

The Multifaceted Role of p53 in Cancer Molecular Biology: Insights for Precision Diagnosis and Therapeutic Breakthroughs

 and
and
Abstract
1. Introduction
2. Molecular Mechanisms of p53
2.1. Transcriptional Regulation
2.2. Post-Translational Modifications
2.3. Non-Transcriptional Functions
3. p53 in Tumor Microenvironment and Immune Modulation
3.1. Angiogenesis Regulation
3.2. Immune Surveillance
3.3. Inflammation Within the Tumor Microenvironment
4. p53 and Cancer Metabolism
4.1. Glucose Metabolism
4.1.1. Glycolysis
4.1.2. Gluconeogenesis
4.1.3. Tricarboxylic Acid Cycle
4.1.4. The Insulin–p53 Axis in Metabolic Dysfunction and Cancer Risk
4.2. Lipid Metabolism
4.3. Amino Acid Metabolism
4.4. Nucleotide Metabolism
4.5. Iron Metabolism
5. p53 in Cancer Stem Cells and Therapeutic Resistance
5.1. p53 and Cancer Stem Cell (CSC) Stemness Maintenance
5.2. p53 Mutations and Resistance Mechanisms
6. Diagnostic and Therapeutic Implications
6.1. Diagnostic Biomarker Potential
6.2. Therapeutic Strategy
6.2.1. Restoring Wild-Type Activity
6.2.2. Degradation of Mutp53
6.2.3. Anti-MDM2 Agents
6.2.4. Targeting p53-Immune Axis for Cancer Therapy
6.2.5. Disrupting Protein Interactions
6.3. p53-Independent Therapeutic Strategy
7. Challenges and Future Directions
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AKT | Protein kinase B |
| AML | Acute myeloid leukemia |
| APCs | Antigen-presenting cells |
| BCCS | Breast cancer-corrected survival |
| CIITA | Class II transactivator |
| CAFs | Cancer-associated fibroblasts |
| CMF | Cyclophosphamide, methotrexate, and 5-fluorouracil |
| COX-2 | Cyclooxygenase-2 |
| CSCs | Cancer stem cells |
| CTL | Cytotoxic T lymphocytes |
| DAMPs | Damage-associated molecular patterns |
| DDR | DNA damage response |
| DN | Dominant-negative |
| ECM | Extracellular matrix |
| EMT | Epithelial–mesenchymal transition |
| FISH | Fluorescence in situ hybridization |
| GBM | Glioblastoma |
| GOF | Gain-of-function |
| GSH | Glutathione |
| HCC | Hepatocellular carcinoma |
| HIF-1α | Hypoxia-inducible factor-1α |
| IHC | Immunohistochemistry |
| IMPDH | Inosine monophosphate dehydrogenase |
| LOF | Loss-of-function |
| MDR1 | Multidrug resistance protein 1 |
| MDM2 | Mouse double minute 2 homolog |
| MHC-I | Major histocompatibility complex class I |
| miRNA/miR | MicroRNA |
| MOMP | Mitochondrial outer membrane permeabilization |
| MVA | Mevalonate |
| NAFLD | Non-alcoholic fatty liver disease |
| NK | Natural killer |
| NGS | Next-generation sequencing |
| OS | Overall survival |
| OXPHOS | Oxidative phosphorylation |
| PDH | Pyruvate dehydrogenase |
| PD-L1 | Programmed death-ligand 1 |
| PI3K | Phosphoinositide 3-kinase |
| PUMA | p53 upregulated modulator of apoptosis |
| RFS | Recurrence-free survival |
| ROS | Reactive oxygen species |
| SIRT1 | Sirtuin 1 |
| TAP | Transporter associated with antigen processing |
| TAD | Terminal transactivation domain |
| TCA | Tricarboxylic acid cycle |
| TIGAR | TP53-induced glycolysis and apoptosis regulator |
| TME | Tumor microenvironment |
| TRAIL | Tumor necrosis factor-related apoptosis-inducing ligand |
| VEGF | Vascular endothelial growth factor |
| WT | Wild-type |
| MHC-II | Major Histocompatibility Complex II |
| IL-6 | Interleukin-6 |
| IL-8 | Interleukin-8 |
| NF-κB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| PTMs | Post-translational modifications |
| SREBP | Sterol regulatory element-binding protein |
| STING | Stimulator of interferon genes |
| TNF-α | Tumor necrosis factor alpha |
References
- Hafner, A.; Bulyk, M.L.; Jambhekar, A.; Lahav, G. The multiple mechanisms that regulate p53 activity and cell fate. Nat. Rev. Mol. Cell Biol. 2019, 20, 199–210. [Google Scholar] [CrossRef]
