
Combinatorial Control of mRNA Fates by RNA-Binding Proteins and Non-Coding RNAs



Abstract
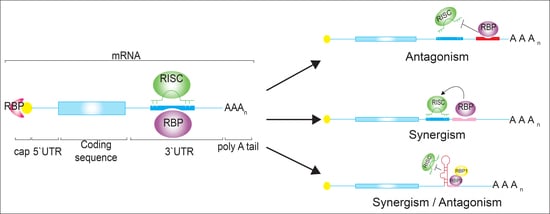
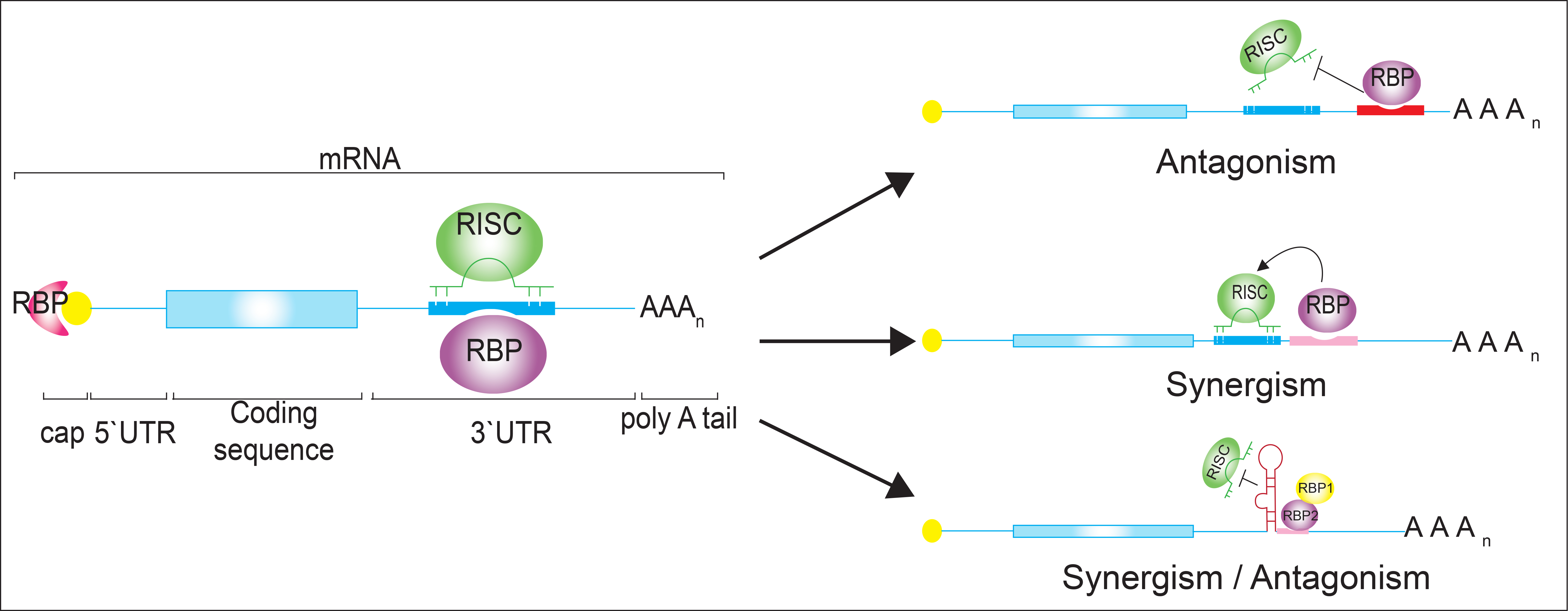
:
{kind=link}
{kind=link}
1. Introduction
2. Cross-Talk between RBPs and miRNAs on Cytoplasmic mRNAs
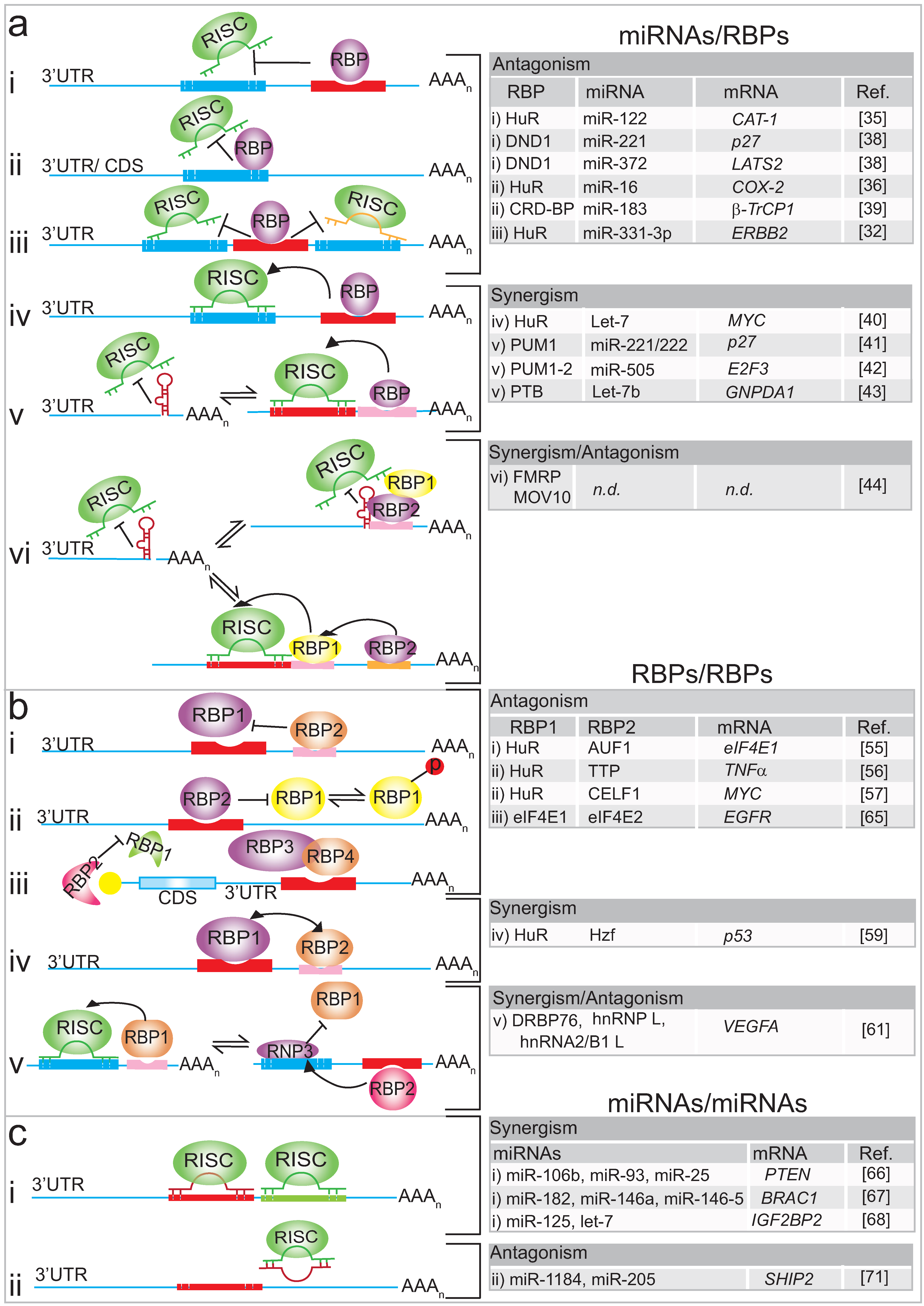
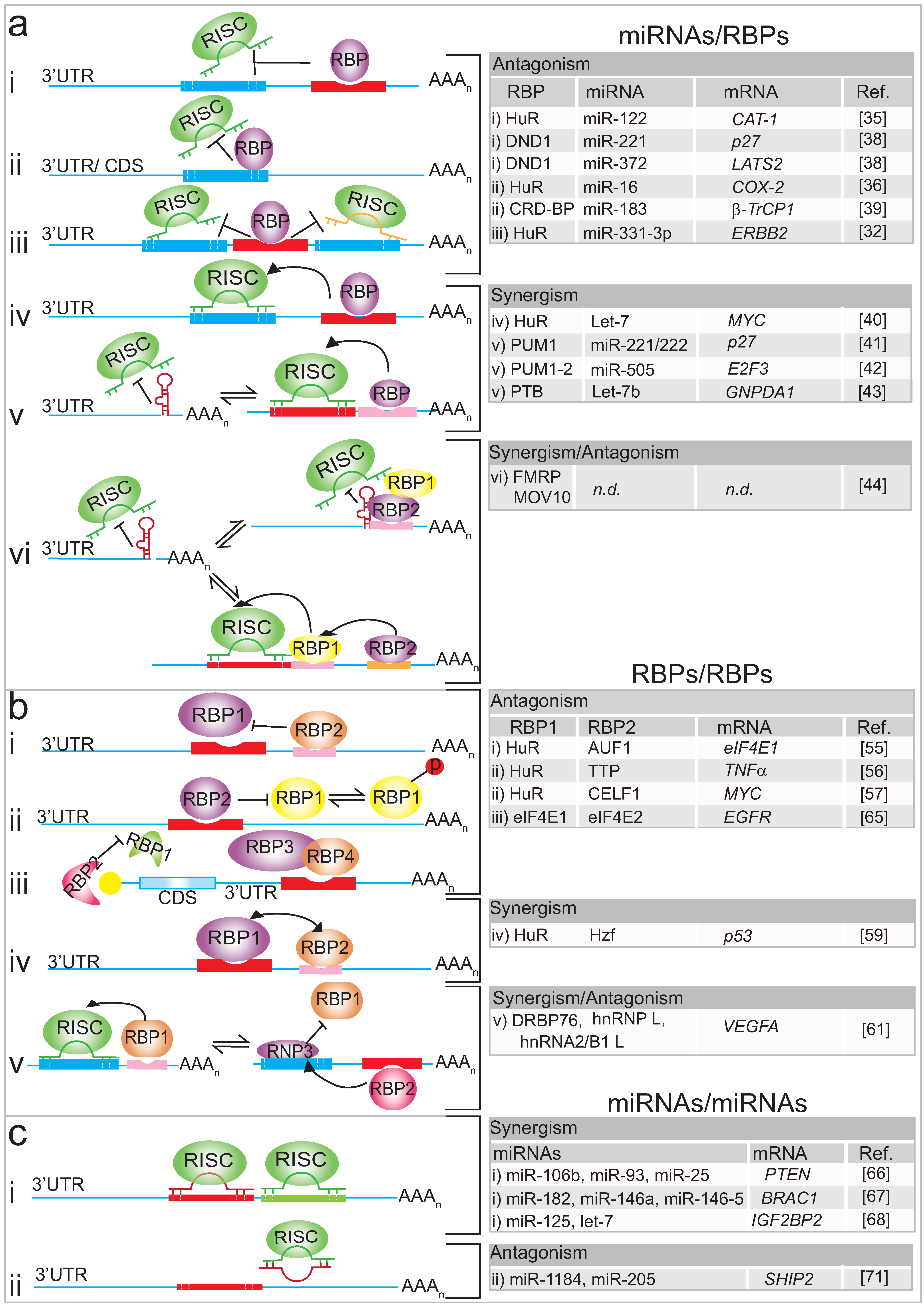
3. Interplay of RBPs on mRNA Targets
4. Cross-Talk among miRNAs
5. Conclusions and Final Remarks
Acknowledgments
Conflicts of Interest
References
- Cooper, T.A.; Wan, L.; Dreyfuss, G. RNA and disease. Cell 2009, 136, 777–793. [Google Scholar] [CrossRef] [PubMed]
- Castello, A.; Fischer, B.; Hentze, M.W.; Preiss, T. RNA-binding proteins in Mendelian disease. Trends Genet. 2013, 29, 318–327. [Google Scholar] [CrossRef] [PubMed]
- Wurth, L.; Gebauer, F. RNA-binding proteins, multifaceted translational regulators in cancer. Biochim. Biophys. Acta 2014, 1849, 881–886. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, S.F.; Parker, R. Principles and properties of eukaryotic mRNPs. Mol. Cell 2014, 54, 547–558. [Google Scholar] [CrossRef] [PubMed]
- Gerstberger, S.; Hafner, M.; Tuschl, T. A census of human RNA-binding proteins. Nat. Rev. Genet. 2014, 15, 829–845. [Google Scholar] [CrossRef] [PubMed]
- Darnell, J.E., Jr. Reflections on the history of pre-mRNA processing and highlights of current knowledge: A unified picture. RNA 2013, 19, 443–460. [Google Scholar] [CrossRef] [PubMed]
- St Johnston, D. Moving messages: The intracellular localization of mRNAs. Nat. Rev. Mol. Cell Biol. 2005, 6, 363–375. [Google Scholar] [CrossRef] [PubMed]
- Sonenberg, N.; Hinnebusch, A.G. Regulation of translation initiation in eukaryotes: Mechanisms and biological targets. Cell 2009, 136, 731–745. [Google Scholar] [CrossRef] [PubMed]
- Kishore, S.; Luber, S.; Zavolan, M. Deciphering the role of RNA-binding proteins in the post-transcriptional control of gene expression. Brief. Funct. Genomics 2010, 9, 391–404. [Google Scholar] [CrossRef] [PubMed]
- Imig, J.; Kanitz, A.; Gerber, A.P. RNA regulons and the RNA-protein interaction network. Biomol. Concepts 2012, 3, 403–414. [Google Scholar] [CrossRef] [PubMed]
- Cook, K.B.; Hughes, T.R.; Morris, Q.D. High-throughput characterization of protein-RNA interactions. Brief. Funct. Genomics 2015, 14, 74–89. [Google Scholar] [CrossRef] [PubMed]
