
Microenvironmental Drivers of Bone Disease in Multiple Myeloma: Oxidative Stress, Sterile Inflammation, Autophagy–Lysosomal Remodeling, and the Iron–Lipid Peroxidation Axis

 , ,
, ,
Abstract
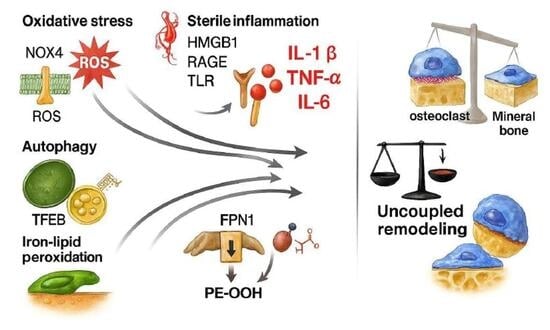
1. Introduction
1.1. Canonical Mechanisms of Bone Remodelling Imbalance
1.2. Stress-Adaption Programs Shaping the Osteolytic Niche
1.3. Osteocyte Dysfunction and Microarchitectural Deterioration
2. From Coupled to Uncoupled Remodeling in MM
2.1. Drivers of Uncoupling in Myeloma Microenvironment
2.2. Derailment of Coupling Signals—Osteocyte Mechanics, Redox Tone and Perilacunar Remodeling: Cytokine-Danger Signal Integration
2.3. Potential Diagnostic Assessments of Cytokine-Induced Uncoupling and Possible Therapeutic Interventions
3. ROS-Dependent Osteoclastogenic Pathways—Role of Ferroptosis
4. Sterile Inflammation and Osteoimmunology
5. Autophagy, Proteostasis, and Lysosomal Control
6. Lipid Remodeling, and Ferroptotic Thresholds in Bone and Tumor Compartments
7. Optimizing Early-Phase Assessment of Myeloma Bone Disease Through Standardized Biochemical Markers, Redox Signatures, and Imaging-Based Response Metrics
8. Therapeutic Implications and Roadmap
Operationalization and Sequencing
9. In Vivo Models of Multiple Myeloma Bone Disease Supporting Redox, Inflammatory, and Autophagy–Iron Pathways
9.1. Syngeneic Immunocompetent Models
9.2. Human Xenograft and Intrabone Models
9.3. Genetically Engineered and Pathway-Focused Mouse Models
9.4. Iron Handling and Ferroptosis-Related Models
10. Translational Outlook
11. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| MAPK | Mitogen-Activated Protein Kinase |
| IOF | International Osteoporosis Foundation |
| IMWG | International Myeloma Working Group |
| TLR | Toll-Like Receptor |
| CLEAR | Coordinated Lysosomal Expression and Regulation network |
| DHODH | Dihydroorotate Dehydrogenase |
| WNT | Wingless-type Signaling Pathway |
| RANK | Receptor Activator of Nuclear Factor-κB |
| IFCC | International Federation of Clinical Chemistry |
| MM | Multiple Myeloma |
| NADPH | Nicotinamide Adenine Dinucleotide Phosphate |
| OB | Osteoblast |
| WBCT | Whole-Body Computed Tomography |
| BTM/BTMs | Bone Turnover Marker(s) |
| LD-WBCT | Low-Dose Whole-Body CT |
| ER | Endoplasmic Reticulum |
| EB | Earliest Binding |
| MDR | Multidrug Resistance |
| PRR | Pattern-Recognition Receptor |
| BAFF | B-cell Activating Factor |
| MRI | Magnetic Resonance Imaging |
| OC | Osteoclast |
| CREB | cAMP Response Element-Binding protein |
| FDG | Fluorodeoxyglucose |
| ROS | Reactive Oxygen Species |
| CTX | C-Terminal Telopeptide of Type I Collagen |
| OBD | Osteolytic Bone Disease |
| CLL | Chronic Lymphocytic Leukemia |
| SRE | Skeletal-Related Event |
| MIP (MIP-1α) | Macrophage Inflammatory Protein-1 alpha |
| PET | Positron Emission Tomography |
| NOX/NOX4 | NADPH Oxidase/NADPH Oxidase 4 |
| MRD | Minimal Residual Disease |
| CT | Computed Tomography |
| PINP | Procollagen Type I N-Terminal Propeptide |
| TFEB | Transcription Factor EB |
| RAGE | Receptor for Advanced Glycation End-products |
| APRIL | A Proliferation-Inducing Ligand |
| AOPP | Advanced Oxidation Protein Products |
| WB | Whole Body |
| DAMP | Damage-Associated Molecular Pattern |
| RANKL | Receptor Activator of NF-κB Ligand |
| OPG | Osteoprotegerin |
| NF (NF-κB) | Nuclear Factor Kappa-Light-Chain-Enhancer of Activated B Cells |
| TGF (TGF-β) | Transforming Growth Factor-β |
| TNF (TNF-α) | Tumor Necrosis Factor-α |
| IL (IL-1β, IL-6 ecc.) | Interleukin |
| ATG | Autophagy-Related Gene/Protein |
References
- Terpos, E.; Ntanasis-Stathopoulos, I.; Dimopoulos, M.A. Myeloma bone disease: From biology findings to treatment approaches. Blood 2019, 133, 1534–1539. [Google Scholar] [CrossRef]
- Terpos, E.; Ntanasis-Stathopoulos, I.; Gavriatopoulou, M.; Dimopoulos, M.A. Pathogenesis of bone disease in multiple myeloma: From bench to bedside. Blood Cancer J. 2018, 8, 7. [Google Scholar] [CrossRef] [PubMed]