- Roszkowska, K.A.; Gizinski, S.; Sady, M.; Gajewski, Z.; Olszewski, M.B. Gain-of-Function Mutations in p53 in Cancer Invasiveness and Metastasis. Int. J. Mol. Sci. 2020, 21, 1334. [Google Scholar] [CrossRef]
- Zhang, C.; Liu, J.; Xu, D.; Zhang, T.; Hu, W.; Feng, Z. Gain-of-function mutant p53 in cancer progression and therapy. J. Mol. Cell Biol. 2020, 12, 674–687. [Google Scholar] [CrossRef]
- Temaj, G.; Chichiarelli, S.; Telkoparan-Akillilar, P.; Saha, S.; Nuhii, N.; Hadziselimovic, R.; Saso, L. P53: A key player in diverse cellular processes including nuclear stress and ribosome biogenesis, highlighting potential therapeutic compounds. Biochem. Pharmacol. 2024, 226, 116332. [Google Scholar] [CrossRef]
- Cordani, M.; Garufi, A.; Benedetti, R.; Tafani, M.; Aventaggiato, M.; D’Orazi, G.; Cirone, M. Recent Advances on Mutant p53: Unveiling Novel Oncogenic Roles, Degradation Pathways, and Therapeutic Interventions. Biomolecules 2024, 14, 649. [Google Scholar] [CrossRef]
- Carlsen, L.; Zhang, S.; Tian, X.; De La Cruz, A.; George, A.; Arnoff, T.E.; El-Deiry, W.S. The role of p53 in anti-tumor immunity and response to immunotherapy. Front. Mol. Biosci. 2023, 10, 1148389. [Google Scholar] [CrossRef] [PubMed]
- Xiong, S.; Chachad, D.; Zhang, Y.; Gencel-Augusto, J.; Sirito, M.; Pant, V.; Yang, P.; Sun, C.; Chau, G.; Qi, Y.; et al. Differential Gain-of-Function Activity of Three p53 Hotspot Mutants In Vivo. Cancer Res. 2022, 82, 1926–1936. [Google Scholar] [CrossRef] [PubMed]
- Ding, Z.; Wang, N.; Ji, N.; Chen, Z.S. Proteomics technologies for cancer liquid biopsies. Mol. Cancer 2022, 21, 53. [Google Scholar] [CrossRef] [PubMed]
- Iwatate, Y.; Hoshino, I.; Yokota, H.; Ishige, F.; Itami, M.; Mori, Y.; Chiba, S.; Arimitsu, H.; Yanagibashi, H.; Nagase, H.; et al. Radiogenomics for predicting p53 status, PD-L1 expression, and prognosis with machine learning in pancreatic cancer. Br. J. Cancer 2020, 123, 1253–1261. [Google Scholar] [CrossRef]
- Hassin, O.; Oren, M. Drugging p53 in cancer: One protein, many targets. Nat. Rev. Drug Discov. 2023, 22, 127–144. [Google Scholar] [CrossRef]
- Sammons, M.A.; Nguyen, T.T.; McDade, S.S.; Fischer, M. Tumor suppressor p53: From engaging DNA to target gene regulation. Nucleic Acids Res. 2020, 48, 8848–8869. [Google Scholar] [CrossRef]
- Fischer, M. Gene regulation by the tumor suppressor p53—The omics era. Biochim. Biophys. Acta Rev. Cancer 2024, 1879, 189111. [Google Scholar] [CrossRef]
- Kearns, S.; Lurz, R.; Orlova, E.V.; Okorokov, A.L. Two p53 tetramers bind one consensus DNA response element. Nucleic Acids Res. 2016, 44, 6185–6199. [Google Scholar] [CrossRef]
- Monti, P.; Menichini, P.; Speciale, A.; Cutrona, G.; Fais, F.; Taiana, E.; Neri, A.; Bomben, R.; Gentile, M.; Gattei, V.; et al. Heterogeneity of TP53 Mutations and P53 Protein Residual Function in Cancer: Does It Matter? Front. Oncol. 2020, 10, 593383. [Google Scholar] [CrossRef] [PubMed]
- Coronel, L.; Riege, K.; Schwab, K.; Forste, S.; Hackes, D.; Semerau, L.; Bernhart, S.H.; Siebert, R.; Hoffmann, S.; Fischer, M. Transcription factor RFX7 governs a tumor suppressor network in response to p53 and stress. Nucleic Acids Res. 2021, 49, 7437–7456. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Su, Z.; Tavana, O.; Gu, W. Understanding the complexity of p53 in a new era of tumor suppression. Cancer Cell 2024, 42, 946–967. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Thorne, R.F.; Zhang, X.D.; Wu, M.; Liu, L. Non-coding RNAs, guardians of the p53 galaxy. Semin. Cancer Biol. 2021, 75, 72–83. [Google Scholar] [CrossRef]
- Boutelle, A.M.; Attardi, L.D. p53 and Tumor Suppression: It Takes a Network. Trends Cell Biol. 2021, 31, 298–310. [Google Scholar] [CrossRef]
- Sullivan, K.D.; Galbraith, M.D.; Andrysik, Z.; Espinosa, J.M. Mechanisms of transcriptional regulation by p53. Cell Death Differ. 2018, 25, 133–143. [Google Scholar] [CrossRef]