- Tenenbaum, S.A.; Carson, C.C.; Lager, P.J.; Keene, J.D. Identifying mRNA subsets in messenger ribonucleoprotein complexes by using cDNA arrays. Proc. Natl. Acad. Sci. USA 2000, 97, 14085–14090. [Google Scholar] [CrossRef] [PubMed]
- Keene, J.D. RNA regulons: Coordination of post-transcriptional events. Nat. Rev. Genet. 2007, 8, 533–543. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [PubMed]
- Chekulaeva, M.; Filipowicz, W. Mechanisms of miRNA-mediated post-transcriptional regulation in animal cells. Curr. Opin. Cell Biol. 2009, 21, 452–460. [Google Scholar] [CrossRef] [PubMed]
- Kaboli, P.J.; Rahmat, A.; Ismail, P.; Ling, K.H. MicroRNA-based therapy and breast cancer: A comprehensive review of novel therapeutic strategies from diagnosis to treatment. Pharmacol. Res. 2015, 97, 104–121. [Google Scholar] [CrossRef] [PubMed]
- Adams, B.D.; Kasinski, A.L.; Slack, F.J. Aberrant regulation and function of microRNAs in cancer. Curr. Biol. 2014, 24, R762–R776. [Google Scholar] [CrossRef] [PubMed]
- Ha, M.; Kim, V.N. Regulation of microRNA biogenesis. Nat. Rev. Mol. Cell Biol. 2014, 15, 509–524. [Google Scholar] [CrossRef] [PubMed]
- Agami, R. MicroRNAs, RNA binding proteins and cancer. Eur. J. Clin. Invest. 2010, 40, 370–374. [Google Scholar] [CrossRef] [PubMed]
- Eom, T.; Muslimov, I.A.; Tsokas, P.; Berardi, V.; Zhong, J.; Sacktor, T.C.; Tiedge, H. Neuronal BC RNAs cooperate with eIF4B to mediate activity-dependent translational control. J. Cell Biol. 2014, 207, 237–252. [Google Scholar] [CrossRef] [PubMed]
- Kozomara, A.; Griffiths-Jones, S. MiRBase: Annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res 2014, 42, D68–D73. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Singh, M.; Coller, H.A. Computational assessment of the cooperativity between RNA binding proteins and microRNAs in Transcript Decay. PLoS Comput. Biol. 2013, 9, e1003075. [Google Scholar] [CrossRef] [PubMed]
- Van Kouwenhove, M.; Kedde, M.; Agami, R. MicroRNA regulation by RNA-binding proteins and its implications for cancer. Nat. Rev. Cancer 2011, 11, 644–656. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Coller, H. Functional interactions between microRNAs and RNA binding proteins. MicroRNA 2012, 1, 70–79. [Google Scholar] [CrossRef] [PubMed]