- Giuliani, N.; Morandi, F.; Tagliaferri, S.; Lazzaretti, M.; Donofrio, G.; Bonomini, S.; Sala, R.; Mangoni, M.; Rizzoli, V. Production of Wnt inhibitors by myeloma cells: Potential effects on the bone microenvironment. Cancer Res. 2007, 67, 7665–7674. [Google Scholar] [CrossRef]
- Sezer, O.; Heider, U.; Zavrski, I.; Kühne, C.A.; Hofbauer, L.C. RANK ligand and osteoprotegerin in myeloma bone disease. Blood 2003, 101, 2094–2098. [Google Scholar] [CrossRef]
- Colucci, S.; Brunetti, G.; Oranger, A.; Mori, G.; Sardone, F.; Specchia, G.; Rinaldi, E.; Curci, P.; Liso, V.; Passeri, G.; et al. Myeloma cells suppress osteoblasts through sclerostin secretion. Blood Cancer J. 2011, 1, e27. [Google Scholar] [CrossRef]
- Sun, W.; Sun, J.; Hu, W.; Luo, C.; Lu, Z.; He, F.; Zhao, H.; Zeng, X.; Cao, D.; Li, J.; et al. Sulforaphane inhibits multiple myeloma cell-induced osteoclast differentiation and macrophage proliferation by elevating ferroportin1. Cancer Chemother. Pharmacol. 2024, 95, 3. [Google Scholar] [CrossRef]
- Iantomasi, T.; Romagnoli, C.; Palmini, G.; Donati, S.; Falsetti, I.; Miglietta, F.; Aurilia, C.; Marini, F.; Giusti, F.; Brandi, M.L. Oxidative stress and inflammation in osteoporosis: Molecular mechanisms involved and the relationship with microRNAs. Int. J. Mol. Sci. 2023, 24, 3772. [Google Scholar] [CrossRef] [PubMed]
- Anloague, A.; Sabol, H.M.; Kaur, J.; Khan, S.; Ashby, C.; Schinke, C.; Barnes, C.L.; Alturkmani, F.; Ambrogini, E.; Gundesen, M.T.; et al. A novel CCL3-HMGB1 signaling axis regulating osteocyte RANKL expression in multiple myeloma. Haematologica 2025, 110, 952–966. [Google Scholar] [CrossRef]
- Mukkamalla, S.K.R.; Malipeddi, D. Myeloma bone disease: A comprehensive review. Int. J. Mol. Sci. 2021, 22, 6208. [Google Scholar] [CrossRef]
- Zhang, R.; Yang, X.; Shi, X.; Xing, E.; Wang, L.; Hao, C.; Zhang, Z. Bortezomib modulated the autophagy-lysosomal pathway in a TFEB-dependent manner in multiple myeloma. Leuk. Res. 2024, 138, 107455. [Google Scholar] [CrossRef] [PubMed]
- Kozalak, G.; Koşar, A. Autophagy-related mechanisms for treatment of multiple myeloma. Cancer Drug Resist. 2023, 6, 838–857. [Google Scholar] [CrossRef]
- Melaccio, A.; Reale, A.; Saltarella, I.; Desantis, V.; Lamanuzzi, A.; Cicco, S.; Frassanito, M.A.; Vacca, A.; Ria, R. Pathways of angiogenic and inflammatory cytokines in multiple myeloma: Role in plasma cell clonal expansion and drug resistance. J. Clin. Med. 2022, 11, 6491. [Google Scholar] [CrossRef] [PubMed]
- Gu, Z.; Wang, H.; Xia, J.; Yang, Y.; Jin, Z.; Xu, H.; Shi, J.; De Domenico, I.; Tricot, G.; Zhan, F. Decreased ferroportin promotes myeloma cell growth and osteoclast differentiation. Cancer Res. 2015, 75, 2211–2221. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, Y.; Li, Q.; Zuo, L.; Zhang, B.; Zhao, F.; Fan, F.; Luo, S.; Hu, Y.; Sun, C. ACSL4: A double-edged sword target in multiple myeloma, promotes cell proliferation and sensitizes cells to ferroptosis. Carcinogenesis 2023, 44, 242–251. [Google Scholar] [CrossRef]
- Liu, J.; Tang, D.; Kang, R. Targeting GPX4 in ferroptosis and cancer: Chemical strategies and challenges. Trends Pharmacol. Sci. 2024, 45, 666–670. [Google Scholar] [CrossRef]
- Evans, H.; Andrews, R.; Abedi, F.A.; Sprules, A.; Trend, J.; Lovric, G.; Green, A.; Chantry, A.; Clarkin, C.; Brown, J.; et al. Evidence for peri-lacunar remodeling and altered osteocyte lacuno-canalicular network in mouse models of myeloma-induced bone disease. JBMR Plus 2024, 8, ziae093. [Google Scholar] [CrossRef] [PubMed]
- Yee, C.S.; Schurman, C.A.; White, C.R.; Alliston, T. Investigating osteocytic perilacunar/canalicular remodeling. Curr. Osteoporos. Rep. 2019, 17, 157–168. [Google Scholar] [CrossRef]
- Fotiou, D.; Katodritou, E. From biology to clinical practice: The bone marrow microenvironment in multiple myeloma. J. Clin. Med. 2025, 14, 327. [Google Scholar] [CrossRef] [PubMed]
- Kamrani, S.; Naseramini, R.; Khani, P.; Razavi, Z.S.; Afkhami, H.; Atashzar, M.R.; Nasri, F.; Alavimanesh, S.; Saeidi, F.; Ronaghi, H. Mesenchymal stromal cells in bone marrow niche of patients with multiple myeloma: A double-edged sword. Cancer Cell Int. 2025, 25, 117. [Google Scholar] [CrossRef]
