
Fluorinated Carbohydrates as Lectin Ligands: 19F-Based Direct STD Monitoring for Detection of Anomeric Selectivity


Abstract
:
1. Introduction
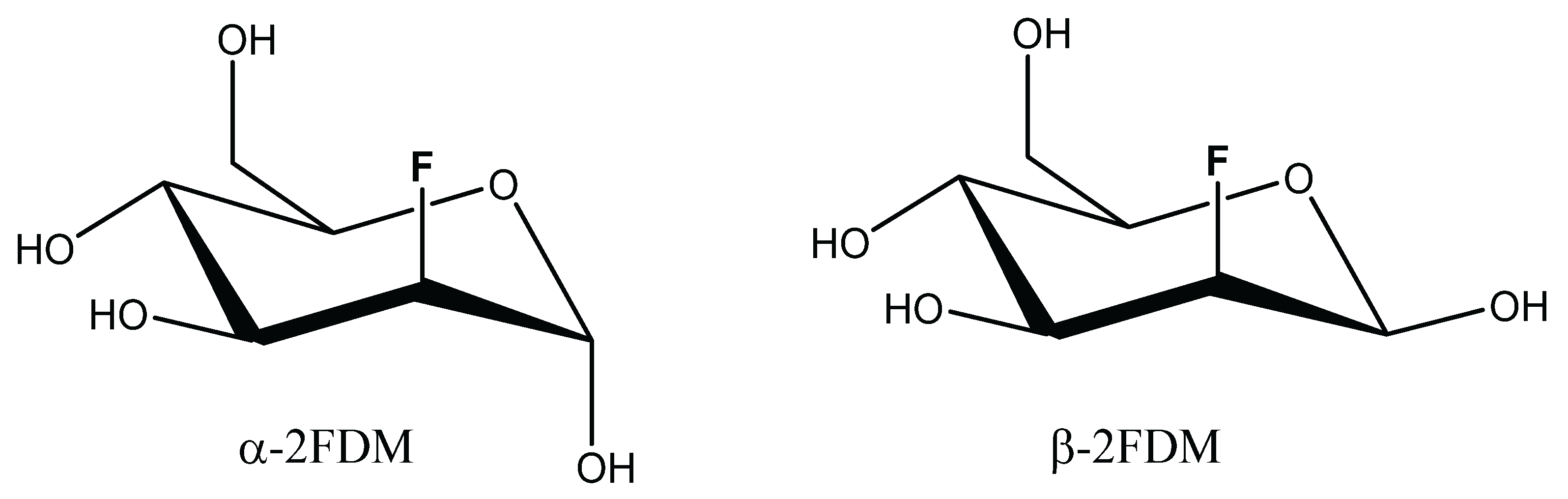
2. Material and Methods
2.1. Chemicals and Reagents
2.2. Purification of ConA
2.3. NMR Experiments
2.4. Kinetic Model
2.4.1. Components
2.4.2. Equations
| R* + α ⇆ R*α | koffα/kon = KDα (konα ≡ konβ ≡ kon) | (1) |
| R* + β ⇆ R*β | koffβ/kon = KDβ (konβ ≡ konα ≡ kon) | (2) |
| R*α → R*α* | km (kmα ≡ kmβ) | (3) |
| R*β → R*β* | km (kmβ ≡ kmα) | (4) |
| α* → α | krα | (5) |
| β* → β | krβ | (6) |
| R* + α* ⇆ R*α* | koffα/kon (KDα* ≡ KDα) | (7) |
| R* + β* ⇆ R*β* | koffβ/kon (KDβ* ≡ KDβ) | (8) |
2.4.3. Derived Differential Equations Used in Dynafit Fitting Proccedure
| d[R*]/dt = −kon[R*][α] + koffα[R*α] − kon[R*][α*] + koffα[R*α*] − kon[R*][β] + koffβ[R*β] − kon[R*][β*] + koffβ[R*β*] | (9) |
| d[α]/dt = −kon[R*][α] + koffα[R*α] + krα[α*] | (10) |
| d[α*]/dt = −kon[R*][α*] + koffα[R*α*] − krα[α*] | (11) |
| d[β]/dt = −kon[R*][β] + koffβ[R*β] + krβ[β*] | (12) |
| d[β*]/dt = −kon[R*][β*] + koffβ[R*β*] − krβ[β*] | (13) |
| d[R*α]/dt = +kon[R*][α] − koffα[R*α] − km[R*α] | (14) |
| d[R*β]/dt = +kon[R*][β] − koffβ[R*β] − km[R*β] | (15) |
| d[R*α*]/dt = +kon[R*][α*] − koffα[R*α*] + km[R*α] | (16) |
| d[R*β*]/dt = +kon [R*][β*] − koffβ[R*β*] + km[R*β] | (17) |
2.4.4. Constants
2.4.5. Concentrations
2.4.6. Experimental Data Sets
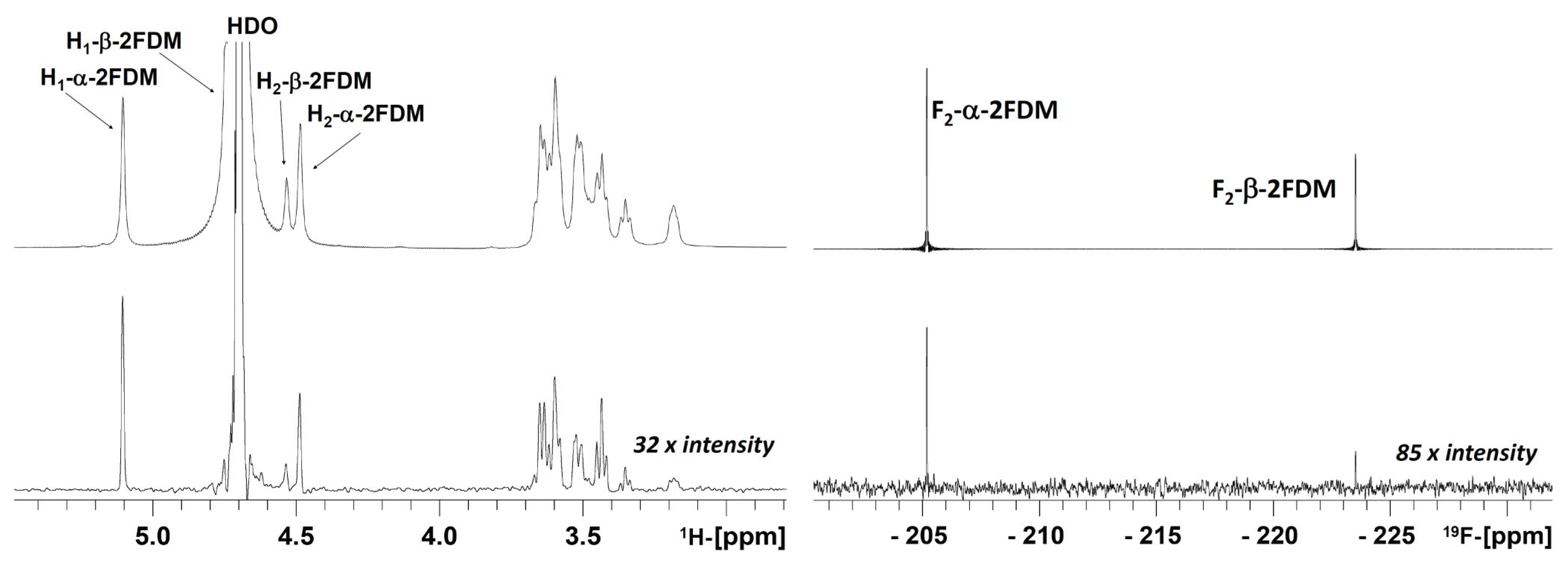
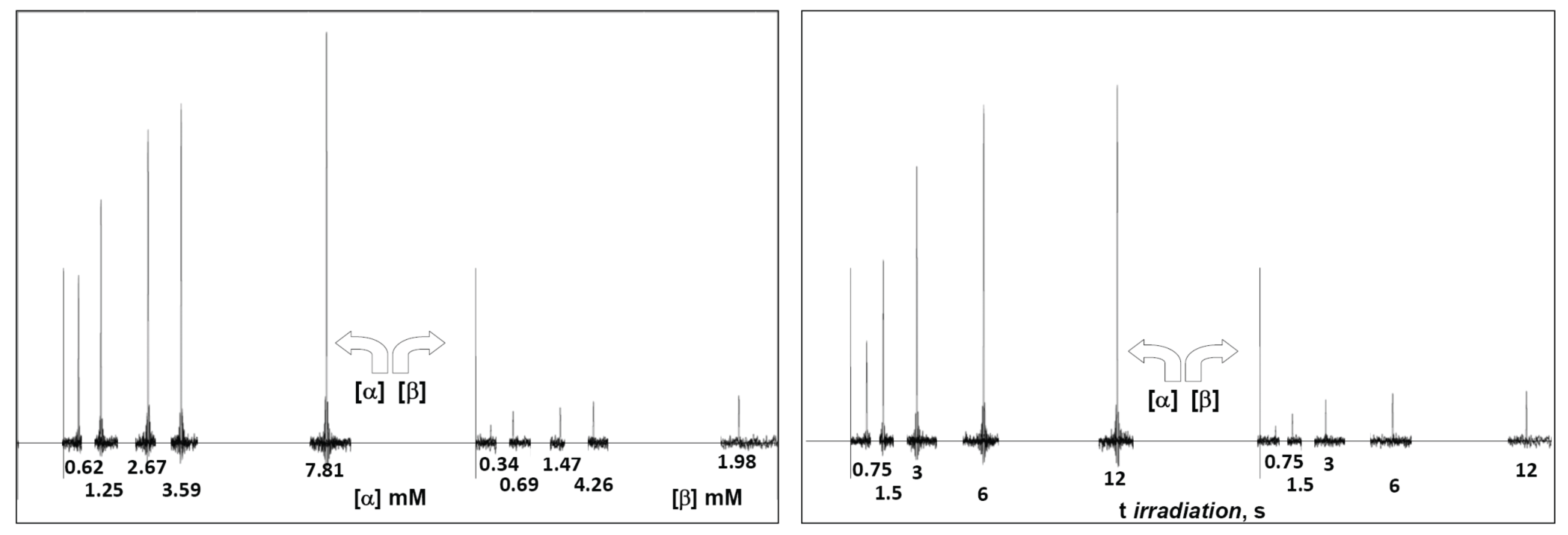
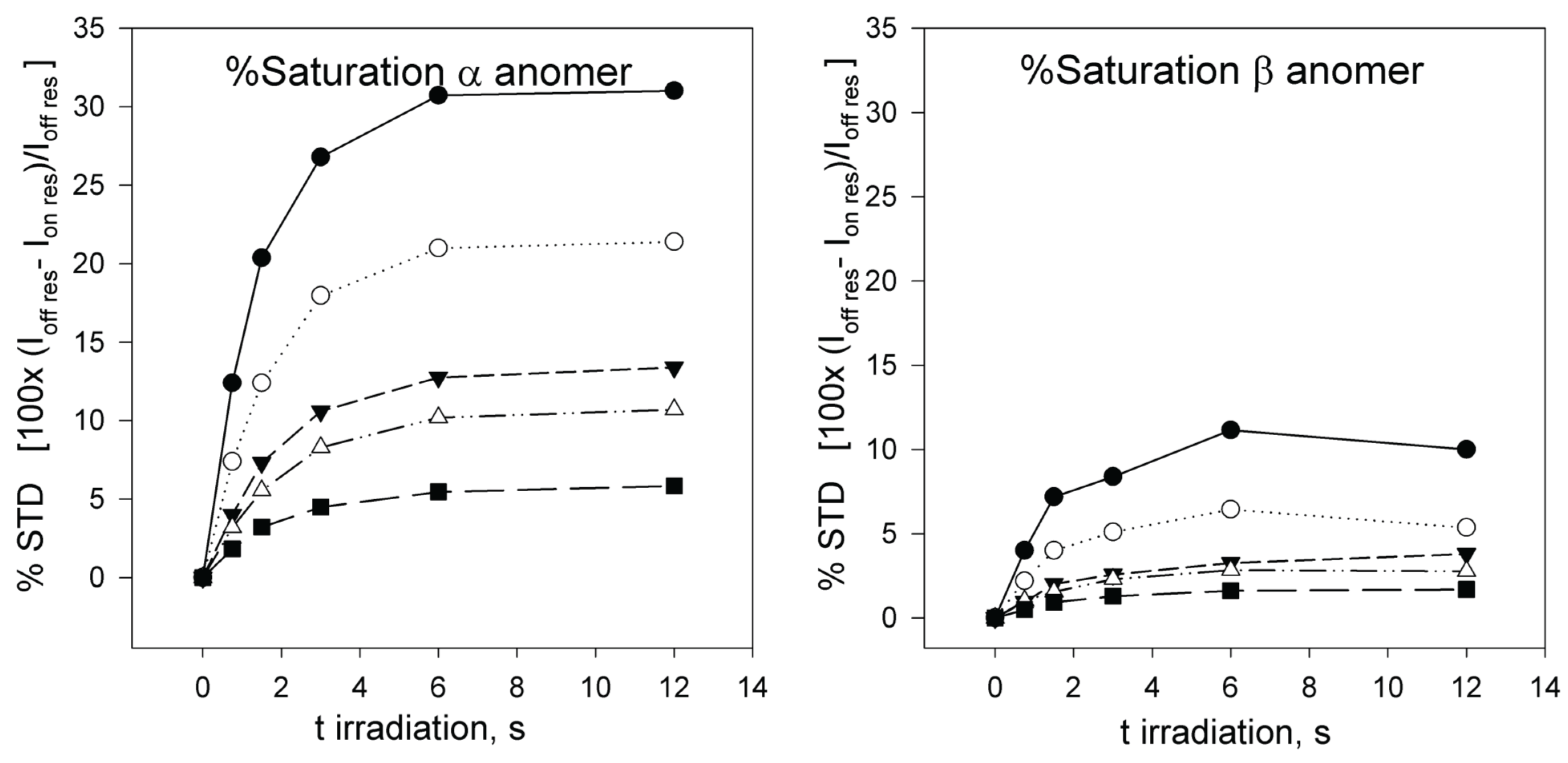
3. Results and Discussions
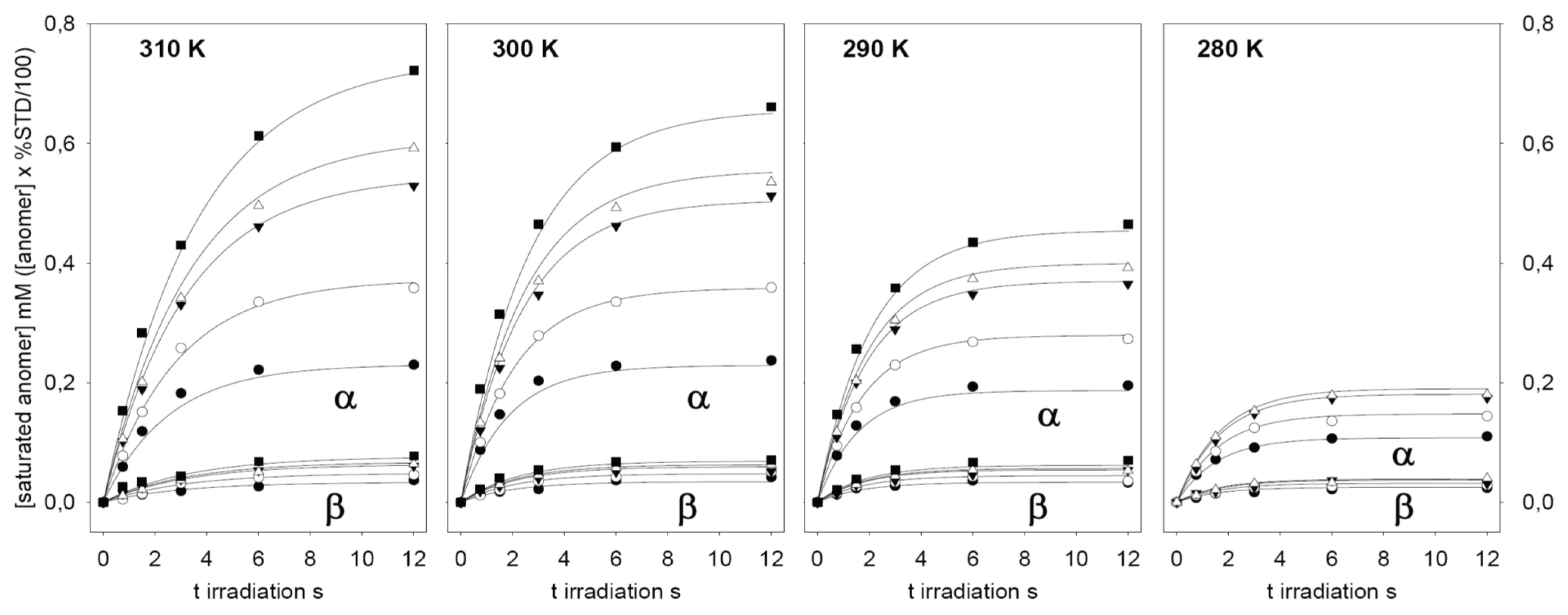

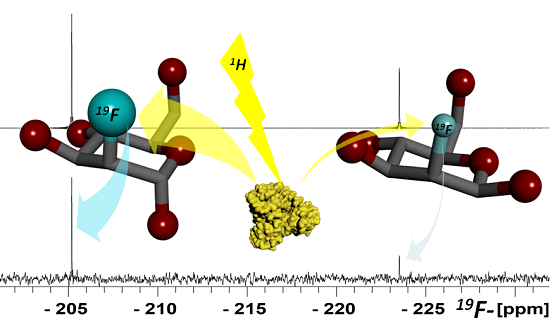{kind=link}
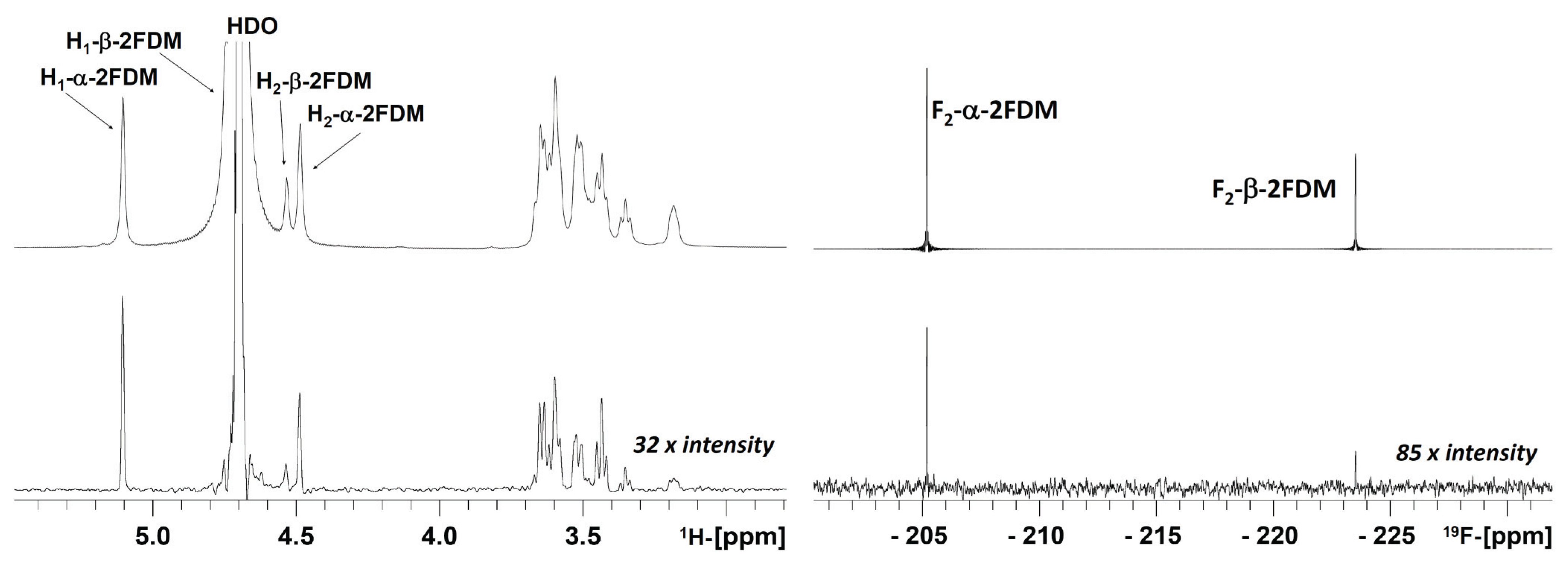{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Temperature K | 280 | 290 | 300 | 310 |
|---|---|---|---|---|
| KDα (mM) | 0.43 ± 0.04 | 0.59 ± 0.04 | 0.68 ± 0.05 | 0.88 ± 0.05 |
| KDβ (mM) | 1.15 ± 0.2 | 2.1 ± 0.24 | 3.1 ± 0.5 | 4.7 ± 0.7 |
| KD (mM) ª | 0.54 | 0.78 | 0.94 | 1.15 |
| krα (s−1) | 0.57 ± 0.02 | 0.46 ± 0.01 | 0.35 ± 0.01 | 0.25 ± 0.01 |
