
The Impact of Non-Enzymatic Reactions and Enzyme Promiscuity on Cellular Metabolism during (Oxidative) Stress Conditions


Abstract
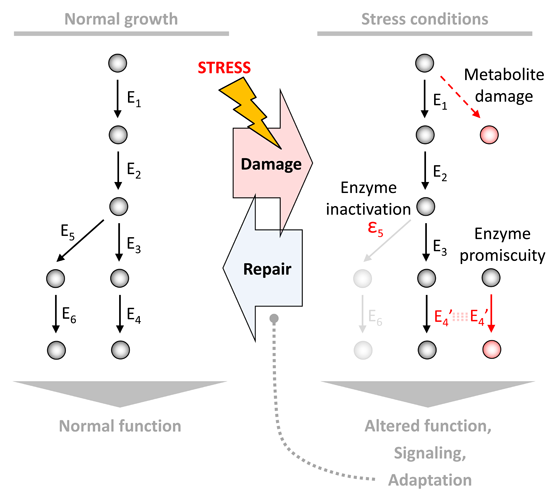
: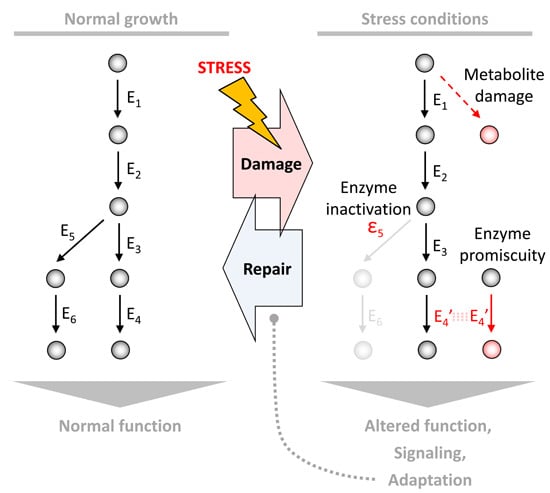
1. Introduction
2. Chemical Damage to Metabolites: Oxidants and Antioxidants
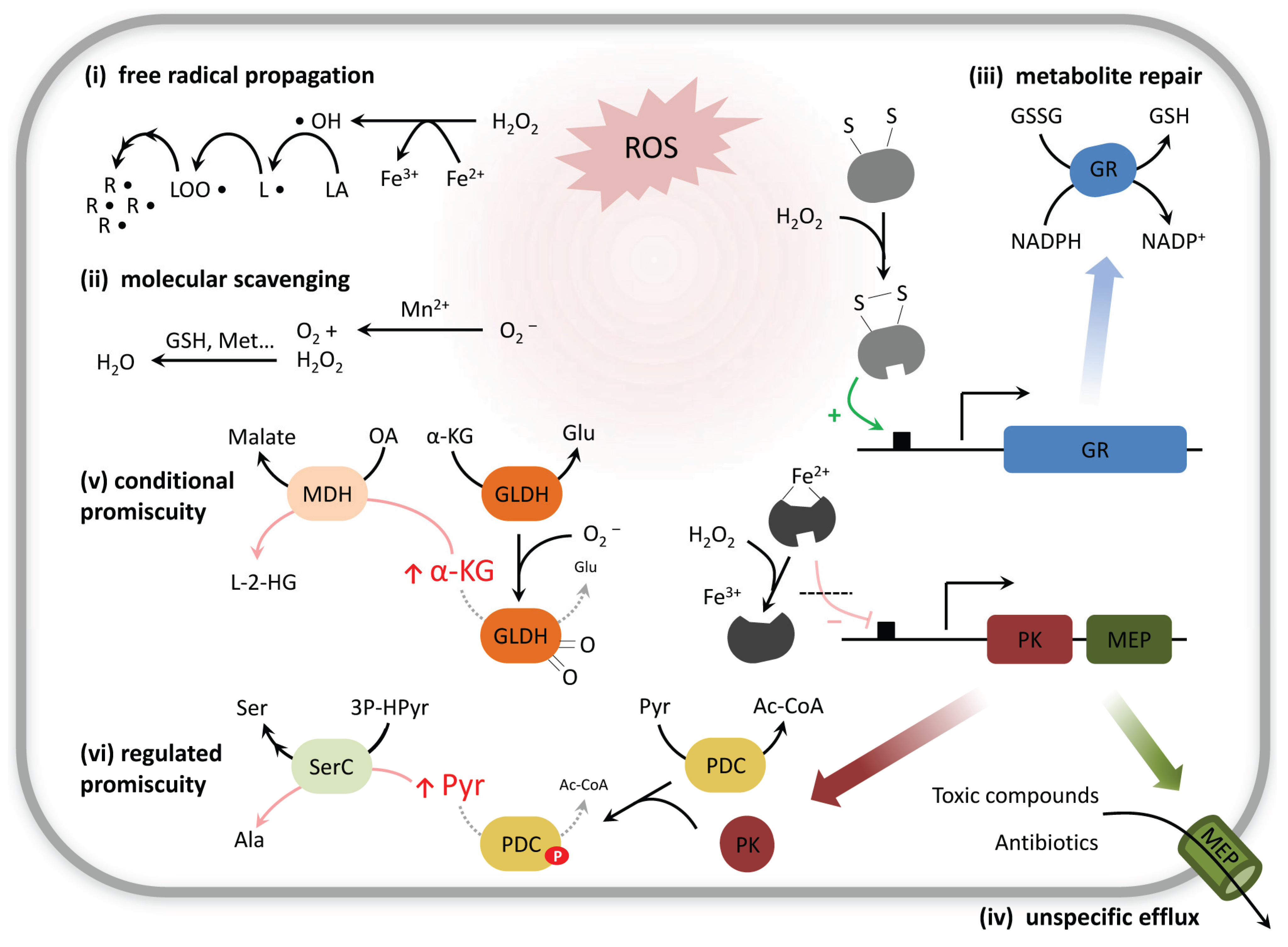
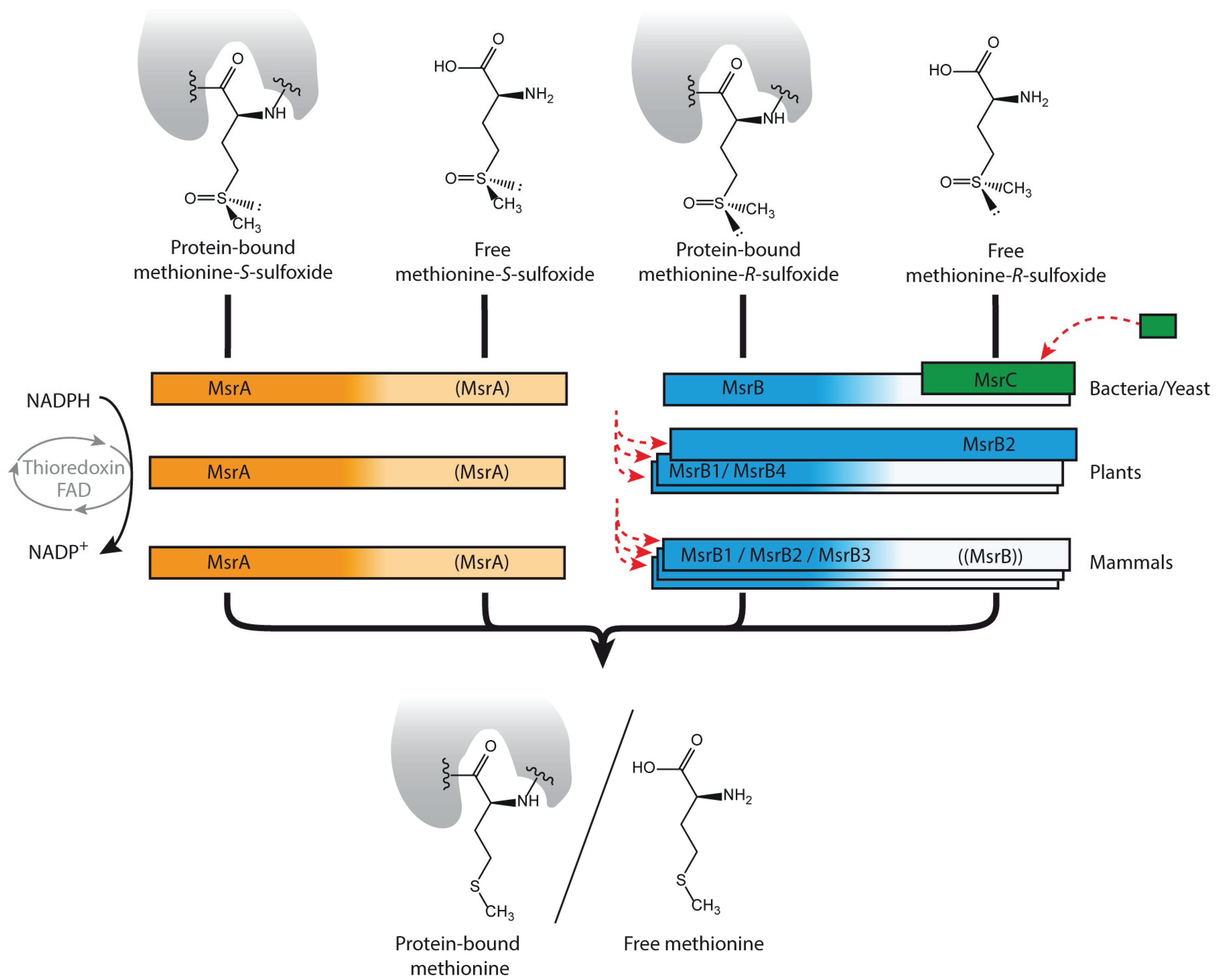
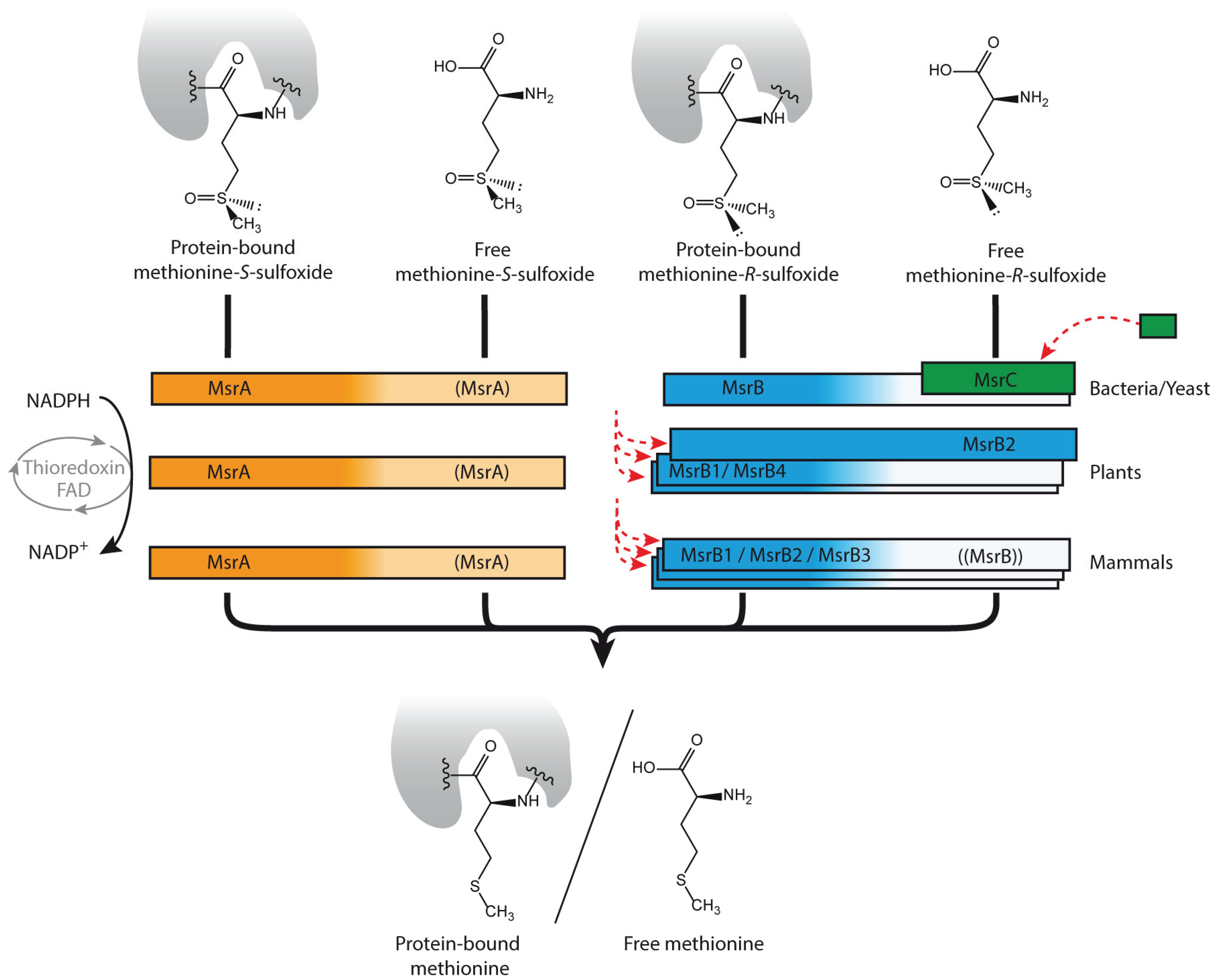
2.1. Not only Protein and DNA Damage, but also Metabolites Can Be Repaired
2.2. Other Forms of Metabolite Damage: Examples of Nucleotide and Cofactor Metabolism
3. Non-Enzymatic Metabolic Reactions Are Affected by Stress Conditions
4. Enzymes That Change Substrate Specificity during Stress Conditions

Adaptive Promiscuity: When Altered Substrate Specificity Becomes Advantageous

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Stress | Enzyme | Induced Activity | Alleged Function | Reference |
|---|---|---|---|---|
| Oxidative stress | Mammalian MetRS | Mismethionylation of non-cognate tRNAs | Protect enzymes from inactivation | [110] |
| Biotic interaction | SerRS/LeuRS from Candida | Misleucinylation of Ser-tRNA | Increased invasivity, immune invisibility | [113] |
| Biotic interaction, anaerobiosis, starvation... | Bacterial transaminases | Redundancy in Ala & Lys biosynthesis | Robust peptidoglycan biosynthesis | [114] |
| Oxidative stress | Bacterial Pentosephosphate epimerase (Rpe) | Mismetallation with Mn(II) | Protect enzymes from inactivation | [102] |
5. Changing Activity and Metabolic Switches: Stress Situations That Affect Flux Distribution
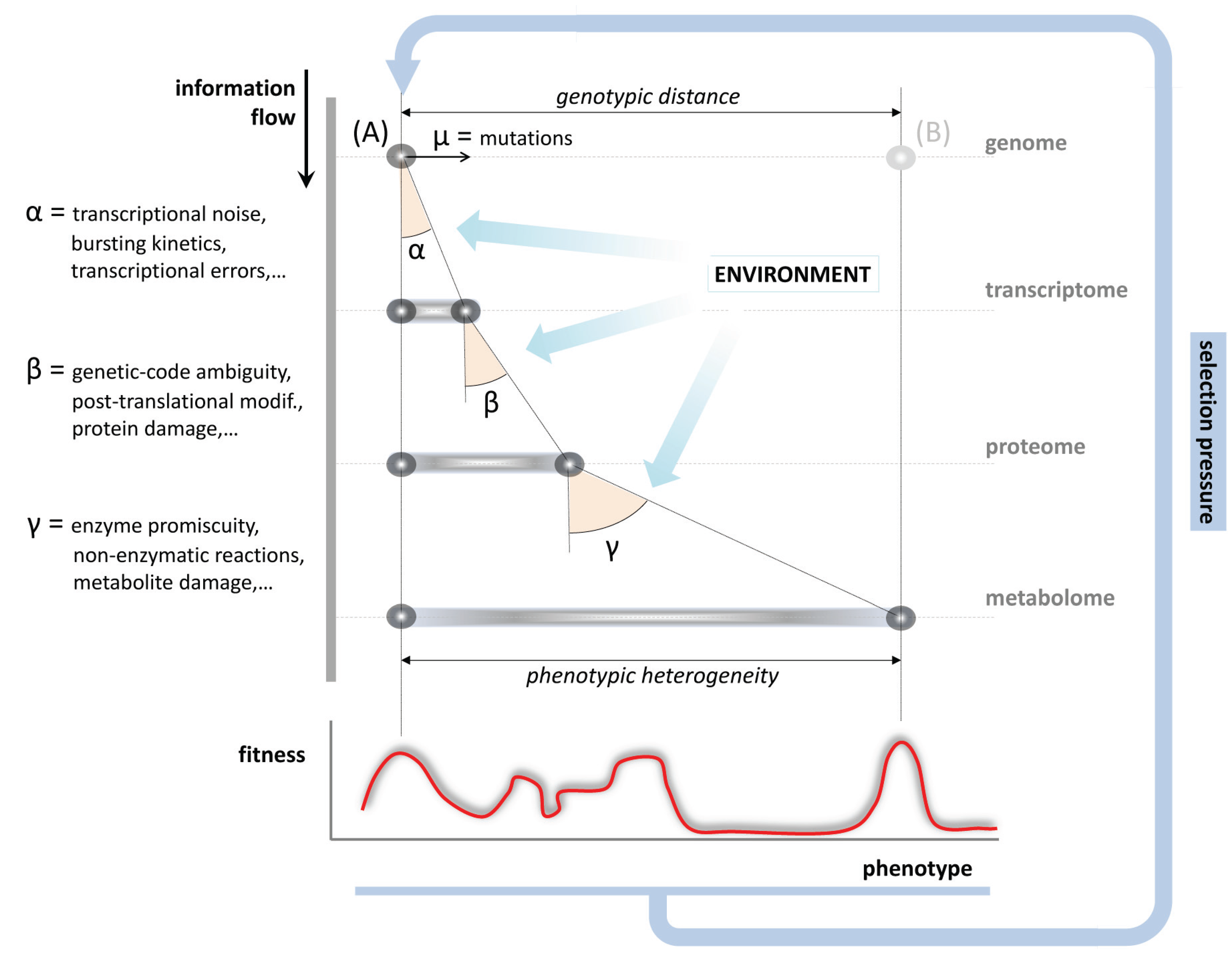
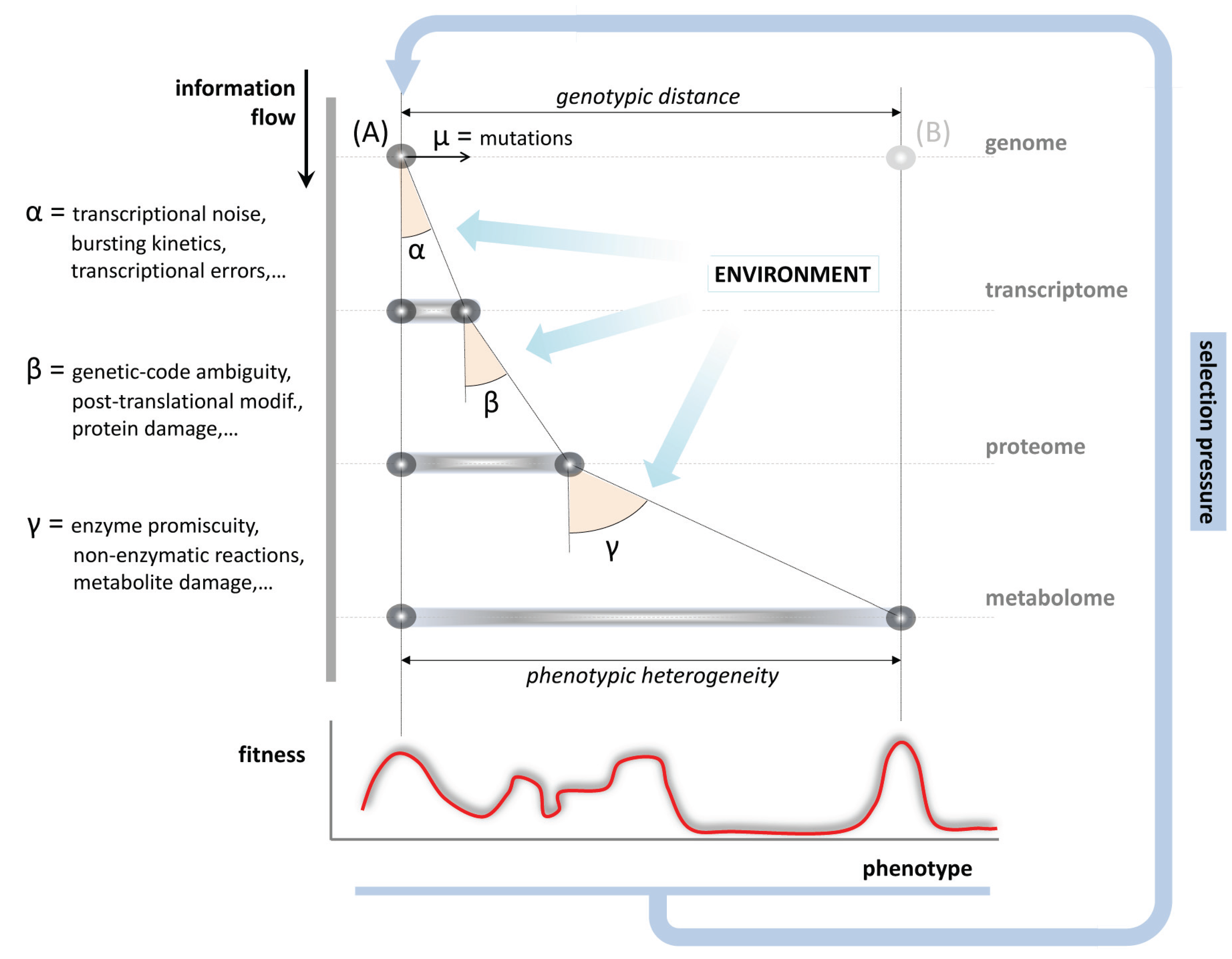
6. Evolution and the Impact of Metabolic Inaccuracy on the Genotype to Phenotype Relationship
7. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Van Houten, B. Nucleotide excision repair in Escherichia coli. Microbiol. Rev. 1990, 54, 18–51. [Google Scholar] [PubMed]
- Mary, J.; Vougier, S.; Picot, C.R.; Perichon, M.; Petropoulos, I.; Friguet, B. Enzymatic reactions involved in the repair of oxidized proteins. Exp. Gerontol. 2004, 39, 1117–1123. [Google Scholar] [CrossRef] [PubMed]
- Perrone, G.G.; Tan, S.-X.; Dawes, I.W. Reactive oxygen species and yeast apoptosis. Biochim. Biophys. Acta 2008, 1783, 1354–1368. [Google Scholar] [CrossRef] [PubMed]
- Morano, K.A.; Grant, C.M.; Moye-Rowley, W.S. The response to heat shock and oxidative stress in Saccharomyces cerevisiae. Genetics 2012, 190, 1157–1195. [Google Scholar] [CrossRef] [PubMed]
- Harman, D. Aging: A theory based on free radical and radiation chemistry. J. Gerontol. 1956, 11, 298–300. [Google Scholar] [CrossRef] [PubMed]
- Sies, H. Biochemistry of oxidative stress. Angew. Chem. Int. Ed. Engl. 1986, 25, 1058–1071. [Google Scholar] [CrossRef]
- Liu, J.; Litt, L.; Segal, M.R.; Kelly, M.J.S.; Pelton, J.G.; Kim, M. Metabolomics of oxidative stress in recent studies of endogenous and exogenously administered intermediate metabolites. Int. J. Mol. Sci. 2011, 12, 6469–6501. [Google Scholar] [CrossRef] [PubMed]
- Noctor, G.; Lelarge-Trouverie, C.; Mhamdi, A. The metabolomics of oxidative stress. Phytochemistry 2014, 112, 33–53. [Google Scholar] [CrossRef] [PubMed]
- Ott, M.; Gogvadze, V.; Orrenius, S.; Zhivotovsky, B. Mitochondria, oxidative stress and cell death. Apoptosis 2007, 12, 913–922. [Google Scholar] [CrossRef] [PubMed]
- D’Autréaux, B.; Toledano, M.B. ROS as signalling molecules: Mechanisms that generate specificity in ROS homeostasis. Nat. Rev. Mol. Cell Biol. 2007, 8, 813–824. [Google Scholar] [CrossRef] [PubMed]
- Sies, H. Oxidative stress: A concept in redox biology and medicine. Redox Biol. 2015, 4C, 180–183. [Google Scholar] [CrossRef] [PubMed]
- Valko, M.; Morris, H.; Cronin, M.T.D. Metals, toxicity and oxidative stress. Curr. Med. Chem. 2005, 12, 1161–1208. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Gebicki, J.M. Proteins are major initial cell targets of hydroxyl free radicals. Int. J. Biochem. Cell Biol. 2004, 36, 2334–2343. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.-C.; Gebicki, J.M. Intracellular GSH and ascorbate inhibit radical-induced protein chain peroxidation in HL-60 cells. Free Radic. Biol. Med. 2012, 52, 420–426. [Google Scholar] [CrossRef] [PubMed]