- Qiang, Y.W.; Chen, Y.; Stephens, O.; Brown, N.; Chen, B.; Epstein, J.; Barlogie, B.; Shaughnessy, J.D., Jr. Myeloma-derived Dickkopf-1 disrupts Wnt-regulated osteoprotegerin and RANKL production by osteoblasts: A potential mechanism underlying osteolytic bone lesions in multiple myeloma. Blood 2008, 112, 196–207. [Google Scholar] [CrossRef]
- Takahashi, N.; Maeda, K.; Ishihara, A.; Uehara, S.; Kobayashi, Y. Regulatory mechanism of osteoclastogenesis by RANKL and Wnt signals. Front. Biosci. 2011, 16, 21–30. [Google Scholar] [CrossRef]
- Merico, F.; Bergui, L.; Gregoretti, M.G.; Ghia, P.; Aimo, G.; Lindley, I.J.; Caligaris-Cappio, F. Cytokines involved in the progression of multiple myeloma. Clin. Exp. Immunol. 1993, 92, 27–31. [Google Scholar] [CrossRef] [PubMed]
- Giuliani, N.; Ferretti, M.; Bolzoni, M.; Storti, P.; Lazzaretti, M.; Dalla Palma, B.; Bonomini, S.; Martella, E.; Agnelli, L.; Neri, A.; et al. Increased osteocyte death in multiple myeloma patients: Role in myeloma-induced osteoclast formation. Leukemia 2012, 26, 1391–1401. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Qian, J.; Li, J.; Su, T.; Deng, X.; Fu, Y.; Liang, X.; Cui, H. From death to birth: How osteocyte death promotes osteoclast formation. Front. Immunol. 2025, 16, 1551542. [Google Scholar] [CrossRef]
- Zhou, Z.; Han, J.Y.; Xi, C.X.; Xie, J.X.; Feng, X.; Wang, C.Y.; Mei, L.; Xiong, W.C. HMGB1 regulates RANKL-induced osteoclastogenesis in a manner dependent on RAGE. J. Bone Miner. Res. 2008, 23, 1084–1096. [Google Scholar] [CrossRef]
- O’Brien, C.A.; Gubrij, I.; Lin, S.C.; Saylors, R.L.; Manolagas, S.C. STAT3 activation in stromal/osteoblastic cells is required for induction of the receptor activator of NF-κB ligand and stimulation of osteoclastogenesis by gp130-utilizing cytokines or interleukin-1 but not 1,25-dihydroxyvitamin D3 or parathyroid hormone. J. Biol. Chem. 1999, 274, 19301–19308. [Google Scholar] [CrossRef]
- Jiang, Z.; Jin, L.; Jiang, C.; Yan, Z.; Cao, Y. IL-1β contributes to the secretion of sclerostin by osteocytes, and targeting sclerostin promotes spinal fusion at early stages. J. Orthop. Surg. Res. 2023, 18, 162. [Google Scholar] [CrossRef]
- Nagata, Y.; Miyagawa, K.; Ohata, Y.; Petrusca, D.N.; Pagnotti, G.M.; Mohammad, K.S.; Guise, T.A.; Windle, J.J.; Roodman, G.D.; Kurihara, N. Increased S1P expression in osteoclasts enhances bone formation in an animal model of Paget’s disease. J. Cell. Biochem. 2021, 122, 335–348. [Google Scholar] [CrossRef]
- Maiso, P.; Mogollón, P.; Ocio, E.M.; Garayoa, M. Bone marrow mesenchymal stromal cells in multiple myeloma: Their role as active contributors to myeloma progression. Cancers 2021, 13, 2542. [Google Scholar] [CrossRef]
- Gangemi, S.; Allegra, A.; Alonci, A.; Cristani, M.; Russo, S.; Speciale, A.; Penna, G.; Spatari, G.; Cannavò, A.; Bellomo, G.; et al. Increase of novel biomarkers for oxidative stress in patients with plasma cell disorders and in multiple myeloma patients with bone lesions. Inflamm. Res. 2012, 61, 1063–1067. [Google Scholar] [CrossRef] [PubMed]
- Mulé, S.; Reizine, E.; Blanc-Durand, P.; Baranes, L.; Zerbib, P.; Burns, R.; Nouri, R.; Itti, E.; Luciani, A. Whole-body functional MRI and PET/MRI in multiple myeloma. Cancers 2020, 12, 3155. [Google Scholar] [CrossRef]
- Hillengass, J.; Usmani, S.; Rajkumar, S.V.; Durie, B.G.M.; Mateos, M.V.; Lonial, S.; Joao, C.; Anderson, K.C.; García-Sanz, R.; Riva, E.; et al. International Myeloma Working Group consensus recommendations on imaging in monoclonal plasma cell disorders. Lancet Oncol. 2019, 20, e302–e312. [Google Scholar] [CrossRef]
- Goettsch, C.; Babelova, A.; Trummer, O.; Erben, R.G.; Rauner, M.; Rammelt, S.; Weissmann, N.; Weinberger, V.; Benkhoff, S.; Kampschulte, M.; et al. NADPH oxidase 4 limits bone mass by promoting osteoclastogenesis. J. Clin. Investig. 2013, 123, 4731–4738. [Google Scholar] [CrossRef] [PubMed]
- Dzubanova, M.; Bond, J.M.; Craige, S.M.; Tencerova, M. NOX4-reactive oxygen species axis: Critical regulators of bone health and metabolism. Front. Cell Dev. Biol. 2024, 12, 1432668. [Google Scholar] [CrossRef]
