
Splicing Regulation of Pro-Inflammatory Cytokines and Chemokines: At the Interface of the Neuroendocrine and Immune Systems

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
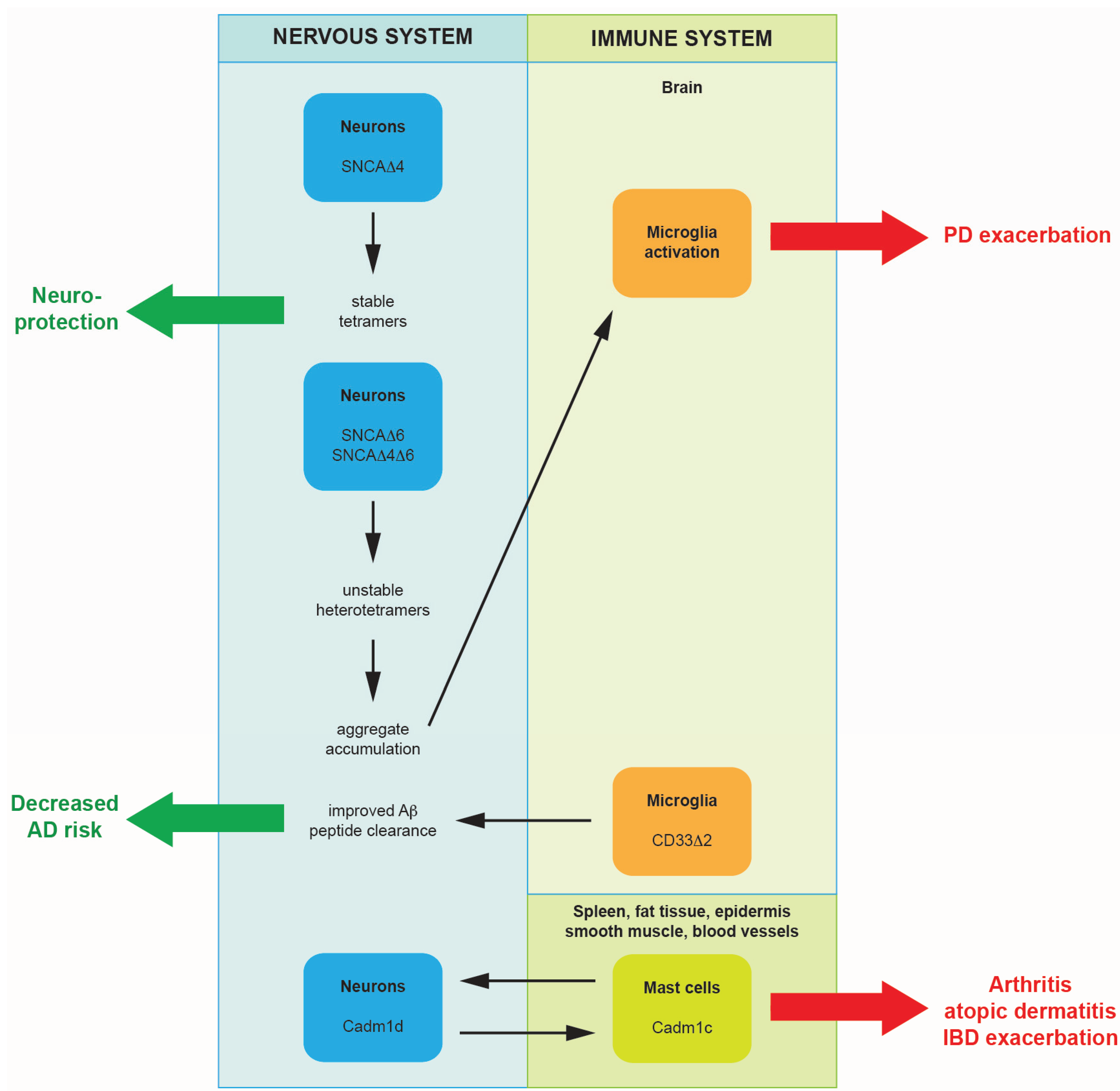
2. Alternative Splicing Promoting Neurodegeneration or Providing Neuroprotection
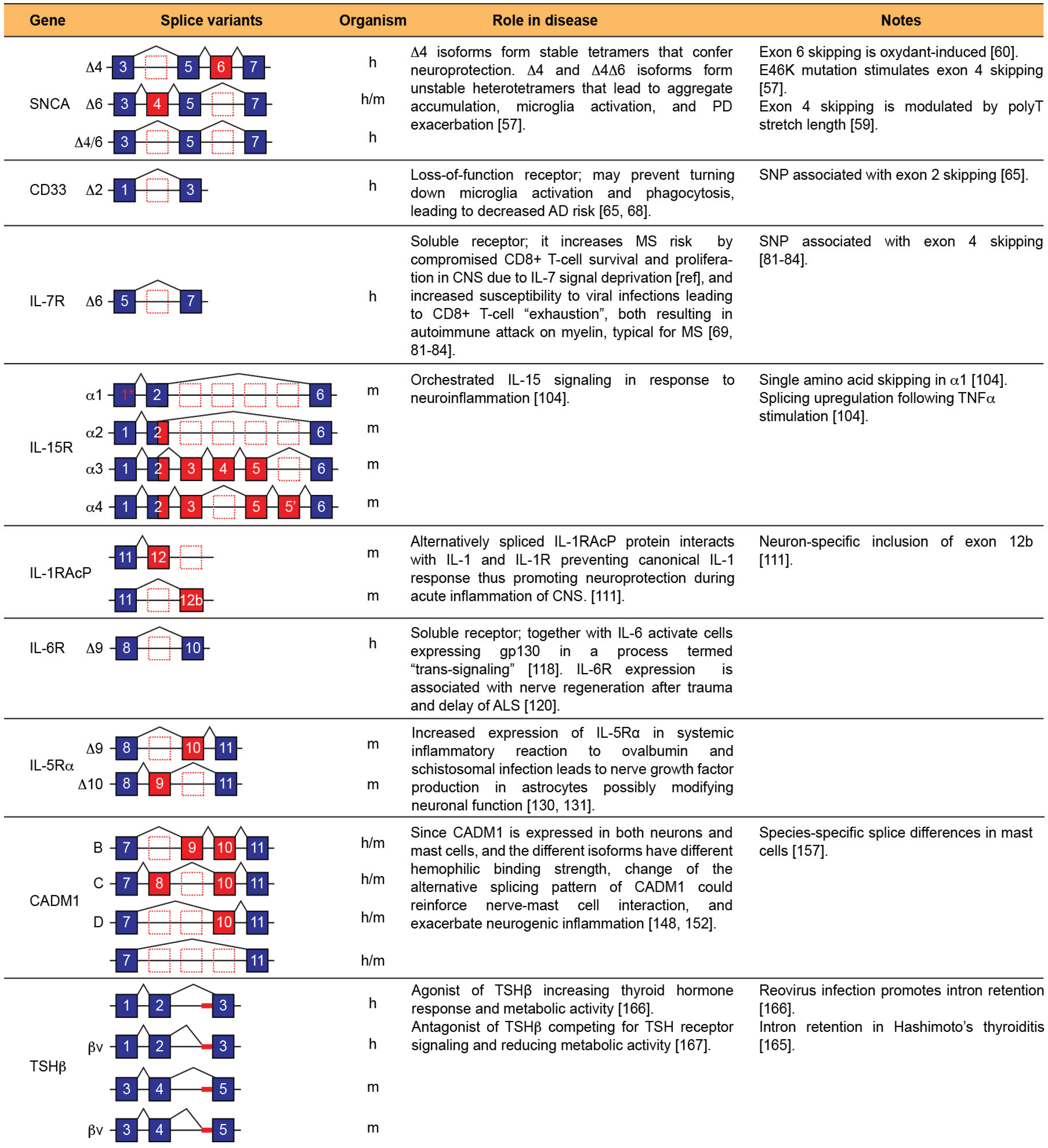
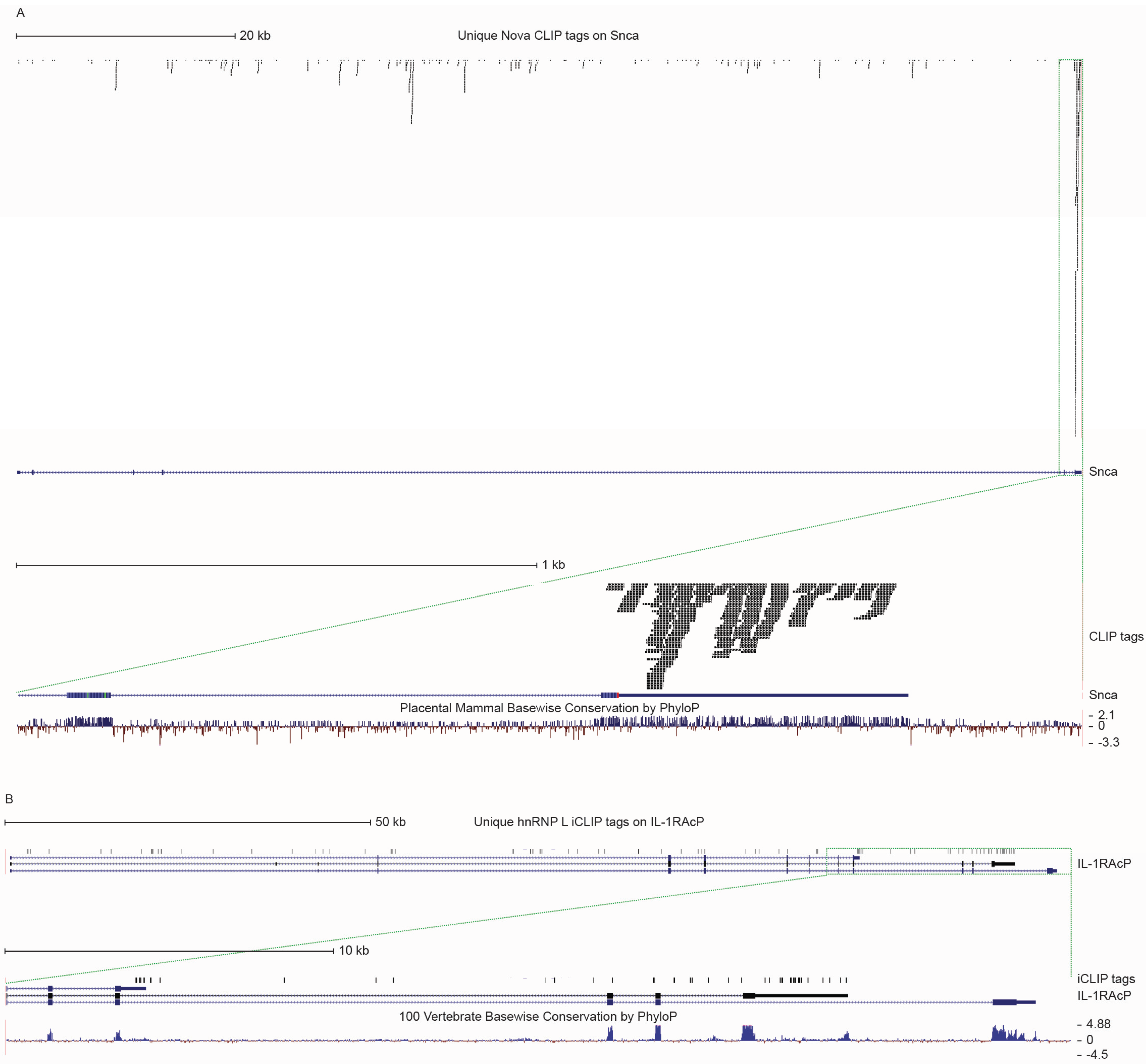
2.1. Alternatively Spliced Isoforms of Alpha-Synuclein Promote the Progression of Parkinson’s Disease
2.2. A Single Nucleotide Polymorphism Affecting Alternative Splicing Decreases Levels of Functional CD33 in the Brain: A Mechanism for Providing Protection from Alzheimer’s Disease
2.3. A Soluble Isoform of the Interleukin-7 Receptor Increases the Risk of Developing Multiple Sclerosis
2.4. Alternative Splicing of Interleukin-15 Receptor Modulates Interleukin-15 Signaling in Experimentally Induced Neuroinflammation
2.5. Alternatively Spliced Isoforms of the Interleukin-1 Receptor Accessory Protein Promote Neuroprotection during Acute Inflammation of the Central Nervous System
2.6. A Soluble Form of the Interleukin-6 Receptor Accelerates Nerve Regeneration after Trauma
3. Changes in Alternative Splicing of the Neuroendocrine System as a Response to Inflammation Elsewhere in the Body
3.1. Alternative Splicing of Interleukin-5 Receptor α Changes in Systemic Inflammation, Possibly Leading to Alterations in Neuronal Growth and Function
3.2. Alternatively Spliced Isoforms of Cell Adhesion Molecule-1 Could Reinforce Nerve-Mast Cell Interaction and May Exacerbate Neurogenic Inflammation
3.3. An Alternatively Spliced Isoform of the Thyroid Stimulating Hormone Produced during Acute and Chronic Inflammation May Alter Metabolism by Interfering with the Native Hormone Signaling
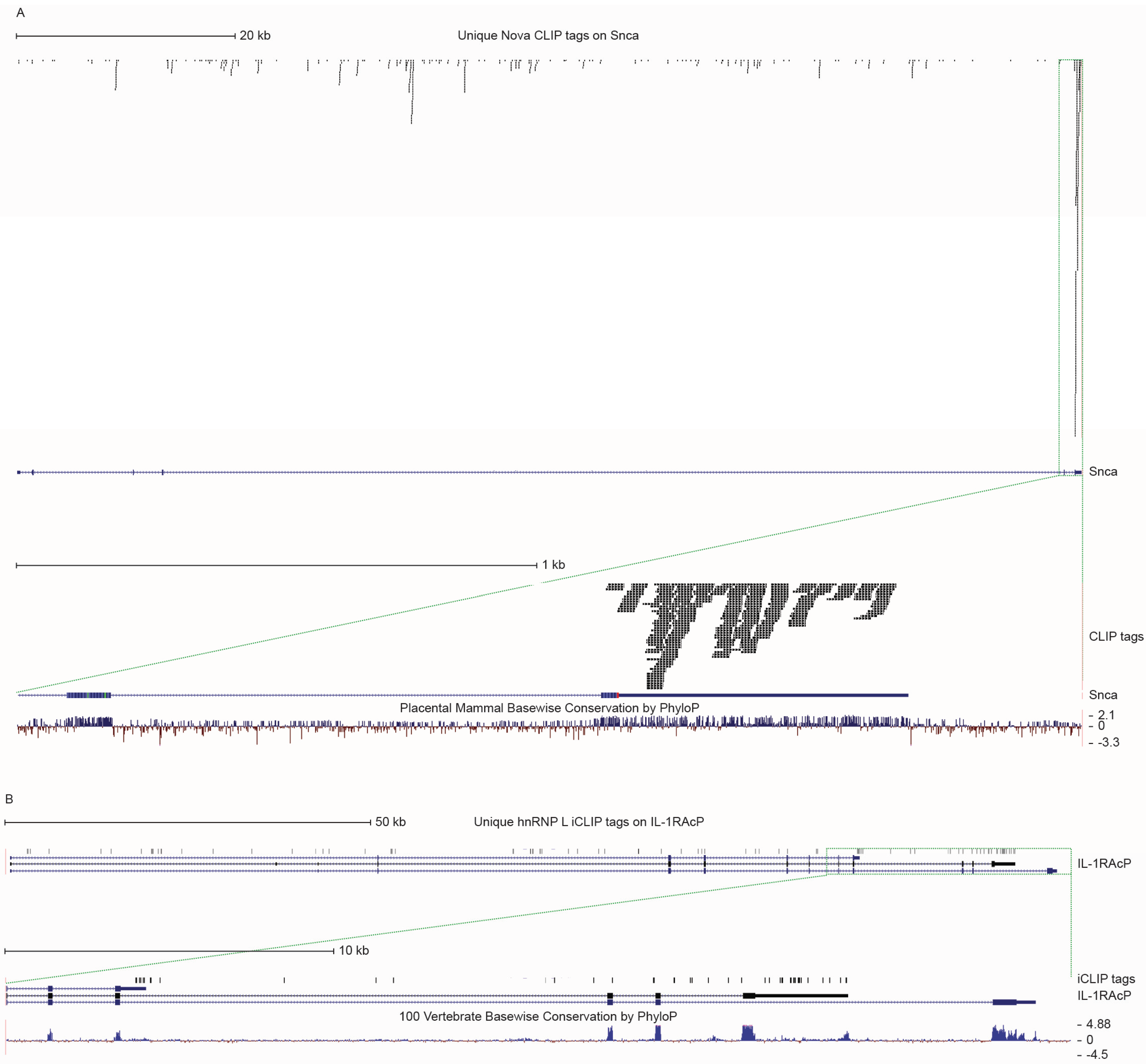
4. Regulation of Alternative Splicing at the Interface of the Immune and Neuroendocrine Systems during Inflammation
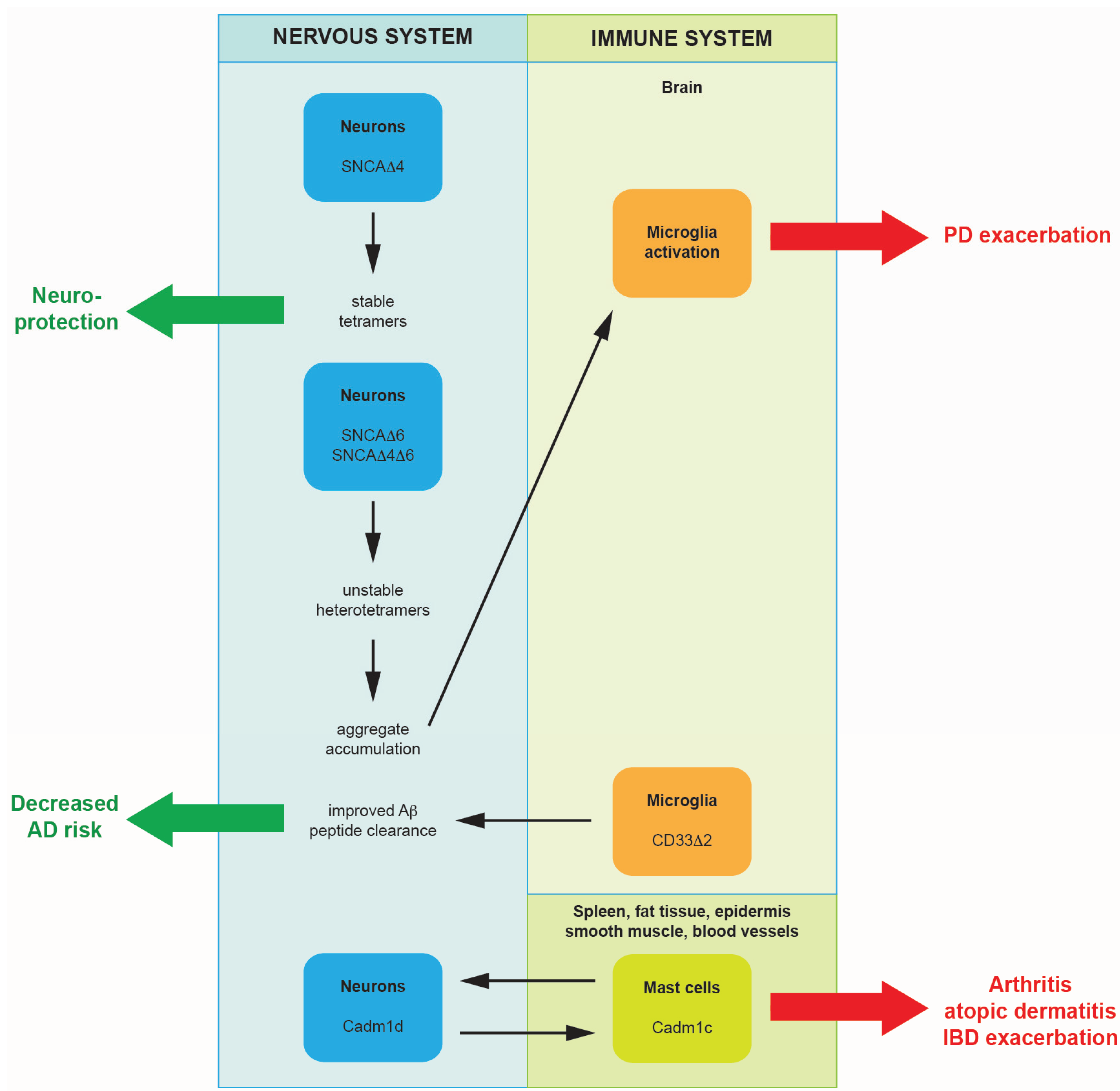
5. Conclusions
Author Contributions
Conflicts of Interest
References
- Kelemen, O.; Convertini, P.; Zhang, Z.; Wen, Y.; Shen, M.; Falaleeva, M.; Stamm, S. Function of alternative splicing. Gene 2013, 514, 1–30. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.K.; Cooper, T.A. Pre-mRNA splicing in disease and therapeutics. Trends Mol. Med. 2012, 18, 472–482. [Google Scholar] [CrossRef] [PubMed]
- López-Bigas, N.; Audit, B.; Ouzounis, C.; Parra, G.; Guigó, R. Are splicing mutations the most frequent cause of hereditary disease? FEBS Lett. 2005, 579, 1900–1903. [Google Scholar] [CrossRef] [PubMed]
- Grabowski, P.J.; Black, D.L. Alternative RNA splicing in the nervous system. Prog. Neurobiol. 2001, 65, 289–308. [Google Scholar] [CrossRef]
- Modrek, B.; Resch, A.; Grasso, C.; Lee, C. Genome-wide detection of alternative splicing in expressed sequences of human genes. Nucleic Acids Res. 2001, 29, 2850–2859. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.M.; Castle, J.; Garrett-Engele, P.; Kan, Z.; Loerch, P.M.; Armour, C.D.; Santos, R.; Schadt, E.E.; Stoughton, R.; Shoemaker, D.D. Genome-wide survey of human alternative pre-mRNA splicing with exon junction microarrays. Science 2003, 302, 2141–2144. [Google Scholar] [CrossRef] [PubMed]
- Lynch, K.W. Consequences of regulated pre-mRNA splicing in the immune system. Nat. Rev. Immunol. 2004, 4, 931–940. [Google Scholar] [CrossRef] [PubMed]
- Martinez, N.M.; Pan, Q.; Cole, B.S.; Yarosh, C.A.; Babcock, G.A.; Heyd, F.; Zhu, W.; Ajith, S.; Blencowe, B.J.; Lynch, K.W. Alternative splicing networks regulated by signaling in human T cells. RNA 2012, 18, 1029–1040. [Google Scholar] [CrossRef] [PubMed]
- Martinez, N.M.; Lynch, K.W. Control of alternative splicing in immune responses: Many regulators, many predictions, much still to learn. Immunol. Rev. 2013, 253, 216–236. [Google Scholar] [CrossRef] [PubMed]
- Yap, K.; Makeyev, E.V. Regulation of gene expression in mammalian nervous system through alternative pre-mRNA splicing coupled with RNA quality control mechanisms. Mol. Cell. Neurosci. 2013, 56, 420–428. [Google Scholar] [CrossRef] [PubMed]
- Popp, M.W.; Maquat, L.E. The dharma of nonsense-mediated mRNA decay in mammalian cells. Mol. Cells 2014, 37, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Ge, Y.; Porse, B.T. The functional consequences of intron retention: Alternative splicing coupled to NMD as a regulator of gene expression. BioEssays 2014, 36, 236–243. [Google Scholar] [CrossRef] [PubMed]
- Sahoo, A.; Im, S.H. Interleukin and interleukin receptor diversity: Role of alternative splicing. Int. Rev. Immunol. 2010, 29, 77–109. [Google Scholar] [CrossRef] [PubMed]
- Ehlers, M.D.; Tingley, W.G.; Huganir, R.L. Regulated subcellular distribution of the NR1 subunit of the NMDA receptor. Science 1995, 269, 1734–1737. [Google Scholar] [CrossRef] [PubMed]
- Zukin, R.S.; Bennett, M.V. Alternatively spliced isoforms of the NMDARI receptor subunit. Trends Neurosci. 1995, 18, 306–313. [Google Scholar] [CrossRef]
- Ruggiu, M.; Herbst, R.; Kim, N.; Jevsek, M.; Fak, J.J.; Mann, M.A.; Fischbach, G.; Burden, S.J.; Darnell, R.B. Rescuing Z+ agrin splicing in Nova null mice restores synapse formation and unmasks a physiologic defect in motor neuron firing. Proc. Natl. Acad. Sci. USA 2009, 106, 3513–3518. [Google Scholar] [CrossRef] [PubMed]
- Wilusz, J.E.; Sunwoo, H.; Spector, D.L. Long noncoding RNAs: Functional surprises from the RNA world. Genes Dev. 2009, 23, 1494–1504. [Google Scholar] [CrossRef] [PubMed]
- Guennewig, B.; Cooper, A.A. The central role of noncoding RNA in the brain. Int. Rev. Neurobiol. 2014, 116, 153–194. [Google Scholar] [PubMed]
- Maniatis, T.; Tasic, B. Alternative pre-mRNA splicing and proteome expansion in metazoans. Nature 2002, 418, 236–243. [Google Scholar] [CrossRef] [PubMed]
- Black, D.L. Mechanisms of alternative pre-messenger RNA splicing. Annu. Rev. Biochem. 2003, 72, 291–336. [Google Scholar] [CrossRef] [PubMed]
- Stamm, S.; Ben-Ari, S.; Rafalska, I.; Tang, Y.; Zhang, Z.; Toiber, D.; Thanaraj, T.A.; Soreq, H. Function of alternative splicing. Gene 2005, 344, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Braunschweig, U.; Gueroussov, S.; Plocik, A.M.; Graveley, B.R.; Blencowe, B.J. Dynamic integration of splicing within gene regulatory pathways. Cell 2013, 152, 1252–1269. [Google Scholar] [CrossRef] [PubMed]