- Yang, J.; Jin, A.; Han, J.; Chen, X.; Zheng, J.; Zhang, Y. MDMX Recruits UbcH5c to Facilitate MDM2 E3 Ligase Activity and Subsequent p53 Degradation In Vivo. Cancer Res. 2021, 81, 898–909. [Google Scholar] [CrossRef]
- Sanford, J.D.; Yang, J.; Han, J.; Tollini, L.A.; Jin, A.; Zhang, Y. MDMX is essential for the regulation of p53 protein levels in the absence of a functional MDM2 C-terminal tail. BMC Mol. Cell Biol. 2021, 22, 46. [Google Scholar] [CrossRef]
- Zhang, Y.; Xiong, Y. Control of p53 ubiquitination and nuclear export by MDM2 and ARF. Cell Growth Differ. 2001, 12, 175–186. [Google Scholar] [PubMed]
- Mayo, L.D.; Donner, D.B. A phosphatidylinositol 3-kinase/Akt pathway promotes translocation of Mdm2 from the cytoplasm to the nucleus. Proc. Natl. Acad. Sci. USA 2001, 98, 11598–11603. [Google Scholar] [CrossRef]
- He, F.; Borcherds, W.; Song, T.; Wei, X.; Das, M.; Chen, L.; Daughdrill, G.W.; Chen, J. Interaction between p53 N terminus and core domain regulates specific and nonspecific DNA binding. Proc. Natl. Acad. Sci. USA 2019, 116, 8859–8868. [Google Scholar] [CrossRef] [PubMed]
- Magnussen, H.M.; Ahmed, S.F.; Sibbet, G.J.; Hristova, V.A.; Nomura, K.; Hock, A.K.; Archibald, L.J.; Jamieson, A.G.; Fushman, D.; Vousden, K.H.; et al. Structural basis for DNA damage-induced phosphoregulation of MDM2 RING domain. Nat. Commun. 2020, 11, 2094. [Google Scholar] [CrossRef] [PubMed]
- Dornan, D.; Shimizu, H.; Perkins, N.D.; Hupp, T.R. DNA-dependent acetylation of p53 by the transcription coactivator p300. J. Biol. Chem. 2003, 278, 13431–13441. [Google Scholar] [CrossRef]
- Datta, D.; Navalkar, A.; Sakunthala, A.; Paul, A.; Patel, K.; Masurkar, S.; Gadhe, L.; Manna, S.; Bhattacharyya, A.; Sengupta, S.; et al. Nucleo-cytoplasmic environment modulates spatiotemporal p53 phase separation. Sci. Adv. 2024, 10, eads0427. [Google Scholar] [CrossRef]
- Petronilho, E.C.; Pedrote, M.M.; Marques, M.A.; Passos, Y.M.; Mota, M.F.; Jakobus, B.; de Sousa, G.D.S.; Pereira da Costa, F.; Felix, A.L.; Ferretti, G.D.S.; et al. Phase separation of p53 precedes aggregation and is affected by oncogenic mutations and ligands. Chem. Sci. 2021, 12, 7334–7349. [Google Scholar] [CrossRef]
- Menendez, D.; Inga, A.; Resnick, M.A. The expanding universe of p53 targets. Nat. Rev. Cancer 2009, 9, 724–737. [Google Scholar] [CrossRef]
- Thayer, K.M.; Stetson, S.; Caballero, F.; Chiu, C.; Han, I.S.M. Navigating the complexity of p53-DNA binding: Implications for cancer therapy. Biophys. Rev. 2024, 16, 479–496. [Google Scholar] [CrossRef]
- Luzhin, A.; Rajan, P.; Safina, A.; Leonova, K.; Stablewski, A.; Wang, J.; Robinson, D.; Isaeva, N.; Kantidze, O.; Gurova, K. Comparison of cell response to chromatin and DNA damage. Nucleic Acids Res. 2023, 51, 11836–11855. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Xu, D.; Trefts, E.; Lv, M.; Inuzuka, H.; Song, G.; Liu, M.; Lu, J.; Liu, J.; Chu, C.; et al. Metabolic orchestration of cell death by AMPK-mediated phosphorylation of RIPK1. Science 2023, 380, 1372–1380. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.K. Emerging roles of long non-coding RNAs in the p53 network. Rna Biol. 2020, 17, 1648–1656. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Guo, M.; Wei, H.; Chen, Y. Targeting p53 pathways: Mechanisms, structures, and advances in therapy. Signal Transduct. Target. Ther. 2023, 8, 92. [Google Scholar] [CrossRef]
- Fischer, M.; Riege, K.; Hoffmann, S. The landscape of human p53-regulated long non-coding RNAs reveals critical host gene co-regulation. Mol. Oncol. 2023, 17, 1263–1279. [Google Scholar] [CrossRef]
