1. Introduction
In recent years, interest in plant-derived substances as potential therapeutic agents has grown, particularly in extracellular vesicles (EVs) [
1]. Furthermore, exosomes, a subtype of EVs, have gained attention due to their ability to mediate intercellular communication by transferring various bioactive molecules, including proteins, lipids, microRNAs (miRNAs), messenger RNA (mRNA), DNA, and other nucleic acids. These nanometer-sized vesicles, typically ranging from 50 to 200 nm, are generated within cells and secreted into extracellular space. They possess a stable structure characterized by a phospholipid bilayer and play a crucial role in cellular signaling by fusing with recipient cells and delivering their contents [
2]. Recent studies have highlighted the therapeutic potential of plant-derived vesicles (PDVs), which exhibit bioactive properties that contribute to the treatment of various diseases and enhance human health. Thus, recognition of the value of PDVs in the development of novel therapeutic and cosmetic applications is growing [
3].
miRNAs are short non-coding RNA molecules that play a critical role in regulating gene expression by interacting with messenger RNA (mRNA). By binding to the target mRNA, miRNAs inhibit gene translation or destabilize the mRNA, thereby modulating protein expression levels [
4]. Typically transcribed within cells, miRNAs specifically bind to mRNA that encodes certain proteins, resulting in translational repression or mRNA degradation, which, in turn, suppresses protein synthesis. Regulatory functions of miRNAs are essential for maintaining normal cellular functions and supporting growth and development. However, the dysregulation of miRNA activity has been implicated in various diseases, particularly cancer [
5,
6]. In recent years, miRNAs have emerged as valuable biomarkers of various diseases. Their altered expression patterns, which often change in response to disease conditions, make them promising diagnostic and prognostic tools for biomedical research. A biomarker is an objective indicator of biological or pathogenic processes or responses to therapeutic interventions. The potential of miRNAs as biomarkers was first recognized when Lawrie et al. (2008) demonstrated their utility in detecting diffuse large B-cell lymphoma in patient serum samples [
7]. Since then, miRNAs have been extensively studied and cited in the literature for their potential application as biomarkers of numerous diseases, supporting their relevance in advancing diagnostic precision and therapeutic monitoring.
Similarly to animals, plant miRNAs also function as post-transcriptional regulators. Current research on plant miRNAs spans various aspects of plant biology, including biological processes, growth, development, immune responses, stress adaptation, and the regulation of gene expression [
2,
8]. Plant miRNAs play a crucial role in plant development and growth, particularly in regulating processes, including cell proliferation, differentiation, and tissue formation in the roots, stems, and leaves. Additionally, plant miRNAs are central to plant environmental stress responses. These miRNAs enable plants to adapt by regulating gene expression in response to factors, such as climate change, heat, drought, and salinity, which are critical for their survival and ecological functions [
9]. miRNA408 (miR408), identified in plants, is known for its significant physiological functions across various plant species. Predominantly expressed in chloroplasts, miR408 regulates several key processes, including photosynthesis and chlorophyll biosynthesis, processes essential for plants to maintain optimal photosynthetic efficiency, especially under stress conditions induced by high-intensity light. These processes enable plants to mitigate damage from environmental stressors, enhancing their photosynthetic resilience [
10]. Apart from photosynthesis, miR408 is actively involved in plant responses to other environmental stresses, such as extreme heat. miR408 regulates the expression of specific genes that help plants activate survival mechanisms, increasing their stress tolerance and adaptability to challenging environments. These attributes have made exploring the potential effects of miR408 beyond plant biology an emerging area of interest.
Despite the well-established roles of miRNAs in plants, research on the interactions between plant-derived miRNAs from EVs and human skin cells is limited. Hence, this study aimed to explore the therapeutic potential of these plant-derived miRNAs from EVs for skin health applications in skin health. Understanding the interactions between plant-derived miR408 from EVs and human skin cells may offer novel insights into the molecular pathways associated with skin aging [
11]. The discovery of cross-kingdom regulatory mechanisms mediated by miR408 may pave the way for therapeutic applications and innovative skincare strategies that utilize plant-derived miRNAs from EVs [
12].
In this study, we isolated extracellular vesicles from the callus of Camellia japonica L. (Cj-callus EVs) and investigated their in vitro biological effects on human skin cells, with a focus on wound healing and anti-inflammatory properties. Small RNA sequencing revealed that miR408 was highly expressed in Cj-callus EVs. Subsequent experiments involved the transfection of human skin cells with miR408 and assessing various biological markers to determine their impact on skin cell functions. This study suggests the involvement of plant-derived miR408 from Cj-callus EVs in the promotion of wound healing and modulation of inflammatory processes in human skin cells. These findings suggest potential implications for future research and development in the field of skin care.
2. Materials and Methods
2.1. Callus Induction of Camellia japonica L. and Suspension Culture
For the induction of callus from the leaves of Camellia japonica L. (Cj-callus), leaves were surface sterilized by soaking in 95% ethanol for 60 s, followed by treatment with a disinfectant solution (50% bleach + 0.1% Tween-20) for 20 min. The sterilized leaves were washed at least three times with sterile distilled water, supplemented with 500 mg/L of cefotaxime sodium (Duchefa, Haarlem, The Netherlands) to prevent contamination from soil pathogens. Sterilized leaves were cut into 5 × 5 mm pieces and placed on a Murashige and Skoog (MS) medium containing 3% (w/v) sucrose and 0.35% (w/v) gelatin, along with 0.5 mg/L of 2,4-dichlorophenoxyacetic acid (2,4-D, Duchefa, Haarlem, The Netherlands), which was used as a callus induction medium (CIM). The cultures were maintained in the dark to promote callus formation. The induced callus was subcultured every 4 weeks for 2–3 times on a solid medium. Subsequently, the callus was transferred to liquid medium and cultured under suspension conditions for 3–4 weeks at a shaker speed of 100–300 rpm to promote further growth.