- Davies, K.J. Oxidative stress, antioxidant defenses, and damage removal, repair, and replacement systems. IUBMB Life 2000, 50, 279–289. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B.; Gutteridge, J. Free Radicals in Biology and Medicine, 4th ed.; Oxford University Press: Oxford, OX, USA, 2007. [Google Scholar]
- Breen, A.P.; Murphy, J.A. Reactions of oxyl radicals with DNA. Free Radic. Biol. Med. 1995, 18, 1033–1077. [Google Scholar] [CrossRef]
- Gilbert, B.C.; King, D.M.; Thomas, C.B. Radical reactions of carbohydrates. Part 2. An electron spin resonance study of the oxidation of d-glucose and related compounds with the hydroxyl radical. J. Chem. Soc. Perkin Trans. 1981, 2, 1186–1199. [Google Scholar] [CrossRef]
- Manini, P.; la Pietra, P.; Panzella, L.; Napolitano, A.; d’Ischia, M. Glyoxal formation by Fenton-induced degradation of carbohydrates and related compounds. Carbohydr. Res. 2006, 341, 1828–1833. [Google Scholar] [CrossRef] [PubMed]
- Arteel, G.E.; Briviba, K.; Sies, H. Protection against peroxynitrite. FEBS Lett. 1999, 445, 226–230. [Google Scholar] [CrossRef]
- Sawa, T.; Akaike, T.; Maeda, H. Tyrosine nitration by peroxynitrite formed from nitric oxide and superoxide generated by xanthine oxidase. J. Biol. Chem. 2000, 275, 32467–32474. [Google Scholar] [CrossRef] [PubMed]
- Klotz, L.-O.; Schroeder, P.; Sies, H. Peroxynitrite signaling: Receptor tyrosine kinases and activation of stress-responsive pathways. Free Radic. Biol. Med. 2002, 33, 737–743. [Google Scholar] [CrossRef]
- Klotz, L.-O.; Sies, H. Defenses against peroxynitrite: Selenocompounds and flavonoids. Toxicol. Lett. 2003, 140–141, 125–132. [Google Scholar] [CrossRef]
- Williams, T.I.; Lynn, B.C.; Markesbery, W.R.; Lovell, M.A. Increased levels of 4-hydroxynonenal and acrolein, neurotoxic markers of lipid peroxidation, in the brain in Mild Cognitive Impairment and early Alzheimer’s disease. Neurobiol. Aging 2006, 27, 1094–1099. [Google Scholar] [CrossRef] [PubMed]
- Pamplona, R. Membrane phospholipids, lipoxidative damage and molecular integrity: A causal role in aging and longevity. Biochim. Biophys. Acta 2008, 1777, 1249–1262. [Google Scholar] [CrossRef] [PubMed]
- Thanan, R.; Oikawa, S.; Hiraku, Y.; Ohnishi, S.; Ma, N.; Pinlaor, S.; Yongvanit, P.; Kawanishi, S.; Murata, M. Oxidative stress and its significant roles in neurodegenerative diseases and cancer. Int. J. Mol. Sci. 2015, 16, 193–217. [Google Scholar] [CrossRef] [PubMed]
- Cremers, C.M.; Jakob, U. Oxidant sensing by reversible disulfide bond formation. J. Biol. Chem. 2013, 288, 26489–26496. [Google Scholar] [CrossRef] [PubMed]
- Moskovitz, J. Methionine sulfoxide reductases: Ubiquitous enzymes involved in antioxidant defense, protein regulation, and prevention of aging-associated diseases. Biochim. Biophys. Acta 2005, 1703, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Stadtman, E.R.; van Remmen, H.; Richardson, A.; Wehr, N.B.; Levine, R.L. Methionine oxidation and aging. Biochim. Biophys. Acta 2005, 1703, 135–140. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell-Tormey, J.; Nathan, C.F.; Lanks, K.; DeBoer, C.J.; de la Harpe, J. Secretion of pyruvate. An antioxidant defense of mammalian cells. J. Exp. Med. 1987, 165, 500–514. [Google Scholar] [CrossRef] [PubMed]
- Troxell, B.; Zhang, J.-J.; Bourret, T.J.; Zeng, M.Y.; Blum, J.; Gherardini, F.; Hassan, H.M.; Yang, X.F. Pyruvate protects pathogenic spirochetes from H2O2 killing. PLoS ONE 2014, 9, e84625. [Google Scholar] [CrossRef] [PubMed]
- Rose, R.C.; Bode, A.M. Biology of free radical scavengers: An evaluation of ascorbate. FASEB J. 1993, 7, 1135–1142. [Google Scholar] [PubMed]
- Levine, R.L.; Mosoni, L.; Berlett, B.S.; Stadtman, E.R. Methionine residues as endogenous antioxidants in proteins. Proc. Natl. Acad. Sci. USA 1996, 93, 15036–15040. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.; Levine, R.L. Methionine in proteins defends against oxidative stress. FASEB J. 2009, 23, 464–472. [Google Scholar] [CrossRef] [PubMed]
- Rzem, R.; Vincent, M.-F.; van Schaftingen, E.; Veiga-da-Cunha, M. l-2-hydroxyglutaric aciduria, a defect of metabolite repair. J. Inherit. Metab. Dis. 2007, 30, 681–689. [Google Scholar] [CrossRef] [PubMed]