- Ciafre, S.A.; Galardi, S. MicroRNAs and RNA-binding proteins: A complex network of interactions and reciprocal regulations in cancer. RNA Biol. 2013, 10, 935–942. [Google Scholar] [CrossRef]
- Srikantan, S.; Tominaga, K.; Gorospe, M. Functional interplay between RNA-binding protein HuR and microRNAs. Curr. Protein Pept. Sci. 2012, 13, 372–379. [Google Scholar] [CrossRef] [PubMed]
- De Silanes, I.L.; Lal, A.; Gorospe, M. HuR: Post-transcriptional paths to malignancy. RNA Biol. 2005, 2, 11–13. [Google Scholar] [CrossRef]
- Saunus, J.M.; French, J.D.; Edwards, S.L.; Beveridge, D.J.; Hatchell, E.C.; Wagner, S.A.; Stein, S.R.; Davidson, A.; Simpson, K.J.; Francis, G.D.; et al. Posttranscriptional regulation of the breast cancer susceptibility gene BRCA1 by the RNA binding protein HuR. Cancer Res. 2008, 68, 9469–9478. [Google Scholar] [CrossRef] [PubMed]
- Erkinheimo, T.L.; Lassus, H.; Sivula, A.; Sengupta, S.; Furneaux, H.; Hla, T.; Haglund, C.; Butzow, R.; Ristimaki, A. Cytoplasmic HuR expression correlates with poor outcome and with cyclooxygenase 2 expression in serous ovarian carcinoma. Cancer Res. 2003, 63, 7591–7594. [Google Scholar] [PubMed]
- Lim, S.J.; Lee, S.H.; Joo, S.H.; Song, J.Y.; Choi, S.I. Cytoplasmic expression of HuR is related to cyclooxygenase-2 expression in colon cancer. Cancer Res. Treat. 2009, 41, 87–92. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, B.; Bi, J.; Zhang, C. Cytoplasmic HuR expression correlates with angiogenesis, lymphangiogenesis, and poor outcome in lung cancer. Med. Oncol. 2011, 28, 577–585. [Google Scholar] [CrossRef] [PubMed]
- Epis, M.R.; Barker, A.; Giles, K.M.; Beveridge, D.J.; Leedman, P.J. The RNA-binding protein HuR opposes the repression of ERBB-2 gene expression by microRNA miR-331–3p in prostate cancer cells. J. Biol. Chem. 2011, 286, 41442–41454. [Google Scholar] [CrossRef] [PubMed]
- Stoppoloni, D.; Cardillo, I.; Verdina, A.; Vincenzi, B.; Menegozzo, S.; Santini, M.; Sacchi, A.; Baldi, A.; Galati, R. Expression of the embryonic lethal abnormal vision-like protein HuR in human mesothelioma: Association with cyclooxygenase-2 and prognosis. Cancer 2008, 113, 2761–2769. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Guo, Y.; Chu, H.; Guan, Y.; Bi, J.; Wang, B. Multiple Functions of the RNA-Binding Protein HuR in Cancer Progression, Treatment Responses and Prognosis. Int. J. Mol. Sci. 2013, 14, 10015–10041. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, S.N.; Habermacher, R.; Martine, U.; Closs, E.I.; Filipowicz, W. Relief of microRNA-mediated translational repression in human cells subjected to stress. Cell 2006, 125, 1111–1124. [Google Scholar] [CrossRef] [PubMed]