- Rappsilber, J.; Ryder, U.; Lamond, A.I.; Mann, M. Large-scale proteomic analysis of the human spliceosome. Genome Res. 2002, 12, 1231–1245. [Google Scholar] [CrossRef] [PubMed]
- Barbosa-Morais, N.L.; Carmo-Fonseca, M.; Aparício, S. Systematic genome-wide annotation of spliceosomal proteins reveals differential gene family expansion. Genome Res. 2006, 16, 66–77. [Google Scholar] [CrossRef] [PubMed]
- De la Grange, P.; Gratadou, L.; Delord, M.; Dutertre, M.; Auboeuf, D. Splicing factor and exon profiling across human tissues. Nucleic Acids Res. 2010, 38, 2825–2838. [Google Scholar] [CrossRef] [PubMed]
- Jelen, N.; Ule, J.; Zivin, M.; Darnell, R.B. Evolution of Nova-dependent splicing regulation in the brain. PLoS Genet. 2007, 3, 1838–1847. [Google Scholar] [CrossRef] [PubMed]
- Fogel, B.L.; Wexler, E.; Wahnich, A.; Friedrich, T.; Vijayendran, C.; Gao, F.; Parikshak, N.; Konopka, G.; Geschwind, D.H. Rbfox1 regulates both splicing and transcriptional networks in human neuronal development. Hum. Mol. Genet. 2012, 21, 4171–4186. [Google Scholar] [CrossRef] [PubMed]
- Gehman, L.T.; Meera, P.; Stoilov, P.; Shiue, L.; O’Brien, J.E.; Meisler, M.H.; Ares, M., Jr.; Otis, T.S.; Black, D.L. The splicing regulator Rbfox2 is required for both cerebellar development and mature motor function. Genes Dev. 2012, 26, 445–460. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.K.; Nam, J.; Mukouyama, Y.S.; Kawamoto, S. Rbfox3-regulated alternative splicing of Numb promotes neuronal differentiation during development. J. Cell Biol. 2013, 200, 443–458. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Muñoz, M.D.; Bell, S.E.; Fairfax, K.; Monzon-Casanova, E.; Cunningham, A.F.; Gonzalez-Porta, M.; Andrews, S.R.; Bunik, V.I.; Zarnack, K.; Curk, T.; et al. The RNA-binding protein HuR is essential for the B cell antibody response. Nat. Immunol. 2015, 16, 415–425. [Google Scholar] [CrossRef] [PubMed]
- Katsumoto, A.; Lu, H.; Miranda, A.S.; Ransohoff, R.M. Ontogeny and functions of central nervous system macrophages. J. Immunol. 2014, 193, 2615–2621. [Google Scholar] [CrossRef] [PubMed]
- Polfliet, M.M.; van de Veerdonk, F.; Döpp, E.A.; van Kesteren-Hendrikx, E.M.; van Rooijen, N.; Dijkstra, C.D.; van den Berg, T.K. The role of perivascular and meningeal macrophages in experimental allergic encephalomyelitis. J. Neuroimmunol. 2002, 122, 1–8. [Google Scholar] [CrossRef]
- Arac, A.; Grimbaldeston, M.A.; Nepomuceno, A.R.; Olayiwola, O.; Pereira, M.P.; Nishiyama, Y.; Tsykin, A.; Goodall, G.J.; Schlecht, U.; Vogel, H.; et al. Evidence that meningeal mast cells can worsen stroke pathology in mice. Am. J. Pathol. 2014, 184, 2493–2504. [Google Scholar] [CrossRef] [PubMed]
- Lawson, L.J.; Perry, V.H.; Dri, P.; Gordon, S. Heterogeneity in the distribution and morphology of microglia in the normal adult mouse brain. Neuroscience 1990, 39, 151–170. [Google Scholar] [CrossRef]
- Von Bredow, J.D.; Adams, N.L.; Groff, W.A.; Vick, J.A. Effectiveness of oral pyridostigmine pretreatment and cholinolytic-oxime therapy against soman intoxication in nonhuman primates. Fundam. Appl. Toxicol. 1991, 17, 761–770. [Google Scholar] [CrossRef]
- Schafer, D.P.; Lehrman, E.K.; Kautzman, A.G.; Koyama, R.; Mardinly, A.R.; Yamasaki, R.; Ransohoff, R.M.; Greenberg, M.E.; Barres, B.A.; Stevens, B. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 2012, 74, 691–705. [Google Scholar] [CrossRef] [PubMed]
- Zhan, Y.; Paolicelli, R.C.; Sforazzini, F.; Weinhard, L.; Bolasco, G.; Pagani, F.; Vyssotski, A.L.; Bifone, A.; Gozzi, A.; Ragozzino, D.; et al. Deficient neuron-microglia signaling results in impaired functional brain connectivity and social behavior. Nat. Neurosci. 2014, 17, 400–406. [Google Scholar] [CrossRef] [PubMed]
- Hatterer, E.; Davoust, N.; Didier-Bazes, M.; Vuaillat, C.; Malcus, C.; Belin, M.F.; Nataf, S. How to drain without lymphatics? Dendritic cells migrate from the cerebrospinal fluid to the B-cell follicles of cervical lymph nodes. Blood 2006, 107, 806–812. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.Y.; Chow, N.H.; Ho, T.S.; Lei, H.Y. Intracerebral injection of myelin basic protein (MBP) induces inflammation in brain and causes paraplegia in MBP-sensitized B6 mice. Clin. Exp. Immunol. 1997, 109, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, K.; Bean, A.; Shah, S.; Schutten, E.; Huseby, P.G.; Peters, B.; Shen, Z.T.; Vanguri, V.; Liggitt, D.; Huseby, E.S. Relapsing-remitting central nervous system autoimmunity mediated by GFAP-specific CD8 T cells. J. Immunol. 2014, 192, 3029–3042. [Google Scholar] [CrossRef] [PubMed]
- Debnath, M.; Berk, M. Th17 pathway-mediated immunopathogenesis of schizophrenia: Mechanisms and implications. Schizophr. Bull. 2014, 40, 1412–1421. [Google Scholar] [CrossRef] [PubMed]
- Heinhuis, B.; Koenders, M.I.; van de Loo, F.A.; Netea, M.G.; van den Berg, W.B.; Joosten, L.A. Inflammation-dependent secretion and splicing of IL-32{gamma} in rheumatoid arthritis. Proc. Natl. Acad. Sci. USA 2011, 108, 4962–4967. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, S.; Ricci, E.P.; Mercier, B.C.; Moore, M.J.; Fitzgerald, K.A. Post-transcriptional regulation of gene expression in innate immunity. Nat. Rev. Immunol. 2014, 14, 361–376. [Google Scholar] [CrossRef] [PubMed]
- Kamer, A.R.; Craig, R.G.; Dasanayake, A.P.; Brys, M.; Glodzik-Sobanska, L.; de Leon, M.J. Inflammation and Alzheimer’s disease: Possible role of periodontal diseases. Alzheimers Dement. 2008, 4, 242–250. [Google Scholar] [CrossRef] [PubMed]
- Herz, J.; Zipp, F.; Siffrin, V. Neurodegeneration in autoimmune CNS inflammation. Exp. Neurol. 2010, 225, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Su, X.; Federoff, H.J. Immune responses in Parkinson’s disease: Interplay between central and peripheral immune systems. Biomed. Res. Int. 2014. [Google Scholar] [CrossRef] [PubMed]
- Cappellano, G.; Carecchio, M.; Fleetwood, T.; Magistrelli, L.; Cantello, R.; Dianzani, U.; Comi, C. Immunity and inflammation in neurodegenerative diseases. Am. J. Neurodegener. Dis. 2013, 2, 89–107. [Google Scholar] [PubMed]
- O’Callaghan, J.P.; Sriram, K.; Miller, D.B. Defining “neuroinflammation”. Ann. NY Acad. Sci. 2008, 1139, 318–330. [Google Scholar] [CrossRef] [PubMed]