- Linster, C.L.; van Schaftingen, E.; Hanson, A.D. Metabolite damage and its repair or pre-emption. Nat. Chem. Biol. 2013, 9, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Galperin, M.Y.; Moroz, O.V.; Wilson, K.S.; Murzin, A.G. House cleaning, a part of good housekeeping. Mol. Microbiol. 2006, 59, 5–19. [Google Scholar] [CrossRef] [PubMed]
- Van Schaftingen, E.; Rzem, R.; Marbaix, A.; Collard, F.; Veiga-da-Cunha, M.; Linster, C.L. Metabolite proofreading, a neglected aspect of intermediary metabolism. J. Inherit. Metab. Dis. 2013, 36, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Ursini, F.; Maiorino, M.; Brigelius-Flohé, R.; Aumann, K.D.; Roveri, A.; Schomburg, D.; Flohé, L. Diversity of glutathione peroxidases. Methods Enzymol. 1995, 252, 38–53. [Google Scholar] [PubMed]
- Labunskyy, V.M.; Hatfield, D.L.; Gladyshev, V.N. Selenoproteins: Molecular pathways and physiological roles. Physiol. Rev. 2014, 94, 739–777. [Google Scholar] [CrossRef] [PubMed]
- Galano, A.; Alvarez-Idaboy, J.R. Glutathione: Mechanism and kinetics of its non-enzymatic defense action against free radicals. RSC Adv. 2011, 1, 1763–1771. [Google Scholar] [CrossRef]
- Haenen, G.R.M.M.; Bast, A. Glutathione revisited: A better scavenger than previously thought. Front. Pharmacol. 2014. [Google Scholar] [CrossRef] [PubMed]
- Stadtman, E.R. Oxidation of free amino acids and amino acid residues in proteins by radiolysis and by metal-catalyzed reactions. Annu. Rev. Biochem. 1993, 62, 797–821. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.C.; Gladyshev, V.N. The biological significance of methionine sulfoxide stereochemistry. Free Radic. Biol. Med. 2011, 50, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Boschi-Muller, S.; Olry, A.; Antoine, M.; Branlant, G. The enzymology and biochemistry of methionine sulfoxide reductases. Biochim. Biophys. Acta 2005, 1703, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Johnson, L.C.; Weissbach, H.; Brot, N.; Lively, M.O.; Lowther, W.T. Free methionine-(R)-sulfoxide reductase from Escherichia coli reveals a new GAF domain function. Proc. Natl. Acad. Sci. USA 2007, 104, 9597–9602. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Lee, B.C.; Marino, S.M.; Zhang, Y.; Fomenko, D.E.; Kaya, A.; Hacioglu, E.; Kwak, G.-H.; Koc, A.; Kim, H.-Y.; et al. Functional analysis of free methionine-R-sulfoxide reductase from Saccharomyces cerevisiae. J. Biol. Chem. 2009, 284, 4354–4364. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Tarrago, L.; Watanabe, Y.; Kaya, A.; Lee, B.C.; Tran, U.; Nishiyama, R.; Fomenko, D.E.; Gladyshev, V.N.; Tran, L.-S.P. Diversity of plant methionine sulfoxide reductases B and evolution of a form specific for free methionine sulfoxide. PLoS ONE 2013, 8, e65637. [Google Scholar] [CrossRef] [PubMed]
- Heurlier, K.; Vendeville, A.; Halliday, N.; Green, A.; Winzer, K.; Tang, C.M.; Hardie, K.R. Growth deficiencies of Neisseria meningitidis pfs and luxS mutants are not due to inactivation of quorum sensing. J. Bacteriol. 2009, 191, 1293–1302. [Google Scholar] [CrossRef] [PubMed]
- Lyu, L.-D.; Tang, B.-K.; Fan, X.-Y.; Ma, H.; Zhao, G.-P. Mycobacterial MazG safeguards genetic stability via housecleaning of 5-OH-dCTP. PLoS Pathog. 2013, 9, e1003814. [Google Scholar] [CrossRef] [PubMed]
- Van Schaftingen, E.; Rzem, R.; Veiga-da-Cunha, M. l-2-Hydroxyglutaric aciduria, a disorder of metabolite repair. J. Inherit. Metab. Dis. 2009, 32, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Nakabeppu, Y. Cellular levels of 8-oxoguanine in either DNA or the nucleotide pool play pivotal roles in carcinogenesis and survival of cancer cells. Int. J. Mol. Sci. 2014, 15, 12543–12557. [Google Scholar] [CrossRef] [PubMed]
- Marinaki, A.M.; Duley, J.A.; Arenas, M.; Ansari, A.; Sumi, S.; Lewis, C.M.; Shobowale-Bakre, M.; Fairbanks, L.D.; Sanderson, J. Mutation in the ITPA gene predicts intolerance to azathioprine. Nucleosides. Nucleotides Nucleic Acids 2004, 23, 1393–1397. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, T.; Maebayashi, K.; Nakagawa, Y.; Imai, H. Deletion of the four phospholipid hydroperoxide glutathione peroxidase genes accelerates aging in Caenorhabditis elegans. Genes Cells 2014, 19, 778–792. [Google Scholar] [CrossRef] [PubMed]
- Inoué, S.; Honda, K.; Komoda, Y. Sleep as neuronal detoxification and restitution. Behav. Brain Res. 1995, 69, 91–96. [Google Scholar] [CrossRef]