2.2. Isolation of Extracellular Vesicles (EVs) from C. japonica L.
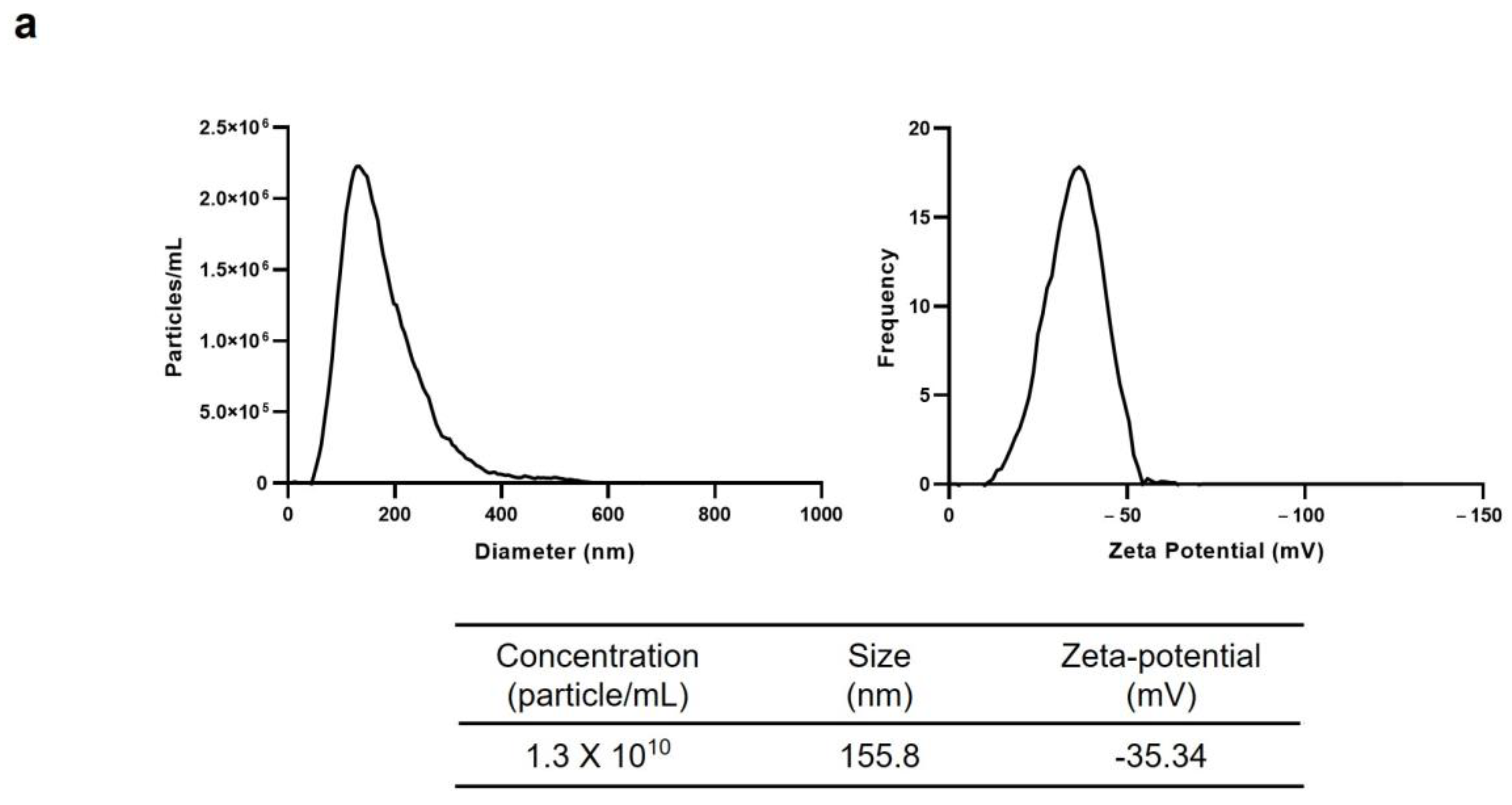
EVs from C. japonica callus (Cj-callus EVs) were isolated using a liquid suspension culture system. Four-week-old Cj-callus cultures from a bioreactor were first separated into callus clusters and conditioned medium using 100- and 300-mesh filters. The medium was then centrifuged at 3000–4000× g for 20 min to remove cell debris, fibers, and large particles. Only the supernatant was collected, and this step was repeated to ensure the removal of any residual impurities. The cleared supernatant was further subjected to ultracentrifugation at 100,000–150,000× g for 2 h using an ultracentrifuge (Hitachi, Tokyo, Japan), which allowed EVs to settle into the pellet. The supernatants were carefully discarded, and the EV pellet was resuspended in a small volume of deionized water (DIW). The EV suspension was filtered through a 0.22 μm syringe filter (Advantec, Tokyo, Japan) for sterilization and stored at −80 °C until further use. The size distribution and the particle concentration of EVs were analyzed using Nanoparticle Tracking Analysis (NTA), and the zeta potential of the EVs was measured using a ZetaView (Particle Metrix, Inning am Ammersee, Starnberg, Germany).