- Kim, J.M.; Lin, C.; Stavre, Z.; Greenblatt, M.B.; Shim, J.H. Osteoblast-osteoclast communication and bone homeostasis. Cells 2020, 9, 2073. [Google Scholar] [CrossRef]
- Lee, N.K.; Choi, Y.G.; Baik, J.Y.; Han, S.Y.; Jeong, D.W.; Bae, Y.S.; Kim, N.; Lee, S.Y. A crucial role for reactive oxygen species in RANKL-induced osteoclast differentiation. Blood 2005, 106, 852–859. [Google Scholar] [CrossRef] [PubMed]
- Riegger, J.; Schoppa, A.; Ruths, L.; Haffner-Luntzer, M.; Ignatius, A. Oxidative stress as a key modulator of cell fate decision in osteoarthritis and osteoporosis: A narrative review. Cell. Mol. Biol. Lett. 2023, 28, 76. [Google Scholar] [CrossRef]
- Mao, C.; Liu, X.; Zhang, Y.; Lei, G.; Yan, Y.; Lee, H.; Koppula, P.; Wu, S.; Zhuang, L.; Fang, B.; et al. DHODH-mediated ferroptosis defence is a targetable vulnerability in cancer. Nature 2021, 593, 586–590. [Google Scholar] [CrossRef]
- Doll, S.; Freitas, F.P.; Shah, R.; Aldrovandi, M.; da Silva, M.C.; Ingold, I.; Goya Grocin, A.; Xavier da Silva, T.N.; Panzilius, E.; Scheel, C.H.; et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature 2019, 575, 693–698. [Google Scholar] [CrossRef] [PubMed]
- Hofbauer, D.; Mougiakakos, D.; Broggini, L.; Zaiss, M.; Büttner-Herold, M.; Bach, C.; Spriewald, B.; Neumann, F.; Bisht, S.; Nolting, J.; et al. β2-Microglobulin triggers NLRP3 inflammasome activation in tumor-associated macrophages to promote multiple myeloma progression. Immunity 2021, 54, 1772–1787.e9. [Google Scholar] [CrossRef]
- Harmer, D.; Falank, C.; Reagan, M.R. Interleukin-6 interweaves the bone marrow microenvironment, bone loss, and multiple myeloma. Front. Endocrinol. 2019, 9, 788. [Google Scholar] [CrossRef]
- Garbers, C.; Jänner, N.; Chalaris, A.; Moss, M.L.; Floss, D.M.; Meyer, D.; Koch-Nolte, F.; Rose-John, S.; Scheller, J. Species specificity of ADAM10 and ADAM17 proteins in interleukin-6 trans-signaling and novel role of ADAM10 in inducible IL-6 receptor shedding. J. Biol. Chem. 2011, 286, 14804–14811. [Google Scholar] [CrossRef]
- Tominari, T.; Matsumoto, C.; Tanaka, Y.; Shimizu, K.; Takatoya, M.; Sugasaki, M.; Karouji, K.; Kasuga, U.; Miyaura, C.; Miyata, S.; et al. Roles of Toll-like receptor signaling in inflammatory bone resorption. Biology 2024, 13, 692. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Yu, L.; Liu, F.; Wan, L.; Deng, Z. The effect of cytokines on osteoblasts and osteoclasts in bone remodeling in osteoporosis: A review. Front. Immunol. 2023, 14, 1222129. [Google Scholar] [CrossRef]
- Kitaura, H.; Marahleh, A.; Ohori, F.; Noguchi, T.; Shen, W.R.; Qi, J.; Nara, Y.; Pramusita, A.; Kinjo, R.; Mizoguchi, I. Osteocyte-related cytokines regulate osteoclast formation and bone resorption. Int. J. Mol. Sci. 2020, 21, 5169. [Google Scholar] [CrossRef] [PubMed]
- Sapir-Koren, R.; Livshits, G. Osteocyte control of bone remodeling: Is sclerostin a key molecular coordinator of the balanced bone resorption-formation cycles? Osteoporos. Int. 2014, 25, 2685–2700. [Google Scholar] [CrossRef] [PubMed]
- Robling, A.G.; Bonewald, L.F. The osteocyte: New insights. Annu. Rev. Physiol. 2020, 82, 485–506. [Google Scholar] [CrossRef]
- Zhu, S.; Yan, M.Q.; Masson, A.; Chen, W.; Li, Y.P. Cell signaling and transcriptional regulation of osteoclast lineage commitment, differentiation, bone resorption and diseases. Cell Discov. 2026, 12, 6. [Google Scholar] [CrossRef]
- Zhao, C.; Liu, P.; Kong, K.; Chen, X.; Wu, J.; Wu, W.; Wang, X.; Wang, L. Therapeutic targeting of STING–IL6/STAT3 axis to inhibit osteoclastic niche formation and breast cancer bone metastasis. Cell Death Discov. 2025, 11, 483. [Google Scholar] [CrossRef]
- Rose-John, S.; Jenkins, B.J.; Garbers, C.; Moll, J.M.; Scheller, J. Targeting IL-6 trans-signalling: Past, present and future prospects. Nat. Rev. Immunol. 2023, 23, 666–681. [Google Scholar] [CrossRef]
- Wolf, J.; Waetzig, G.H.; Chalaris, A.; Reinheimer, T.M.; Wege, H.; Rose-John, S.; Garbers, C. Different soluble forms of the interleukin-6 family signal transducer gp130 fine-tune the blockade of interleukin-6 trans-signaling. J. Biol. Chem. 2016, 291, 16186–16196. [Google Scholar] [CrossRef] [PubMed]
