


Oral Pharmacokinetic Evaluation of a Microemulsion-Based Delivery System for Novel A190 Prodrugs

 , ,
, ,  , and
, and 
Abstract
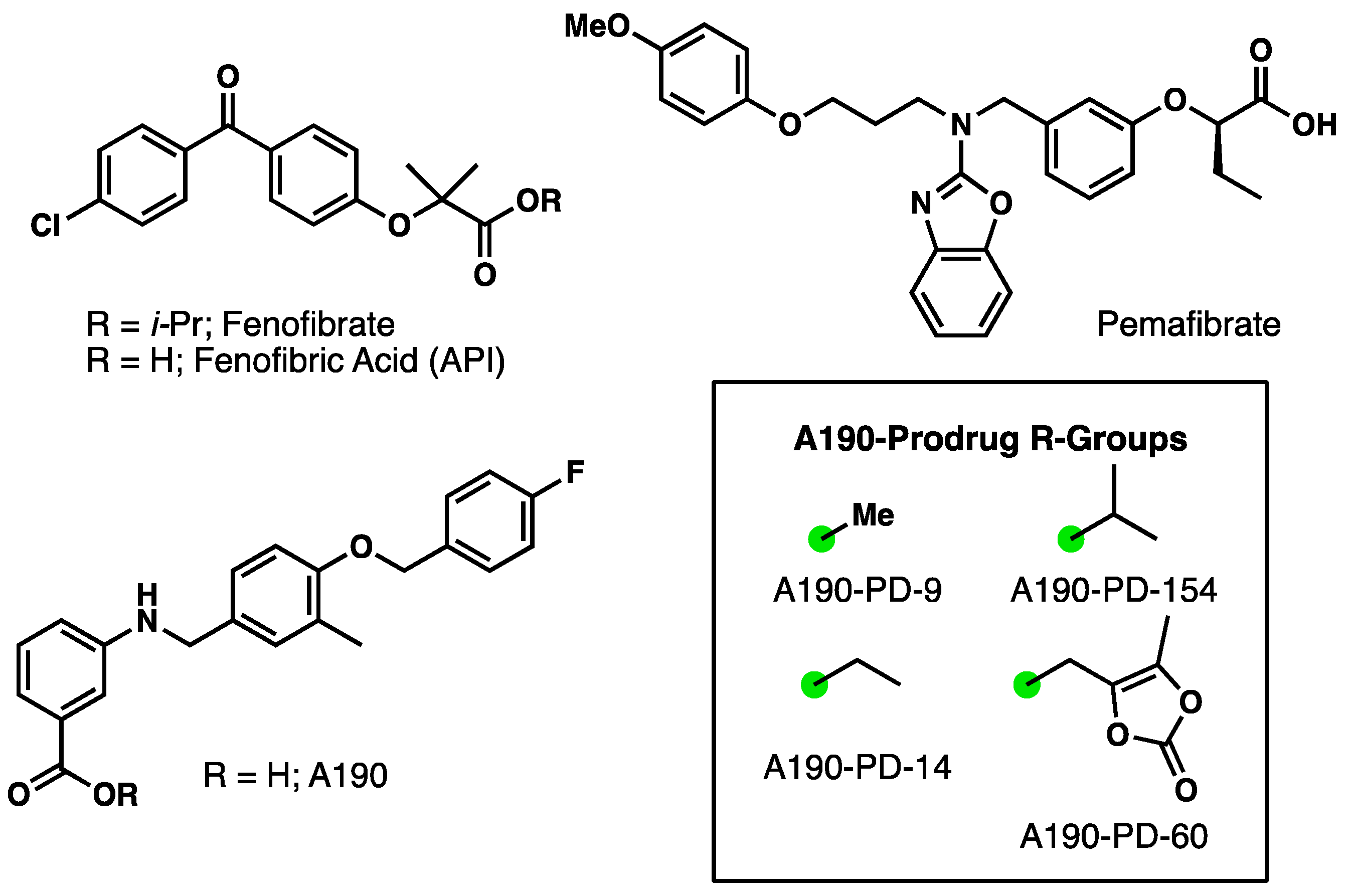
1. Introduction
2. Materials and Reagents
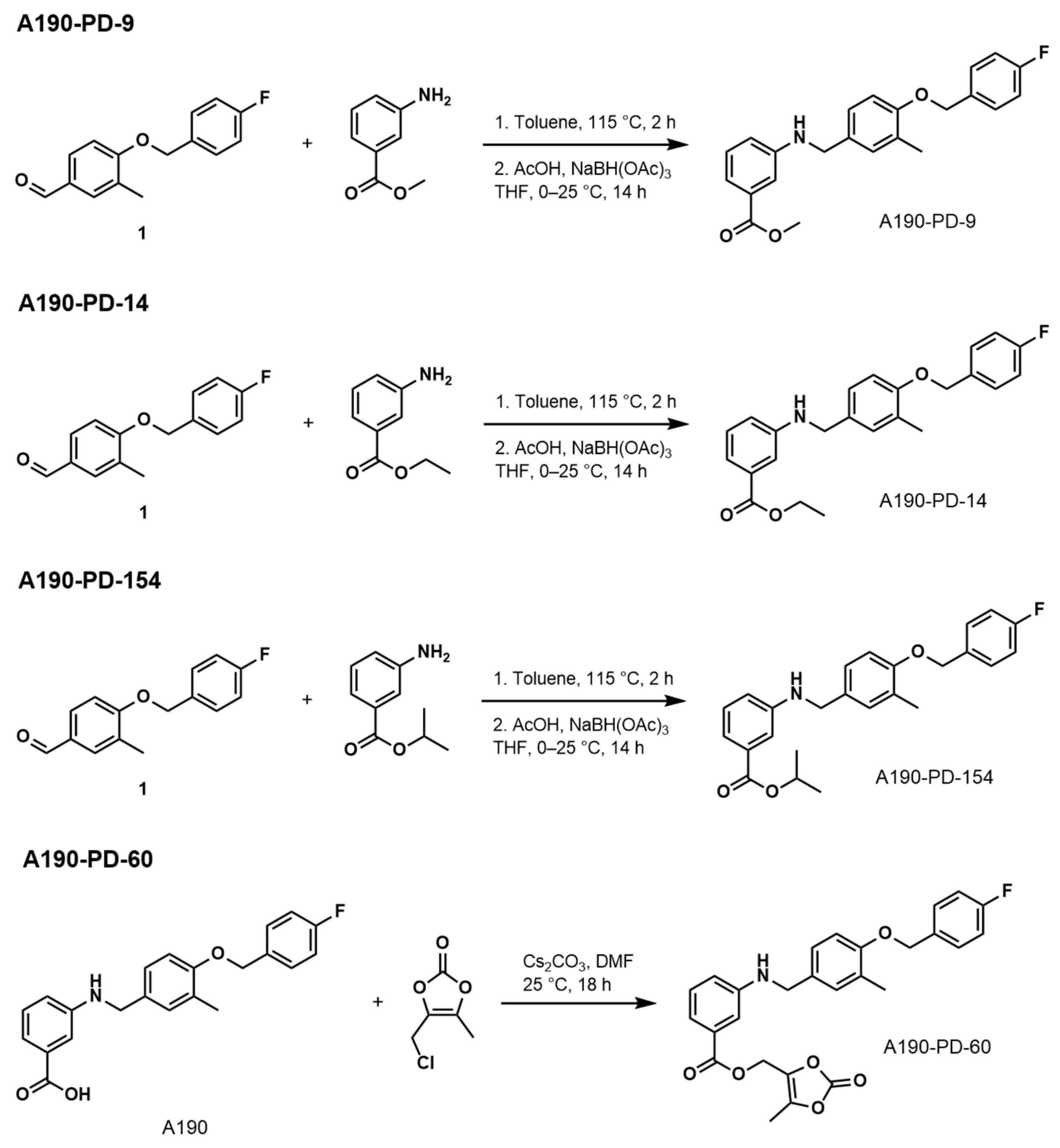
2.1. Prodrug Synthesis and Characterization
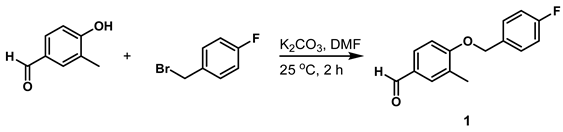
- 4-((4-Fluorobenzyl)oxy)-3-Methylbenzaldehyde (Compound 1)
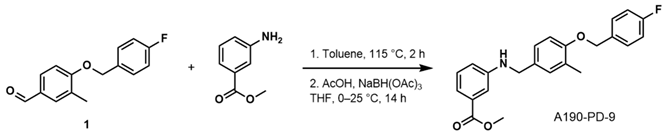
- Methyl 3-((4-((4-Fluorobenzyl)oxy)-3-Methylbenzyl)amino)benzoate (A190-PD-9)
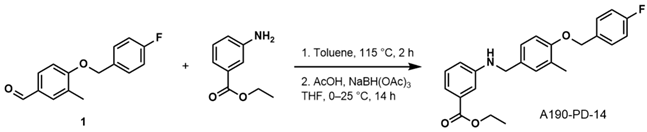
- Ethyl 3-((4-((4-Fluorobenzyl)oxy)-3-Methylbenzyl)amino)benzoate (A190-PD-14)
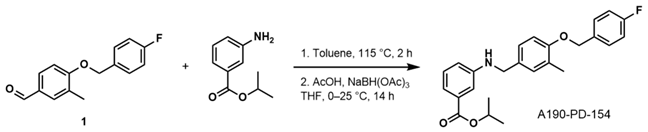
- Isopropyl 3-((4-((4-Fluorobenzyl)oxy)-3-Methylbenzyl)amino)benzoate (A190-PD-154)
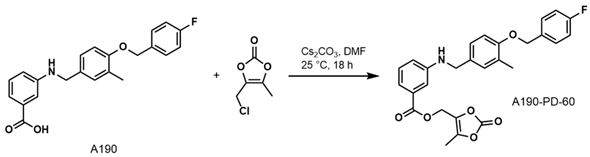
- (5-Methyl-2-oxo-1,3-dioxol-4-yl)methyl3-((4-((4-Fluorobenzyl)oxy)-3-methylbenzyl) amino)benzoate (A190-PD-60)
2.2. Aqueous Solubility of Prodrugs
2.3. Bioconversion of Prodrugs in Plasma
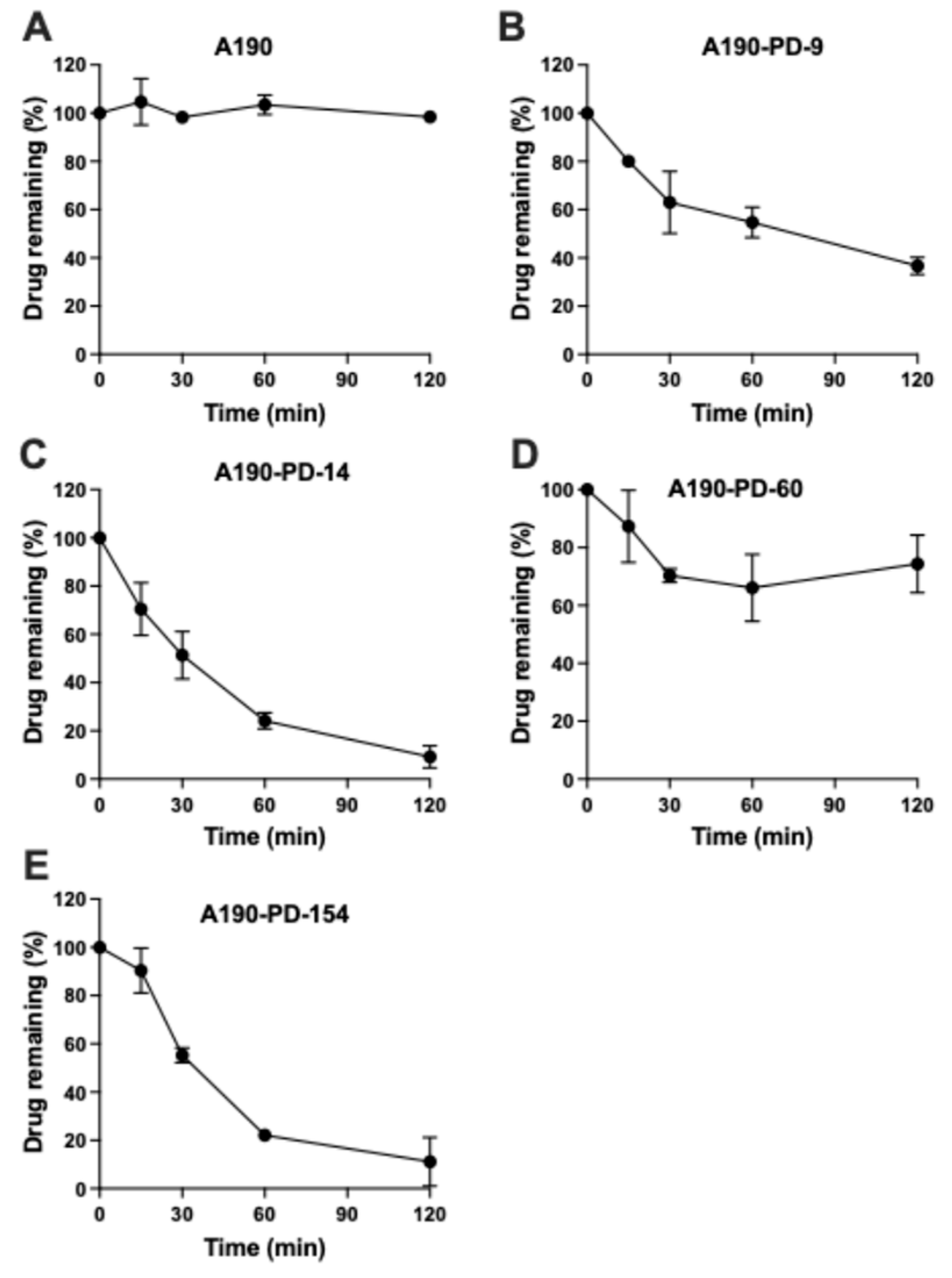
2.4. Chemical Hydrolysis Stability of Prodrugs
2.5. Preparation and Characterization of A190-PD-60 Microemulsion
2.5.1. Solubility Based Selection of Microemulsion Components
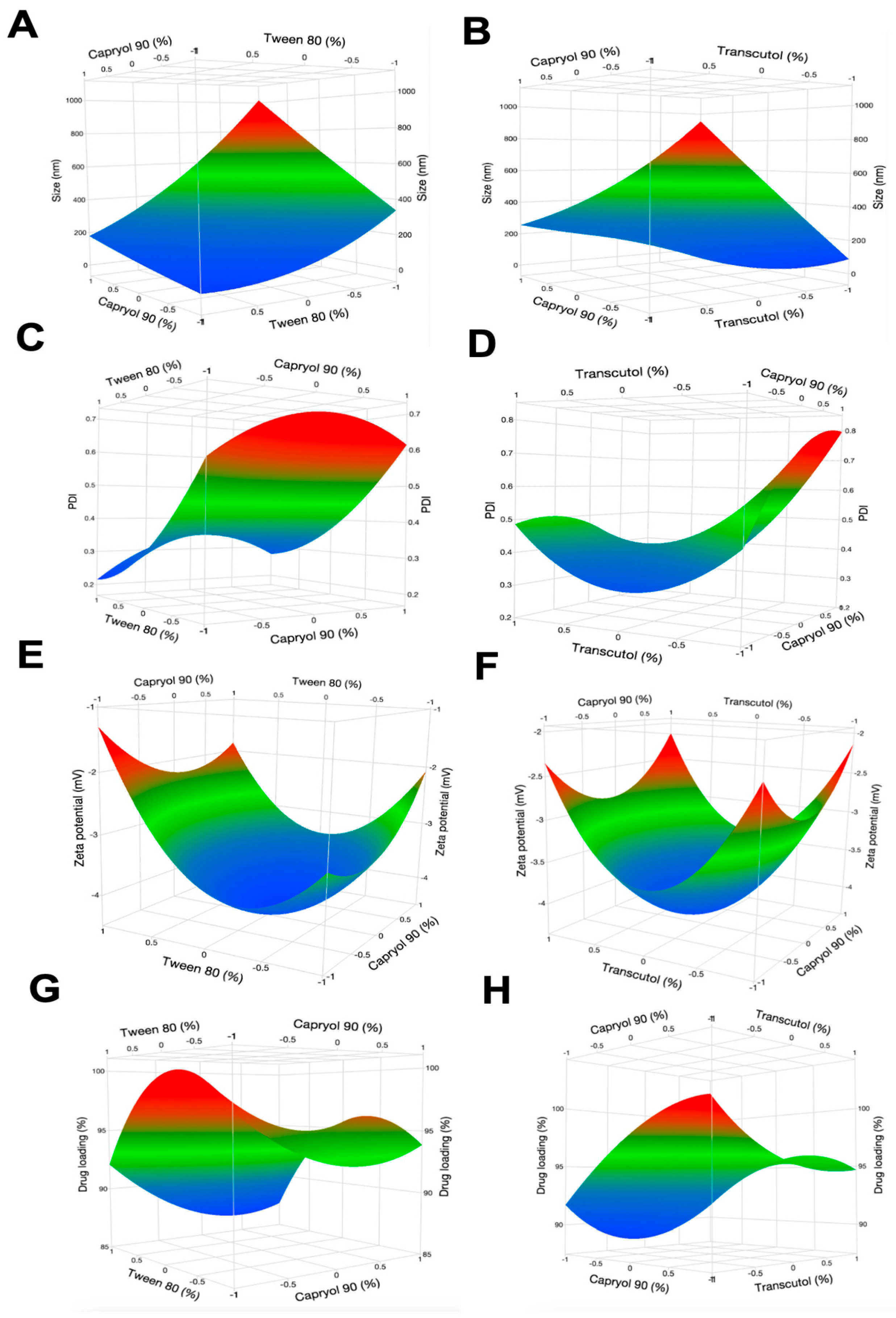
2.5.2. Formulation Optimization with Box-Behnken Experimental Design
2.5.3. Formulation of A190-PD-60-ME
2.6. In-Vitro Permeability Assay
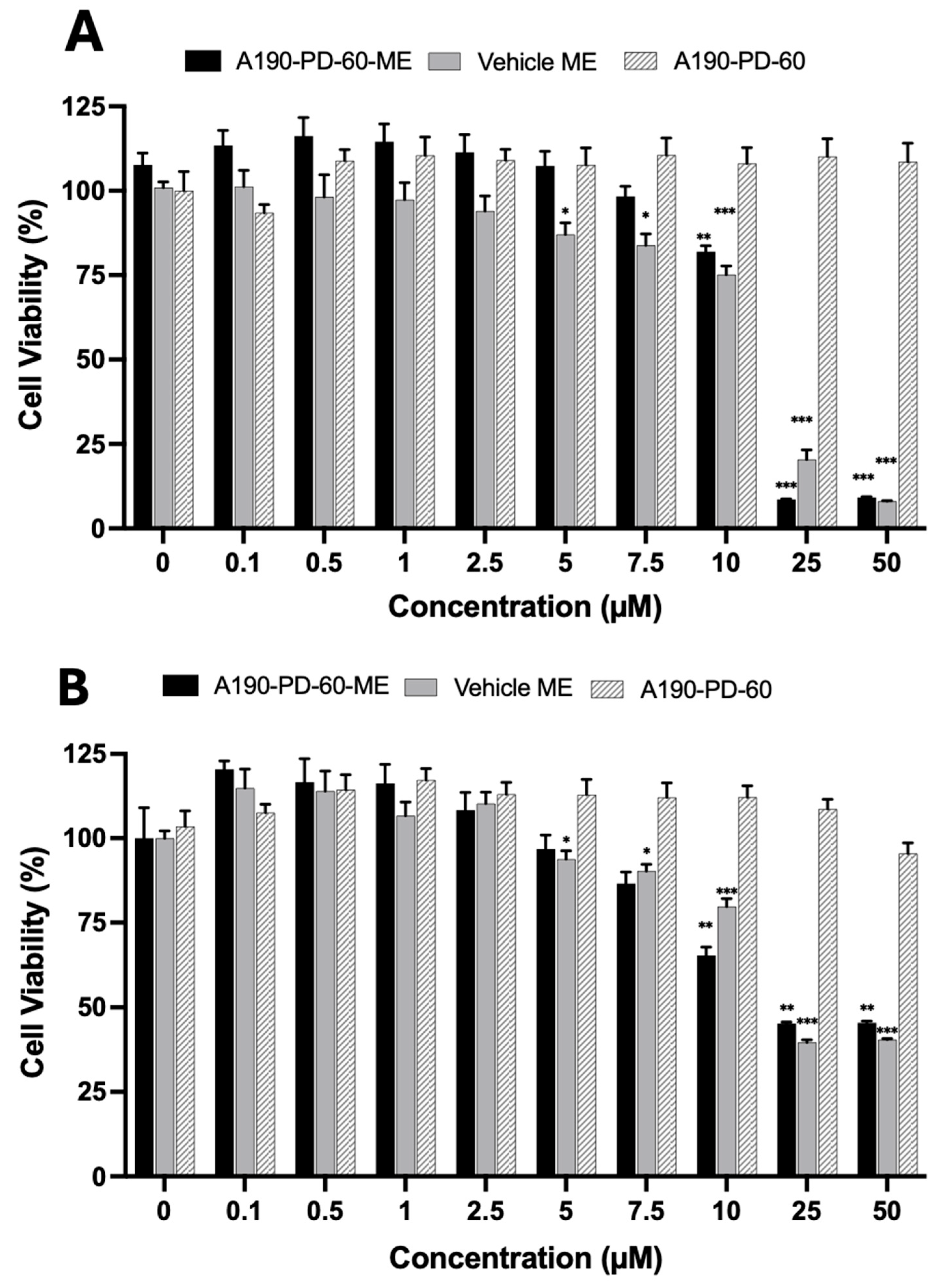
2.7. Cell Viability Studies
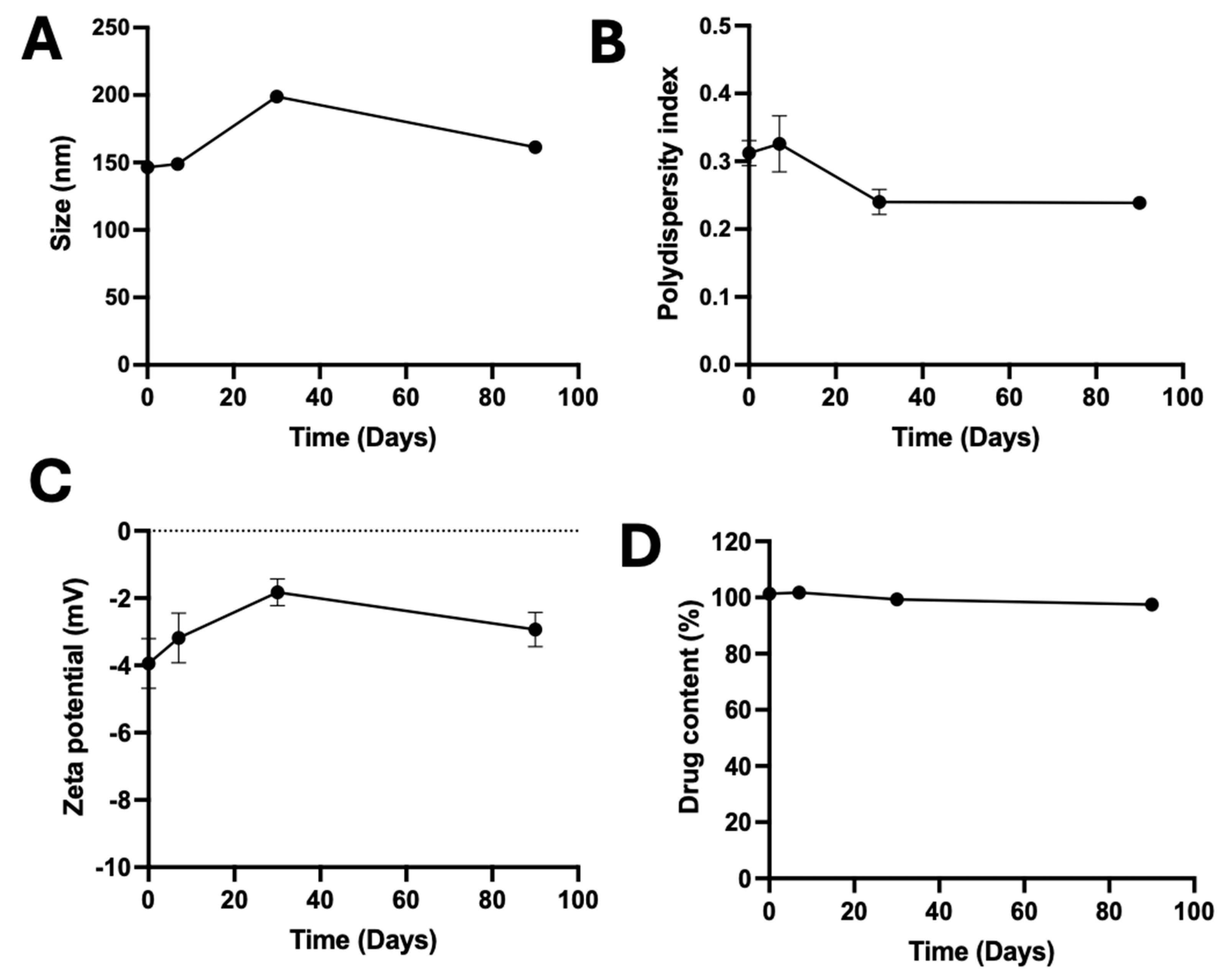
2.8. Stability Studies
2.9. Animals
2.10. In-Vivo Oral Pharmacokinetics of A190-PD-60-ME
2.11. Statistical Analysis
3. Results
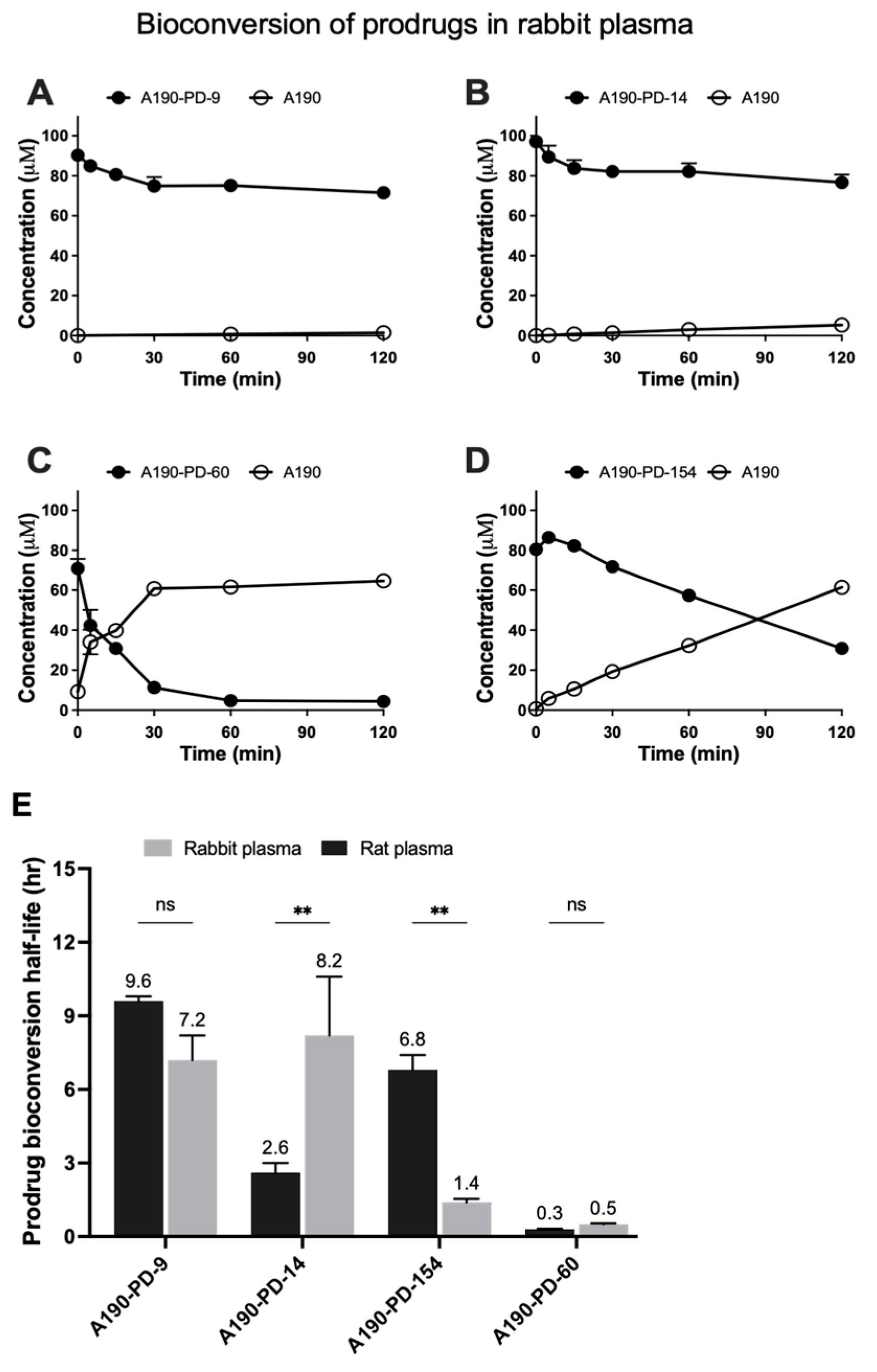
3.1. Prodrugs Plasma Bioconversion
3.2. Chemical Hydrolysis of Prodrugs
3.3. Formulation Optimization of A190-PD-60-ME
3.3.1. Solubility Analysis of Microemulsion Components
3.3.2. Box-Behnken Experimental Design
3.4. In-Vitro Parallel Artificial Membrane Permeability
3.5. Cell Cytotoxicity Analysis
3.6. Stability of A190-PD-60-ME
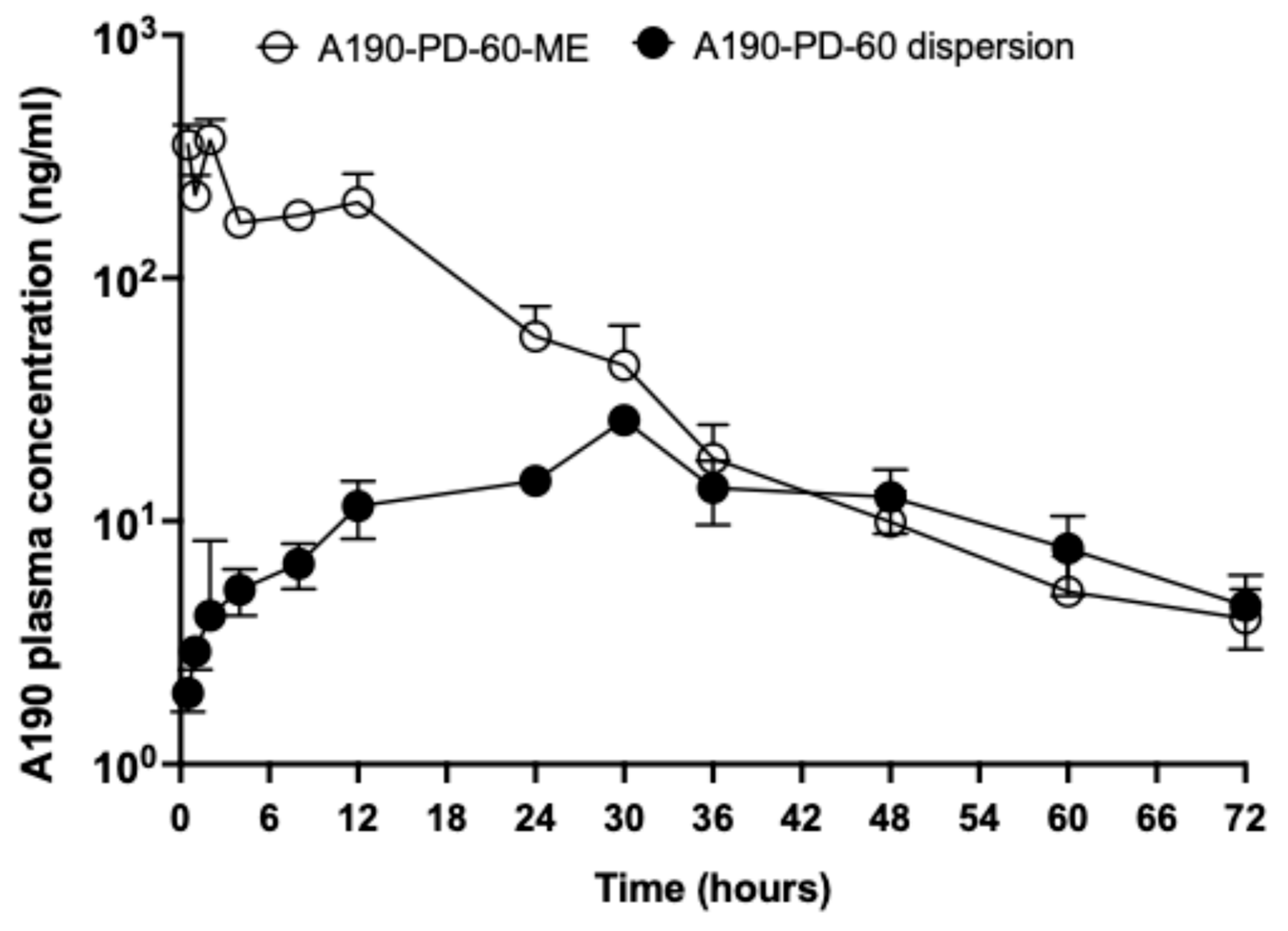
3.7. In-Vivo Oral Pharmacokinetic Study
4. Discussion
5. Conclusions and Future Directions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tomita, Y.; Lee, D.; Tsubota, K.; Kurihara, T. PPARalpha Agonist Oral Therapy in Diabetic Retinopathy. Biomedicines 2020, 8, 433. [Google Scholar] [CrossRef]