2.3. Transmission Electron Microscope (TEM) Imaging
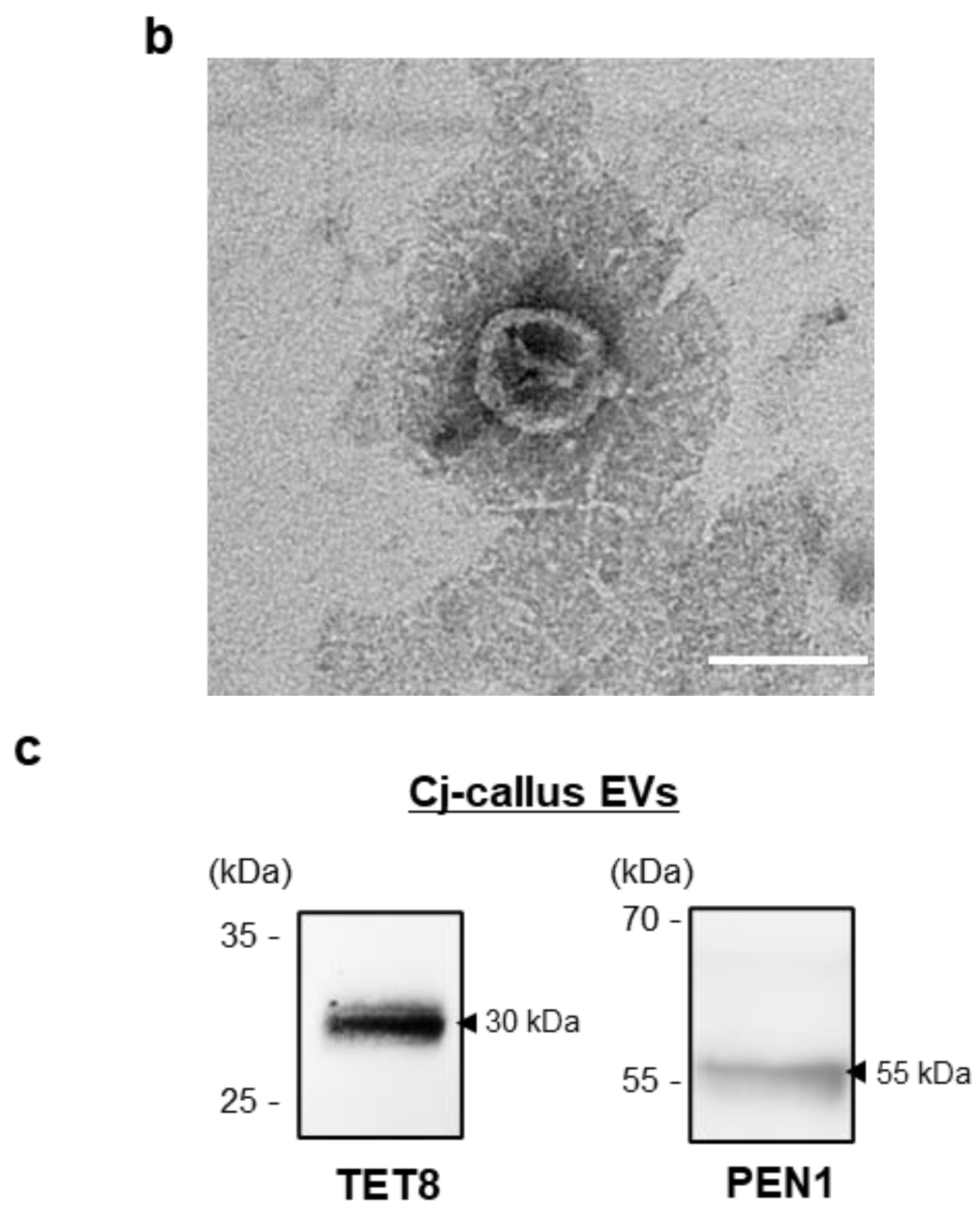
EVs were placed onto carbon-coated grids (EMS, Houston, TX, USA) at room temperature for 3–5 min. Excess sample was carefully removed, and the grids were allowed to air dry. The grids were then stained with 2% uranyl acetate solution (EMS, Houston, TX, USA) at room temperature for less than 10 s. Nanoparticle imaging was conducted using a transmission electron microscope (JEOL, Tokyo, Japan) [
13].
2.4. Western Blot Analysis
Protein was extracted from EVs using RIPA buffer and quantified with the Bradford reagent (Thermo Fisher Scientific, Waltham, MA, USA). Equal amounts of protein were separated on SDS-polyacrylamide gels and transferred onto polyvinylidene fluoride (PVDF, Invitrogen, Carlsbad, CA, USA) membranes. Transferred membranes were blocked with 5% BSA and then incubated with the following primary antibodies: anti-TET8 (PHY1490A, PhytoAB, San Jose, CA, USA) or anti-PEN1 (CSB-PA875527XA01DOA, Cusabio, Houston, TX, USA) overnight at 4 °C. Following washing with TBST, membranes were incubated with horseradish peroxidase (HRP)-conjugated secondary antibody (PHY6000, PhytoAB, San Jose, CA, USA) for 1 h at room temperature. The membranes were developed using ECL (Bio-Rad, Hercules, CA, USA) on a chemiluminescence imaging system (Bio-Rad, Hercules, CA, USA) [
14].
2.5. Cell Culture
The human keratinocyte (HaCaT) cell line and human fibroblast (HFF) cell line were cultured in Dulbecco’s modified Eagle medium (DMEM, Welgene, Gyeongsan, Gyeongbuk, Republic of Korea), supplemented with 10% fetal bovine serum (FBS) (Gibco, Grand Island, NY, USA) and 1% of a penicillin–streptomycin solution (Gibco, Grand Island, NY, USA). Cells were maintained at 37 °C in a humidified incubator with 5% CO2.
2.6. Cell Viability Assay
HaCaT and HFF cells were seeded at a density of 1.0 × 104 cells/well in 96-well plates and cultured for 24 h. Cultured cells were treated with EVs at specified concentrations or transfected with a microRNA mimic (RNA double-strand oligonucleotides) for each experiment and incubated for 48 h. Cell viability was assessed using the WST-1 cell proliferation and cytotoxicity detection kit (DoGENBIO, Seoul, Republic of Korea) following the manufacturer’s protocol. After adding the WST-1 reagent, the mixture was incubated for 2 h, and absorbance was then measured at 450 nm using a microplate reader (Bio Tek, Shoreline, WA, USA).