- Young, L.E.; Moore, A.E.; Sokol, L.; Meisner-Kober, N.; Dixon, D.A. The mRNA stability factor HuR inhibits microRNA-16 targeting of COX-2. Mol. Cancer Res. 2012, 10, 167–180. [Google Scholar] [CrossRef] [PubMed]
- Ng, K.; Meyerhardt, J.A.; Chan, A.T.; Sato, K.; Chan, J.A.; Niedzwiecki, D.; Saltz, L.B.; Mayer, R.J.; Benson, A.B., 3rd; Schaefer, P.L.; et al. Aspirin and COX-2 inhibitor use in patients with stage III colon cancer. J. Natl. CancerInst. 2015. [Google Scholar] [CrossRef] [PubMed]
- Kedde, M.; Strasser, M.J.; Boldajipour, B.; Vrielink, J.A.F.O.; Slanchev, K.; le Sage, C.; Nagel, R.; Voorhoeve, P.M.; van Duijse, J.; Orom, U.A.; et al. RNA-binding protein Dnd1 inhibits microRNA access to target mRNA. Cell 2007, 131, 1273–1286. [Google Scholar] [CrossRef] [PubMed]
- Elcheva, I.; Goswami, S.; Noubissi, F.K.; Spiegelman, V.S. CRD-BP protects the coding region of betaTrCP1 mRNA from miR-183-mediated degradation. Mol. Cell 2009, 35, 240–246. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.H.; Kuwano, Y.; Srikantan, S.; Lee, E.K.; Martindale, J.L.; Gorospe, M. HuR recruits let-7/RISC to repress c-Myc expression. Genes Dev. 2009, 23, 1743–1748. [Google Scholar] [CrossRef] [PubMed]
- Kedde, M.; van Kouwenhove, M.; Zwart, W.; Vrielink, J.A.F.O.; Elkon, R.; Agami, R. A Pumilio-induced RNA structure switch in p27-3' UTR controls miR-221 and miR-222 accessibility. Nat. Cell Biol. 2010, 12, 1014–1020. [Google Scholar] [CrossRef] [PubMed]
- Miles, W.O.; Tschop, K.; Herr, A.; Ji, J.Y.; Dyson, N.J. Pumilio facilitates miRNA regulation of the E2F3 oncogene. Genes Dev. 2012, 26, 356–368. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.; Ouyang, K.; Huang, J.; Zhou, Y.; Ouyang, H.; Li, H.; Wang, G.; Wu, Q.; Wei, C.; Bi, Y.; et al. Direct conversion of fibroblasts to neurons by reprogramming PTB-regulated microRNA circuits. Cell 2013, 152, 82–96. [Google Scholar] [CrossRef] [PubMed]