| 1/T1α (s−1) | 0.84 | 0.66 | 0.50 | 0.42 |
| krβ (s−1) | 0.60 ± 0.09 | 0.53 ± 0.07 | 0.45 ± 0.09 | 0.30 ± 0.06 |
| 1/T1β (s−1) | 0.95 | 0.79 | 0.59 | 0.50 |
| km (s−1) | 1.6 ± 0.06 | 3.1 ± 0.07 | 3.4 ± 0.01 | 2.84 ± 0.07 |
| %α %β | 65.9% 34.1% | 65.8% 34.2% | 64.5% 35.5% | 63.3% 36.7% |
4. Conclusions
Acknowledgments
Author Contributions
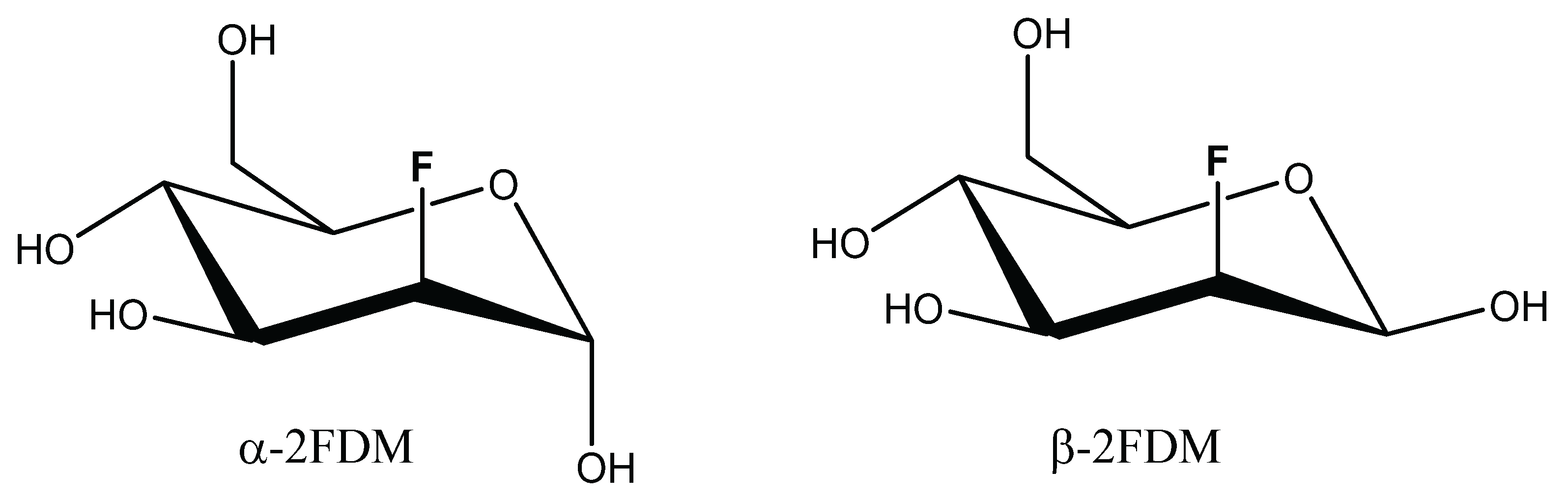
Abbreviations
| 2FDM | 2-fluoro-2-deoxy- d-mannose |
| 2α-FDM | 2-fluoro-2-deoxy-α- d-mannose |
| 2β-FDM | 2-fluoro-2-deoxy-β- d-mannose |
| ConA | concanavalin A |
| STD | Saturation Transfer Difference |
| NMR | Nuclear Magnetic Resonance |
| 1H → 1H STDre19F-NMR | H,H-Saturation Transfer Difference (STD) experiment with relay to 19F |
Conflicts of Interest
References
- Roseman, S. Reflections on glycobiology. J. Biol. Chem. 2001, 276, 41527–41542. [Google Scholar] [CrossRef] [PubMed]
- Gabius, H.-J. The Sugar Code: Fundamentals of Glycosciences; Wiley-VCH: Weinheim, Germany, 2009. [Google Scholar]
- Jiménez-Barbero, J.; Peters, T. NMR Spectroscopy of Glycoconjugates; Wiley-VCH: Weinheim, Germany, 2002. [Google Scholar]
- Kamerling, J.P.; Vliegenthart, J.F.G. High-resolution 1H-nuclear magnetic resonance spectroscopy of oligosaccharide-alditols released from mucin-type O-glycoproteins. In Carbohydrates and Nucleic Acids; Berliner, L., Reuben, J., Eds.; Springer: New York, NY, USA, 1992; Volume 10, pp. 1–194. [Google Scholar]
- Vliegenthart, J.F.G.; Dorland, L.; VanHalbeek, H. High-resolution, 1H-nuclear magnetic resonance spectroscopy as a tool in the structural analysis of carbohydrates related to glycoproteins. Adv. Carbohydr. Chem. Biochem. 1983, 41, 209–374. [Google Scholar]
- Vliegenthart, J.F.G.; van Halbeek, H.; Dorland, L. The applicability of 500 MHz high-resolution 1H-NMR spectroscopy for the structure determination of carbohydrates derived from glycoproteins. Pure Appl. Chem. 1981, 53, 45–77. [Google Scholar] [CrossRef] [Green Version]
- Poveda, A.; Jiménez-Barbero, J. NMR studies of carbohydrate-protein interactions in solution. Chem. Soc. Rev. 1998, 27, 133–143. [Google Scholar] [CrossRef]