- Oo, J.A.; Warwick, T.; Palfi, K.; Lam, F.; McNicoll, F.; Prieto-Garcia, C.; Gunther, S.; Cao, C.; Zhou, Y.; Gavrilov, A.A.; et al. Long non-coding RNAs direct the SWI/SNF complex to cell type-specific enhancers. Nat. Commun. 2025, 16, 131. [Google Scholar] [CrossRef]
- Segeren, H.A.; van Liere, E.A.; Riemers, F.M.; de Bruin, A.; Westendorp, B. Oncogenic RAS sensitizes cells to drug-induced replication stress via transcriptional silencing of P53. Oncogene 2022, 41, 2719–2733. [Google Scholar] [CrossRef]
- Zhou, Y.; Hu, T.; Zeng, H.; Lin, L.; Xie, H.; Lin, R.; Huang, M. Naringenin Inhibits Ferroptosis in Renal Tubular Epithelial Cells of Diabetic Nephropathy Through SIRT1/FOXO3a Signaling Pathway. Drug Dev. Res. 2025, 86, e70044. [Google Scholar] [CrossRef]
- Zhong, S.; Wang, Z.; Yang, J.; Jiang, D.; Wang, K. Ferroptosis-related oxaliplatin resistance in multiple cancers: Potential roles and therapeutic Implications. Heliyon 2024, 10, e37613. [Google Scholar] [CrossRef]
- Kume, S.; Haneda, M.; Kanasaki, K.; Sugimoto, T.; Araki, S.; Isono, M.; Isshiki, K.; Uzu, T.; Kashiwagi, A.; Koya, D. Silent information regulator 2 (SIRT1) attenuates oxidative stress-induced mesangial cell apoptosis via p53 deacetylation. Free Radic. Biol. Med. 2006, 40, 2175–2182. [Google Scholar] [CrossRef]
- Zeng, Y.; He, Y.; Wang, L.; Xu, H.; Zhang, Q.; Wang, Y.; Zhang, J.; Wang, L. Dihydroquercetin improves experimental acute liver failure by targeting ferroptosis and mitochondria-mediated apoptosis through the SIRT1/p53 axis. Phytomedicine 2024, 128, 155533. [Google Scholar] [CrossRef]
- Dancy, B.M.; Cole, P.A. Protein lysine acetylation by p300/CBP. Chem. Rev. 2015, 115, 2419–2452. [Google Scholar] [CrossRef]
- Zhang, H.; Xu, J.; Long, Y.; Maimaitijiang, A.; Su, Z.; Li, W.; Li, J. Unraveling the Guardian: p53’s Multifaceted Role in the DNA Damage Response and Tumor Treatment Strategies. Int. J. Mol. Sci. 2024, 25, 12928. [Google Scholar] [CrossRef]
- Kloet, S.L.; Whiting, J.L.; Gafken, P.; Ranish, J.; Wang, E.H. Phosphorylation-dependent regulation of cyclin D1 and cyclin A gene transcription by TFIID subunits TAF1 and TAF7. Mol. Cell Biol. 2012, 32, 3358–3369. [Google Scholar] [CrossRef]
- Li, H.H.; Li, A.G.; Sheppard, H.M.; Liu, X. Phosphorylation on Thr-55 by TAF1 mediates degradation of p53: A role for TAF1 in cell G1 progression. Mol. Cell 2004, 13, 867–878. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Lin, J.C.; Piluso, L.G.; Dhahbi, J.M.; Bobadilla, S.; Spindler, S.R.; Liu, X. Phosphorylation of p53 by TAF1 inactivates p53-dependent transcription in the DNA damage response. Mol. Cell 2014, 53, 63–74. [Google Scholar] [CrossRef] [PubMed]
- Allende-Vega, N.; Saville, M.K.; Meek, D.W. Transcription factor TAFII250 promotes Mdm2-dependent turnover of p53. Oncogene 2007, 26, 4234–4242. [Google Scholar] [CrossRef] [PubMed]
- Enari, M.; Matsushima-Hibiya, Y.; Miyazaki, M.; Otomo, R. Studies of ATM Kinase Activity Using Engineered ATM Sensitive to ATP Analogues (ATM-AS). Methods Mol. Biol. 2017, 1599, 145–156. [Google Scholar] [CrossRef]
- Koo, N.; Sharma, A.K.; Narayan, S. Therapeutics Targeting p53-MDM2 Interaction to Induce Cancer Cell Death. Int. J. Mol. Sci. 2022, 23, 5005. [Google Scholar] [CrossRef]
- Wen, W.; Zhang, W.L.; Tan, R.; Zhong, T.T.; Zhang, M.R.; Fang, X.S. Progress in deciphering the role of p53 in diffuse large B-cell lymphoma: Mechanisms and therapeutic targets. Am. J. Cancer Res. 2024, 14, 3280–3293. [Google Scholar] [CrossRef]
- Yee-Lin, V.; Pooi-Fong, W.; Soo-Beng, A.K. Nutlin-3, A p53-Mdm2 Antagonist for Nasopharyngeal Carcinoma Treatment. Mini Rev. Med. Chem. 2018, 18, 173–183. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Wang, X.; Liu, J.; Zheng, J.; Liu, Y.; Li, Y.; Su, F.; Ou, W.; Wang, R. Nutlin-3 reverses the epithelial-mesenchymal transition in gemcitabine-resistant hepatocellular carcinoma cells. Oncol. Rep. 2016, 36, 1325–1332. [Google Scholar] [CrossRef]