- Członkowska, A.; Kohutnicka, M.; Kurkowska-Jastrzebska, I.; Członkowski, A. Microglial reaction in MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) induced Parkinson’s disease mice model. Neurodegeneration 1996, 5, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Block, M.L.; Zecca, L.; Hong, J.S. Microglia-mediated neurotoxicity: Uncovering the molecular mechanisms. Nat. Rev. Neurosci. 2007, 8, 57–69. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, D.K.; Basu, A. A friend in need may not be a friend indeed: Role of microglia in neurodegenerative diseases. CNS Neurol. Disord. Drug Targets 2013, 12, 726–740. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Guajardo, V.; Barnum, C.J.; Tansey, M.G.; Romero-Ramos, M. Neuroimmunological processes in Parkinson’s disease and their relation to α-synuclein: Microglia as the referee between neuronal processes and peripheral immunity. ASN Neuro. 2013, 5, 113–139. [Google Scholar] [CrossRef] [PubMed]
- Su, X.; Maguire-Zeiss, K.A.; Giuliano, R.; Prifti, L.; Venkatesh, K.; Federoff, H.J. Synuclein activates microglia in a model of Parkinson’s disease. Neurobiol. Aging 2008, 29, 1690–1701. [Google Scholar] [CrossRef] [PubMed]
- Klegeris, A.; Pelech, S.; Giasson, B.I.; Maguire, J.; Zhang, H.; McGeer, E.G.; McGeer, P.L. Alpha-synuclein activates stress signaling protein kinases in THP-1 cells and microglia. Neurobiol. Aging 2008, 29, 739–752. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Wang, T.; Pei, Z.; Miller, D.S.; Wu, X.; Block, M.L.; Wilson, B.; Zhang, W.; Zhou, Y.; Hong, J.S.; et al. Aggregated alpha-synuclein activates microglia: A process leading to disease progression in Parkinson’s disease. FASEB J. 2005, 19, 533–542. [Google Scholar] [CrossRef] [PubMed]
- Alieva, A.K.; Shadrina, M.I.; Filatova, E.V.; Karabanov, A.V.; Illarioshkin, S.N.; Limborska, S.A.; Slominsky, P.A. Involvement of endocytosis and alternative splicing in the formation of the pathological process in the early stages of Parkinson’s disease. Biomed. Res. Int. 2014. [Google Scholar] [CrossRef] [PubMed]
- Beyer, K.; Ariza, A. α-Synuclein posttranslational modification and alternative splicing as a trigger for neurodegeneration. Mol. Neurobiol. 2013, 47, 509–524. [Google Scholar] [CrossRef] [PubMed]
- Bisaglia, M.; Trolio, A.; Bellanda, M.; Bergantino, E.; Bubacco, L.; Mammi, S. Structure and topology of the non-amyloid-beta component fragment of human alpha-synuclein bound to micelles: Implications for the aggregation process. Protein Sci. 2006, 15, 1408–1416. [Google Scholar] [CrossRef] [PubMed]
- Beyer, K.; Humbert, J.; Ferrer, A.; Lao, J.I.; Latorre, P.; Lopez, D.; Tolosa, E.; Ferrer, I.; Ariza, A. A variable poly-T sequence modulates alpha-synuclein isoform expression and is associated with aging. J. Neurosci. Res. 2007, 85, 1538–1546. [Google Scholar] [CrossRef] [PubMed]
- Kalivendi, S.V.; Yedlapudi, D.; Hillard, C.J.; Kalyanaraman, B. Oxidants induce alternative splicing of α-synuclein: Implications for Parkinson’s disease. Free Radic. Biol. Med. 2010, 48, 377–383. [Google Scholar] [CrossRef] [PubMed]
- Bungeroth, M.; Appenzeller, S.; Regulin, A.; Völker, W.; Lorenzen, I.; Grötzinger, J.; Pendziwiat, M.; Kuhlenbäumer, G. Differential aggregation properties of α-synuclein isoforms. Neurobiol. Aging 2014, 35, 1913–1919. [Google Scholar] [CrossRef] [PubMed]
- Pasinetti, G.M. Cyclooxygenase and inflammation in Alzheimer’s disease: Experimental approaches and clinical interventions. J. Neurosci. Res. 1998, 54, 1–6. [Google Scholar] [CrossRef]
- González-Scarano, F.; Baltuch, G. Microglia as mediators of inflammatory and degenerative diseases. Annu. Rev. Neurosci. 1999, 22, 219–240. [Google Scholar] [CrossRef] [PubMed]
- Julien, J.P. Amyotrophic lateral sclerosis. unfolding the toxicity of the misfolded. Cell 2001, 104, 581–591. [Google Scholar] [CrossRef]
- Malik, M.; Simpson, J.F.; Parikh, I.; Wilfred, B.R.; Fardo, D.W.; Nelson, P.T.; Estus, S. CD33 Alzheimer’s risk-altering polymorphism, CD33 expression, and exon 2 splicing. J. Neurosci. 2013, 33, 13320–13325. [Google Scholar] [CrossRef] [PubMed]
- Crocker, P.R.; Paulson, J.C.; Varki, A. Siglecs and their roles in the immune system. Nat. Rev. Immunol. 2007, 7, 255–266. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Yu, J.T.; Hu, N.; Tan, M.S.; Zhu, X.C.; Tan, L. CD33 in Alzheimer’s disease. Mol. Neurobiol. 2014, 49, 529–535. [Google Scholar] [CrossRef] [PubMed]
- Linnartz, B.; Neumann, H. Microglial activatory (immunoreceptor tyrosine-based activation motif)- and inhibitory (immunoreceptor tyrosine-based inhibition motif)-signaling receptors for recognition of the neuronal glycocalyx. Glia 2013, 61, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Evsyukova, I.; Somarelli, J.A.; Gregory, S.G.; Garcia-Blanco, M.A. Alternative splicing in multiple sclerosis and other autoimmune diseases. RNA Biol. 2010, 7, 462–473. [Google Scholar] [CrossRef] [PubMed]
- Henney, C.S. Interleukin 7: Effects on early events in lymphopoiesis. Immunol. Today 1989, 10, 170–173. [Google Scholar] [CrossRef]
- Namen, A.E.; Lupton, S.; Hjerrild, K.; Wignall, J.; Mochizuki, D.Y.; Schmierer, A.; Mosley, B.; March, C.J.; Urdal, D.; Gillis, S. Stimulation of B-cell progenitors by cloned murine interleukin-7. Nature 1988, 333, 571–573. [Google Scholar] [CrossRef] [PubMed]
- Namen, A.E.; Schmierer, A.E.; March, C.J.; Overell, R.W.; Park, L.S.; Urdal, D.L.; Mochizuki, D.Y. B cell precursor growth-promoting activity. Purification and characterization of a growth factor active on lymphocyte precursors. J. Exp. Med. 1988, 167, 988–1002. [Google Scholar] [CrossRef] [PubMed]
- Murray, R.; Suda, T.; Wrighton, N.; Lee, F.; Zlotnik, A. IL-7 is a growth and maintenance factor for mature and immature thymocyte subsets. Int. Immunol. 1989, 1, 526–531. [Google Scholar] [CrossRef] [PubMed]
- Chazen, G.D.; Pereira, G.M.; LeGros, G.; Gillis, S.; Shevach, E.M. Interleukin 7 is a T-cell growth factor. Proc. Natl. Acad. Sci. USA 1989, 86, 5923–5927. [Google Scholar] [CrossRef] [PubMed]
- Michaelson, M.D.; Mehler, M.F.; Xu, H.; Gross, R.E.; Kessler, J.A. Interleukin-7 is trophic for embryonic neurons and is expressed in developing brain. Dev. Biol. 1996, 179, 251–263. [Google Scholar] [CrossRef] [PubMed]
- Wolf, S.S.; Cohen, A. Expression of cytokines and their receptors by human thymocytes and thymic stromal cells. Immunology 1992, 77, 362–368. [Google Scholar] [PubMed]