- Tyagi, S.; Gupta, P.; Saini, A.S.; Kaushal, C.; Sharma, S. The peroxisome proliferator-activated receptor: A family of nuclear receptors role in various diseases. J. Adv. Pharm. Technol. Res. 2011, 2, 236–240. [Google Scholar] [CrossRef] [PubMed]
- Clarke, S.D.; Thuillier, P.; Baillie, R.A.; Sha, X. Peroxisome proliferator-activated receptors: A family of lipid-activated transcription factors. Am. J. Clin. Nutr. 1999, 70, 566–571. [Google Scholar] [CrossRef] [PubMed]
- Grygiel-Gorniak, B. Peroxisome proliferator-activated receptors and their ligands: Nutritional and clinical implications—A review. Nutr. J. 2014, 13, 17. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Hu, Y.; Lin, M.; Jenkins, A.J.; Keech, A.C.; Mott, R.; Lyons, T.J.; Ma, J.X. Therapeutic effects of PPARalpha agonists on diabetic retinopathy in type 1 diabetes models. Diabetes 2013, 62, 261–272. [Google Scholar] [CrossRef]
- Knickelbein, J.E.; Abbott, A.B.; Chew, E.Y. Fenofibrate and Diabetic Retinopathy. Curr. Diab Rep. 2016, 16, 90. [Google Scholar] [CrossRef]
- Cheng, A.Y.; Leiter, L.A. PPAR-alpha: Therapeutic role in diabetes-related cardiovascular disease. Diabetes Obes. Metab. 2008, 10, 691–698. [Google Scholar] [CrossRef]
- Cooreman, M.P.; Vonghia, L.; Francque, S.M. MASLD/MASH and type 2 diabetes: Two sides of the same coin? From single PPAR to pan-PPAR agonists. Diabetes Res. Clin. Pract. 2024, 212, 111688. [Google Scholar] [CrossRef]
- Emami, F.; Hariri, A.; Matinfar, M.; Nematbakhsh, M. Fenofibrate-induced renal dysfunction, yes or no? J. Res. Med. Sci. 2020, 25, 39. [Google Scholar]
- Das Pradhan, A.; Glynn, R.J.; Fruchart, J.C.; MacFadyen, J.G.; Zaharris, E.S.; Everett, B.M.; Campbell, S.E.; Oshima, R.; Amarenco, P.; Blom, D.J.; et al. Triglyceride Lowering with Pemafibrate to Reduce Cardiovascular Risk. N. Engl. J. Med. 2022, 387, 1923–1934. [Google Scholar] [CrossRef]
- Fruchart, J.C.; Hermans, M.P.; Fruchart-Najib, J. Selective Peroxisome Proliferator-Activated Receptor Alpha Modulators (SPPARMalpha): New Opportunities to Reduce Residual Cardiovascular Risk in Chronic Kidney Disease? Curr. Atheroscler. Rep. 2020, 22, 43. [Google Scholar] [CrossRef] [PubMed]
- Balfour, J.A.; McTavish, D.; Heel, R.C. Fenofibrate. A review of its pharmacodynamic and pharmacokinetic properties and therapeutic use in dyslipidaemia. Drugs 1990, 40, 260–290. [Google Scholar] [CrossRef]