- Roldos, V.; Cañada, F.J.; Jiménez-Barbero, J. Carbohydrate-protein interactions: A 3D view by NMR. ChemBioChem 2011, 12, 990–1005. [Google Scholar] [CrossRef] [PubMed]
- Diercks, T.; Coles, M.; Kessler, H. Applications of NMR in drug discovery. Curr. Opin. Chem. Biol. 2001, 5, 285–291. [Google Scholar] [CrossRef]
- Homans, S.W. NMR spectroscopy tools for structure-aided drug design. Angew. Chem. Int. Ed. 2004, 43, 290–300. [Google Scholar] [CrossRef] [PubMed]
- Dwek, R.A.; Kent, P.W.; Xavier, A.V. N-Fluoroacetyl-d-glucosamine as a molecular probe of lysozyme structure using 19F fluorine nuclear magnetic resonance techniques. Eur. J. Biochem. 1971, 23, 343–348. [Google Scholar] [CrossRef] [PubMed]
- Millett, F.; Raftery, M.A. 19F Nuclear magnetic resonance study of binding of trifluoroacetylglucosamine oligomers to lysozyme. Biochemistry 1972, 11, 1639–1643. [Google Scholar] [CrossRef] [PubMed]
- Midoux, P.; Grivet, J.P.; Monsigny, M. Lectin-sugar interactions: The binding of 1-O-methyl-di-N-trifluoroacetyl-b-chitobioside to wheat germ agglutinin. FEBS Lett. 1980, 120, 29–32. [Google Scholar] [CrossRef]
- Jordan, F.; Bahr, H.; Patrick, J.; Woo, P.W. Nuclear magnetic resonance studies on wheat germ agglutinin-monomeric amino sugar interactions. Arch. Biochem. Biophys. 1981, 207, 81–86. [Google Scholar] [CrossRef]
- Alter, G.M.; Magnuson, J.A. Characterization of concanavalin A sugar binding site by 19F nuclear magnetic resonance. Biochemistry 1974, 13, 4038–4045. [Google Scholar] [CrossRef] [PubMed]
- Dalvit, C. Ligand- and substrate-based 19F-NMR screening: Principles and applications to drug discovery. Prog. Nucl. Magn. Reson. Spectrosc. 2007, 51, 243–271. [Google Scholar] [CrossRef]
- Cobb, S.L.; Murphy, C.D. 19F-NMR applications in chemical biology. J. Fluor. Chem. 2009, 130, 132–143. [Google Scholar] [CrossRef]
