221, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Kruszewski, M.; Wojewódzka, M.; Iwaneńko, T.; Szumiel, I.; Okuyama, A. Differential inhibitory effect of OK-1035 on DNA repair in L5178Y murine lymphoma sublines with functional or defective repair of double strand breaks. Mut. Res. 1998, 409, 31–36. [Google Scholar] [CrossRef]
- Stockley, M.; Clegg, W.; Fontana, G.; Golding, B.T.; Martin, N.; Rigoreau, L.J.M.; Smith, G.C.M.; Griffin, R.J. Synthesis, crystal structure determination, and biological properties of the DNA-dependent protein kinase (DNA-PK) inhibitor 3-cyano-6-hydrazonomethyl-5-(4-pyridyl)pyrid-[1h]-2-one (OK-1035). Bioorg. Med. Chem. Lett. 2001, 11, 2837–2841. [Google Scholar] [CrossRef]
- Ismail, I.H.; Martensson, S.; Moshinsky, D.; Rice, A.; Tang, C.; Howlett, A.; McMahon, G.; Hammarsten, O. SU11752 inhibits the DNA-dependent protein kinase and DNA double-strand break repair resulting in ionizing radiation sensitization. Oncogene 2003, 23, 873–882. [Google Scholar] [CrossRef] [PubMed]
- Ohta, T. Modification of genotoxicity by naturally occurring flavorings and their derivatives. Crit. Rev. Toxicol. 1993, 23, 127–146. [Google Scholar] [CrossRef] [PubMed]
- Durant, S.; Karran, P. Vanillins—A novel family of DNA-PK inhibitors. Nucl. Acids Res. 2003, 31, 5501–5512. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.Y.; Lai, P.Y.; Chen, T.Y.; Chung, B.C. NR5A1 prevents centriole splitting by inhibiting centrosomal DNA-PK activation and β-catenin accumulation. Cell Commun. Signal 2014. [Google Scholar] [CrossRef]
- Guo, L.; Liu, X.; Nishikawa, K.; Plunkett, W. Inhibition of topoisomerase IIα and G2 cell cycle arrest by NK314, a novel benzo[c]phenanthridine currently in clinical trials. Mol. Cancer Ther. 2007, 6, 1501–1508. [Google Scholar] [CrossRef] [PubMed]
- Onda, T.; Toyoda, E.; Miyazaki, O.; Seno, C.; Kagaya, S.; Okamoto, K.; Nishikawa, K. NK314, a novel topoisomerase ii inhibitor, induces rapid DNA double-strand breaks and exhibits superior antitumor effects against tumors resistant to other topoisomerase ii inhibitors. Cancer Lett. 2008, 259, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Toyoda, E.; Kagaya, S.; Cowell, I.G.; Kurosawa, A.; Kamoshita, K.; Nishikawa, K.; Iiizumi, S.; Koyama, H.; Austin, C.A.; Adachi, N. NK314, a topoisomerase II inhibitor that specifically targets the α isoform. J. Biol. Chem. 2008, 283, 23711–23720. [Google Scholar] [CrossRef] [PubMed]
- Hisatomi, T.; Sueoka-Aragane, N.; Sato, A.; Tomimasu, R.; Ide, M.; Kurimasa, A.; Okamoto, K.; Kimura, S.; Sueoka, E. NK314 potentiates antitumor activity with adult T-cell leukemia-lymphoma cells by inhibition of dual targets on topoisomerase IIα and DNA-dependent protein kinase. Blood 2011, 117, 3575–3584. [Google Scholar] [CrossRef] [PubMed]
- Mortensen, D.S.; Perrin-Ninkovic, S.M.; Shevlin, G.; Elsner, J.; Zhao, J.; Whitefield, B.; Tehrani, L.; Sapienza, J.; Riggs, J.R.; Parnes, J.S.; et al. Optimization of a series of triazole containing mammalian target of rapamycin (mTOR) kinase inhibitors and the discovery of CC-115. J. Med. Chem. 2015, 58, 5599–5608. [Google Scholar] [CrossRef] [PubMed]
- Shawi, M.; Chu, T.W.; Martinez-Marignac, V.; Yu, Y.; Gryaznov, S.M.; Johnston, J.B.; Lees-Miller, S.P.; Assouline, S.E.; Autexier, C.; Aloyz, R. Telomerase contributes to fludarabine resistance in primary human leukemic lymphocytes. PLoS ONE 2013, 8, e70428. [Google Scholar] [CrossRef] [PubMed]