- Korbecki, J.; Bosiacki, M.; Stelmach, R.; Barczak, K. The role of rarely studied chemokines in tumor progression in multiple myeloma (MM). Cancers 2026, 18, 673. [Google Scholar] [CrossRef]
- Vallet, S.; Pozzi, S.; Patel, K.; Vaghela, N.; Fulciniti, M.T.; Veiby, P.; Hideshima, T.; Santo, L.; Cirstea, D.; Scadden, D.T.; et al. A novel role for CCL3 (MIP-1α) in myeloma-induced bone disease via osteocalcin downregulation and inhibition of osteoblast function. Leukemia 2011, 25, 1174–1181. [Google Scholar] [CrossRef]
- DeSelm, C.J.; Miller, B.C.; Zou, W.; Beatty, W.L.; van Meel, E.; Takahata, Y.; Klumperman, J.; Tooze, S.A.; Teitelbaum, S.L.; Virgin, H.W. Autophagy proteins regulate the secretory component of osteoclastic bone resorption. Dev. Cell 2011, 21, 966–974. [Google Scholar] [CrossRef] [PubMed]
- Park, H.J.; Yoon, S.Y.; Park, J.N.; Suh, J.H.; Choi, H.S. Doxorubicin induces bone loss by increasing autophagy through a mitochondrial ROS/TRPML1/TFEB axis in osteoclasts. Antioxidants 2022, 11, 1476. [Google Scholar] [CrossRef]
- Obeng, E.A.; Carlson, L.M.; Gutman, D.M.; Harrington, W.J., Jr.; Lee, K.P.; Boise, L.H. Proteasome inhibitors induce a terminal unfolded protein response in multiple myeloma cells. Blood 2006, 107, 4907–4916. [Google Scholar] [CrossRef]
- Tang, P.; Yu, Z.; Sun, H.; Liu, L.; Gong, L.; Fang, T.; Sun, X.; Xie, S.; An, G.; Xu, Z.; et al. CRIP1 involves the pathogenesis of multiple myeloma via dual regulation of proteasome and autophagy. eBioMedicine 2024, 100, 104961. [Google Scholar] [CrossRef]
- Settembre, C.; Medina, D.L. TFEB and the CLEAR network. Methods Cell Biol. 2015, 126, 45–62. [Google Scholar] [CrossRef]
- Palmieri, M.; Impey, S.; Kang, H.; di Ronza, A.; Pelz, C.; Sardiello, M.; Ballabio, A. Characterization of the CLEAR network reveals an integrated control of cellular clearance pathways. Hum. Mol. Genet. 2011, 20, 3852–3866. [Google Scholar] [CrossRef] [PubMed]
- Aronson, L.I.; Davenport, E.L.; Mirabella, F.; Morgan, G.J.; Davies, F.E. Understanding the interplay between the proteasome pathway and autophagy in response to dual PI3K/mTOR inhibition in myeloma cells is essential for their effective clinical application. Leukemia 2013, 27, 2397–2403. [Google Scholar] [CrossRef]
- Li, C.; Wang, X.; Li, X.; Qiu, K.; Jiao, F.; Liu, Y.; Kong, Q.; Liu, Y.; Wu, Y. Proteasome inhibition activates autophagy–lysosome pathway associated with TFEB dephosphorylation and nuclear translocation. Front. Cell Dev. Biol. 2019, 7, 170. [Google Scholar] [CrossRef]
- Salimi, A.; Schroeder, K.M.; Schemionek-Reinders, M.; Vieri, M.; Maletzke, S.; Gezer, D.; Masouleh, B.K.; Appelmann, I. Targeting autophagy increases the efficacy of proteasome inhibitor treatment in multiple myeloma by induction of apoptosis and activation of JNK. BMC Cancer 2022, 22, 735. [Google Scholar] [CrossRef]
- Leng, H.; Simon, A.K.; Horwood, N.J. Blocking glycosphingolipid production alters autophagy in osteoclasts and improves myeloma bone disease. Autophagy 2024, 20, 930–932. [Google Scholar] [CrossRef]
- James, A.; Hendrixson, J.A.; Kadhim, I.; Marques-Carvalho, A.; Laster, J.; Crawford, J.; Thostenson, J.; Wanchai, V.; Sato, A.Y.; Nookaew, I.; et al. Elevation of master autophagy regulator Tfeb in osteoblast lineage cells increases bone mass and strength. JCI Insight 2025, 10, e191688. [Google Scholar] [CrossRef]
- Huang, T.; Wang, Y.; Yu, Z.; Miao, X.; Jiang, Z.; Yu, K.; Fu, M.; Lai, K.; Wang, Y.; Yang, G. Effect of mitophagy in the formation of osteomorphs derived from osteoclasts. iScience 2023, 26, 106682. [Google Scholar] [CrossRef]
- Ballard, A.; Zeng, R.; Zarei, A.; Shao, C.; Cox, L.; Yan, H.; Franco, A.; Dorn, G.W., 2nd; Faccio, R.; Veis, D.J. The tethering function of mitofusin-2 controls osteoclast differentiation by modulating the Ca2+-NFATc1 axis. J. Biol. Chem. 2020, 295, 6629–6640. [Google Scholar] [CrossRef]
- Liu, M.Z.; Kong, N.; Zhang, G.Y.; Xu, Q.; Xu, Y.; Ke, P.; Liu, C. The critical role of ferritinophagy in human disease. Front. Pharmacol. 2022, 13, 933732. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Wu, W.; Sun, X.; Zhang, P. Glucocorticoids enhance osteoclast autophagy through the PI3K/Akt/mTOR signaling pathway. Calcif. Tissue Int. 2020, 107, 60–71. [Google Scholar] [CrossRef] [PubMed]