- Page, T.H.; Willcocks, J.L.; Taylor-Fishwick, D.A.; Foxwell, B.M. Characterization of a novel high affinity human IL-7 receptor. Expression on T cells and association with IL-7 driven proliferation. J. Immunol. 1993, 151, 4753–4763. [Google Scholar] [PubMed]
- Benjamin, D.; Sharma, V.; Knobloch, T.J.; Armitage, R.J.; Dayton, M.A.; Goodwin, R.G. B cell IL-7. Human B cell lines constitutively secrete IL-7 and express IL-7 receptors. J. Immunol. 1994, 152, 4749–4757. [Google Scholar] [PubMed]
- Moors, M.; Vudattu, N.K.; Abel, J.; Krämer, U.; Rane, L.; Ulfig, N.; Ceccatelli, S.; Seyfert-Margolies, V.; Fritsche, E.; Maeurer, M.J.; et al. Interleukin-7 (IL-7) and IL-7 splice variants affect differentiation of human neural progenitor cells. Genes Immun. 2010, 11, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Nunnari, G.; Xu, Y.; Acheampong, E.A.; Fang, J.; Daniel, R.; Zhang, C.; Zhang, H.; Mukhtar, M.; Pomerantz, R.J. Exogenous IL-7 induces Fas-mediated human neuronal apoptosis: Potential effects during human immunodeficiency virus type 1 infection. J. Neurovirol. 2005, 11, 319–328. [Google Scholar] [CrossRef] [PubMed]
- The International Multiple Sclerosis Genetics Consortium; Hafler, D.A.; Compston, A.; Sawcer, S.; Lander, E.S.; Daly, M.J.; de Jager, P.L.; de Bakker, P.I.; Gabriel, S.B.; Mirel, D.B.; et al. Risk alleles for multiple sclerosis identified by a genomewide study. N. Engl. J. Med. 2007, 357, 851–862. [Google Scholar] [PubMed]
- Lundmark, F.; Duvefelt, K.; Iacobaeus, E.; Kockum, I.; Wallström, E.; Khademi, M.; Oturai, A.; Ryder, L.P.; Saarela, J.; Harbo, H.F.; et al. Variation in interleukin 7 receptor alpha chain (IL7R) influences risk of multiple sclerosis. Nat. Genet. 2007, 39, 1108–1113. [Google Scholar] [CrossRef] [PubMed]
- Gregory, S.G.; Schmidt, S.; Seth, P.; Oksenberg, J.R.; Hart, J.; Prokop, A.; Caillier, S.J.; Ban, M.; Goris, A.; Barcellos, L.F.; et al. Interleukin 7 receptor alpha chain (IL7R) shows allelic and functional association with multiple sclerosis. Nat. Genet. 2007, 39, 1083–1091. [Google Scholar] [CrossRef] [PubMed]
- Majdinasab, N.; Behbahani, M.H.; Galehdari, H.; Mohaghegh, M. Association of interleukin 7 receptor gene polymorphism rs6897932 with multiple sclerosis patients in Khuzestan. Iran. J. Neurol. 2014, 13, 168–171. [Google Scholar] [PubMed]
- Goodwin, R.G.; Friend, D.; Ziegler, S.F.; Jerzy, R.; Falk, B.A.; Gimpel, S.; Cosman, D.; Dower, S.K.; March, C.J.; Namen, A.E.; et al. Cloning of the human and murine interleukin-7 receptors: Demonstration of a soluble form and homology to a new receptor superfamily. Cell 1990, 60, 941–951. [Google Scholar] [CrossRef]
- Rose, T.; Lambotte, O.; Pallier, C.; Delfraissy, J.F.; Colle, J.H. Identification and biochemical characterization of human plasma soluble IL-7R: Lower concentrations in HIV-1-infected patients. J. Immunol. 2009, 182, 7389–7397. [Google Scholar] [CrossRef] [PubMed]
- Heaney, M.L.; Golde, D.W. Soluble cytokine receptors. Blood 1996, 87, 847–857. [Google Scholar] [PubMed]
- Xing, Y.; Xu, Q.; Lee, C. Widespread production of novel soluble protein isoforms by alternative splicing removal of transmembrane anchoring domains. FEBS Lett. 2003, 555, 572–578. [Google Scholar] [CrossRef]
- Haegele, K.F.; Stueckle, C.A.; Malin, J.P.; Sindern, E. Increase of CD8+ T-effector memory cells in peripheral blood of patients with relapsing-remitting multiple sclerosis compared to healthy controls. J. Neuroimmunol. 2007, 183, 168–174. [Google Scholar] [CrossRef] [PubMed]
- Jacobsen, M.; Schweer, D.; Ziegler, A.; Gaber, R.; Schock, S.; Schwinzer, R.; Wonigeit, K.; Lindert, R.B.; Kantarci, O.; Schaefer-Klein, J.; et al. A point mutation in PTPRC is associated with the development of multiple sclerosis. Nat. Genet. 2000, 26, 495–499. [Google Scholar] [PubMed]
- Ballerini, C.; Rosati, E.; Salvetti, M.; Ristori, G.; Cannoni, S.; Biagioli, T.; Massacesi, L.; Sorbi, S.; Vergelli, M. Protein tyrosine phosphatase receptor-type C exon 4 gene mutation distribution in an Italian multiple sclerosis population. Neurosci. Lett. 2002, 328, 325–327. [Google Scholar] [CrossRef]
- Jacobsen, M.; Hoffmann, S.; Cepok, S.; Stei, S.; Ziegler, A.; Sommer, N.; Hemmer, B. A novel mutation in PTPRC interferes with splicing and alters the structure of the human CD45 molecule. Immunogenetics 2002, 54, 158–163. [Google Scholar] [CrossRef] [PubMed]
- Vyshkina, T.; Leist, T.P.; Shugart, Y.Y.; Kalman, B. CD45 (PTPRC) as a candidate gene in multiple sclerosis. Mult. Scler. 2004, 10, 614–617. [Google Scholar] [CrossRef] [PubMed]
- Hermiston, M.L.; Xu, Z.; Weiss, A. CD45: A critical regulator of signaling thresholds in immune cells. Annu. Rev. Immunol. 2003, 21, 107–137. [Google Scholar] [CrossRef] [PubMed]
- Lynch, K.W.; Weiss, A. A CD45 polymorphism associated with multiple sclerosis disrupts an exonic splicing silencer. J. Biol. Chem. 2001, 276, 24341–24347. [Google Scholar] [CrossRef] [PubMed]
- Tong, A.; Nguyen, J.; Lynch, K.W. Differential expression of CD45 isoforms is controlled by the combined activity of basal and inducible splicing-regulatory elements in each of the variable exons. J. Biol. Chem. 2005, 280, 38297–38304. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.Y.; Xu, X.; Ding, J.H.; Bermingham, J.R.; Fu, X.D. SC35 plays a role in T cell development and alternative splicing of CD45. Mol. Cell 2001, 7, 331–342. [Google Scholar] [CrossRef]
- Oberdoerffer, S.; Moita, L.F.; Neems, D.; Freitas, R.P.; Hacohen, N.; Rao, A. Regulation of CD45 alternative splicing by heterogeneous ribonucleoprotein, hnRNPLL. Science 2008, 321, 686–691. [Google Scholar] [CrossRef] [PubMed]
- Topp, J.D.; Jackson, J.; Melton, A.A.; Lynch, K.W. A cell-based screen for splicing regulators identifies hnRNP LL as a distinct signal-induced repressor of CD45 variable exon 4. RNA 2008, 14, 2038–2049. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wu, Z.; Jia, X.; de la Cruz, L.; Su, X.C.; Marzolf, B.; Troisch, P.; Zak, D.; Hamilton, A.; Whittle, B.; Yu, D.; et al. Memory T cell RNA rearrangement programmed by heterogeneous nuclear ribonucleoprotein hnRNPLL. Immunity 2008, 29, 863–875. [Google Scholar] [CrossRef] [PubMed]