- Delaney, S.; Jarem, D.A.; Volle, C.B.; Yennie, C.J. Chemical and biological consequences of oxidatively damaged guanine in DNA. Free Radic. Res. 2012, 46, 420–441. [Google Scholar] [CrossRef] [PubMed]
- Kamiya, H. Mutagenic potentials of damaged nucleic acids produced by reactive oxygen/nitrogen species: Approaches using synthetic oligonucleotides and nucleotides: Survey and summary. Nucleic Acids Res. 2003, 31, 517–531. [Google Scholar] [CrossRef] [PubMed]
- Bessman, M.J.; Frick, D.N.; O’Handley, S.F. The MutT proteins or “Nudix” hydrolases, a family of versatile, widely distributed, “housecleaning” enzymes. J. Biol. Chem. 1996, 271, 25059–25062. [Google Scholar] [CrossRef] [PubMed]
- Sekiguchi, M.; Tsuzuki, T. Oxidative nucleotide damage: Consequences and prevention. Oncogene 2002, 21, 8895–8904. [Google Scholar] [CrossRef] [PubMed]
- McLennan, A.G. The Nudix hydrolase superfamily. Cell. Mol. Life Sci. 2006, 63, 123–143. [Google Scholar] [CrossRef] [PubMed]
- Persson, R.; Cedergren-Zeppezauer, E.S.; Wilson, K.S. Homotrimeric dUTPases; structural solutions for specific recognition and hydrolysis of dUTP. Curr. Protein Pept. Sci. 2001, 2, 287–300. [Google Scholar] [CrossRef] [PubMed]
- Hemsworth, G.R.; González-Pacanowska, D.; Wilson, K.S. On the catalytic mechanism of dimeric dUTPases. Biochem. J. 2013, 456, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Marbaix, A.Y.; Noël, G.; Detroux, A.M.; Vertommen, D.; van Schaftingen, E.; Linster, C.L. Extremely conserved ATP- or ADP-dependent enzymatic system for nicotinamide nucleotide repair. J. Biol. Chem. 2011, 286, 41246–41252. [Google Scholar] [CrossRef] [PubMed]
- Oppenheimer, N.J.; Kaplan, N.O. Glyceraldehyde-3-phosphate dehydrogenase catalyzed hydration of the 5–6 double bond of reduced β-nicotinamide adenine dinucleotide (βNADH). Formation of β-6-hydroxy-1,4,5,6-tetrahydronicotinamide adenine dinucleotide. Biochemistry 1974, 13, 4685–4694. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, A.; Dave, V. Inhibition of NADP-dependent dehydrogenases by modified products of NADPH. Arch. Biochem. Biophys. 1975, 169, 298–303. [Google Scholar] [CrossRef]
- Niehaus, T.D.; Richardson, L.G.L.; Gidda, S.K.; ElBadawi-Sidhu, M.; Meissen, J.K.; Mullen, R.T.; Fiehn, O.; Hanson, A.D. Plants utilize a highly conserved system for repair of NADH and NADPH hydrates. Plant Physiol. 2014, 165, 52–61. [Google Scholar] [CrossRef] [PubMed]
- Colinas, M.; Shaw, H.V.; Loubéry, S.; Kaufmann, M.; Moulin, M.; Fitzpatrick, T.B. A pathway for repair of NAD(P)H in plants. J. Biol. Chem. 2014, 289, 14692–14706. [Google Scholar] [CrossRef] [PubMed]
- Marbaix, A.Y.; Tyteca, D.; Niehaus, T.D.; Hanson, A.D.; Linster, C.L.; van Schaftingen, E. Occurrence and subcellular distribution of the NADPHX repair system in mammals. Biochem. J. 2014, 460, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Keller, M.A.; Piedrafita, G.; Ralser, M. The widespread role of non-enzymatic reactions in cellular metabolism. Curr. Opin. Biotechnol. 2015, 34, 153–161. [Google Scholar] [CrossRef] [PubMed]
- D’Ari, R.; Casadesús, J. Underground metabolism. Bioessays 1998, 20, 181–186. [Google Scholar] [CrossRef]
- Keller, M.A.; Turchyn, A.V.; Ralser, M. Non-enzymatic glycolysis and pentose phosphate pathway-like reactions in a plausible Archean ocean. Mol. Syst. Biol. 2014. [Google Scholar] [CrossRef] [PubMed]
- Weinert, B.T.; Iesmantavicius, V.; Moustafa, T.; Schölz, C.; Wagner, S.A.; Magnes, C.; Zechner, R.; Choudhary, C. Acetylation dynamics and stoichiometry in Saccharomyces cerevisiae. Mol. Syst. Biol. 2014. [Google Scholar] [CrossRef] [PubMed]
- Wagner, G.R.; Payne, R.M. Widespread and enzyme-independent Nε-acetylation and Nε-succinylation of proteins in the chemical conditions of the mitochondrial matrix. J. Biol. Chem. 2013, 288, 29036–29045. [Google Scholar] [CrossRef] [PubMed]
- Wagner, G.R.; Hirschey, M.D. Nonenzymatic protein acylation as a carbon stress regulated by sirtuin deacylases. Mol. Cell 2014, 54, 5–16. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Xiao, M.; Horiyama, T.; Zhang, Y.; Li, X.; Nishino, K.; Yan, A. The multidrug efflux pump MdtEF protects against nitrosative damage during the anaerobic respiration in Escherichia coli. J. Biol. Chem. 2011, 286, 26576–26584. [Google Scholar] [CrossRef] [PubMed]