- Kenny, P.J.; Zhou, H.; Kim, M.; Skariah, G.; Khetani, R.S.; Drnevich, J.; Arcila, M.L.; Kosik, K.S.; Ceman, S. MOV10 and FMRP regulate AGO2 association with microRNA recognition elements. Cell Rep. 2014, 9, 1729–1741. [Google Scholar] [CrossRef] [PubMed]
- Maurin, T.; Zongaro, S.; Bardoni, B. Fragile X Syndrome: From molecular pathology to therapy. Neurosci. Biobehav. Rev. 2014, 46, 242–255. [Google Scholar] [CrossRef] [PubMed]
- Jacobsen, A.; Wen, J.; Marks, D.S.; Krogh, A. Signatures of RNA binding proteins globally coupled to effective microRNA target sites. Genome Res. 2010, 20, 1010–1019. [Google Scholar] [CrossRef] [PubMed]
- Galgano, A.; Forrer, M.; Jaskiewicz, L.; Kanitz, A.; Zavolan, M.; Gerber, A.P. Comparative analysis of mRNA targets for human PUF-family proteins suggests extensive interaction with the miRNA regulatory system. PLoS ONE 2008, 3, e3164. [Google Scholar] [CrossRef] [PubMed]
- Incarnato, D.; Neri, F.; Diamanti, D.; Oliviero, S. MREdictor: A two-step dynamic interaction model that accounts for mRNA accessibility and Pumilio binding accurately predicts microRNA targets. Nucleic Acids Res. 2013, 41, 8421–8433. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, N.; Corcoran, D.L.; Nusbaum, J.D.; Reid, D.W.; Georgiev, S.; Hafner, M.; Ascano, M., Jr.; Tuschl, T.; Ohler, U.; Keene, J.D. Integrative regulatory mapping indicates that the RNA-binding protein HuR couples pre-mRNA processing and mRNA stability. Mol. Cell 2011, 43, 327–339. [Google Scholar] [CrossRef] [PubMed]
- Doyle, F.; Tenenbaum, S.A. Trans-regulation of RNA-binding protein motifs by microRNA. Front. Genet. 2014. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, Y.; Vigilante, A.; Darbo, E.; Zirra, A.; Militti, C.; D’Ambrogio, A.; Luscombe, N.M.; Ule, J. hiCLIP reveals the in vivo atlas of mRNA secondary structures recognized by Staufen 1. Nature 2015, 519, 491–494. [Google Scholar] [CrossRef] [PubMed]
- Pandit, S.; Zhou, Y.; Shiue, L.; Coutinho-Mansfield, G.; Li, H.; Qiu, J.; Huang, J.; Yeo, G.W.; Ares, M., Jr.; Fu, X.D. Genome-wide analysis reveals SR protein cooperation and competition in regulated splicing. Mol. Cell 2013, 50, 223–235. [Google Scholar] [CrossRef] [PubMed]
- Zarnack, K.; Konig, J.; Tajnik, M.; Martincorena, I.; Eustermann, S.; Stevant, I.; Reyes, A.; Anders, S.; Luscombe, N.M.; Ule, J. Direct competition between hnRNP C and U2AF65 protects the transcriptome from the exonization of Alu elements. Cell 2013, 152, 453–466. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.D.; Ares, M., Jr. Context-dependent control of alternative splicing by RNA-binding proteins. Nat. Rev. Genet. 2014, 15, 689–701. [Google Scholar] [CrossRef] [PubMed]