- He, Y.; Qin, M.Z.; Chen, Y.W. Liver injury caused by fenofibrate within 48 h after first administration: A case report. BMC Gastroenterol. 2021, 21, 298. [Google Scholar] [CrossRef] [PubMed]
- Dou, X.; Nath, D.; Shin, H.; Nurmemmedov, E.; Bourne, P.C.; Ma, J.X.; Duerfeldt, A.S. Evolution of a 4-Benzyloxy-benzylamino Chemotype to Provide Efficacious, Potent, and Isoform Selective PPARalpha Agonists as Leads for Retinal Disorders. J. Med. Chem. 2020, 63, 2854–2876. [Google Scholar] [CrossRef]
- Cui, Y.; Poudel, S.; Xu, N.; Zhou, K.; Cheng, R.; Liang, W.; Yuan, T.; Zhao, L.; Qin, C.; Stevens, K.G.; et al. Sustained release of a novel non-fibrate PPARalpha agonist from microparticles for neuroprotection in murine models of age-related macular degeneration. J. Control Release 2025, 380, 910–926. [Google Scholar] [CrossRef]
- Pangeni, R.; Poudel, S.; Herz, S.M.; Berkbigler, G.; Duerfeldt, A.S.; Damaj, M.I.; Xu, Q. New PPARalpha Agonist A190-Loaded Microemulsion for Chemotherapy-Induced Peripheral Neuropathy. Mol. Pharm. 2025, 22, 1641–1656. [Google Scholar] [CrossRef] [PubMed]
- Homayun, B.; Lin, X.; Choi, H.J. Challenges and Recent Progress in Oral Drug Delivery Systems for Biopharmaceuticals. Pharmaceutics 2019, 11, 129. [Google Scholar] [CrossRef]
- Lou, J.; Duan, H.; Qin, Q.; Teng, Z.; Gan, F.; Zhou, X.; Zhou, X. Advances in Oral Drug Delivery Systems: Challenges and Opportunities. Pharmaceutics 2023, 15, 484. [Google Scholar] [CrossRef]
- Kim, Y.H.; Kim, S.B.; Choi, S.H.; Nguyen, T.T.; Ahn, S.H.; Moon, K.S.; Cho, K.H.; Sim, T.Y.; Heo, E.J.; Kim, S.T.; et al. Development and Evaluation of Self-Microemulsifying Drug Delivery System for Improving Oral Absorption of Poorly Water-Soluble Olaparib. Pharmaceutics 2023, 15, 1669. [Google Scholar] [CrossRef]
- Prakash, S. Nano-based drug delivery system for therapeutics: A comprehensive review. Biomed. Phys. Eng. Express 2023, 9, 052002. [Google Scholar] [CrossRef]
- Silberstein, S.; Spierings, E.L.H.; Kunkel, T. Celecoxib Oral Solution and the Benefits of Self-Microemulsifying Drug Delivery Systems (SMEDDS) Technology: A Narrative Review. Pain. Ther. 2023, 12, 1109–1119. [Google Scholar] [CrossRef]
- Sharma, A.K.; Garg, T.; Goyal, A.K.; Rath, G. Role of microemuslsions in advanced drug delivery. Artif. Cells Nanomed. Biotechnol. 2016, 44, 1177–1185. [Google Scholar] [CrossRef]