- Zhou, K.; Zou, R.; Stephanopoulos, G.; Too, H.-P. Metabolite profiling identified methylerythritol cyclodiphosphate efflux as a limiting step in microbial isoprenoid production. PLoS ONE 2012, 7, e47513. [Google Scholar] [CrossRef] [PubMed]
- Fung, D.K.C.; Lau, W.Y.; Chan, W.T.; Yan, A. Copper efflux is induced during anaerobic amino acid limitation in Escherichia coli to protect iron-sulfur cluster enzymes and biogenesis. J. Bacteriol. 2013, 195, 4556–4568. [Google Scholar] [CrossRef] [PubMed]
- Piddock, L.J.V. Clinically relevant chromosomally encoded multidrug resistance efflux pumps in bacteria. Clin. Microbiol. Rev. 2006, 19, 382–402. [Google Scholar] [CrossRef] [PubMed]
- Nikaido, H.; Zgurskaya, H.I. AcrAB and related multidrug efflux pumps of Escherichia coli. J. Mol. Microbiol. Biotechnol. 2001, 3, 215–218. [Google Scholar] [PubMed]
- Li, X.-Z.; Nikaido, H. Efflux-mediated drug resistance in bacteria: An update. Drugs 2009, 69, 1555–1623. [Google Scholar] [CrossRef] [PubMed]
- Bleuel, C.; Grosse, C.; Taudte, N.; Scherer, J.; Wesenberg, D.; Krauss, G.J.; Nies, D.H.; Grass, G. TolC is involved in enterobactin efflux across the outer membrane of Escherichia coli. J. Bacteriol. 2005, 187, 6701–6707. [Google Scholar] [CrossRef] [PubMed]
- Du, D.; Wang, Z.; James, N.R.; Voss, J.E.; Klimont, E.; Ohene-Agyei, T.; Venter, H.; Chiu, W.; Luisi, B.F. Structure of the AcrAB-TolC multidrug efflux pump. Nature 2014, 509, 512–515. [Google Scholar] [CrossRef] [PubMed]
- Takatsuka, Y.; Chen, C.; Nikaido, H. Mechanism of recognition of compounds of diverse structures by the multidrug efflux pump AcrB of Escherichia coli. Proc. Natl. Acad. Sci. USA 2010, 107, 6559–6565. [Google Scholar] [CrossRef] [PubMed]
- Poole, K. Bacterial multidrug efflux pumps serve other functions. Microbe 2008, 3, 179–185. [Google Scholar]
- Deininger, K.N.W.; Horikawa, A.; Kitko, R.D.; Tatsumi, R.; Rosner, J.L.; Wachi, M.; Slonczewski, J.L. A requirement of TolC and MDR efflux pumps for acid adaptation and GadAB induction in Escherichia coli. PLoS ONE 2011, 6, e18960. [Google Scholar] [CrossRef] [PubMed]
- Tatsumi, R.; Wachi, M. TolC-dependent exclusion of porphyrins in Escherichia coli. J. Bacteriol. 2008, 190, 6228–6233. [Google Scholar] [CrossRef] [PubMed]
- Rosner, J.L.; Martin, R.G. Reduction of cellular stress by TolC-dependent efflux pumps in Escherichia coli indicated by BaeSR and CpxARP activation of spy in efflux mutants. J. Bacteriol. 2013, 195, 1042–1050. [Google Scholar] [CrossRef] [PubMed]
- Nagy, P.; Lechte, T.P.; Das, A.B.; Winterbourn, C.C. Conjugation of glutathione to oxidized tyrosine residues in peptides and proteins. J. Biol. Chem. 2012, 287, 26068–26076. [Google Scholar] [CrossRef] [PubMed]
- Bar-Even, A.; Noor, E.; Savir, Y.; Liebermeister, W.; Davidi, D.; Tawfik, D.S.; Milo, R. The moderately efficient enzyme: Evolutionary and physicochemical trends shaping enzyme parameters. Biochemistry 2011, 50, 4402–4410. [Google Scholar] [CrossRef] [PubMed]
- Khersonsky, O.; Tawfik, D.S. Enzyme promiscuity: A mechanistic and evolutionary perspective. Annu. Rev. Biochem. 2010, 79, 471–505. [Google Scholar] [PubMed]
- Hult, K.; Berglund, P. Enzyme promiscuity: Mechanism and applications. Trends Biotechnol. 2007, 25, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Copley, S.D. Enzymes with extra talents: Moonlighting functions and catalytic promiscuity. Curr. Opin. Chem. Biol. 2003, 7, 265–272. [Google Scholar] [CrossRef]
- Bensaad, K.; Tsuruta, A.; Selak, M.A.; Vidal, M.N.C.; Nakano, K.; Bartrons, R.; Gottlieb, E.; Vousden, K.H. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell 2006, 126, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Gerin, I.; Noël, G.; Bolsée, J.; Haumont, O.; van Schaftingen, E.; Bommer, G.T. Identification of TP53-induced glycolysis and apoptosis regulator (TIGAR) as the phosphoglycolate-independent 2,3-bisphosphoglycerate phosphatase. Biochem. J. 2014, 458, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Yebra, M.J.; Bhagwat, A.S. A cytosine methyltransferase converts 5-methylcytosine in DNA to thymine. Biochemistry 1995, 34, 14752–14757. [Google Scholar] [CrossRef] [PubMed]