- Bartusik, D.; Tomanek, B. Detection of 19F-labeled biopharmaceuticals in cell cultures with magnetic resonance. Adv. Drug Deliv. Rev. 2013, 65, 1056–1064. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Viel, S.; Ziarelli, F.; Peng, L. 19F-NMR: A valuable tool for studying biological events. Chem. Soc. Rev. 2013, 42, 7971–7982. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.X.; Hallac, R.R.; Chiguru, S.; Mason, R.P. New frontiers and developing applications in 19F-NMR. Prog. Nucl. Magn. Reson. Spectrosc. 2013, 70, 25–49. [Google Scholar] [CrossRef] [PubMed]
- Gabius, H.-J.; Kaltner, H.; Kopitz, J.; André, S. The glycobiology of the CD system: A dictionary for translating marker designations into glycan/lectin structure and function. Trends Biochem. Sci. 2015, 40, 360–376. [Google Scholar] [CrossRef] [PubMed]
- Gabius, H.-J. The magic of the sugar code. Trends Biochem. Sci. 2015. [Google Scholar] [CrossRef] [PubMed]
- André, S.; Kaltner, H.; Manning, J.C.; Murphy, P.V.; Gabius, H.-J. Lectins: Getting familiar with translators of the sugar code. Molecules 2015, 20, 1788–1823. [Google Scholar] [CrossRef] [PubMed]
- Solís, D.; Jiménez-Barbero, J.; Martín-Lomas, M.; Díaz-Mauriño, T. Probing hydrogen-bonding interactions of bovine heart galectin-1 and methyl β-lactoside by use of engineered ligands. Eur. J. Biochem. 1994, 223, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Solís, D.; Fernández, P.; Díaz-Mauriño, T.; Jiménez-Barbero, J.; Martín-Lomas, M. Hydrogen-bonding pattern of methyl β-lactoside binding to the Ricinus communis lectins. Eur. J. Biochem. 1993, 214, 677–683. [Google Scholar] [CrossRef] [PubMed]
- Lemieux, R.U. The origin of the specificity in the recognition of oligosaccharides by proteins. Chem. Soc. Rev. 1989, 18, 347–374. [Google Scholar] [CrossRef]
- Solís, D.; Romero, A.; Kaltner, H.; Gabius, H.-J.; Diaz-Mauriño, T. Different architecture of the combining site of the two chicken galectins revealed by chemical mapping studies with synthetic ligand derivatives. J. Biol. Chem. 1996, 271, 12744–12748. [Google Scholar] [PubMed]