- Pan, S.; Chen, R. Pathological implication of protein post-translational modifications in cancer. Mol. Asp. Med. 2022, 86, 101097. [Google Scholar] [CrossRef]
- Wen, J.; Wang, D. Deciphering the PTM codes of the tumor suppressor p53. J. Mol. Cell Biol. 2022, 13, 774–785. [Google Scholar] [CrossRef]
- Bruno, P.S.; Arshad, A.; Gogu, M.-R.; Waterman, N.; Flack, R.; Dunn, K.; Darie, C.C.; Neagu, A.-N. Post-Translational Modifications of Proteins Orchestrate All Hallmarks of Cancer. Life 2025, 15, 126. [Google Scholar] [CrossRef]
- Ho, T.; Tan, B.X.; Lane, D. How the Other Half Lives: What p53 Does When It Is Not Being a Transcription Factor. Int. J. Mol. Sci. 2019, 21, 13. [Google Scholar] [CrossRef]
- Zheng, J.H.; Viacava Follis, A.; Kriwacki, R.W.; Moldoveanu, T. Discoveries and controversies in BCL-2 protein-mediated apoptosis. FEBS J. 2016, 283, 2690–2700. [Google Scholar] [CrossRef]
- Schweighofer, S.V.; Jans, D.C.; Keller-Findeisen, J.; Folmeg, A.; Ilgen, P.; Bates, M.; Jakobs, S. Endogenous BAX and BAK form mosaic rings of variable size and composition on apoptotic mitochondria. Cell Death Differ. 2024, 31, 469–478. [Google Scholar] [CrossRef]
- Han, J.; Goldstein, L.A.; Hou, W.; Gastman, B.R.; Rabinowich, H. Regulation of mitochondrial apoptotic events by p53-mediated disruption of complexes between antiapoptotic Bcl-2 members and Bim. J. Biol. Chem. 2010, 285, 22473–22483. [Google Scholar] [CrossRef] [PubMed]
- Speidel, D.; Helmbold, H.; Deppert, W. Dissection of transcriptional and non-transcriptional p53 activities in the response to genotoxic stress. Oncogene 2006, 25, 940–953. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Thomas, H.R.; Li, Z.; Yeo, N.C.F.; Scott, H.E.; Dang, N.; Hossain, M.I.; Andrabi, S.A.; Parant, J.M. Puma, noxa, p53, and p63 differentially mediate stress pathway induced apoptosis. Cell Death Dis. 2021, 12, 659. [Google Scholar] [CrossRef] [PubMed]
- Castrogiovanni, C.; Waterschoot, B.; De Backer, O.; Dumont, P. Serine 392 phosphorylation modulates p53 mitochondrial translocation and transcription-independent apoptosis. Cell Death Differ. 2018, 25, 190–203. [Google Scholar] [CrossRef] [PubMed]
- Zavileyskiy, L.; Bunik, V. Regulation of p53 Function by Formation of Non-Nuclear Heterologous Protein Complexes. Biomolecules 2022, 12, 327. [Google Scholar] [CrossRef] [PubMed]
- Krois, A.S.; Park, S.; Martinez-Yamout, M.A.; Dyson, H.J.; Wright, P.E. Mapping Interactions of the Intrinsically Disordered C-Terminal Regions of Tetrameric p53 by Segmental Isotope Labeling and NMR. Biochemistry 2022, 61, 2709–2719. [Google Scholar] [CrossRef]
- Chipuk, J.E.; Green, D.R. PUMA cooperates with direct activator proteins to promote mitochondrial outer membrane permeabilization and apoptosis. Cell Cycle 2009, 8, 2692–2696. [Google Scholar] [CrossRef]
- Lacroix, M.; Riscal, R.; Arena, G.; Linares, L.K.; Le Cam, L. Metabolic functions of the tumor suppressor p53: Implications in normal physiology, metabolic disorders, and cancer. Mol. Metab. 2020, 33, 2–22. [Google Scholar] [CrossRef]
- Liu, Y.; Gu, W. p53 in ferroptosis regulation: The new weapon for the old guardian. Cell Death Differ. 2022, 29, 895–910. [Google Scholar] [CrossRef]
- Gupta, S.; Cassel, S.L.; Sutterwala, F.S.; Dagvadorj, J. Regulation of the NLRP3 inflammasome by autophagy and mitophagy. Immunol. Rev. 2025, 329, e13410. [Google Scholar] [CrossRef]
- Pathak, A.; Pal, A.K.; Roy, S.; Nandave, M.; Jain, K. Role of Angiogenesis and Its Biomarkers in Development of Targeted Tumor Therapies. Stem Cells Int. 2024, 2024, 9077926. [Google Scholar] [CrossRef]
- Men, H.; Cai, H.; Cheng, Q.; Zhou, W.; Wang, X.; Huang, S.; Zheng, Y.; Cai, L. The regulatory roles of p53 in cardiovascular health and disease. Cell Mol. Life Sci. 2021, 78, 2001–2018. [Google Scholar] [CrossRef]