2.7. Extracellular Vesicles Uptake Assay
HaCaT cells were seeded at a density of 1.0 × 10
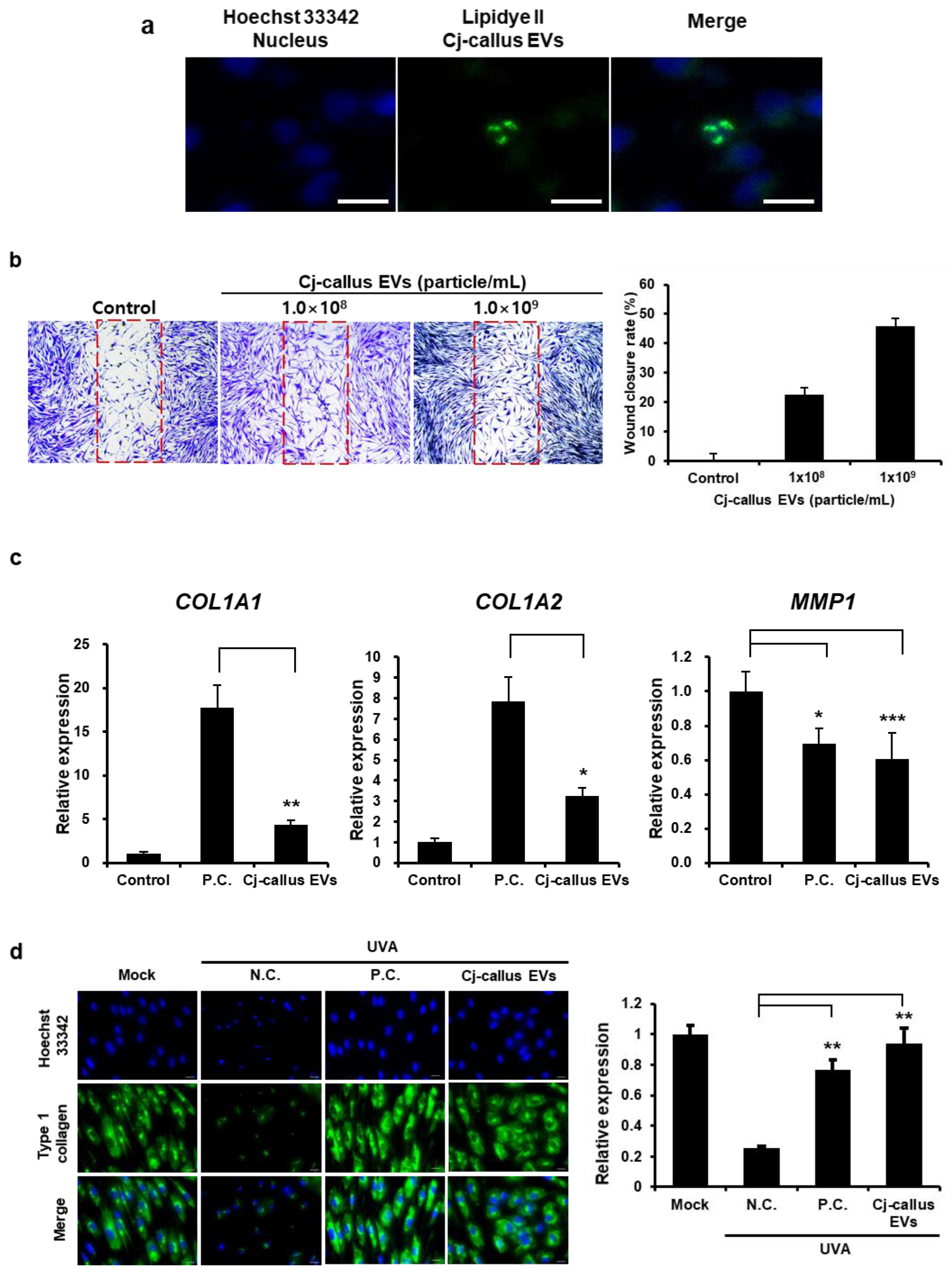
5 cells/well in black 24-well plates and cultured for 24 h. EVs were labeled with LipidyeII (Funakoshi, Morioka, Japan) according to the manufacturer’s protocol. Hoechst 33342 was applied to cultured cells for nuclear counterstaining, and subsequently, the labeled EVs were added to each well, followed by an additional incubation for 6 h. After treatment, the wells were washed 3 times with DPBS to remove any unbound EVs in the medium, and cells were fixed in 4% paraformaldehyde (PFA) solution for 15 min at room temperature. Fluorescence images were obtained using a fluorescence microscope (Ts2, Nikon, Tokyo, Japan) to assess EV uptake [
15].
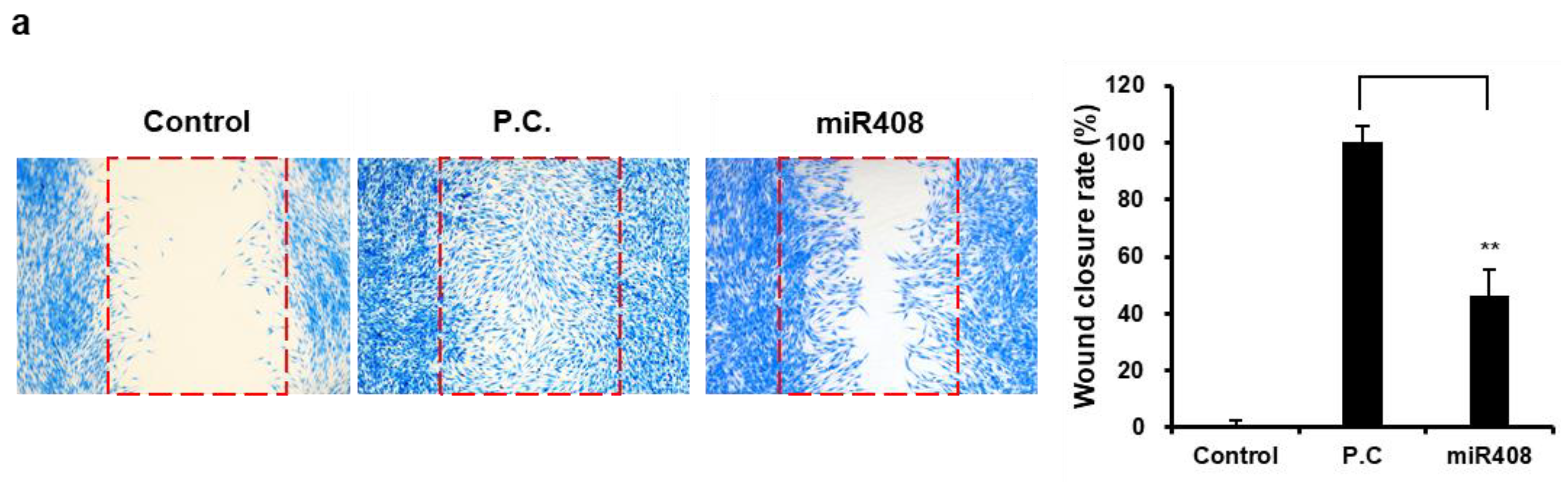
2.8. In Vitro Wound Healing Assay
HFF cells were seeded into a 24-well plate at a density of 5.0 × 104 cells/well using Wound Healing Insert (Cellbiolabs, San Diego, CA, USA) and allowed to adhere and culture for 24 h. Following incubation, the inserts were carefully removed, and the wells were washed with DPBS to remove debris and non-adherent cells. The cells were then treated with EVs at an optimized concentration in a serum-free medium for an additional 48 h. The size of the cell-free gaps was visualized and detected under a microscope equipped with a digital camera (Nikon, Tokyo, Japan) and measured using Image J software v 1.54g. It was expressed as a percentage of the control interval.