- Ghosh, P.K.; Majithiya, R.J.; Umrethia, M.L.; Murthy, R.S. Design and development of microemulsion drug delivery system of acyclovir for improvement of oral bioavailability. AAPS PharmSciTech 2006, 7, 77. [Google Scholar] [CrossRef]
- Narang, A.S.; Delmarre, D.; Gao, D. Stable drug encapsulation in micelles and microemulsions. Int. J. Pharm. 2007, 345, 9–25. [Google Scholar] [CrossRef] [PubMed]
- Dash, R.P.; Tichy, T.; Veeravalli, V.; Lam, J.; Alt, J.; Wu, Y.; Tenora, L.; Majer, P.; Slusher, B.S.; Rais, R. Enhanced Oral Bioavailability of 2-(Phosphonomethyl)-pentanedioic Acid (2-PMPA) from its (5-Methyl-2-oxo-1,3-dioxol-4-yl)methyl (ODOL)-Based Prodrugs. Mol. Pharm. 2019, 16, 4292–4301. [Google Scholar] [CrossRef]
- Fedi, A.; Vitale, C.; Ponschin, G.; Ayehunie, S.; Fato, M.; Scaglione, S. In vitro models replicating the human intestinal epithelium for absorption and metabolism studies: A systematic review. J. Control Release 2021, 335, 247–268. [Google Scholar] [CrossRef]
- Zhang, Y.; Huo, M.; Zhou, J.; Xie, S. PKSolver: An add-in program for pharmacokinetic and pharmacodynamic data analysis in Microsoft Excel. Comput. Methods Programs Biomed. 2010, 99, 306–314. [Google Scholar] [CrossRef]
- Bahar, F.G.; Ohura, K.; Ogihara, T.; Imai, T. Species difference of esterase expression and hydrolase activity in plasma. J. Pharm. Sci. 2012, 101, 3979–3988. [Google Scholar] [CrossRef] [PubMed]
- Cook, C.S.; Karabatsos, P.J.; Schoenhard, G.L.; Karim, A. Species dependent esterase activities for hydrolysis of an anti-HIV prodrug glycovir and bioavailability of active SC-48334. Pharm. Res. 1995, 12, 1158–1164. [Google Scholar] [CrossRef]
- Zheng, Y.; Zi, Y.; Shi, C.; Gong, H.; Zhang, H.; Wang, X.; Zhong, J. Tween emulsifiers improved alginate-based dispersions and ionic crosslinked milli-sized capsules. NPJ Sci. Food 2023, 7, 33. [Google Scholar] [CrossRef] [PubMed]
- Xie, B.; Liu, Y.; Li, X.; Yang, P.; He, W. Solubilization techniques used for poorly water-soluble drugs. Acta Pharm. Sin. B 2024, 14, 4683–4716. [Google Scholar] [CrossRef]
- Ait-Touchente, Z.; Zine, N.; Jaffrezic-Renault, N.; Errachid, A.; Lebaz, N.; Fessi, H.; Elaissari, A. Exploring the Versatility of Microemulsions in Cutaneous Drug Delivery: Opportunities and Challenges. Nanomaterials 2023, 13, 1688. [Google Scholar] [CrossRef]
- Sarheed, O.; Dibi, M.; Ramesh, K. Studies on the Effect of Oil and Surfactant on the Formation of Alginate-Based O/W Lidocaine Nanocarriers Using Nanoemulsion Template. Pharmaceutics 2020, 12, 1223. [Google Scholar] [CrossRef]