- Søe, K.; Delaissé, J.M. Glucocorticoids maintain human osteoclasts in the active mode of their resorption cycle. J. Bone Miner. Res. 2010, 25, 2184–2192. [Google Scholar] [CrossRef] [PubMed]
- Di Paola, S.; Medina, D.L. TRPML1-/TFEB-dependent regulation of lysosomal exocytosis. Methods Mol. Biol. 2019, 1925, 143–144. [Google Scholar] [CrossRef]
- Erkhembaatar, M.; Gu, D.R.; Lee, S.H.; Yang, Y.M.; Park, S.; Muallem, S.; Shin, D.M.; Kim, M.S. Lysosomal Ca2+ signaling is essential for osteoclastogenesis and bone remodeling. J. Bone Miner. Res. 2017, 32, 385–396. [Google Scholar] [CrossRef]
- Zhou, J.; Ye, S.; Fujiwara, T.; Manolagas, S.C.; Zhao, H. Steap4 plays a critical role in osteoclastogenesis in vitro by regulating cellular iron/reactive oxygen species (ROS) levels and CREB activation. J. Biol. Chem. 2013, 288, 30064–30074. [Google Scholar] [CrossRef]
- Campanella, A.; Santambrogio, P.; Fontana, F.; Frenquelli, M.; Cenci, S.; Marcatti, M.; Sitia, R.; Tonon, G.; Camaschella, C. Iron increases the susceptibility of multiple myeloma cells to bortezomib. Haematologica 2013, 98, 971–979. [Google Scholar] [CrossRef]
- Bordini, J.; Morisi, F.; Cerruti, F.; Cascio, P.; Camaschella, C.; Ghia, P.; Campanella, A. Iron causes lipid oxidation and inhibits proteasome function in multiple myeloma cells: A proof of concept for novel combination therapies. Cancers 2020, 12, 970. [Google Scholar] [CrossRef]
- Zhang, H.L.; Hu, B.X.; Li, Z.L.; Du, T.; Shan, J.L.; Ye, Z.P.; Peng, X.D.; Li, X.; Huang, Y.; Zhu, X.Y.; et al. PKCβII phosphorylates ACSL4 to amplify lipid peroxidation and induce ferroptosis. Nat. Cell Biol. 2022, 24, 88–98. [Google Scholar] [CrossRef] [PubMed]
- D’Herde, K.; Krysko, D.V. Ferroptosis: Oxidized PEs trigger death. Nat. Chem. Biol. 2017, 13, 4–5. [Google Scholar] [CrossRef]
- Merkel, M.; Goebel, B.; Boll, M.; Adhikari, A.; Maurer, V.; Steinhilber, D.; Culmsee, C. Mitochondrial reactive oxygen species formation determines ACSL4/LPCAT2-mediated ferroptosis. Antioxidants 2023, 12, 1590. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Gong, G.; Song, J.; Chen, J.; Wang, S.; Li, J.; Wang, G. Ferroptosis-mediated osteoclast–osteoblast crosstalk: Signaling pathways governing bone remodeling in osteoporosis. J. Orthop. Surg. Res. 2025, 20, 888. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, X.; Zhu, Y.; Cao, Y.; Zhang, G.; Li, Y.; Zhang, Z.; Chen, J.; Chen, J.; Ding, M. Ferroptosis targeting: A novel therapeutic armamentarium in multiple myeloma. Cancer Cell Int. 2026, 26, 187. [Google Scholar] [CrossRef]
- Mansour, G.K.; Hajjar, A.W.; Sajid, M.R. Therapeutic targeting of the hepcidin-ferroportin axis and erythropoietic modulators: A narrative review. Front. Med. 2025, 12, 1726337. [Google Scholar] [CrossRef] [PubMed]
- Alves, F.; Lane, D.; Nguyen, T.P.M.; Bush, A.I.; Ayton, S. In defence of ferroptosis. Signal Transduct. Target Ther. 2025, 10, 2. [Google Scholar] [CrossRef]
- Guo, C.; Peng, J.; Cheng, P.; Yang, C.; Gong, S.; Zhang, L.; Zhang, T.; Peng, J. Mechanistic elucidation of ferroptosis and ferritinophagy: Implications for advancing our understanding of arthritis. Front. Physiol. 2024, 15, 1290234. [Google Scholar] [CrossRef]
- Szulc, P.; Naylor, K.; Hoyle, N.R.; Eastell, R.; Leary, E.T. Use of CTX-I and PINP as bone turnover markers: National Bone Health Alliance recommendations to standardize sample handling and patient preparation to reduce pre-analytical variability. Osteoporos. Int. 2017, 28, 2541–2556. [Google Scholar] [CrossRef] [PubMed]
- Eastell, R.; Szulc, P. Use of bone turnover markers in postmenopausal osteoporosis. Lancet Diabetes Endocrinol. 2017, 5, 908–923. [Google Scholar] [CrossRef] [PubMed]
- Melough, M.M.; Sun, X.; Chun, O.K. The role of AOPP in age-related bone loss and the potential benefits of berry anthocyanins. Nutrients 2017, 9, 789. [Google Scholar] [CrossRef]