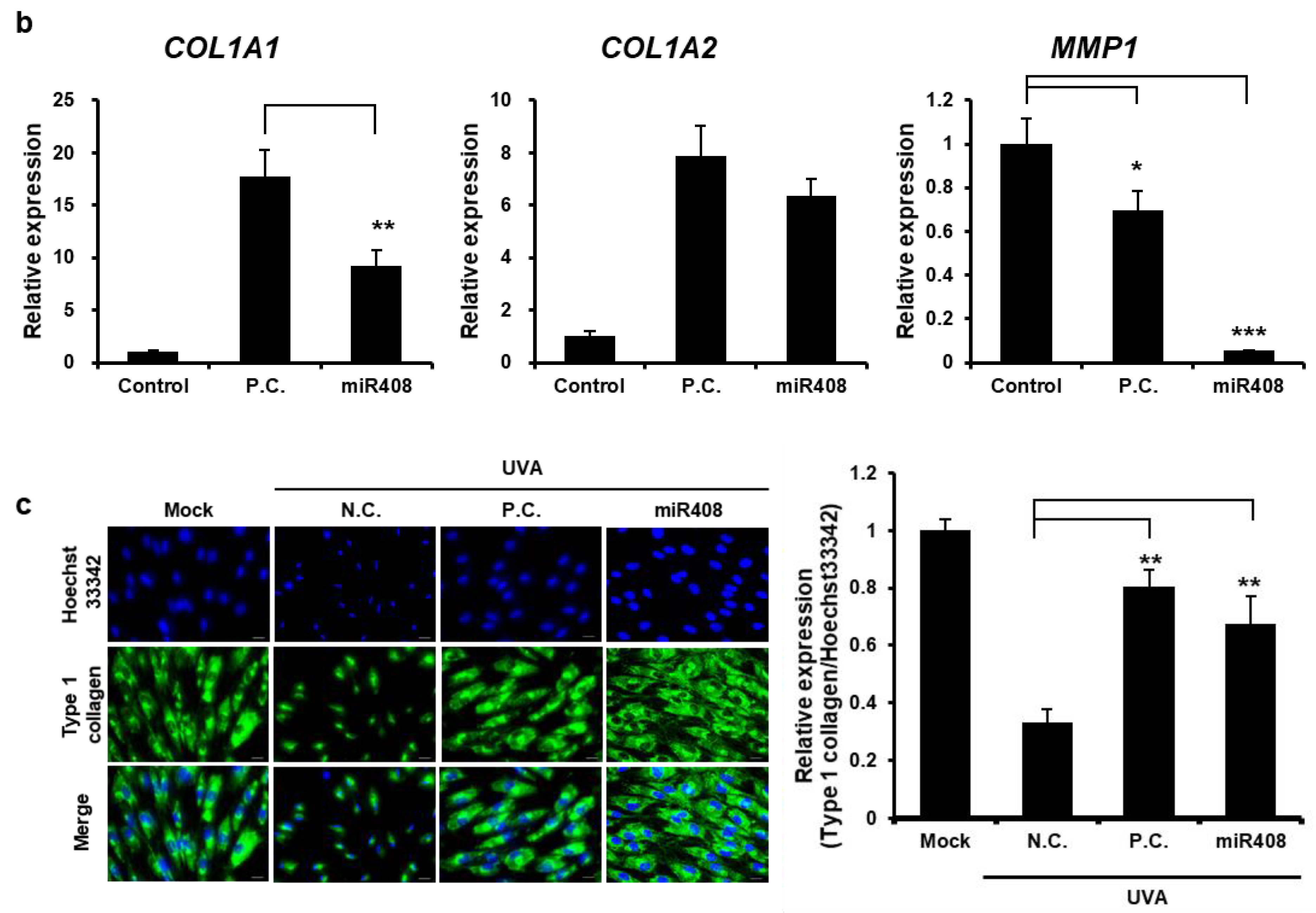
2.9. Immunocytochemistry (ICC)
HFF cells were seeded at 2.0 × 10
5 cells/well in 24-well plates and cultured for 24 h. After the initial culture, the cells were exposed to 1.5 J/cm
2 UVA radiation using a UV illuminator (Bioteck, Seoul, Republic of Korea). Following UVA exposure, cells were either treated with EVs or transfected with a microRNA mimic, as specified for each experimental condition, and then incubated for an additional 48 h. After incubation, supernatants were discarded, and the cells were gently washed with DPBS before fixation in 4% (
w/
v) paraformaldehyde (PFA) for 20 min at room temperature. Cells were washed 3 times in DPBS, permeabilized with 0.1% Triton-X 100 in DPBS for 20 min, and washed again 3 times. Subsequently, a 5% bovine serum albumin (BSA, BOVOGEN, Keilor East, VIC, Australia) in DPBS for blocking solution was applied to prevent non-specific antibody binding and then incubated with collagen type I A1 (COL1A1) primary antibody (ab316222, Abcam, Cambridge, UK) overnight at 4 °C. The next day, cells were incubated with goat anti-rabbit IgG antibody conjugated to Alexa Fluor 488 (ab150077, Abcam, Cambridge, UK) as a secondary antibody for 1 h at room temperature, followed by staining with 1 μg/mL DAPI for 15 min. Finally, cells were washed with DPBS, and the cell images were taken using a fluorescence microscope [
16].