- Topisirovic, I.; Siddiqui, N.; Orolicki, S.; Skrabanek, L.A.; Tremblay, M.; Hoang, T.; Borden, K.L. Stability of eukaryotic translation initiation factor 4E mRNA is regulated by HuR, and this activity is dysregulated in cancer. Mol. Cell. Biol. 2009, 29, 1152–1162. [Google Scholar] [CrossRef] [PubMed]
- Tiedje, C.; Ronkina, N.; Tehrani, M.; Dhamija, S.; Laass, K.; Holtmann, H.; Kotlyarov, A.; Gaestel, M. The p38/MK2-driven exchange between tristetraprolin and HuR regulates AU-rich element-dependent translation. PLoS Genet. 2012, 8, e1002977. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Ouyang, M.; Rao, J.N.; Zou, T.; Xiao, L.; Chung, H.K.; Wu, J.; Donahue, J.M.; Gorospe, M.; Wang, J.Y. Competition between RNA-binding proteins CELF1 and HuR modulates MYC translation and intestinal epithelium renewal. Mol. Biol. Cell 2015, 26, 1797–1810. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, N.; Jacobs, N.C.; Hafner, M.; Kennington, E.A.; Nusbaum, J.D.; Tuschl, T.; Blackshear, P.J.; Ohler, U. Global target mRNA specification and regulation by the RNA-binding protein ZFP36. Genome Biol. 2014. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, H.; Kawagishi, H.; Watanabe, A.; Sugimoto, K.; Maruyama, M.; Sugimoto, M. Cooperative role of the RNA-binding proteins Hzf and HuR in p53 activation. Mol. Cell. Biol. 2011, 31, 1997–2009. [Google Scholar] [CrossRef] [PubMed]
- Ray, P.S.; Jia, J.; Yao, P.; Majumder, M.; Hatzoglou, M.; Fox, P.L. A stress-responsive RNA switch regulates VEGFA expression. Nature 2009, 457, 915–919. [Google Scholar] [CrossRef] [PubMed]
- Jafarifar, F.; Yao, P.; Eswarappa, S.M.; Fox, P.L. Repression of VEGFA by CA-rich element-binding microRNAs is modulated by hnRNP L. EMBO J. 2011, 30, 1324–1334. [Google Scholar] [CrossRef] [PubMed]
- Goldberg-Cohen, I.; Furneauxb, H.; Levy, A.P. A 40-bp RNA element that mediates stabilization of vascular endothelial growth factor mRNA by HuR. J. Biol. Chem. 2002, 277, 13635–13640. [Google Scholar] [CrossRef] [PubMed]