- Griggio, V.; Vitale, C.; Todaro, M.; Riganti, C.; Kopecka, J.; Salvetti, C.; Bomben, R.; Bo, M.D.; Magliulo, D.; Rossi, D.; et al. HIF-1alpha is over-expressed in leukemic cells from TP53-disrupted patients and is a promising therapeutic target in chronic lymphocytic leukemia. Haematologica 2020, 105, 1042–1054. [Google Scholar] [CrossRef]
- Babaei, G.; Aliarab, A.; Asghari Vostakolaei, M.; Hotelchi, M.; Neisari, R.; Gholizadeh-Ghaleh Aziz, S.; Bazl, M.R. Crosslink between p53 and metastasis: Focus on epithelial-mesenchymal transition, cancer stem cell, angiogenesis, autophagy, and anoikis. Mol. Biol. Rep. 2021, 48, 7545–7557. [Google Scholar] [CrossRef]
- Li, A.M.; Boichard, A.; Kurzrock, R. Mutated TP53 is a marker of increased VEGF expression: Analysis of 7,525 pan-cancer tissues. Cancer Biol. Ther. 2020, 21, 95–100. [Google Scholar] [CrossRef]
- Janani, S.K.; Dhanabal, S.P.; Sureshkumar, R. Guardian of genome on the tract: Wild type p53-mdm2 complex inhibition in healing the breast cancer. Gene 2021, 786, 145616. [Google Scholar] [CrossRef]
- Wu, K.K. Cytoguardin: A Tryptophan Metabolite against Cancer Growth and Metastasis. Int. J. Mol. Sci. 2021, 22, 4490. [Google Scholar] [CrossRef]
- Nakayama, M.; Saito, H.; Murakami, K.; Oshima, H.; Oshima, M. Missense Mutant p53 Transactivates Wnt/beta-Catenin Signaling in Neighboring p53-Destabilized Cells through the COX-2/PGE2 Pathway. Cancer Res. Commun. 2025, 5, 13–23. [Google Scholar] [CrossRef]
- Dameron, K.M.; Volpert, O.V.; Tainsky, M.A.; Bouck, N. Control of angiogenesis in fibroblasts by p53 regulation of thrombospondin-1. Science 1994, 265, 1582–1584. [Google Scholar] [CrossRef] [PubMed]
- Ou, A.; Zhao, X.; Lu, Z. The potential roles of p53 signaling reactivation in pancreatic cancer therapy. Biochim. Biophys. Acta Rev. Cancer 2022, 1877, 188662. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Wang, Q.; Mao, Y.; Gao, W.; Duan, S. Targeting the p53 signaling pathway in cancers: Molecular mechanisms and clinical studies. MedComm 2023, 4, e288. [Google Scholar] [CrossRef] [PubMed]
- Wilson, K.; Shiuan, E.; Brantley-Sieders, D.M. Oncogenic functions and therapeutic targeting of EphA2 in cancer. Oncogene 2021, 40, 2483–2495. [Google Scholar] [CrossRef]
- Huang, Z.; Bao, S.D. Roles of main pro- and anti-angiogenic factors in tumor angiogenesis. World J. Gastroenterol. 2004, 10, 463–470. [Google Scholar] [CrossRef]
- Blagih, J.; Buck, M.D.; Vousden, K.H.; Lennon-Duménil, A.-M. p53, cancer and the immune response. J. Cell Sci. 2020, 133. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Niu, D.; Lai, L.; Ren, E.C. p53 increases MHC class I expression by upregulating the endoplasmic reticulum aminopeptidase ERAP1. Nat. Commun. 2013, 4, 2359. [Google Scholar] [CrossRef] [PubMed]
- Agupitan, A.D.; Neeson, P.; Williams, S.; Howitt, J.; Haupt, S.; Haupt, Y. P53: A Guardian of Immunity Becomes Its Saboteur through Mutation. Int. J. Mol. Sci. 2020, 21, 3452. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Chen, Q.; Liu, Q.; Luo, C. Master regulator: p53’s pivotal role in steering NK-cell tumor patrol. Front. Immunol. 2024, 15, 1428653. [Google Scholar] [CrossRef]
- Shi, D.; Jiang, P. A Different Facet of p53 Function: Regulation of Immunity and Inflammation During Tumor Development. Front. Cell Dev. Biol. 2021, 9, 762651. [Google Scholar] [CrossRef]
- Pan, W.; Chai, B.; Li, L.; Lu, Z.; Ma, Z. p53/MicroRNA-34 axis in cancer and beyond. Heliyon 2023, 9, e15155. [Google Scholar] [CrossRef]
- Liu, Y.; Gu, W. The complexity of p53-mediated metabolic regulation in tumor suppression. Semin. Cancer Biol. 2022, 85, 4–32. [Google Scholar] [CrossRef]