- Qi, D.; Hu, Y.; Zhang, Y.; Peng, T.; Ji, W. Effect of KU70 expression on radiosensitivity in renal carcinoma 786-O cells. Cancer Cell Int. 2014, 14, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhong, S.; Zhu, X.; Dziegielewska, B.; Ellenberger, T.; Wilson, G.M.; MacKerell, A.D.; Tomkinson, A.E. Rational design of human DNA ligase inhibitors that target cellular DNA replication and repair. Cancer Res. 2008, 68, 3169–3177. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, M.; Nambiar, M.; Sharma, S.; Karki, S.S.; Goldsmith, G.; Hegde, M.; Kumar, S.; Pandey, M.; Singh, R.K.; Ray, P.; et al. An inhibitor of nonhomologous end-joining abrogates double-strand break repair and impedes cancer progression. Cell 2012, 151, 1474–1487. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.F.; Chen, H.Y.; Tsai, F.J.; Liu, S.H.; Chen, C.Y.; Chen, C.Y.C. Search for novel remedies to augment radiation resistance of inhabitants of fukushima and chernobyl disasters: Identifying DNA repair protein XRCC4 inhibitors. J. Biomol. Struct. Dyn. 2011, 29, 325–337. [Google Scholar] [CrossRef] [PubMed]
- Tobin, L.A.; Robert, C.; Rapoport, A.P.; Gojo, I.; Baer, M.R.; Tomkinson, A.E.; Rassool, F.V. Targeting abnormal DNA double strand break repair in tyrosine kinase inhibitor-resistant chronic myeloid leukemias. Oncogene 2013, 32, 1784–1793. [Google Scholar] [CrossRef] [PubMed]
- Tobin, L.A.; Robert, C.; Nagaria, P.; Chumsri, S.; Twaddell, W.; Ioffe, O.B.; Greco, G.E.; Brodie, A.H.; Tomkinson, A.E.; Rassool, F.V. Targeting abnormal DNA repair in therapy-resistant breast cancers. Mol. Cancer Res. 2012, 10, 96–107. [Google Scholar] [CrossRef] [PubMed]
- Gelmon, K.A.; Tischkowitz, M.; Mackay, H.; Swenerton, K.; Robidoux, A.; Tonkin, K.; Hirte, H.; Huntsman, D.; Clemons, M.; Gilks, B.; et al. Olaparib in patients with recurrent high-grade serous or poorly differentiated ovarian carcinoma or triple-negative breast cancer: A phase 2, multicentre, open-label, non-randomised study. Lancet Oncol. 2011, 12, 852–861. [Google Scholar] [CrossRef]
- Lord, C.J.; McDonald, S.; Swift, S.; Turner, N.C.; Ashworth, A. A high-throughput RNA interference screen for DNA repair determinants of PARP inhibitor sensitivity. DNA repair 2008, 7, 2010–2019. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.C.; Lord, C.J.; Iorns, E.; Brough, R.; Swift, S.; Elliott, R.; Rayter, S.; Tutt, A.N.; Ashworth, A. A synthetic lethal sirna screen identifying genes mediating sensitivity to a PARP inhibitor. EMBO J. 2008, 27, 1368–1377. [Google Scholar] [CrossRef] [PubMed]
- Fang, Z.; Kozlov, S.; McKay, M.J.; Woods, R.; Birrell, G.; Sprung, C.N.; Murrell, D.F.; Wangoo, K.; Teng, L.; Kearsley, J.H.; et al. Low levels of ATM in breast cancer patients with clinical radiosensitivity. Genome Integr. 2010. [Google Scholar] [CrossRef] [PubMed]
- Tommiska, J.; Bartkova, J.; Heinonen, M.; Hautala, L.; Kilpivaara, O.; Eerola, H.; Aittomaki, K.; Hofstetter, B.; Lukas, J.; von Smitten, K.; et al. The DNA damage signalling kinase ATM is aberrantly reduced or lost in BRCA1/BRCA2-deficient and ER/PR/ERBB2-triple-negative breast cancer. Oncogene 2008, 27, 2501–2506. [Google Scholar] [CrossRef] [PubMed]