- Bhattacharyya, L.; Brewer, C.F. Lectin-carbohydrate interactions—Studies of the nature of hydrogen-bonding between d-galactose and certain d-galactose-specific lectins, and between d-mannose and concanavalin A. Eur. J. Biochem. 1988, 176, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Bittiger, H.; Schnebli, H.P. Concanavalin A as a Tool; Wiley: London, UK, 1976; p. 639. [Google Scholar]
- Goldstein, I.J.; Hollerman, C.E.; Smith, E.E. Protein-carbohydrate interaction. II. Inhibition studies on the interaction of concanavalin A with polysaccharides. Biochemistry 1965, 4, 876–883. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.E.; Goldstein, I.J. Protein-carbohydrate interaction. V. Further inhibition studies directed toward defining the stereochemical requirements of the reactive sites of concanavalin A. Arch. Biochem. Biophys. 1967, 121, 88–95. [Google Scholar] [PubMed]
- Schwarz, F.P.; Puri, K.D.; Bhat, R.G.; Surolia, A. Thermodynamics of monosaccharide binding to concanavalin A, pea (Pisum sativum) lectin, and lentil (Lens culinaris) lectin. J. Biol. Chem. 1993, 268, 7668–7677. [Google Scholar] [PubMed]
- Schwarz, F.P.; Misquith, S.; Surolia, A. Effect of substituent on the thermodynamics of d-glucopyranoside binding to concanavalin A, pea (Pisum sativum) lectin and lentil (Lens culinaris) lectin. Biochem. J. 1996, 316, 123–129. [Google Scholar] [CrossRef] [PubMed]
- Benkovic, S.J. Anomeric specificity of carbohydrate-utilizing enzymes. Methods Enzymol. 1979, 63, 370–379. [Google Scholar] [PubMed]
- Salas, J.; Salas, M.; Viñuela, E.; Sols, A. Glucokinase of rabbit liver. J. Biol. Chem. 1965, 240, 1014–1018. [Google Scholar] [PubMed]
- Gabius, H.-J.; André, S.; Jiménez-Barbero, J.; Romero, A.; Solís, D. From lectin structure to functional glycomics: Principles of the sugar code. Trends Biochem. Sci. 2011, 36, 298–313. [Google Scholar] [CrossRef] [PubMed]
- Dahlquist, F.W.; Raftery, M.A. A nuclear magnetic resonance study of association equilibria and enzyme-boud environments of N-acetyl-d-glucosamine anomers and lysozyme. Biochemistry 1968, 7, 3269–3276. [Google Scholar] [CrossRef] [PubMed]