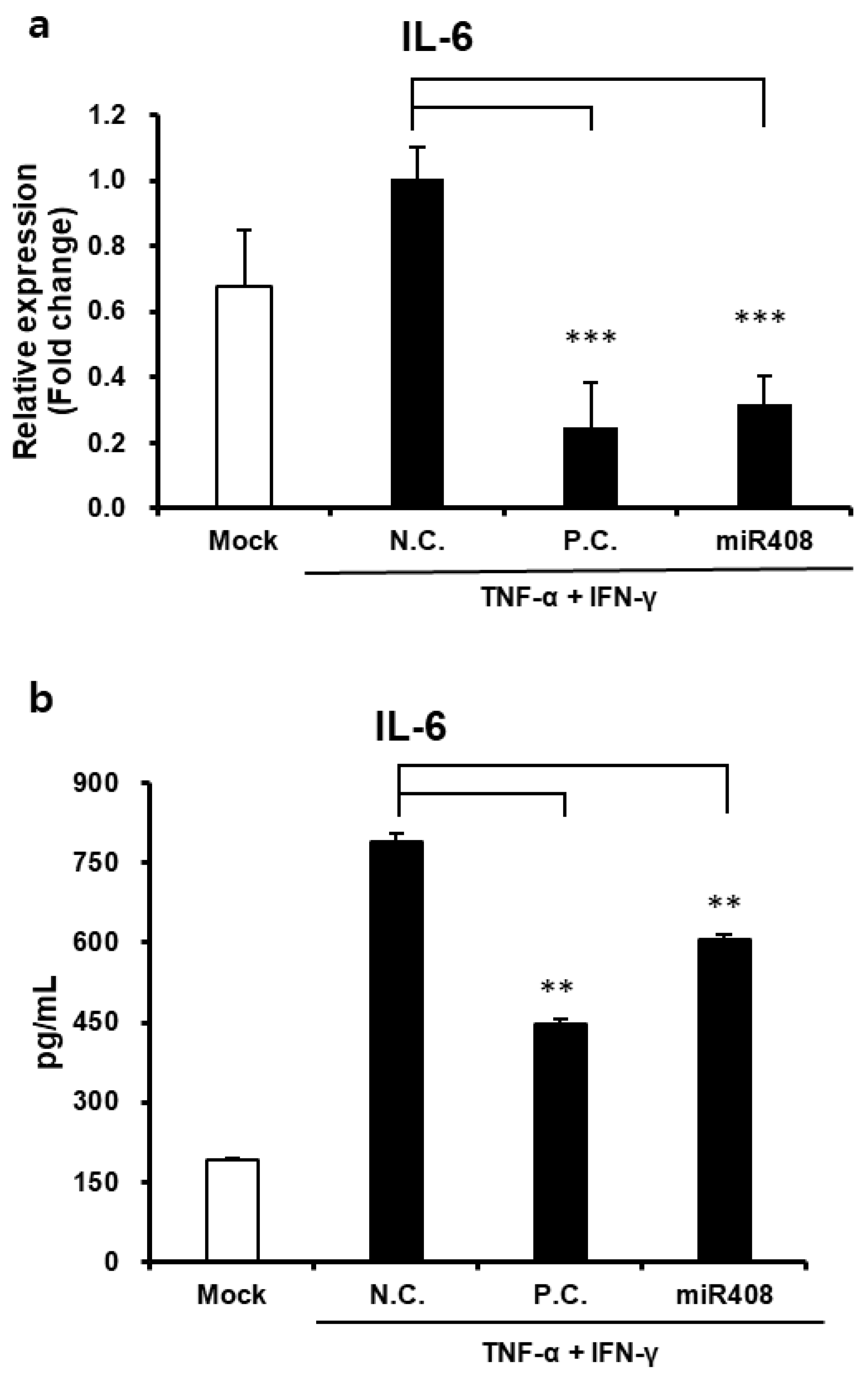
2.10. ELISA
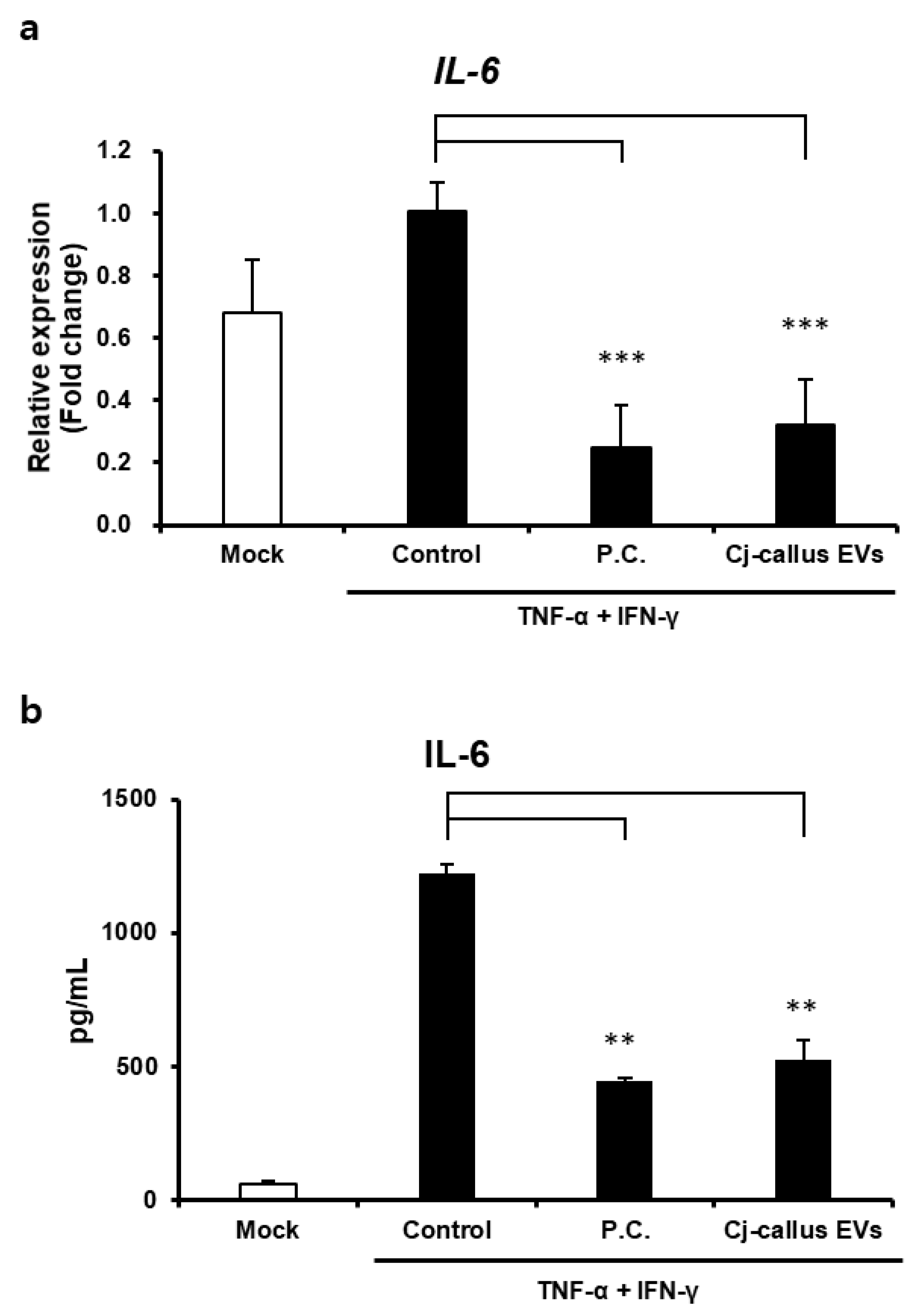
HaCaT cells were seeded at a density of 5.0 × 104 cells/well in 24-well plates and cultured for 24 h. Following initial culture, the medium was replaced with a fresh medium containing EVs or miRNA mimics at varying concentrations, and cells were incubated for an additional 24 h. After incubation, supernatants were collected and centrifuged, then stored at −20 °C until further use. IL-6 expression levels in the supernatants were quantified using a human IL-6 ELISA kit (#430501, BioLegend, San Diego, CA, USA) according to the manufacturer’s instructions. Absorbance was measured at 450 nm to determine IL-6 concentrations.