- Som, I.; Bhatia, K.; Yasir, M. Status of surfactants as penetration enhancers in transdermal drug delivery. J. Pharm. Bioallied Sci. 2012, 4, 2–9. [Google Scholar] [CrossRef]
- Aguirre-Ramirez, M.; Silva-Jimenez, H.; Banat, I.M.; Diaz De Rienzo, M.A. Surfactants: Physicochemical interactions with biological macromolecules. Biotechnol. Lett. 2021, 43, 523–535. [Google Scholar] [CrossRef] [PubMed]
- De Stefani, C.; Vasarri, M.; Salvatici, M.C.; Grifoni, L.; Quintela, J.C.; Bilia, A.R.; Degl’Innocenti, D.; Bergonzi, M.C. Microemulsions Enhance the In Vitro Antioxidant Activity of Oleanolic Acid in RAW 264.7 Cells. Pharmaceutics 2022, 14, 2232. [Google Scholar] [CrossRef] [PubMed]
- Markovic, M.; Ben-Shabat, S.; Dahan, A. Prodrugs for Improved Drug Delivery: Lessons Learned from Recently Developed and Marketed Products. Pharmaceutics 2020, 12, 1031. [Google Scholar] [CrossRef] [PubMed]
- Bhalani, D.V.; Nutan, B.; Kumar, A.; Singh Chandel, A.K. Bioavailability Enhancement Techniques for Poorly Aqueous Soluble Drugs and Therapeutics. Biomedicines 2022, 10, 2055. [Google Scholar] [CrossRef]
- Ukai, H.; Iwasa, K.; Deguchi, T.; Morishita, M.; Katsumi, H.; Yamamoto, A. Enhanced Intestinal Absorption of Insulin by Capryol 90, a Novel Absorption Enhancer in Rats: Implications in Oral Insulin Delivery. Pharmaceutics 2020, 12, 462. [Google Scholar] [CrossRef]
- Tartaro, G.; Mateos, H.; Schirone, D.; Angelico, R.; Palazzo, G. Microemulsion Microstructure(s): A Tutorial Review. Nanomaterials 2020, 10, 1657. [Google Scholar] [CrossRef]
- Censi, R.; Martena, V.; Hoti, E.; Malaj, L.; Di Martino, P. Permeation and skin retention of quercetin from microemulsions containing Transcutol(R) P. Drug Dev. Ind. Pharm. 2012, 38, 1128–1133. [Google Scholar] [CrossRef]
- Virani, A.; Dholaria, N.; Matharoo, N.; Michniak-Kohn, B. A Study of Microemulsion Systems for Transdermal Delivery of Risperidone Using Penetration Enhancers. J. Pharm. Sci. 2023, 112, 3109–3119. [Google Scholar] [CrossRef]
- Irizarry, L.D.; Luu, T.H.; McKoy, J.M.; Samaras, A.T.; Fisher, M.J.; Carias, E.E.; Raisch, D.W.; Calhoun, E.A.; Bennett, C.L. Cremophor EL-containing paclitaxel-induced anaphylaxis: A call to action. Community Oncol. 2009, 6, 132–134. [Google Scholar] [CrossRef]