- Pavlakis, E.; Stiewe, T. p53’s Extended Reach: The Mutant p53 Secretome. Biomolecules 2020, 10, 307. [Google Scholar] [CrossRef]
- Zheng, Y.; Yao, Y.; Ge, T.; Ge, S.; Jia, R.; Song, X.; Zhuang, A. Amino acid metabolism reprogramming: Shedding new light on T cell anti-tumor immunity. J. Exp. Clin. Cancer Res. 2023, 42, 291. [Google Scholar] [CrossRef]
- Liu, N.; Jiang, X.; Guo, L.; Zhang, C.; Jiang, M.; Sun, Z.; Zhang, Y.; Mi, W.; Li, J.; Fu, Y.; et al. Mutant p53 achieved Gain-of-Function by promoting tumor growth and immune escape through PHLPP2/AKT/PD-L1 pathway. Int. J. Biol. Sci. 2022, 18, 2419–2438. [Google Scholar] [CrossRef]
- Kennedy, M.C.; Lowe, S.W. Mutant p53: It’s not all one and the same. Cell Death Differ. 2022, 29, 983–987. [Google Scholar] [CrossRef]
- Chauhan, S.; Jaiswal, S.; Jakhmola, V.; Singh, B.; Bhattacharya, S.; Garg, M.; Sengupta, S. Potential role of p53 deregulation in modulating immune responses in human malignancies: A paradigm to develop immunotherapy. Cancer Lett. 2024, 588, 216766. [Google Scholar] [CrossRef]
- Diepstraten, S.T.; Yuan, Y.; La Marca, J.E.; Young, S.; Chang, C.; Whelan, L.; Ross, A.M.; Fischer, K.C.; Pomilio, G.; Morris, R.; et al. Putting the STING back into BH3-mimetic drugs for TP53-mutant blood cancers. Cancer Cell 2024, 42, 850–868. [Google Scholar] [CrossRef] [PubMed]
- Asl, E.R.; Rostamzadeh, D.; Duijf, P.H.G.; Mafi, S.; Mansoori, B.; Barati, S.; Cho, W.C.; Mansoori, B. Mutant P53 in the formation and progression of the tumor microenvironment: Friend or foe. Life Sci. 2023, 315, 121361. [Google Scholar] [CrossRef] [PubMed]
- Bejarano, L.; Jordao, M.J.C.; Joyce, J.A. Therapeutic Targeting of the Tumor Microenvironment. Cancer Discov. 2021, 11, 933–959. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Wu, L.; Yan, G.; Chen, Y.; Zhou, M.; Wu, Y.; Li, Y. Inflammation and tumor progression: Signaling pathways and targeted intervention. Signal Transduct. Target. Ther. 2021, 6, 263. [Google Scholar] [CrossRef]
- Uehara, I.; Tanaka, N. Role of p53 in the Regulation of the Inflammatory Tumor Microenvironment and Tumor Suppression. Cancers 2018, 10, 219. [Google Scholar] [CrossRef]
- Carra, G.; Lingua, M.F.; Maffeo, B.; Taulli, R.; Morotti, A. P53 vs NF-kappaB: The role of nuclear factor-kappa B in the regulation of p53 activity and vice versa. Cell Mol. Life Sci. 2020, 77, 4449–4458. [Google Scholar] [CrossRef]
- Li, R.; Mukherjee, M.B.; Lin, J. Coordinated Regulation of Myeloid-Derived Suppressor Cells by Cytokines and Chemokines. Cancers 2022, 14, 1236. [Google Scholar] [CrossRef]
- Gudkov, A.V.; Komarova, E.A. p53 and the Carcinogenicity of Chronic Inflammation. Cold Spring Harb. Perspect. Med. 2016, 6, a026161. [Google Scholar] [CrossRef]
- Capaci, V.; Mantovani, F.; Del Sal, G. Amplifying Tumor-Stroma Communication: An Emerging Oncogenic Function of Mutant p53. Front. Oncol. 2020, 10, 614230. [Google Scholar] [CrossRef] [PubMed]
- Pei, L.; Liu, Y.; Liu, L.; Gao, S.; Gao, X.; Feng, Y.; Sun, Z.; Zhang, Y.; Wang, C. Roles of cancer-associated fibroblasts (CAFs) in anti- PD-1/PD-L1 immunotherapy for solid cancers. Mol. Cancer 2023, 22, 29. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Cai, Q.; Chen, Y.; Shi, T.; Liu, W.; Mao, L.; Deng, B.; Ying, Z.; Gao, Y.; Luo, H.; et al. CAFs shape myeloid-derived suppressor cells to promote stemness of intrahepatic cholangiocarcinoma through 5-lipoxygenase. Hepatology 2022, 75, 28–42. [Google Scholar] [CrossRef] [PubMed]
- Maddalena, M.; Mallel, G.; Nataraj, N.B.; Shreberk-Shaked, M.; Hassin, O.; Mukherjee, S.; Arandkar, S.; Rotkopf, R.; Kapsack, A.; Lambiase, G.; et al. TP53 missense mutations in PDAC are associated with enhanced fibrosis and an immunosuppressive microenvironment. Proc. Natl. Acad. Sci. USA 2021, 118, e2025631118. [Google Scholar] [CrossRef]