2.11. qRT-PCR (Quantitative Reverse-Transcription–PCR)
Each cell line was seeded in a 24-well plate at a density of 1.0 × 10
5 cells/well and cultured at 37 °C with 5% CO
2 for 24 h. After incubation, the medium was discarded, and cells were washed with DPBS. Cells were then treated with appropriately diluted EVs in serum-free medium and incubated for another 24 h. Total RNA was isolated using the NucleoSpin RNA kit (Macherey-Nagel, Düren, Germany) according to the manufacturer’s instructions, and RNA quantity was assessed using a Nanodrop ND-1000 spectrophotometer(Thermo Fisher Scientific, Waltham, MA, USA). cDNA was synthesized using a cDNA synthesis kit (GenDEPOT, Katy, TX, USA) following the manufacturer’s protocol. Target gene expression was detected with SYBR Green Supermix (Bio-Rad, Hercules, CA, USA) using a real-time PCR machine (Bio-Rad, Hercules, CA, USA). Expression levels were normalized to β-actin as an internal control, and relative gene expression was calculated. Primer sequences used in this study are listed in
Supplementary Table S2.
2.12. Small RNA Isolation
Small RNA was isolated from Cj-callus and Cj-callus EVs using a XENOPURE Plant Small RNA Purification kit (Xenohelix, Incheon, Republic of Korea) and XENO-EVARI Kit (Xenohelix, Incheon, Republic of Korea), respectively, following the manufacturer’s instructions. Purified RNA samples were further cleaned using the XENOPURE Small RNA clean-up kit, as instructed. Isolated RNA was eluted in 20 μL of RNase-free water and stored at −80 °C until further use. The concentration of RNA was quantified with a nanodrop spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA) and analyzed for quality and quantity using the 2100 Bioanalyzer (Agilent® Technologies, Santa Clara, CA, USA) with the RNA 6000 Pico chip. All RNA samples were stored at −80 °C until subsequent analysis.
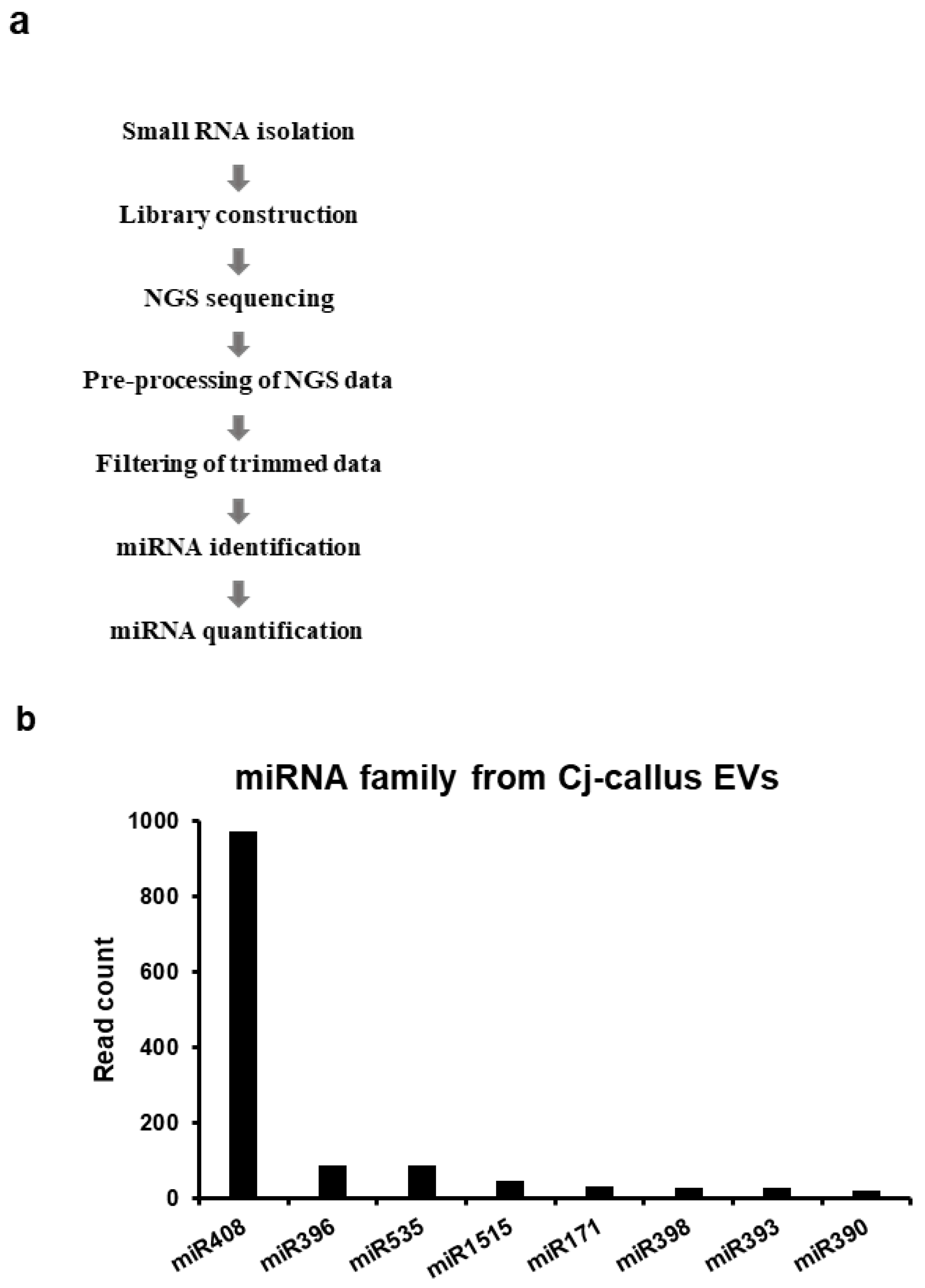
2.13. Small RNA Library Construction and Analysis
Small RNA libraries were generated using the XENO-LIBERA library kit (Xenohelix, Incheon, Republic of Korea) according to the manufacturer’s instructions. Sequencing was conducted on an Illumina NextSeq 500 platform with a 76-cycle single-end read configuration. The quality of raw miRNA-seq data was assessed using FastQC v0.11.9 [
17], and adaptor sequences were removed using Cutadapt v4.4 [
18]. Only reads of 18–30 nucleotides were retained, and the 3′ bases with a quality score below 20 were trimmed. Trimmed data were further processed to collect reads aligned with non-coding RNA sequences using Bowtie v1.1.2 [
19]. miRNA prediction was conducted using the miRkwood tool [
20] with a score range of 0 to 5 alongside BrumiR v3.0 [
21]. To quantify miRNA expression levels, clean miRNA-seq reads were mapped to the predicted miRNAs, which were subsequently compared against the miRBase database [
22] for validation. Predicted miRNA families were further identified using BLASTn v2.2.29 in the NCBI-BLAST package [
23].