- Steel, J.C.; Waldmann, T.A.; Morris, J.C. Interleukin-15 biology and its therapeutic implications in cancer. Trends Pharmacol. Sci. 2012, 33, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Budagian, V.; Bulanova, E.; Paus, R.; Bulfone-Paus, S. IL-15/IL-15 receptor biology: A guided tour through an expanding universe. Cytokine Growth Factor Rev. 2006, 17, 259–280. [Google Scholar] [CrossRef] [PubMed]
- Pan, W.; Wu, X.; He, Y.; Hsuchou, H.; Huang, E.Y.; Mishra, P.K.; Kastin, A.J. Brain interleukin-15 in neuroinflammation and behavior. Neurosci. Biobehav. Rev. 2013, 37, 184–192. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Pan, W.; Stone, K.P.; Zhang, Y.; Hsuchou, H.; Kastin, A.J. Expression and signaling of novel IL15Ralpha splicing variants in cerebral endothelial cells of the blood-brain barrier. J. Neurochem. 2010, 114, 122–129. [Google Scholar] [PubMed]
- Dubois, S.; Magrangeas, F.; Lehours, P.; Raher, S.; Bernard, J.; Boisteau, O.; Leroy, S.; Minvielle, S.; Godard, A.; Jacques, Y. Natural splicing of exon 2 of human interleukin-15 receptor alpha-chain mRNA results in a shortened form with a distinct pattern of expression. J. Biol. Chem. 1999, 274, 26978–26984. [Google Scholar] [CrossRef] [PubMed]
- Bulanova, E.; Budagian, V.; Orinska, Z.; Krause, H.; Paus, R.; Bulfone-Paus, S. Mast cells express novel functional IL-15 receptor alpha isoforms. J. Immunol. 2003, 170, 5045–5055. [Google Scholar] [CrossRef] [PubMed]
- Anderson, K.M.; Olson, K.E.; Estes, K.A.; Flanagan, K.; Gendelman, H.E.; Mosley, R.L. Dual destructive and protective roles of adaptive immunity in neurodegenerative disorders. Transl. Neurodegener. 2014. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Trapp, B.D. Microglia and neuroprotection. J. Neurochem. 2015. [Google Scholar] [CrossRef] [PubMed]
- Dinarello, C.A. Biologic basis for interleukin-1 in disease. Blood 1996, 87, 2095–2147. [Google Scholar] [PubMed]
- Sims, J.E. IL-1 and IL-18 receptors, and their extended family. Curr. Opin. Immunol. 2002, 14, 117–122. [Google Scholar] [CrossRef]
- Smith, D.E.; Lipsky, B.P.; Russell, C.; Ketchem, R.R.; Kirchner, J.; Hensley, K.; Huang, Y.; Friedman, W.J.; Boissonneault, V.; Plante, M.M.; et al. A central nervous system-restricted isoform of the interleukin-1 receptor accessory protein modulates neuronal responses to interleukin-1. Immunity 2009, 30, 817–831. [Google Scholar] [CrossRef] [PubMed]
- Gosselin, D.; Bellavance, M.A.; Rivest, S. IL-1RAcPb signaling regulates adaptive mechanisms in neurons that promote their long-term survival following excitotoxic insults. Front. Cell. Neurosci. 2013. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.L.; Yang, C.Y.; Chen, H.C.; Hung, C.S.; Chiang, Y.C.; Ting, L.P. A novel alternatively spliced interleukin-1 receptor accessory protein mIL-1RAcP687. Mol. Immunol. 2008, 45, 1374–1384. [Google Scholar] [CrossRef] [PubMed]
- Scheller, J.; Garbers, C.; Rose-John, S. Interleukin-6: From basic biology to selective blockade of pro-inflammatory activities. Semin. Immunol. 2014, 26, 2–12. [Google Scholar] [CrossRef] [PubMed]
- Müllberg, J.; Schooltink, H.; Stoyan, T.; Günther, M.; Graeve, L.; Buse, G.; Mackiewicz, A.; Heinrich, P.C.; Rose-John, S. The soluble interleukin-6 receptor is generated by shedding. Eur. J. Immunol. 1993, 23, 473–480. [Google Scholar] [CrossRef] [PubMed]
- Lust, J.A.; Donovan, K.A.; Kline, M.P.; Greipp, P.R.; Kyle, R.A.; Maihle, N.J. Isolation of an mRNA encoding a soluble form of the human interleukin-6 receptor. Cytokine 1992, 4, 96–100. [Google Scholar] [CrossRef]
- Taga, T.; Hibi, M.; Hirata, Y.; Yamasaki, K.; Yasukawa, K.; Matsuda, T.; Hirano, T.; Kishimoto, T. Interleukin-6 triggers the association of its receptor with a possible signal transducer, gp130. Cell 1989, 58, 573–581. [Google Scholar] [CrossRef]
- Rose-John, S.; Heinrich, P.C. Soluble receptors for cytokines and growth factors: Generation and biological function. Biochem. J. 1994, 300, 281–290. [Google Scholar] [CrossRef] [PubMed]
- März, P.; Otten, U.; Rose-John, S. Neural activities of IL-6-type cytokines often depend on soluble cytokine receptors. Eur. J. Neurosci. 1999, 11, 2995–3004. [Google Scholar] [CrossRef] [PubMed]
- Hirota, H.; Kiyama, H.; Kishimoto, T.; Taga, T. Accelerated Nerve Regeneration in Mice by upregulated expression of interleukin (IL) 6 and IL-6 receptor after trauma. J. Exp. Med. 1996, 183, 2627–2634. [Google Scholar] [CrossRef] [PubMed]
- Moser, J.M.; Bigini, P.; Schmitt-John, T. The wobbler mouse, an ALS animal model. Mol. Genet. Genomics 2013, 288, 207–229. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, K.; Kinoshita, M.; Tagaya, N.; Shiojima, T.; Taga, T.; Yasukawa, K.; Suzuki, H.; Okano, A. Coadministration of interleukin-6 (IL-6) and soluble IL-6 receptor delays progression of wobbler mouse motor neuron disease. Brain Res. 1996, 726, 91–97. [Google Scholar] [CrossRef]
- Pennica, D.; Wood, W.I.; Chien, K.R. Cardiotrophin-1: A multifunctional cytokine that signals via LIF receptor-gp 130 dependent pathways. Cytokine Growth Factor Rev. 1996, 7, 81–91. [Google Scholar] [CrossRef]
- Yao, G.L.; Kato, H.; Khalil, M.; Kiryu, S.; Kiyama, H. Selective upregulation of cytokine receptor subchain and their intracellular signalling molecules after peripheral nerve injury. Eur. J. Neurosci. 1997, 9, 1047–1054. [Google Scholar] [CrossRef] [PubMed]
- Mazin, P.; Xiong, J.; Liu, X.; Yan, Z.; Zhang, X.; Li, M.; He, L.; Somel, M.; Yuan, Y.; Phoebe Chen, Y.P.; et al. Widespread splicing changes in human brain development and aging. Mol. Syst. Biol. 2013. [Google Scholar] [CrossRef]
- Rock, K.L.; Latz, E.; Ontiveros, F.; Kono, H. The sterile inflammatory response. Annu. Rev. Immunol. 2010, 28, 321–342. [Google Scholar] [CrossRef] [PubMed]
- Sanderson, C.J. Interleukin-5, eosinophils, and disease. Blood 1992, 79, 3101–3109. [Google Scholar] [PubMed]
- Iwama, A.; Pan, J.; Zhang, P.; Reith, W.; Mach, B.; Tenen, D.G.; Sun, Z. Dimeric RFX proteins contribute to the activity and lineage specificity of the interleukin-5 receptor alpha promoter through activation and repression domains. Mol. Cell. Biol. 1999, 19, 3940–3950. [Google Scholar] [PubMed]
- Tuypens, T.; Plaetinck, G.; Baker, E.; Sutherland, G.; Brusselle, G.; Fiers, W.; Devos, R.; Tavernier, J. Organization and chromosomal localization of the human interleukin 5 receptor alpha-chain gene. Eur. Cytokine Netw. 1992, 3, 451–459. [Google Scholar] [PubMed]