- Engqvist, M.K.M.; Eßer, C.; Maier, A.; Lercher, M.J.; Maurino, V.G. Mitochondrial 2-hydroxyglutarate metabolism. Mitochondrion 2014, 19, 275–281. [Google Scholar] [CrossRef] [PubMed]
- Ward, P.S.; Patel, J.; Wise, D.R.; Abdel-Wahab, O.; Bennett, B.D.; Coller, H.A.; Cross, J.R.; Fantin, V.R.; Hedvat, C.V.; Perl, A.E.; et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell 2010, 17, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Struys, E.A.; Salomons, G.S.; Achouri, Y.; van Schaftingen, E.; Grosso, S.; Craigen, W.J.; Verhoeven, N.M.; Jakobs, C. Mutations in the d-2-hydroxyglutarate dehydrogenase gene cause d-2-hydroxyglutaric aciduria. Am. J. Hum. Genet. 2005, 76, 358–360. [Google Scholar] [CrossRef] [PubMed]
- Kranendijk, M.; Struys, E.A.; van Schaftingen, E.; Gibson, K.M.; Kanhai, W.A.; van der Knaap, M.S.; Amiel, J.; Buist, N.R.; Das, A.M.; de Klerk, J.B.; et al. IDH2 mutations in patients with d-2-hydroxyglutaric aciduria. Science 2010. [Google Scholar] [CrossRef] [PubMed]
- Sobota, J.M.; Imlay, J.A. Iron enzyme ribulose-5-phosphate 3-epimerase in Escherichia coli is rapidly damaged by hydrogen peroxide but can be protected by manganese. Proc. Natl. Acad. Sci. USA 2011, 108, 5402–5407. [Google Scholar] [CrossRef] [PubMed]
- Anjem, A.; Imlay, J.A. Mononuclear iron enzymes are primary targets of hydrogen peroxide stress. J. Biol. Chem. 2012, 287, 15544–15556. [Google Scholar] [CrossRef] [PubMed]
- Cotruvo, J.A.; Stubbe, J. Metallation and mismetallation of iron and manganese proteins in vitro and in vivo: The class I ribonucleotide reductases as a case study. Metallomics 2012, 4, 1020–1036. [Google Scholar] [CrossRef] [PubMed]
- Frawley, E.R.; Fang, F.C. The ins and outs of bacterial iron metabolism. Mol. Microbiol. 2014, 93, 609–616. [Google Scholar] [CrossRef] [PubMed]
- Barnese, K.; Gralla, E.B.; Valentine, J.S.; Cabelli, D.E. Biologically relevant mechanism for catalytic superoxide removal by simple manganese compounds. Proc. Natl. Acad. Sci. USA 2012, 109, 6892–6897. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, R.J.; Srinivasan, C.; Munroe, W.H.; Wallace, M.A.; Martins, J.; Kao, T.Y.; Le, K.; Gralla, E.B.; Valentine, J.S. Exogenous manganous ion at millimolar levels rescues all known dioxygen-sensitive phenotypes of yeast lacking CuZnSOD. J. Biol. Inorg. Chem. 2005, 10, 913–923. [Google Scholar] [CrossRef] [PubMed]
- Copley, S.D. Toward a systems biology perspective on enzyme evolution. J. Biol. Chem. 2012, 287, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Söll, D. Enter a new amino acid. Nature 1988, 331, 662–663. [Google Scholar] [CrossRef] [PubMed]
- Prat, L.; Heinemann, I.U.; Aerni, H.R.; Rinehart, J.; O’Donoghue, P.; Söll, D. Carbon source-dependent expansion of the genetic code in bacteria. Proc. Natl. Acad. Sci. USA 2012, 109, 21070–21075. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Boniecki, M.T.; Jaffe, J.D.; Imai, B.S.; Yau, P.M.; Luthey-Schulten, Z.A.; Martinis, S.A. Naturally occurring aminoacyl-tRNA synthetases editing-domain mutations that cause mistranslation in Mycoplasma parasites. Proc. Natl. Acad. Sci. USA 2011, 108, 9378–9383. [Google Scholar] [CrossRef] [PubMed]
- Netzer, N.; Goodenbour, J.M.; David, A.; Dittmar, K.A.; Jones, R.B.; Schneider, J.R.; Boone, D.; Eves, E.M.; Rosner, M.R.; Gibbs, J.S.; et al. Innate immune and chemically triggered oxidative stress modifies translational fidelity. Nature 2009, 462, 522–526. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Kim, D.G.; Kim, B.-G.; Yang, W.S.; Hong, J.; Kang, T.; Oh, Y.S.; Kim, K.R.; Han, B.W.; Hwang, B.J.; et al. Promiscuous methionyl-tRNA synthetase mediates adaptive mistranslation to protect cells against oxidative stress. J. Cell Sci. 2014, 127, 4234–4245. [Google Scholar] [CrossRef] [PubMed]
- Bender, A.; Hajieva, P.; Moosmann, B. Adaptive antioxidant methionine accumulation in respiratory chain complexes explains the use of a deviant genetic code in mitochondria. Proc. Natl. Acad. Sci. USA 2008, 105, 16496–16501. [Google Scholar] [CrossRef] [PubMed]
- Miranda, I.; Silva-Dias, A.; Rocha, R.; Teixeira-Santos, R.; Coelho, C.; Gonçalves, T.; Santos, M.A.S.; Pina-Vaz, C.; Solis, N.V.; Filler, S.G.; et al. Candida albicans CUG mistranslation is a mechanism to create cell surface variation. MBio 2013, 4. [Google Scholar] [CrossRef] [PubMed]