- Fu, Y.; Xiao, S.; Liu, S.; Chang, Y.; Ma, R.; Zhang, Z.; He, J. Atomistic Insights into the Droplet Size Evolution during Self-Microemulsification. Langmuir 2022, 38, 3129–3138. [Google Scholar] [CrossRef] [PubMed]
- Kumari, L.; Choudhari, Y.; Patel, P.; Gupta, G.D.; Singh, D.; Rosenholm, J.M.; Bansal, K.K.; Kurmi, B.D. Advancement in Solubilization Approaches: A Step towards Bioavailability Enhancement of Poorly Soluble Drugs. Life 2023, 13, 1099. [Google Scholar] [CrossRef] [PubMed]
- Chan, H.W.; Chow, S.; Zhang, X.; Kwok, P.C.L.; Chow, S.F. Role of Particle Size in Translational Research of Nanomedicines for Successful Drug Delivery: Discrepancies and Inadequacies. J. Pharm. Sci. 2023, 112, 2371–2384. [Google Scholar] [CrossRef]
- Ray, D.; Jangid, L.; Joshi, D.; Prakash, S.; Ojha, K.; Manor, O.; Mandal, A. Formulation of Polymer-Augmented Surfactant-Based Oil-Water Microemulsions for Application in Enhanced Oil Recovery. ACS Omega 2024, 9, 50024–50040. [Google Scholar] [CrossRef] [PubMed]
- Hippalgaonkar, K.; Adelli, G.R.; Hippalgaonkar, K.; Repka, M.A.; Majumdar, S. Indomethacin-loaded solid lipid nanoparticles for ocular delivery: Development, characterization, and in vitro evaluation. J. Ocul. Pharmacol. Ther. 2013, 29, 216–228. [Google Scholar] [CrossRef]
- Singh, G.; Pai, R.S. Trans-resveratrol self-nano-emulsifying drug delivery system (SNEDDS) with enhanced bioavailability potential: Optimization, pharmacokinetics and in situ single pass intestinal perfusion (SPIP) studies. Drug Deliv. 2015, 22, 522–530. [Google Scholar] [CrossRef] [PubMed]
- Subongkot, T. Development and mechanistic study of a microemulsion containing vitamin E TPGS for the enhancement of oral absorption of celecoxib. Int. J. Nanomedicine 2019, 14, 3087–3102. [Google Scholar] [CrossRef]
- Porter, C.J.; Trevaskis, N.L.; Charman, W.N. Lipids and lipid-based formulations: Optimizing the oral delivery of lipophilic drugs. Nat. Rev. Drug Discov. 2007, 6, 231–248. [Google Scholar] [CrossRef] [PubMed]
- Pouton, C.W. Lipid formulations for oral administration of drugs: Non-emulsifying, self-emulsifying and ‘self-microemulsifying’ drug delivery systems. Eur. J. Pharm. Sci. 2000, 11 (Suppl. 2), S93–S98. [Google Scholar] [CrossRef] [PubMed]
- Yanez, J.A.; Remsberg, C.M.; Sayre, C.L.; Forrest, M.L.; Davies, N.M. Flip-flop pharmacokinetics—Delivering a reversal of disposition: Challenges and opportunities during drug development. Ther. Deliv. 2011, 2, 643–672. [Google Scholar] [CrossRef]