- Lins, C.; Borojevic, R. Interleukin-5 receptor alpha chain expression and splicing during brain development in mice. Growth Factors 2001, 19, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Awatsuji, H.; Furukawa, Y.; Hirota, M.; Murakami, Y.; Nii, S.; Furukawa, S.; Hayashi, K. Interleukin-4 and -5 as modulators of nerve growth factor synthesis/secretion in astrocytes. J. Neurosci. Res. 1993, 34, 539–545. [Google Scholar] [CrossRef] [PubMed]
- Kotsimbos, A.T.; Hamid, Q. IL-5 and IL-5 receptor in asthma. Mem. Inst. Oswaldo Cruz 1997, 92, 75–91. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, Y.; Migita, M.; Takaki, S.; Tominaga, A.; Takatsu, K. Biochemical and functional characterization of soluble form of IL-5 receptor alpha (sIL-5R α). Development of ELISA system for detection of sIL-5R α. J. Immunol. Methods 1994, 167, 289–298. [Google Scholar] [CrossRef]
- Takatsu, K.; Takaki, S.; Hitoshi, Y. Interleukin-5 and its receptor system: Implications in the immune system and inflammation. Adv. Immunol. 1994, 57, 145–190. [Google Scholar] [PubMed]
- Imamura, F.; Takaki, S.; Akagi, K.; Ando, M.; Yamamura, K.; Takatsu, K.; Tominaga, A. The murine interleukin-5 receptor alpha-subunit gene: Characterization of the gene structure and chromosome mapping. DNA Cell Biol. 1994, 13, 283–292. [Google Scholar] [CrossRef] [PubMed]
- Karras, J.G.; McKay, R.A.; Dean, N.M.; Monia, B.P. Deletion of individual exons and induction of soluble murine interleukin-5 receptor-α chain expression through antisense oligonucleotide-mediated redirection of pre-mRNA splicing. Mol. Pharmacol. 2000, 58, 380–387. [Google Scholar] [PubMed]
- Tavernier, J.; Tuypens, T.; Plaetinck, G.; Verhee, A.; Fiers, W.; Devos, R. Molecular basis of the membrane-anchored and two soluble isoforms of the human interleukin 5 receptor alpha subunit. Proc. Natl. Acad. Sci. USA 1992, 89, 7041–7045. [Google Scholar] [CrossRef] [PubMed]
- Murata, Y.; Takaki, S.; Migita, M.; Kikuchi, Y.; Tominaga, A.; Takatsu, K. Molecular cloning and expression of the human interleukin 5 receptor. J. Exp. Med. 1992, 175, 341–351. [Google Scholar] [CrossRef] [PubMed]
- Tobin, D.; Nabarro, G.; de la Faille, H.B.; van Vloten, W.A.; van der Putte, S.C.; Schuurman, H.J. Increased number of immunoreactive nerve fibers in atopic dermatitis. J. Allergy Clin. Immunol. 1992, 90, 613–622. [Google Scholar] [CrossRef]
- Ostlere, L.S.; Cowen, T.; Rustin, M.H. Neuropeptides in the skin of patients with atopic dermatitis. Clin. Exp. Dermatol. 1995, 20, 462–467. [Google Scholar] [CrossRef] [PubMed]
- Lourenssen, S.; Wells, R.W.; Blennerhassett, M.G. Differential responses of intrinsic and extrinsic innervation of smooth muscle cells in rat colitis. Exp. Neurol. 2005, 195, 497–507. [Google Scholar] [CrossRef] [PubMed]
- Birch, D.; Knight, G.E.; Boulos, P.B.; Burnstock, G. Analysis of innervation of human mesenteric vessels in non-inflamed and inflamed bowel—A confocal and functional study. Neurogastroenterol. Motil. 2008, 20, 660–670. [Google Scholar] [CrossRef] [PubMed]
- Xia, C.M.; Colomb, D.G.; Akbarali, H.I.; Qiao, L.Y. Prolonged sympathetic innervation of sensory neurons in rat thoracolumbar dorsal root ganglia during chronic colitis. Neurogastroenterol. Motil. 2011. [Google Scholar] [CrossRef] [PubMed]
- Lorton, D.; Lubahn, C.; Sweeney, S.; Major, A.; Lindquist, C.A.; Schaller, J.; Washington, C.; Bellinger, D.L. Differences in the injury/sprouting response of splenic noradrenergic nerves in Lewis rats with adjuvant-induced arthritis compared with rats treated with 6-hydroxydopamine. Brain Behav. Immun. 2009, 23, 276–285. [Google Scholar] [CrossRef] [PubMed]
- Straub, R.H.; Lowin, T.; Klatt, S.; Wolff, C.; Rauch, L. Increased density of sympathetic nerve fibers in metabolically activated fat tissue surrounding human synovium and mouse lymph nodes in arthritis. Arthritis Rheum. 2011, 63, 3234–3242. [Google Scholar] [CrossRef] [PubMed]
- Hasan, W.; Jama, A.; Donohue, T.; Wernli, G.; Onyszchuk, G.; Al-Hafez, B.; Bilgen, M.; Smith, P.G. Sympathetic hyperinnervation and inflammatory cell NGF synthesis following myocardial infarction in rats. Brain Res. 2006, 1124, 142–154. [Google Scholar] [CrossRef] [PubMed]
- Peeker, R.; Aldenborg, F.; Dahlström, A.; Johansson, S.L.; Li, J.Y.; Fall, M. Increased tyrosine hydroxylase immunoreactivity in bladder tissue from patients with classic and nonulcer interstitial cystitis. J. Urol. 2000, 163, 1112–1115. [Google Scholar] [CrossRef]
- Hagiyama, M.; Furuno, T.; Hosokawa, Y.; Iino, T.; Ito, T.; Inoue, T.; Nakanishi, M.; Murakami, Y.; Ito, A. Enhanced nerve-mast cell interaction by a neuronal short isoform of cell adhesion molecule-1. J. Immunol. 2011, 186, 5983–5992. [Google Scholar] [CrossRef] [PubMed]
- Hagiyama, M.; Ichiyanagi, N.; Kimura, K.B.; Murakami, Y.; Ito, A. Expression of a soluble isoform of cell adhesion molecule 1 in the brain and its involvement in directional neurite outgrowth. Am. J. Pathol. 2009, 174, 2278–2289. [Google Scholar] [CrossRef] [PubMed]
- Ito, A.; Jippo, T.; Wakayama, T.; Morii, E.; Koma, Y.; Onda, H.; Nojima, H.; Iseki, S.; Kitamura, Y. SgIGSF: A new mast-cell adhesion molecule used for attachment to fibroblasts and transcriptionally regulated by MITF. Blood 2003, 101, 2601–2608. [Google Scholar] [CrossRef] [PubMed]
- Koma, Y.; Ito, A.; Wakayama, T.; Watabe, K.; Okada, M.; Tsubota, N.; Iseki, S.; Kitamura, Y. Cloning of a soluble isoform of the SgIGSF adhesion molecule that binds the extracellular domain of the membrane-bound isoform. Oncogene 2004, 23, 5687–5692. [Google Scholar] [CrossRef] [PubMed]
- Hosokawa, Y.; Hagiyama, M.; Iino, T.; Murakami, Y.; Ito, A. Noncontact estimation of intercellular breaking force using a femtosecond laser impulse quantified by atomic force microscopy. Proc. Natl. Acad. Sci. USA 2011, 108, 1777–1782. [Google Scholar] [CrossRef] [PubMed]
- Theoharides, T.C. The mast cell: A neuroimmunoendocrine master player. Int. J. Tissue React. 1996, 18, 1–21. [Google Scholar] [PubMed]
- Theoharides, T.C.; Cochrane, D.E. Critical role of mast cells in inflammatory diseases and the effect of acute stress. J. Neuroimmunol. 2004, 146, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Barbara, G.; Stanghellini, V.; de Giorgio, R.; Cremon, C.; Cottrell, G.S.; Santini, D.; Pasquinelli, G.; Morselli-Labate, A.M.; Grady, E.F.; Bunnett, N.W.; et al. Activated mast cells in proximity to colonic nerves correlate with abdominal pain in irritable bowel syndrome. Gastroenterology 2004, 126, 693–702. [Google Scholar] [CrossRef] [PubMed]