- Lal, P.B.; Schneider, B.L.; Vu, K.; Reitzer, L. The redundant aminotransferases in lysine and arginine synthesis and the extent of aminotransferase redundancy in Escherichia coli. Mol. Microbiol. 2014, 94, 843–856. [Google Scholar] [CrossRef] [PubMed]
- Miranda, I.; Rocha, R.; Santos, M.C.; Mateus, D.D.; Moura, G.R.; Carreto, L.; Santos, M.A.S. A genetic code alteration is a phenotype diversity generator in the human pathogen Candida albicans. PLoS ONE 2007, 2, e996. [Google Scholar] [CrossRef] [PubMed]
- Rothstein, M. Aging and the alteration of enzymes: A review. Mech. Ageing Dev. 1975, 4, 325–338. [Google Scholar] [CrossRef]
- Van Montfort, R.L.M.; Congreve, M.; Tisi, D.; Carr, R.; Jhoti, H. Oxidation state of the active-site cysteine in protein tyrosine phosphatase 1B. Nature 2003, 423, 773–777. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-G.; Baek, K.; Soetandyo, N.; Ye, Y. Reversible inactivation of deubiquitinases by reactive oxygen species in vitro and in cells. Nat. Commun. 2013. [Google Scholar] [CrossRef] [PubMed]
- Ralser, M.; Wamelink, M.M.; Kowald, A.; Gerisch, B.; Heeren, G.; Struys, E.A.; Klipp, E.; Jakobs, C.; Breitenbach, M.; Lehrach, H.; et al. Dynamic rerouting of the carbohydrate flux is key to counteracting oxidative stress. J. Biol. 2007. [Google Scholar] [CrossRef] [PubMed]
- Ralser, M.; Wamelink, M.M.C.; Latkolik, S.; Jansen, E.E.W.; Lehrach, H.; Jakobs, C. Metabolic reconfiguration precedes transcriptional regulation in the antioxidant response. Nat. Biotechnol. 2009, 27, 604–605. [Google Scholar] [CrossRef] [PubMed]
- Stincone, A.; Prigione, A.; Cramer, T.; Wamelink, M.M.C.; Campbell, K.; Cheung, E.; Olin-Sandoval, V.; Gruening, N.-M.; Krueger, A.; Tauqeer Alam, M.; et al. The return of metabolism: Biochemistry and physiology of the pentose phosphate pathway. Biol. Rev. Camb. Philos. Soc. 2015, 90, 927–963. [Google Scholar] [CrossRef] [PubMed]
- Peralta, D.; Bronowska, A.K.; Morgan, B.; Dóka, É.; van Laer, K.; Nagy, P.; Gräter, F.; Dick, T.P. A proton relay enhances H2O2 sensitivity of GAPDH to facilitate metabolic adaptation. Nat. Chem. Biol. 2015, 11, 156–163. [Google Scholar] [CrossRef] [PubMed]
- Ralser, M.; Heeren, G.; Breitenbach, M.; Lehrach, H.; Krobitsch, S. Triose phosphate isomerase deficiency is caused by altered dimerization—Not catalytic inactivity—Of the mutant enzymes. PLoS ONE 2006, 1, e30. [Google Scholar] [CrossRef] [PubMed]
- Grüning, N.-M.; Du, D.; Keller, M.A.; Luisi, B.F.; Ralser, M. Inhibition of triosephosphate isomerase by phosphoenolpyruvate in the feedback-regulation of glycolysis. Open Biol. 2014. [Google Scholar] [CrossRef] [PubMed]
- Grüning, N.-M.; Rinnerthaler, M.; Bluemlein, K.; Mülleder, M.; Wamelink, M.M.C.; Lehrach, H.; Jakobs, C.; Breitenbach, M.; Ralser, M. Pyruvate kinase triggers a metabolic feedback loop that controls redox metabolism in respiring cells. Cell Metab. 2011, 14, 415–427. [Google Scholar] [CrossRef] [PubMed]
- Kuehne, A.; Emmert, H.; Soehle, J.; Winnefeld, M.; Fischer, F.; Wenck, H.; Gallinat, S.; Terstegen, L.; Lucius, R.; Hildebrand, J.; et al. Acute activation of oxidative pentose phosphate pathway as first-line response to oxidative stress in human skin cells. Mol. Cell 2015. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Ye, J.; Kamphorst, J.J.; Shlomi, T.; Thompson, C.B.; Rabinowitz, J.D. Quantitative flux analysis reveals folate-dependent NADPH production. Nature 2014, 510, 298–302. [Google Scholar] [CrossRef] [PubMed]
- Miller-Fleming, L.; Olin-Sandoval, V.; Campbell, K.; Ralser, M. Remaining mysteries of molecular biology: The role of polyamines in the cell. J. Mol. Biol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Krüger, A.; Vowinckel, J.; Mülleder, M.; Grote, P.; Capuano, F.; Bluemlein, K.; Ralser, M. Tpo1-mediated spermine and spermidine export controls cell cycle delay and times antioxidant protein expression during the oxidative stress response. EMBO Rep. 2013, 14, 1113–1119. [Google Scholar] [CrossRef] [PubMed]
- Casero, R.A.; Pegg, A.E. Polyamine catabolism and disease. Biochem. J. 2009, 421, 323–338. [Google Scholar] [CrossRef] [PubMed]