- Yao, P.; Potdar, A.A.; Ray, P.S.; Eswarappa, S.M.; Flagg, A.C.; Willard, B.; Fox, P.L. The HILDA complex coordinates a conditional switch in the 3'-untranslated region of the VEGFA mRNA. PLoS Biol. 2013, 11, e1001635. [Google Scholar] [CrossRef] [PubMed]
- Pelletier, J.; Graff, J.; Ruggero, D.; Sonenberg, N. Targeting the eIF4F translation initiation complex: A critical nexus for cancer development. Cancer Res. 2015, 75, 250–263. [Google Scholar] [CrossRef] [PubMed]
- Uniacke, J.; Holterman, C.E.; Lachance, G.; Franovic, A.; Jacob, M.D.; Fabian, M.R.; Payette, J.; Holcik, M.; Pause, A.; Lee, S. An oxygen-regulated switch in the protein synthesis machinery. Nature 2012, 486, 126–129. [Google Scholar] [CrossRef] [PubMed]
- Poliseno, L.; Salmena, L.; Riccardi, L.; Fornari, A.; Song, M.S.; Hobbs, R.M.; Sportoletti, P.; Varmeh, S.; Egia, A.; Fedele, G.; et al. Identification of the miR-106b~25 microRNA cluster as a proto-oncogenic PTEN-targeting intron that cooperates with its host gene MCM7 in transformation. Sci. Signal. 2010. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.; Sharan, S.K. BRCA1 and microRNAs: Emerging networks and potential therapeutic targets. Mol. Cells 2012, 34, 425–432. [Google Scholar] [CrossRef] [PubMed]
- Shenoy, A.; Danial, M.; Blelloch, R.H. Let-7 and miR-125 cooperate to prime progenitors for astrogliogenesis. EMBO J. 2015, 34, 1180–1194. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Gumireddy, K.; Schrier, M.; le Sage, C.; Nagel, R.; Nair, S.; Egan, D.A.; Li, A.; Huang, G.; Klein-Szanto, A.J.; et al. The microRNAs miR-373 and miR-520c promote tumour invasion and metastasis. Nat. Cell Biol. 2008, 10, 202–210. [Google Scholar] [CrossRef] [PubMed]
- Brown, K.K.; Toker, A. The phosphoinositide 3-kinase pathway and therapy resistance in cancer. F1000Prime Rep. 2015. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Ryan, D.G.; Getsios, S.; Oliveira-Fernandes, M.; Fatima, A.; Lavker, R.M. MicroRNA-184 antagonizes microRNA-205 to maintain SHIP2 levels in epithelia. Proc. Natl. Acad. Sci. USA 2008, 105, 19300–19305. [Google Scholar] [CrossRef] [PubMed]
- Cook, K.B.; Kazan, H.; Zuberi, K.; Morris, Q.; Hughes, T.R. RBPDB: A database of RNA-binding specificities. Nucleic Acids Res. 2011, 39, D301–D308. [Google Scholar] [CrossRef] [PubMed]
- A Database of RNA-Binding Specificities. Available online: http://rbpdb.ccbr.utoronto.ca (accessed on 14 September 2015).