- Cavo, M.; Terpos, E.; Nanni, C.; Moreau, P.; Lentzsch, S.; Zweegman, S.; Hillengass, J.; Engelhardt, M.; Usmani, S.Z.; Vesole, D.H.; et al. Role of 18F-FDG PET/CT in the diagnosis and management of multiple myeloma and other plasma cell disorders: A consensus statement by the International Myeloma Working Group. Lancet Oncol. 2017, 18, e206–e217. [Google Scholar] [CrossRef]
- Zamagni, E.; Talarico, M. Fifteen years of use of functional imaging in multiple myeloma: Where we started and where we are going. Blood Adv. 2025, 9, 6252–6266. [Google Scholar] [CrossRef] [PubMed]
- Mesguich, C.; Fardanesh, R.; Tanenbaum, L.; Chari, A.; Jagannath, S.; Kostakoglu, L. State-of-the-art imaging of multiple myeloma: Comparative review of FDG PET/CT imaging in various clinical settings. Eur. J. Radiol. 2014, 83, 2203–2223. [Google Scholar] [CrossRef]
- Jiang, Y.; Zhang, M.; Sun, M. ACSL4 at the helm of the lipid peroxidation ship: A deep-sea exploration towards ferroptosis. Front. Pharmacol. 2025, 16, 1594419. [Google Scholar] [CrossRef]
- Raje, N.; Terpos, E.; Willenbacher, W.; Shimizu, K.; García-Sanz, R.; Durie, B.; Legieć, W.; Krejčí, M.; Laribi, K.; Zhu, L.; et al. Denosumab versus zoledronic acid in bone disease treatment of newly diagnosed multiple myeloma: An international, double-blind, double-dummy, randomised, controlled, phase 3 study. Lancet Oncol. 2018, 19, 370–381. [Google Scholar] [CrossRef]
- Thannickal, V.J.; Jandeleit-Dahm, K.; Szyndralewiez, C.; Török, N.J. Pre-clinical evidence of a dual NADPH oxidase 1/4 inhibitor (setanaxib) in liver, kidney and lung fibrosis. J. Cell. Mol. Med. 2023, 27, 471–481. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Yamaguchi, Y.; Kanzaki, H.; Katsumata, Y.; Itohiya, K.; Fukaya, S.; Miyamoto, Y.; Narimiya, T.; Wada, S.; Nakamura, Y. Dimethyl fumarate inhibits osteoclasts via attenuation of reactive oxygen species signalling by augmented antioxidation. J. Cell. Mol. Med. 2018, 22, 1138–1147. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Che, J.; Yang, X.; Jin, Z.; Xu, C. Nrf2: A promising therapeutic target in bone-related diseases. Biomed. Pharmacother. 2023, 168, 115748. [Google Scholar] [CrossRef] [PubMed]
- Lorenzo, J.A. The Role of Interleukin-6 in Bone. J. Endocr. Soc. 2020, 4, bvaa112. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- McGregor, N.E.; Murat, M.; Elango, J.; Poulton, I.J.; Walker, E.C.; Crimeen-Irwin, B.; Ho, P.W.M.; Gooi, J.H.; Martin, T.J.; Sims, N.A. IL-6 exhibits both cis- and trans-signaling in osteocytes and osteoblasts, but only trans-signaling promotes bone formation and osteoclastogenesis. J. Biol. Chem. 2019, 294, 7850–7863. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Steemers, E.; Talbi, W.M.I.; Hogervorst, J.M.A.; Schoenmaker, T.; de Vries, T.J. IL-1 Receptor Antagonist Anakinra Inhibits the Effect of IL-1β-Mediated Osteoclast Formation by Periodontal Ligament Fibroblasts. Biology 2025, 14, 250. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Tseng, H.W.; Samuel, S.G.; Schroder, K.; Lévesque, J.P.; Alexander, K.A. Inflammasomes and the IL-1 Family in Bone Homeostasis and Disease. Curr. Osteoporos. Rep. 2022, 20, 170–185. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Pullarkat, V.; Meng, Z.; Donohue, C.; Yamamoto, V.N.; Tomassetti, S.; Bhatia, R.; Krishnan, A.; Forman, S.J.; Synold, T.W. Iron chelators induce autophagic cell death in multiple myeloma cells. Leuk. Res. 2014, 38, 988–996. [Google Scholar] [CrossRef] [PubMed]
- Jeffries, N.E.; Sadreyev, D.; Trull, E.C.; Chetal, K.; Yvanovich, E.E.; Mansour, M.K.; Sadreyev, R.I.; Sykes, D.B. Deferasirox, an iron chelator, impacts myeloid differentiation by modulating NF-kB activity via mitochondrial ROS. Br. J. Haematol. 2024, 205, 2000–2007. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Vanderkerken, K.; Asosingh, K.; Willems, A.; De Raeve, H.; Couck, P.; Gorus, F.; Croucher, P.; Van Camp, B. The 5T2MM murine model of multiple myeloma: Maintenance and analysis. Methods Mol. Med. 2005, 113, 191–205. [Google Scholar] [CrossRef] [PubMed]