- Furuno, T.; Ito, A.; Koma, Y.; Watabe, K.; Yokozaki, H.; Bienenstock, J.; Nakanishi, M.; Kitamura, Y. The spermatogenic Ig superfamily/synaptic cell adhesion molecule mast-cell adhesion molecule promotes interaction with nerves. J. Immunol. 2005, 174, 6934–6942. [Google Scholar] [CrossRef] [PubMed]
- Bischoff, S.C. Role of mast cells in allergic and non-allergic immune responses: Comparison of human and murine data. Nat. Rev. Immunol. 2007, 7, 93–104. [Google Scholar] [CrossRef] [PubMed]
- Moiseeva, E.P.; Leyland, M.L.; Bradding, P. CADM1 is expressed as multiple alternatively spliced functional and dysfunctional isoforms in human mast cells. Mol. Immunol. 2013, 53, 345–354. [Google Scholar] [CrossRef] [PubMed]
- Szkudlinski, M.W.; Fremont, V.; Ronin, C.; Weintraub, B.D. Thyroid-stimulating hormone and thyroid-stimulating hormone receptor structure-function relationships. Physiol. Rev. 2002, 82, 473–502. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.M.; Phan, M.; Kruger, T.E.; Coppenhaver, D.H.; Blalock, J.E. Human lymphocyte production of immunoreactive thyrotropin. Proc. Natl. Acad. Sci. USA 1983, 80, 6010–6013. [Google Scholar] [CrossRef] [PubMed]
- Harbour, D.V.; Kruger, T.E.; Coppenhaver, D.; Smith, E.M.; Meyer, W.J. Differential expression and regulation of thyrotropin (TSH) in T cell lines. Mol. Cell. Endocrinol. 1989, 64, 229–241. [Google Scholar] [CrossRef]
- Kelley, K.W.; Weigent, D.A.; Kooijman, R. Protein hormones and immunity. Brain Behav. Immun. 2007, 21, 384–392. [Google Scholar] [CrossRef] [PubMed]
- Klein, J.R.; Wang, H.C. Characterization of a novel set of resident intrathyroidal bone marrow-derived hematopoietic cells: Potential for immune-endocrine interactions in thyroid homeostasis. J. Exp. Biol. 2004, 207, 55–65. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.H.; Koenig, R.J. A locally secreted thyrotropin variant may regulate thyroid function in thyroid inflammatory disorders. Thyroid 2009, 19, 5–6. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Li, L.; Ying, F.; Xu, C.; Zang, X.; Gao, Z. A newly identified TSHβ splice variant is involved in the pathology of Hashimoto’s thyroiditis. Mol. Biol. Rep. 2012, 39, 10019–10030. [Google Scholar] [CrossRef] [PubMed]
- Vincent, B.H.; Montufar-Solis, D.; Teng, B.B.; Amendt, B.A.; Schaefer, J.; Klein, J.R. Bone marrow cells produce a novel TSHbeta splice variant that is upregulated in the thyroid following systemic virus infection. Genes Immun. 2009, 10, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Klein, J.R. Biological impact of the TSHβ splice variant in health and disease. Front. Immunol. 2014. [Google Scholar] [CrossRef] [PubMed]
- Ule, J.; Jensen, K.B.; Ruggiu, M.; Mele, A.; Ule, A.; Darnell, R.B. CLIP identifies Nova-regulated RNA networks in the brain. Science 2003, 302, 1212–1215. [Google Scholar] [CrossRef] [PubMed]
- Lister, R.; O’Malley, R.C.; Tonti-Filippini, J.; Gregory, B.D.; Berry, C.C.; Millar, A.H.; Ecker, J.R. Highly integrated single-base resolution maps of the epigenome in Arabidopsis. Cell 2008, 133, 523–536. [Google Scholar] [CrossRef] [PubMed]
- Nagalakshmi, U.; Wang, Z.; Waern, K.; Shou, C.; Raha, D.; Gerstein, M.; Snyder, M. The transcriptional landscape of the yeast genome defined by RNA sequencing. Science 2008, 320, 1344–1349. [Google Scholar] [CrossRef] [PubMed]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Licatalosi, D.D.; Mele, A.; Fak, J.J.; Ule, J.; Kayikci, M.; Chi, S.W.; Clark, T.A.; Schweitzer, A.C.; Blume, J.E.; Wang, X.; et al. HITS-CLIP yields genome-wide insights into brain alternative RNA processing. Nature 2008, 456, 464–469. [Google Scholar] [CrossRef] [PubMed]
- Chi, S.W.; Zang, J.B.; Mele, A.; Darnell, R.B. Argonaute HITS-CLIP decodes microRNA-mRNA interaction maps. Nature 2009, 460, 479–486. [Google Scholar] [CrossRef] [PubMed]
- Darnell, R.B. HITS-CLIP: Panoramic views of protein-RNA regulation in living cells. Wiley Interdiscip. Rev. RNA 2010, 1, 266–286. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Darnell, R.B. Mapping in vivo protein-RNA interactions at single-nucleotide resolution from HITS-CLIP data. Nat. Biotechnol. 2011, 29, 607–614. [Google Scholar] [CrossRef] [PubMed]
- Yeo, G.W.; Coufal, N.G.; Liang, T.Y.; Peng, G.E.; Fu, X.D.; Gage, F.H. An RNA code for the FOX2 splicing regulator revealed by mapping RNA-protein interactions in stem cells. Nat. Struct. Mol. Biol. 2009, 16, 130–137. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.K.; Yang, Y.; Zhu, J.; Adelstein, R.S.; Kawamoto, S. Rbfox3 controls the biogenesis of a subset of microRNAs. Nat. Struct. Mol. Biol. 2014, 21, 901–910. [Google Scholar] [CrossRef] [PubMed]
- Zarnack, K.; König, J.; Tajnik, M.; Martincorena, I.; Eustermann, S.; Stévant, I.; Reyes, A.; Anders, S.; Luscombe, N.M.; Ule, J. Direct competition between hnRNP C and U2AF65 protects the transcriptome from the exonization of Alu elements. Cell 2013, 152, 453–466. [Google Scholar] [CrossRef] [PubMed]
- Rossbach, O.; Hung, L.H.; Khrameeva, E.; Schreiner, S.; König, J.; Curk, T.; Zupan, B.; Ule, J.; Gelfand, M.S.; Bindereif, A. Crosslinking-immunoprecipitation (iCLIP) analysis reveals global regulatory roles of hnRNP L. RNA Biol. 2014, 11, 146–155. [Google Scholar] [CrossRef] [PubMed]
- Shankarling, G.; Cole, B.S.; Mallory, M.J.; Lynch, K.W. Transcriptome-wide RNA interaction profiling reveals physical and functional targets of hnRNP L in human T cells. Mol. Cell. Biol. 2014, 34, 71–83. [Google Scholar] [CrossRef] [PubMed]
- Ule, J.; Stefani, G.; Mele, A.; Ruggiu, M.; Wang, X.; Taneri, B.; Gaasterland, T.; Blencowe, B.J.; Darnell1, R.B. An RNA map predicting Nova-dependent splicing regulation. Nature 2006, 444, 580–586. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.; Zhou, Y.; Wu, T.; Zhu, T.; Ji, X.; Kwon, Y.S.; Zhang, C.; Yeo, G.; Black, D.L.; Sun, H.; et al. Genome-wide analysis of PTB-RNA interactions reveals a strategy used by the general splicing repressor to modulate exon inclusion or skipping. Mol. Cell 2009, 36, 996–1006. [Google Scholar] [CrossRef] [PubMed]