2.14. miRNA Transfection
Cell lines were transfected with 20 nM miRNA mimic using Lipofectamine RNAiMAX Transfection Reagent (Invitrogen, Carlsbad, CA, USA) following the manufacturer’s protocol. After transfection, the transfected cells or cultured medium were used in other experiments.
2.15. Statistical Analysis
All experiments were performed at least 3 times, and data are presented as the mean ± standard deviation (SD). Statistical significance was determined using ordinary one-way ANOVA, followed by Student’s t-test, with a threshold of p < 0.05 considered statistically significant. All reported data represent the mean ± standard error (SE) unless stated otherwise.
4. Discussion
Camellia japonica L., commonly known as Japanese camellia or Tsubaki, is highly valued in the cosmetic industry because of its beneficial effects on skin health. The oil extracted from camellia seeds is known for its exceptional moisturizing, hydrating, and skin-nourishing properties [
26]. Callus induction and culture from plants, a key technique in plant biotechnology, offer several advantages that make it highly applicable in various fields. One of the primary advantages of callus culture is its ability to efficiently produce large quantities of plant material while ensuring genetic stability. Callus cultures are rich sources of secondary metabolites with diverse applications in medicine, cosmetics, and agriculture [
27]. In this study, we utilized plant biotechnology to derive Cj-callus and established a suspension culture using a bioreactor to produce extracellular vesicles (Cj-callus EVs). This system successfully produced EVs from Cj-callus, suggesting the feasibility of harnessing plant-derived EVs as novel bioactive compounds. Our results may indicate the potential of Cj-callus EVs in various sectors, particularly cosmetics, pharmaceuticals, and agriculture, contributing to the expanding research landscape of plant-derived EVs and their diverse applications.
The NTA results demonstrated a uniform distribution of isolated vesicles, as indicated by their consistent zeta potential values, reflecting the homogeneity of the vesicles. This homogeneity suggests their potential for use in drug delivery systems (DDS). Additionally, the successful detection of TET8 and PEN1, which are established EV markers, confirmed that the isolated vesicles were predominantly EVs.
Our findings also revealed that Cj-callus EVs exhibited notable wound healing and anti-inflammatory effects in human-derived cell lines (
Figure 3 and
Figure 4), supporting their potential for cosmetic and therapeutic applications. In fibroblast cell viability and wound closure assays, treatment with increasing concentrations of Cj-callus EVs resulted in enhanced wound closure, indicating their ability to accelerate healing. This outcome aligns with previous research suggesting that EVs carry bioactive molecules, such as proteins and miRNAs, that can promote cell migration, proliferation, and extracellular matrix (ECM) remodeling during wound healing processes [
28]. A key component of the ECM, COL1A1, plays a crucial role in maintaining tissue integrity and structure, especially in connective tissues such as skin, bone, and tendons. Immunocytochemical results suggested that Cj-callus EV treatment restored type I collagen expression, indicating its potential role in ECM reconstruction and skin barrier reinforcement.
In addition to their regenerative properties, Cj-callus EVs exhibit notable anti-inflammatory effects. In human keratinocytes, Cj-callus EV treatment significantly decreased IL-6 expression at both the gene and protein levels in inflammation-induced cells. This anti-inflammatory effect suggests that Cj-callus EVs may modulate key signaling pathways activated by pro-inflammatory cytokines, such as TNF-α and IFN-γ. Although further research is necessary to elucidate the precise mechanisms involved, these findings indicate the potential of Cj-callus EVs to mitigate inflammation-related processes.
Notably, the reduction in IL-6, a cytokine closely associated with inflammation, aging, and skin damage, might indicate the protective role of Cj-callus EVs in managing inflammatory skin conditions. These findings suggest that Cj-callus EVs could serve as promising agents for therapeutic applications targeting skin inflammation, wound healing, and aging.
Research in 2012 discovered that plant-derived miRNAs regulate gene expression in mammals, which sparked significant interest in cross-kingdom miRNA interactions [
29]. Inspired by this discovery, we focused on miR408 because of its unique properties and discovered that it exerts a novel effect on improving human skin conditions, suggesting potential benefits beyond cross-kingdom interactions. Small-RNA sequencing confirmed a significantly higher concentration of miRNAs in EVs than in callus cells, indicating selective packaging. The known miRNAs detected were miR408, 396, 535, 1515, 171, 398, 393, and 390, along with several novel miRNAs. The enrichment of miR408 in Cj-callus EVs suggests that miRNAs in plant-derived EVs may serve as functional components capable of transferring regulatory signals to recipient cells.
Our findings suggest the impact of miR408 on human skin, indicating a novel bioactive role that extends across species. miR408 independently enhanced wound healing by promoting wound closure, similar to the effects observed with Cj-callus EVs.
COL1A1 expression is tightly regulated at multiple levels, including transcriptional, post-transcriptional, and post-translational levels. An important mechanism in the post-transcriptional regulation of
COL1A1 involves miRNAs. miR29 families are well-known regulators of COL1A1. Downregulation of miR29 is associated with increased collagen expression and fibrosis in different tissues [
30]. In this study, miR408 restored
COL1A1 expression to higher levels than in Cj-callus EVs, with a significant reduction in
MMP1 expression, suggesting that miR408 is involved in collagen formation through MMP-1 inhibition. Additionally, miR408 exhibits notable anti-inflammatory properties by decreasing IL-6 expression in human keratinocytes, similar to the effects of Cj-callus EVs. These results indicate that miR408 plays a crucial role in the wound healing and anti-inflammatory activities of Cj-callus EVs, likely through cross-kingdom regulatory mechanisms. For target gene analysis of miR408, potential genes were predicted using the method described [
31]. However, further research is required to explore the correlation between these genes, wound closure, and their anti-inflammatory effects.


 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}