- Mehdi, S.H.; Morris, C.A.; Lee, J.A.; Yoon, D. An Improved Animal Model of Multiple Myeloma Bone Disease. Cancers 2021, 13, 4277. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Sun, J.; Chen, W.; Li, S.; Yang, S.; Zhang, Y.; Hu, X.; Qiu, H.; Wu, J.; Xu, S.; Chu, T. Nox4 Promotes RANKL-Induced Autophagy and Osteoclastogenesis via Activating ROS/PERK/eIF-2α/ATF4 Pathway. Front. Pharmacol. 2021, 12, 751845. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Jiang, H.; Wang, L.; Zhang, Q.; Wang, S.; Jia, L.; Cheng, H.; Wang, J.; Li, X.; Xie, Y.; Wang, Y.; et al. Bone marrow stromal cells dictate lanosterol biosynthesis and ferroptosis of multiple myeloma. Oncogene 2024, 43, 1644–1653. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Mehdi, S.H.; Nafees, S.; Mehdi, S.J.; Morris, C.A.; Mashouri, L.; Yoon, D. Animal Models of Multiple Myeloma Bone Disease. Front. Genet. 2021, 12, 640954. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Domain | Measure | Rationale | Assay/Acquisition Notes | Endpoints |
|---|---|---|---|---|
| Bone turnover | s-PINP; s-CTX | Standardized monitoring of remodeling dynamics | Follow IOF–IFCC preanalytics and variability control | Primary PD; correlate with imaging/fractures |
| Redox status | AOPP; AGEs; S-nitrosylated proteins | Microenvironmental oxidative/nitrosative load | Batch controls; diet/renal confounders | Secondary PD; link to NOX4/NRF2 strategies |
| Imaging | LD-WBCT; WB-MRI; FDG-PET/CT | Structural + metabolic assessment | IMWG protocols; harmonize PET/CT timing | Structural healing; metabolic remission; SREs |
| Iron–lipid peroxidation | Iron indices; lipid peroxidation markers | Infer ferroptosis/OC iron handling | Standard iron panel + research lipidomics | Exploratory PD; responder enrichment |
| Model | Host Immune Status | Key Pathways Validated In Vivo | Translational/Therapeutic Relevance |
|---|---|---|---|
| 5T2MM/5T33MM | Syngeneic, immunocompetent | RANKL–OPG imbalance; IL-6–driven inflammation; oxidative stress; osteoclastogenesis | Validation of antiresorptives; cytokine modulation; redox-targeted strategies |
| MM.1S intrabone xenograft | SCID/NSG | Proteostasis stress; hypoxia; autophagy dependence; oxidative signaling | Proteasome–autophagy co-targeting; stress-response pathway inhibition |
| U266/RPMI-8226 xenografts | Immunodeficient | Redox imbalance; inflammatory cytokine signaling | Tumor-intrinsic redox and cytokine pathway targeting |
| Conditional NOX4/ATG/TFEB mice | Immunocompetent, lineage-specific | Redox–autophagy–lysosomal coupling in osteoclasts and osteoblasts | Selective NOX4 inhibition; lineage-aware autophagy modulation |
| Ferroptosis-related MM models | Syngeneic or xenograft (context-dependent) | Iron retention; lipid peroxidation; ferroptotic thresholds | Iron modulation; ferroptosis-inducing strategies |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license.
Share and Cite
Nasso, M.E.; Bottaro, A.; Fazio, M.; Stagno, F.; Gangemi, S.; Allegra, A. Microenvironmental Drivers of Bone Disease in Multiple Myeloma: Oxidative Stress, Sterile Inflammation, Autophagy–Lysosomal Remodeling, and the Iron–Lipid Peroxidation Axis. Biomolecules 2026, 16, 710. https://doi.org/10.3390/biom16050710
Nasso ME, Bottaro A, Fazio M, Stagno F, Gangemi S, Allegra A. Microenvironmental Drivers of Bone Disease in Multiple Myeloma: Oxidative Stress, Sterile Inflammation, Autophagy–Lysosomal Remodeling, and the Iron–Lipid Peroxidation Axis. Biomolecules. 2026; 16(5):710. https://doi.org/10.3390/biom16050710
Chicago/Turabian StyleNasso, Maria Elisa, Adele Bottaro, Manlio Fazio, Fabio Stagno, Sebastiano Gangemi, and Alessandro Allegra. 2026. "Microenvironmental Drivers of Bone Disease in Multiple Myeloma: Oxidative Stress, Sterile Inflammation, Autophagy–Lysosomal Remodeling, and the Iron–Lipid Peroxidation Axis" Biomolecules 16, no. 5: 710. https://doi.org/10.3390/biom16050710
APA StyleNasso, M. E., Bottaro, A., Fazio, M., Stagno, F., Gangemi, S., & Allegra, A. (2026). Microenvironmental Drivers of Bone Disease in Multiple Myeloma: Oxidative Stress, Sterile Inflammation, Autophagy–Lysosomal Remodeling, and the Iron–Lipid Peroxidation Axis. Biomolecules, 16(5), 710. https://doi.org/10.3390/biom16050710