- Pledgie, A.; Huang, Y.; Hacker, A.; Zhang, Z.; Woster, P.M.; Davidson, N.E.; Casero, R.A. Spermine oxidase SMO(PAOh1), not N1-acetylpolyamine oxidase PAO, is the primary source of cytotoxic H2O2 in polyamine analogue-treated human breast cancer cell lines. J. Biol. Chem. 2005, 280, 39843–39851. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, M.; Bellini, A.; Cervelli, M.; Degan, P.; Marcocci, L.; Martini, F.; Scatteia, M.; Mariottini, P.; Amendola, R. Chronic sub-lethal oxidative stress by spermine oxidase overactivity induces continuous DNA repair and hypersensitivity to radiation exposure. Biochim. Biophys. Acta 2007, 1773, 774–783. [Google Scholar] [CrossRef] [PubMed]
- Amendola, R.; Cervelli, M.; Fratini, E.; Sallustio, D.E.; Tempera, G.; Ueshima, T.; Mariottini, P.; Agostinelli, E. Reactive oxygen species spermine metabolites generated from amine oxidases and radiation represent a therapeutic gain in cancer treatments. Int. J. Oncol. 2013, 43, 813–820. [Google Scholar] [CrossRef] [PubMed]
- Carbonell, P.; Lecointre, G.; Faulon, J.-L. Origins of specificity and promiscuity in metabolic networks. J. Biol. Chem. 2011, 286, 43994–44004. [Google Scholar] [CrossRef] [PubMed]
- Jensen, R.A. Enzyme recruitment in evolution of new function. Annu. Rev. Microbiol. 1976, 30, 409–425. [Google Scholar] [CrossRef] [PubMed]
- Voordeckers, K.; Brown, C.A.; Vanneste, K.; van der Zande, E.; Voet, A.; Maere, S.; Verstrepen, K.J. Reconstruction of ancestral metabolic enzymes reveals molecular mechanisms underlying evolutionary innovation through gene duplication. PLoS Biol. 2012, 10, e1001446. [Google Scholar] [CrossRef] [PubMed]
- Notebaart, R.A.; Szappanos, B.; Kintses, B.; Pal, F.; Gyorkei, A.; Bogos, B.; Lazar, V.; Spohn, R.; Csorg, B.; Wagner, A.; et al. Network-level architecture and the evolutionary potential of underground metabolism. Proc. Natl. Acad. Sci. USA 2014, 111, 11762–11767. [Google Scholar] [CrossRef] [PubMed]
- Cornish-Bowden, A.; Cardenas, M.L. Self-organization at the origin of life. J. Theor. Biol. 2008, 252, 411–418. [Google Scholar] [CrossRef] [PubMed]
- Piedrafita, G.; Montero, F.; Morán, F.; Cárdenas, M.L.; Cornish-Bowden, A. A simple self-maintaining metabolic system: Robustness, autocatalysis, bistability. PLoS Comput. Biol. 2010. [Google Scholar] [CrossRef] [PubMed]
- Tawfik, D.S. Messy biology and the origins of evolutionary innovations. Nat. Chem. Biol. 2010, 6, 692–696. [Google Scholar] [PubMed]
- Benoit, R.; Auer, M. A direct way of redox sensing. RNA Biol. 2011, 8, 18–23. [Google Scholar] [CrossRef] [PubMed]
- Andersson, D.I. Evolving promiscuously. Proc. Natl. Acad. Sci. USA 2011, 108, 1199–1200. [Google Scholar] [CrossRef] [PubMed]
- Miller, B.G.; Raines, R.T. Identifying latent enzyme activities: Substrate ambiguity within modern bacterial sugar kinases. Biochemistry 2004, 43, 6387–6392. [Google Scholar] [CrossRef] [PubMed]
- Patrick, W.M.; Quandt, E.M.; Swartzlander, D.B.; Matsumura, I. Multicopy suppression underpins metabolic evolvability. Mol. Biol. Evol. 2007, 24, 2716–2722. [Google Scholar] [CrossRef] [PubMed]
- Patrick, W.M.; Matsumura, I. A study in molecular contingency: Glutamine phosphoribosylpyrophosphate amidotransferase is a promiscuous and evolvable phosphoribosylanthranilate isomerase. J. Mol. Biol. 2008, 377, 323–336. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Piedrafita, G.; Keller, M.A.; Ralser, M. The Impact of Non-Enzymatic Reactions and Enzyme Promiscuity on Cellular Metabolism during (Oxidative) Stress Conditions. Biomolecules 2015, 5, 2101-2122. https://doi.org/10.3390/biom5032101
Piedrafita G, Keller MA, Ralser M. The Impact of Non-Enzymatic Reactions and Enzyme Promiscuity on Cellular Metabolism during (Oxidative) Stress Conditions. Biomolecules. 2015; 5(3):2101-2122. https://doi.org/10.3390/biom5032101
Chicago/Turabian StylePiedrafita, Gabriel, Markus A Keller, and Markus Ralser. 2015. "The Impact of Non-Enzymatic Reactions and Enzyme Promiscuity on Cellular Metabolism during (Oxidative) Stress Conditions" Biomolecules 5, no. 3: 2101-2122. https://doi.org/10.3390/biom5032101
APA StylePiedrafita, G., Keller, M. A., & Ralser, M. (2015). The Impact of Non-Enzymatic Reactions and Enzyme Promiscuity on Cellular Metabolism during (Oxidative) Stress Conditions. Biomolecules, 5(3), 2101-2122. https://doi.org/10.3390/biom5032101