- Kang, B.; Guo, R.F.; Tan, X.H.; Zhao, M.; Tang, Z.B.; Lu, Y.Y. Expression status of ataxia-telangiectasia-mutated gene correlated with prognosis in advanced gastric cancer. Mutat. Res. 2008, 638, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Getz, G.; Wheeler, D.A.; Mardis, E.R.; McLellan, M.D.; Cibulskis, K.; Sougnez, C.; Greulich, H.; Muzny, D.M.; Morgan, M.B.; et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature 2008, 455, 1069–1075. [Google Scholar] [CrossRef] [PubMed]
- Boultwood, J. Ataxia telangiectasia gene mutations in leukaemia and lymphoma. J. Clin. Pathol. 2001, 54, 512–516. [Google Scholar] [CrossRef] [PubMed]
- Schaffner, C.; Stilgenbauer, S.; Rappold, G.A.; Dohner, H.; Lichter, P. Somatic ATM mutations indicate a pathogenic role of ATM in B-cell chronic lymphocytic leukemia. Blood 1999, 94, 748–753. [Google Scholar] [PubMed]
- Williamson, C.T.; Kubota, E.; Hamill, J.D.; Klimowicz, A.; Ye, R.; Muzik, H.; Dean, M.; Tu, L.; Gilley, D.; Magliocco, A.M.; et al. Enhanced cytotoxicity of PARP inhibition in mantle cell lymphoma harbouring mutations in both ATM and p53. EMBO Mol. Med. 2012, 4, 515–527. [Google Scholar] [CrossRef] [PubMed]
- Kubota, E.; Williamson, C.T.; Ye, R.; Elegbede, A.; Peterson, L.; Lees-Miller, S.P.; Bebb, D.G. Low ATM protein expression and depletion of p53 correlates with olaparib sensitivity in gastric cancer cell lines. Cell Cycle 2014, 13, 2129–2137. [Google Scholar] [CrossRef] [PubMed]
- Bang, Y.J.; Im, S.A.; Lee, K.W.; Cho, J.Y.; Song, E.K.; Lee, K.H.; Kim, Y.H.; Park, J.O.; Chun, H.G.; Zang, D.Y.; et al. Randomized, double-blind phase II trial with prospective classification by ATM protein level to evaluate the efficacy and tolerability of olaparib plus paclitaxel in patients with recurrent or metastatic gastric cancer. J. Clin. Oncol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Fojo, T.; Bates, S. Mechanisms of resistance to PARP inhibitors—Three and counting. Cancer Discov. 2013, 3, 20–23. [Google Scholar] [CrossRef] [PubMed]
- Mateos-Gomez, P.A.; Gong, F.; Nair, N.; Miller, K.M.; Lazzerini-Denchi, E.; Sfeir, A. Mammalian polymerase θ promotes alternative NHEJ and suppresses recombination. Nature 2015, 518, 254–257. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Velic, D.; Couturier, A.M.; Ferreira, M.T.; Rodrigue, A.; Poirier, G.G.; Fleury, F.; Masson, J.-Y. DNA Damage Signalling and Repair Inhibitors: The Long-Sought-After Achilles’ Heel of Cancer. Biomolecules 2015, 5, 3204-3259. https://doi.org/10.3390/biom5043204
Velic D, Couturier AM, Ferreira MT, Rodrigue A, Poirier GG, Fleury F, Masson J-Y. DNA Damage Signalling and Repair Inhibitors: The Long-Sought-After Achilles’ Heel of Cancer. Biomolecules. 2015; 5(4):3204-3259. https://doi.org/10.3390/biom5043204
Chicago/Turabian StyleVelic, Denis, Anthony M. Couturier, Maria Tedim Ferreira, Amélie Rodrigue, Guy G. Poirier, Fabrice Fleury, and Jean-Yves Masson. 2015. "DNA Damage Signalling and Repair Inhibitors: The Long-Sought-After Achilles’ Heel of Cancer" Biomolecules 5, no. 4: 3204-3259. https://doi.org/10.3390/biom5043204
APA StyleVelic, D., Couturier, A. M., Ferreira, M. T., Rodrigue, A., Poirier, G. G., Fleury, F., & Masson, J.-Y. (2015). DNA Damage Signalling and Repair Inhibitors: The Long-Sought-After Achilles’ Heel of Cancer. Biomolecules, 5(4), 3204-3259. https://doi.org/10.3390/biom5043204