- Borrok, M.J.; Kiessling, L.L.; Forest, K.T. Conformational changes of glucose/galactose-binding protein illuminated by open, unliganded, and ultra-high-resolution ligand-bound structures. Protein Sci. 2007, 16, 1032–1041. [Google Scholar] [CrossRef] [PubMed]
- Gehring, K.; Williams, P.G.; Pelton, J.G.; Morimoto, H.; Wemmer, D.E. Tritium NMR spectroscopy of ligand binding to maltose-binding protein. Biochemistry 1991, 30, 5524–5531. [Google Scholar] [CrossRef] [PubMed]
- Potts, J.R.; Kuchel, P.W. Anomeric preference of fluoroglucose exchange across human red-cell membranes. 19F-NMR studies. Biochem. J. 1992, 281, 753–759. [Google Scholar] [CrossRef] [PubMed]
- Katiyar, S.; Singh, A.; Surolia, A. The interaction of N-trifluoroacetylgalactosamine and its derivatives with winged bean (Psophocarpus tetragonolobus) basic agglutinin reveals differential mechanism of their recognition: A fluorine-19 nuclear magnetic resonance study. Glycoconj. J. 2014, 31, 537–543. [Google Scholar] [CrossRef] [PubMed]
- Diercks, T.; Ribeiro, J.P.; Cañada, F.J.; André, S.; Jiménez-Barbero, J.; Gabius, H.-J. Fluorinated carbohydrates as lectin ligands: Versatile sensors in 19F-detected saturation transfer difference NMR spectroscopy. Chem. Eur. J. 2009, 15, 5666–5668. [Google Scholar] [CrossRef] [PubMed]
- André, S.; Cañada, F.J.; Shiao, T.C.; Largartera, L.; Diercks, T.; Bergeron-Brlek, M.; el Biari, K.; Papadopoulos, A.; Ribeiro, J.P.; Touaibia, M.; et al. Fluorinated carbohydrates as lectin ligands: biorelevant sensors with capacity to monitor anomer affinity in 19F-NMR-based inhibitor screening. Eur. J. Org. Chem. 2012, 2012, 4354–4364. [Google Scholar] [CrossRef]
- Mayer, M.; Meyer, B. Characterization of ligand binding by saturation transfer difference NMR spectroscopy. Angew. Chem. Int. Ed. 1999, 38, 1784–1788. [Google Scholar] [CrossRef]
- Meyer, B.; Peters, T. NMR spectroscopy techniques for screening and identifying ligand binding topProtein receptors. Angew. Chem. Int. Ed. 2003, 42, 864–890. [Google Scholar] [CrossRef] [PubMed]
- Angulo, J.; Nieto, P.M. STD-NMR: Application to transient interactions between biomolecules—A quantitative approach. Eur. Biophys. J. 2011, 40, 1357–1369. [Google Scholar] [CrossRef] [PubMed]