- Ray, D.; Kazan, H.; Cook, K.B.; Weirauch, M.T.; Najafabadi, H.S.; Li, X.; Gueroussov, S.; Albu, M.; Zheng, H.; Yang, A.; et al. A compendium of RNA-binding motifs for decoding gene regulation. Nature 2013, 499, 172–177. [Google Scholar] [CrossRef] [PubMed]
- CIS-BP-RNA, the Online Library of RNA Binding Proteins and Their Motifs. Available online: http://cisbp-rna.ccbr.utoronto.ca (accessed on 14 September 2015).
- Biswas, A.; Brown, C.M. Scan for motifs: A webserver for the analysis of post-transcriptional regulatory elements in the 3' untranslated regions (3' UTRs) of mRNAs. BMC Bioinform. 2014. [Google Scholar] [CrossRef] [PubMed]
- Scan for Motifs. Available online: http://bioanalysis.otago.ac.nz/sfm/sfm_main.pl (accessed on 14 September 2015).
- Agarwal, V.; Bell, G.W.; Nam, J.W.; Bartel, D.P. Predicting effective microRNA target sites in mammalian mRNAs. Elife 2015. [Google Scholar] [CrossRef] [PubMed]
- TargetScan. Available online: http://www.targetscan.org/ (accessed on 14 September 2015).
- Yang, J.H.; Li, J.H.; Shao, P.; Zhou, H.; Chen, Y.Q.; Qu, L.H. StarBase: A database for exploring microRNA-mRNA interaction maps from Argonaute CLIP-Seq and Degradome-Seq data. Nucleic Acids Res. 2011, 39, D202–D209. [Google Scholar] [CrossRef] [PubMed]
- StarBase v2.0 CLIP-Seq, Pan-Cancer. Portal, Visualize, Analyze, Discover. Available online: http://starbase.sysu.edu.cn/ (accessed on 14 September 2015).
- Chen, G.; Wang, Z.; Wang, D.; Qiu, C.; Liu, M.; Chen, X.; Zhang, Q.; Yan, G.; Cui, Q. LncRNADisease: A database for long-non-coding RNA-associated diseases. Nucleic Acids Res. 2013, 41, D983–D986. [Google Scholar] [CrossRef] [PubMed]
- The LncRNADisease Database. Available online: http://www.cuilab.cn/lncrnadisease (accessed on 14 September 2015).
- Kanitz, A.; Gerber, A.P. Circuitry of mRNA regulation. Wiley Interdiscip. Rev. Syst. Biol. Med. 2010, 2, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Pullmann, R., Jr.; Kim, H.H.; Abdelmohsen, K.; Lal, A.; Martindale, J.L.; Yang, X.; Gorospe, M. Analysis of turnover and translation regulatory RNA-binding protein expression through binding to cognate mRNAs. Mol. Cell. Biol. 2007, 27, 6265–6278. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Gregory, R.I. MicroRNA biogenesis pathways in cancer. Nat. Rev. Cancer 2015, 15, 321–333. [Google Scholar] [CrossRef] [PubMed]
- Michlewski, G.; Caceres, J.F. Antagonistic role of hnRNP A1 and KSRP in the regulation of let-7a biogenesis. Nat. Struct. Mol. Biol. 2010, 17, 1011–1018. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iadevaia, V.; Gerber, A.P. Combinatorial Control of mRNA Fates by RNA-Binding Proteins and Non-Coding RNAs. Biomolecules 2015, 5, 2207-2222. https://doi.org/10.3390/biom5042207
Iadevaia V, Gerber AP. Combinatorial Control of mRNA Fates by RNA-Binding Proteins and Non-Coding RNAs. Biomolecules. 2015; 5(4):2207-2222. https://doi.org/10.3390/biom5042207
Chicago/Turabian StyleIadevaia, Valentina, and André P. Gerber. 2015. "Combinatorial Control of mRNA Fates by RNA-Binding Proteins and Non-Coding RNAs" Biomolecules 5, no. 4: 2207-2222. https://doi.org/10.3390/biom5042207
APA StyleIadevaia, V., & Gerber, A. P. (2015). Combinatorial Control of mRNA Fates by RNA-Binding Proteins and Non-Coding RNAs. Biomolecules, 5(4), 2207-2222. https://doi.org/10.3390/biom5042207