- Artursson, P.; Karlsson, J. Correlation between oral drug absorption in humans and apparent drug permeability coefficients in human intestinal epithelial (Caco-2) cells. Biochem. Biophys. Res. Commun. 1991, 175, 880–885. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Independent Variables | Levels | ||
|---|---|---|---|
| Low (−1) | Medium (0) | High (+1) | |
| Factor 1 (X1): Oil (%) | 7.5 | 15 | 22.5 |
| Factor 2 (X2): Surfactant (%) | 5 | 10 | 20 |
| Factor 3 (X3): Co-surfactant (%) | 5 | 10 | 20 |
| Dependent variables | Constraints | ||
| Response 1 (Y1): Mean droplet size | Minimum | ||
| Response 2 (Y2): Polydispersity index | Minimum | ||
| Response 3 (Y3): Zeta potential | In range | ||
| Response 4 (Y4): Drug content | Maximum | ||
| Excipients | Solubility (mg/mL) |
|---|---|
| Oleic acid | 0.92 ± 0.04 |
| Capryol 90 | 4.45 ± 0.31 |
| Maisine oil | 0.32 ± 0.30 |
| Labrafil M | 6.58 ± 0.19 |
| Lauroglycol FCC | 4.59 ± 0.05 |
| Tween 80 | 9.17 ± 0.07 |
| Transcutol HP | 14.1 ± 0.14 |
| Polypropylene glycol | 3.79 ± 0.62 |
| Cremophor EL | 9.05 ± 0.04 |
| Peceol | 2.17 ± 0.07 |
| Run | Independent Variables | Responses | |||||
|---|---|---|---|---|---|---|---|
| X1 | X2 | X3 | Y1 | Y2 | Y3 | Y4 | |
| Capryol 90 (%v/v) | Tween 80 (%v/v) | Transcutol HP (%v/v) | Average Droplet Size (nm) | PDI | Zeta Potential (mV) | Drug Content (%) | |
| 1 | 7.5 | 5 | 10 | 323.6 ± 16.0 | 0.55 ± 0.03 | −3.2 ± 0.2 | 96.3 ± 4.2 |
| 2 | 7.5 | 20 | 10 | 135.5 ± 1.77 | 0.28 ±0.05 | −1.7 ± 0.3 | 90.12 ± 1.2 |
| 3 | 22.5 | 5 | 10 | 914.9 ± 84.3 | 0.45 ±0.35 | −2.09 ± 0.3 | 95.85 ± 1.8 |
| 4 | 22.5 | 20 | 10 | 191.3 ± 3.60 | 0.28 ± 0.016 | −1.67 ± 0.2 | 88.02 ± 3.9 |
| 5 | 15 | 5 | 5 | 807.4 ± 133 | 0.91 ± 0.08 | −1.79 ± 0.1 | 98.43 ± 1.6 |
| 6 | 15 | 5 | 20 | 832.2 ± 161.4 | 0.94 ± 0.07 | −1.86 ± 0.1 | 82.23 ± 2.3 |
| 7 | 15 | 20 | 5 | 143.6 ± 4.34 | 0.51 ±0.01 | −1.85 ± 0.22 | 70.9 ± 0.5 |
| 8 | 15 | 20 | 20 | 124.1 ± 1.34 | 0.32 ± 0.045 | −1.52 ± 0.3 | 101.4 ± 2.9 |
| 9 | 7.5 | 10 | 5 | 167.2 ± 5.25 | 0.36 ± 0.023 | −1.85 ± 0.2 | 93.4 ± 1.01 |
| 10 | 22.5 | 10 | 5 | 1114.0 ± 69.5 | 0.96 ± 0.05 | −1.83 ± 0.9 | 91.1 ± 0.74 |
| 11 | 7.5 | 10 | 20 | 118.8 ± 1.38 | 0.32 ± 0.004 | −1.93 ± 0.4 | 104.0 ± 0.9 |
| 12 | 22.5 | 10 | 20 | 176.5 ± 3.8 | 0.26 ± 0.20 | −2.67 ± 0.9 | 93.3 ± 4.5 |