- Angulo, J.; Enríquez-Navas, P.M.; Nieto, P.M. Ligand-receptor binding affinities from saturation transfer difference (STD) NMR spectroscopy: The binding isotherm of STD initial growth rates. Chem. Eur. J. 2010, 16, 7803–7812. [Google Scholar] [CrossRef] [PubMed]
- Kemper, S.; Patel, M.K.; Errey, J.C.; Davis, B.G.; Jones, J.A.; Claridge, T.D.W. Group epitope mapping considering relaxation of the ligand (GEM-CRL): Including longitudinal relaxation rates in the analysis of saturation transfer difference (STD) experiments. J. Magn. Reson. 2010, 203, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.L.; Kline, A.D.; Mo, H.P.; Shapiro, M.J.; Zartler, E.R. The effect of relaxation on the epitope mapping by saturation transfer difference NMR. J. Magn. Reson. 2003, 163, 270–276. [Google Scholar] [CrossRef]
- Jayalakshmi, V.; Krishna, N.R. Complete relaxation and conformational exchange matrix (CORCEMA) analysis of intermolecular saturation transfer effects in reversibly forming ligand-receptor complexes. J. Magn. Reson. 2002, 155, 106–118. [Google Scholar] [CrossRef] [PubMed]
- Kuzmic, P. DynaFit: A software package for enzymology. Methods Enzymol. 2009, 467, 247–280. [Google Scholar] [PubMed]
- Marcelo, F.; Garcia-Martin, F.; Matsushita, T.; Sardinha, J.; Coelho, H.; Oude-Vrielink, A.; Koller, C.; Andre, S.; Cabrita, E.J.; Gabius, H.-J.; et al. Delineating binding modes of Gal/GalNAc and structural elements of the molecular recognition of tumor-associated mucin glycopeptides by the human macrophage galactose-type lectin. Chem. Eur. J. 2014, 20, 16147–16155. [Google Scholar] [CrossRef] [PubMed]
- Phillips, L.; Wray, V. Stereospecific electronegative effects. Part I. The 19F nuclear magnetic resonance spectra of deoxyfluoro-d-glucopyranoses. J. Chem. Soc. B 1971. [Google Scholar] [CrossRef]
- Tubbs, P.K. Effects of inhibitors on mitochondrial d-α-hydroxy acid dehydrogenase. Biochem. J. 1962, 82, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Pederson, K.; Mitchell, D.A.; Prestegard, J.H. Structural characterization of the DC-SIGN-LewisX complex. Biochemistry 2014, 53, 5700–5709. [Google Scholar] [CrossRef] [PubMed]