- Lebedeva, S.; Jens, M.; Theil, K.; Schwanhäusser, B.; Selbach, M.; Landthaler, M.; Rajewsky, N. Transcriptome-wide analysis of regulatory interactions of the RNA-binding protein HuR. Mol. Cell 2011, 43, 340–352. [Google Scholar] [CrossRef] [PubMed]
- Änkö, M.L.; Müller-McNicoll, M.; Brandl, H.; Curk, T.; Gorup, C.; Henry, I.; Ule, J.; Neugebauer, K.M. The RNA-binding landscapes of two SR proteins reveal unique functions and binding to diverse RNA classes. Genome Biol. 2012. [Google Scholar] [CrossRef] [PubMed]
- Buckanovich, R.J.; Yang, Y.Y.; Darnell, R.B. The onconeural antigen Nova-1 is a neuron-specific RNA-binding protein, the activity of which is inhibited by paraneoplastic antibodies. J. Neurosci. 1996, 16, 1114–1122. [Google Scholar] [PubMed]
- Yang, Y.Y.; Yin, G.L.; Darnell, R.B. The neuronal RNA-binding protein Nova-2 is implicated as the autoantigen targeted in POMA patients with dementia. Proc. Natl. Acad. Sci. USA 1998, 95, 13254–13259. [Google Scholar] [CrossRef] [PubMed]
- Buckanovich, R.J.; Darnell, R.B. The neuronal RNA binding protein Nova-1 recognizes specific RNA targets in vitro and in vivo. Mol. Cell. Biol. 1997, 17, 3194–3201. [Google Scholar] [PubMed]
- Lewis, H.A.; Musunuru, K.; Jensen, K.B.; Edo, C.; Chen, H.; Darnell, R.B.; Burley, S.K. Sequence-specific RNA binding by a Nova KH domain: Implications for paraneoplastic disease and the fragile X syndrome. Cell 2000, 100, 323–332. [Google Scholar] [CrossRef]
- Katsanou, V.; Milatos, S.; Yiakouvaki, A.; Sgantzis, N.; Kotsoni, A.; Alexiou, M.; Harokopos, V.; Aidinis, V.; Hemberger, M.; Kontoyiannis, D.L. The RNA-binding protein Elavl1/HuR is essential for placental branching morphogenesis and embryonic development. Mol. Cell. Biol. 2009, 29, 2762–2776. [Google Scholar] [CrossRef] [PubMed]
- Papadaki, O.; Milatos, S.; Grammenoudi, S.; Mukherjee, N.; Keene, J.D.; Kontoyiannis, D.L. Control of thymic T cell maturation, deletion and egress by the RNA-binding protein HuR. J. Immunol. 2009, 182, 6779–6788. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.G.; Andrews, L.G.; McGowan, K.M.; Pekala, P.H.; Keene, J.D. Ectopic expression of Hel-N1, an RNA-binding protein, increases glucose transporter (GLUT1) expression in 3T3-L1 adipocytes. Mol. Cell. Biol. 1997, 17, 954–962. [Google Scholar] [PubMed]
- Fan, X.C.; Steitz, J.A. Overexpression of HuR, a nuclear-cytoplasmic shuttling protein, increases the in vivo stability of ARE-containing mRNAs. EMBO J. 1998, 17, 3448–3460. [Google Scholar] [CrossRef] [PubMed]
- Peng, S.S.; Chen, C.Y.; Xu, N.; Shyu, A.B. RNA stabilization by the AU-rich element binding protein, HuR, an ELAV protein. EMBO J. 1998, 17, 3461–3470. [Google Scholar] [CrossRef] [PubMed]
- Hafner, M.; Landthaler, M.; Burger, L.; Khorshid, M.; Hausser, J.; Berninger, P.; Rothballer, A.; Ascano, M., Jr.; Jungkamp, A.C.; Munschauer, M.; et al. Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell 2010, 141, 129–141. [Google Scholar] [CrossRef] [PubMed]
- Dorveaux, L.; Twidale, N.; Tonkin, A. Direct identification of parameters in a mathematical model describing conduction through the atrioventricular node. Int. J. Biomed. Comput. 1988, 23, 69–76. [Google Scholar] [CrossRef]
- Spellman, R.; Smith, C.W. Novel modes of splicing repression by PTB. Trends Biochem. Sci. 2006, 31, 73–76. [Google Scholar] [CrossRef] [PubMed]
- Jensen, K.B.; Dredge, B.K.; Stefani, G.; Zhong, R.; Buckanovich, R.J.; Okano, H.J.; Yang, Y.Y.; Darnell, R.B. Nova-1 regulates neuron-specific alternative splicing and is essential for neuronal viability. Neuron 2000, 25, 359–371. [Google Scholar] [CrossRef]
- Möröy, T.; Heyd, F. The impact of alternative splicing in vivo: Mouse models show the way. RNA 2007, 13, 1155–1171. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.A.; Lynch, K.W. Cell-based splicing of minigenes. Methods Mol. Biol. 2014, 1126, 243–255. [Google Scholar] [PubMed]
- Wright, H.L.; Thomas, H.B.; Moots, R.J.; Edwards, S.W. RNA-seq reveals activation of both common and cytokine-specific pathways following neutrophil priming. PLoS ONE 2013, 8, e58598. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Ferguson, J.F.; Xue, C.; Ballantyne, R.L.; Silverman, I.M.; Gosai, S.J.; Serfecz, J.; Morley, M.P.; Gregory, B.D.; Li, M.; et al. Tissue-specific RNA-Seq in human evoked inflammation identifies blood and adipose LincRNA signatures of cardiometabolic diseases. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 902–912. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.H.; De, S.; Srikantan, S.; Abdelmohsen, K.; Grammatikakis, I.; Kim, J.; Kim, K.M.; Noh, J.H.; White, E.J.; Martindale, J.L.; et al. PAR-CLIP analysis uncovers AUF1 impact on target RNA fate and genome integrity. Nat. Commun. 2014, 5, 5248. [Google Scholar] [CrossRef] [PubMed]
- Stoss, O.; Stoilov, P.; Hartmann, A.M.; Nayler, O.; Stamm, S. The in vivo minigene approach to analyze tissue-specific splicing. Brain Res. Brain Res. Protoc. 1999, 4, 383–394. [Google Scholar] [CrossRef]
- Kishore, S.; Khanna, A.; Stamm, S. Rapid generation of splicing reporters with pSpliceExpress. Gene 2008, 427, 104–110. [Google Scholar] [CrossRef] [PubMed]
- Gaildrat, P.; Killian, A.; Martins, A.; Tournier, I.; Frébourg, T.; Tosi, M. Use of splicing reporter minigene assay to evaluate the effect on splicing of unclassified genetic variants. Methods Mol. Biol. 2010, 653, 249–257. [Google Scholar] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shakola, F.; Suri, P.; Ruggiu, M. Splicing Regulation of Pro-Inflammatory Cytokines and Chemokines: At the Interface of the Neuroendocrine and Immune Systems. Biomolecules 2015, 5, 2073-2100. https://doi.org/10.3390/biom5032073
Shakola F, Suri P, Ruggiu M. Splicing Regulation of Pro-Inflammatory Cytokines and Chemokines: At the Interface of the Neuroendocrine and Immune Systems. Biomolecules. 2015; 5(3):2073-2100. https://doi.org/10.3390/biom5032073
Chicago/Turabian StyleShakola, Felitsiya, Parul Suri, and Matteo Ruggiu. 2015. "Splicing Regulation of Pro-Inflammatory Cytokines and Chemokines: At the Interface of the Neuroendocrine and Immune Systems" Biomolecules 5, no. 3: 2073-2100. https://doi.org/10.3390/biom5032073
APA StyleShakola, F., Suri, P., & Ruggiu, M. (2015). Splicing Regulation of Pro-Inflammatory Cytokines and Chemokines: At the Interface of the Neuroendocrine and Immune Systems. Biomolecules, 5(3), 2073-2100. https://doi.org/10.3390/biom5032073