| 13 | 15 | 10 | 10 | 311.9 ± 40.9 | 0.47 ± 0.08 | −2.63 ± 1.4 | 98.19 ± 1.3 |
| 14 | 15 | 10 | 10 | 233.9 ± 15.6 | 0.30 ± 0.015 | −3.14 ± 0.8 | 92.7 ± 2.1 |
| 15 | 15 | 10 | 10 | 330.6 ± 25.5 | 0.55 ± 0.01 | −4.2 ± 1.3 | 89.0 ± 1.8 |
| Variables | Y1 | Y2 | Y3 | Y4 |
|---|---|---|---|---|
| Average Droplet Size (nm) | PDI | Zeta Potential (mV) | Drug Content (%) | |
| Analysis of Variance p-Values | ||||
| X1 = % Oil (Capryol 90) | 0.025 * | 0.688 | 0.618 | 0.204 |
| X2 = % Surfactant (Tween 80) | 0.007 * | 0.061 | 0.047 * | 0.086 |
| X3 = % Co-surfactant (Transcutol HP) | 0.119 | 0.136 | 0.974 | 0.049 * |
| Sample | Effective Permeability Across Artificial Membrane (Pe, ×10−6, cm/s) |
|---|---|
| A190-PD-60 in PBS | BLD |
| A190-PD-60 dispersion | 0.1 ± 0.04 |
| A190-PD-60-ME | 10.8 ± 1.76 *** |
| Formulation | A190-PD-60 Dispersion | A190-PD-60-ME |
|---|---|---|
| Dose (mg/kg) | 20 | 20 |
| tmax (h) | 31.5 ±1.5 | 1.7 ± 0.3 |
| t1/2 (h) | 14.11 ± 1.94 | 9.32 ± 1.27 |
| Cmax (ng/mL) | 26.47 ± 0.55 | 438.85 ± 74.55 |
| AUC0→t (ng/mL × h) | 828.10 ± 102.86 | 4914.33 ± 525.79 |
| AUC0→inf (ng/mL × h) | 930.98 ±130.30 | 4974.52 ±521.22 |
| Relative Bioavailability (%) | 100 | 593 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Poudel, S.; Qin, C.; Pangeni, R.; Hu, Z.; Berkbigler, G.; Gunawardena, M.; Duerfeldt, A.S.; Xu, Q. Oral Pharmacokinetic Evaluation of a Microemulsion-Based Delivery System for Novel A190 Prodrugs. Biomolecules 2025, 15, 1101. https://doi.org/10.3390/biom15081101
Poudel S, Qin C, Pangeni R, Hu Z, Berkbigler G, Gunawardena M, Duerfeldt AS, Xu Q. Oral Pharmacokinetic Evaluation of a Microemulsion-Based Delivery System for Novel A190 Prodrugs. Biomolecules. 2025; 15(8):1101. https://doi.org/10.3390/biom15081101
Chicago/Turabian StylePoudel, Sagun, Chaolong Qin, Rudra Pangeni, Ziwei Hu, Grant Berkbigler, Madeline Gunawardena, Adam S. Duerfeldt, and Qingguo Xu. 2025. "Oral Pharmacokinetic Evaluation of a Microemulsion-Based Delivery System for Novel A190 Prodrugs" Biomolecules 15, no. 8: 1101. https://doi.org/10.3390/biom15081101
APA StylePoudel, S., Qin, C., Pangeni, R., Hu, Z., Berkbigler, G., Gunawardena, M., Duerfeldt, A. S., & Xu, Q. (2025). Oral Pharmacokinetic Evaluation of a Microemulsion-Based Delivery System for Novel A190 Prodrugs. Biomolecules, 15(8), 1101. https://doi.org/10.3390/biom15081101