- Vornholt, W.; Hartmann, M.; Keusgen, M. SPR studies of carbohydrate-lectin interactions as useful tool for screening on lectin sources. Biosens. Bioelectron. 2007, 22, 2983–2988. [Google Scholar] [CrossRef] [PubMed]
- Solís, D.; Bovin, N.V.; Davis, A.P.; Jiménez-Barbero, J.; Romero, A.; Roy, R.; Smetana, K., Jr.; Gabius, H.-J. A guide into glycosciences: How chemistry, biochemistry and biology cooperate to crack the sugar code. Biochim. Biophys. Acta 2015, 1850, 186–235. [Google Scholar] [CrossRef] [PubMed]
- Kaltner, H.; Gabius, H.-J. A toolbox of lectins for translating the sugar code: The galectin network in phylogenesis and tumors. Histol. Histopathol. 2012, 27, 397–416. [Google Scholar] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ribeiro, J.P.; Diercks, T.; Jiménez-Barbero, J.; André, S.; Gabius, H.-J.; Cañada, F.J. Fluorinated Carbohydrates as Lectin Ligands: 19F-Based Direct STD Monitoring for Detection of Anomeric Selectivity. Biomolecules 2015, 5, 3177-3192. https://doi.org/10.3390/biom5043177
Ribeiro JP, Diercks T, Jiménez-Barbero J, André S, Gabius H-J, Cañada FJ. Fluorinated Carbohydrates as Lectin Ligands: 19F-Based Direct STD Monitoring for Detection of Anomeric Selectivity. Biomolecules. 2015; 5(4):3177-3192. https://doi.org/10.3390/biom5043177
Chicago/Turabian StyleRibeiro, João P., Tammo Diercks, Jesús Jiménez-Barbero, Sabine André, Hans-Joachim Gabius, and Francisco Javier Cañada. 2015. "Fluorinated Carbohydrates as Lectin Ligands: 19F-Based Direct STD Monitoring for Detection of Anomeric Selectivity" Biomolecules 5, no. 4: 3177-3192. https://doi.org/10.3390/biom5043177
APA StyleRibeiro, J. P., Diercks, T., Jiménez-Barbero, J., André, S., Gabius, H.-J., & Cañada, F. J. (2015). Fluorinated Carbohydrates as Lectin Ligands: 19F-Based Direct STD Monitoring for Detection of Anomeric Selectivity. Biomolecules, 5(4), 3177-3192. https://doi.org/10.3390/biom5043177