- Finetti, F.; Paradisi, L.; Bernardi, C.; Pannini, M.; Trabalzini, L. Cooperation between Prostaglandin E2 and Epidermal Growth Factor Receptor in Cancer Progression: A Dual Target for Cancer Therapy. Cancers 2023, 15, 2374. [Google Scholar] [CrossRef]
- Levine, A.J. P53 and The Immune Response: 40 Years of Exploration-A Plan for the Future. Int. J. Mol. Sci. 2020, 21, 541. [Google Scholar] [CrossRef]
- Blagih, J.; Zani, F.; Chakravarty, P.; Hennequart, M.; Pilley, S.; Hobor, S.; Hock, A.K.; Walton, J.B.; Morton, J.P.; Gronroos, E.; et al. Cancer-Specific Loss of p53 Leads to a Modulation of Myeloid and T Cell Responses. Cell Rep. 2020, 30, 481–496.e6. [Google Scholar] [CrossRef]
- D’Orazi, G.; Cordani, M.; Cirone, M. Oncogenic pathways activated by pro-inflammatory cytokines promote mutant p53 stability: Clue for novel anticancer therapies. Cell Mol. Life Sci. 2021, 78, 1853–1860. [Google Scholar] [CrossRef]
- Tian, K.; Xu, W.; Chen, M.; Deng, F. miR-155 promotes Th17 differentiation by targeting FOXP3 to aggravate inflammation in MRSA pneumonia. Cytokine 2024, 180, 156662. [Google Scholar] [CrossRef]
- Ma, R.; Su, H.; Jiao, K.; Liu, J. Association Between IL-17 and Chronic Obstructive Pulmonary Disease: A Systematic Review and Meta-Analysis. Int. J. Chron. Obstruct Pulmon Dis. 2023, 18, 1681–1690. [Google Scholar] [CrossRef]
- Mayoux, M.; Roller, A.; Pulko, V.; Sammicheli, S.; Chen, S.; Sum, E.; Jost, C.; Fransen, M.F.; Buser, R.B.; Kowanetz, M.; et al. Dendritic cells dictate responses to PD-L1 blockade cancer immunotherapy. Sci. Transl. Med. 2020, 12, eaav7431. [Google Scholar] [CrossRef]
- Marozzi, M.; Parnigoni, A.; Negri, A.; Viola, M.; Vigetti, D.; Passi, A.; Karousou, E.; Rizzi, F. Inflammation, Extracellular Matrix Remodeling, and Proteostasis in Tumor Microenvironment. Int. J. Mol. Sci. 2021, 22, 8102. [Google Scholar] [CrossRef] [PubMed]
- Madan, E.; Parker, T.M.; Pelham, C.J.; Palma, A.M.; Peixoto, M.L.; Nagane, M.; Chandaria, A.; Tomas, A.R.; Canas-Marques, R.; Henriques, V.; et al. HIF-transcribed p53 chaperones HIF-1alpha. Nucleic Acids Res. 2019, 47, 10212–10234. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhang, C.; Hu, W.; Feng, Z. Tumor suppressor p53 and its mutants in cancer metabolism. Cancer Lett. 2015, 356, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.X.; Choi, S.Y.C.; Niu, X.; Kang, N.; Xue, H.; Killam, J.; Wang, Y. Lactic Acid and an Acidic Tumor Microenvironment suppress Anticancer Immunity. Int. J. Mol. Sci. 2020, 21, 8363. [Google Scholar] [CrossRef]
- Chambers, C.R.; Ritchie, S.; Pereira, B.A.; Timpson, P. Overcoming the senescence-associated secretory phenotype (SASP): A complex mechanism of resistance in the treatment of cancer. Mol. Oncol. 2021, 15, 3242–3255. [Google Scholar] [CrossRef]
- Alvarado-Ortiz, E.; de la Cruz-Lopez, K.G.; Becerril-Rico, J.; Sarabia-Sanchez, M.A.; Ortiz-Sanchez, E.; Garcia-Carranca, A. Mutant p53 Gain-of-Function: Role in Cancer Development, Progression, and Therapeutic Approaches. Front. Cell Dev. Biol. 2020, 8, 607670. [Google Scholar] [CrossRef]
- Mao, Y.; Jiang, P. The crisscross between p53 and metabolism in cancer. Acta Biochim. Et Biophys. Sin. 2023, 55, 914–922. [Google Scholar] [CrossRef]
- Ling, Z.N.; Jiang, Y.F.; Ru, J.N.; Lu, J.H.; Ding, B.; Wu, J. Amino acid metabolism in health and disease. Signal Transduct. Target. Ther. 2023, 8, 345. [Google Scholar] [CrossRef]
- Zhang, J.; Chen, X. p53 tumor suppressor and iron homeostasis. FEBS J. 2019, 286, 620–629. [Google Scholar] [CrossRef]
- Koo, K.Y.; Moon, K.; Song, H.S.; Lee, M.S. Metabolic regulation by p53: Implications for cancer therapy. Mol. Cells 2025, 48, 100198. [Google Scholar] [CrossRef]
- Chen, H.; Zhang, L.F.; Zhang, L.; Miao, Y.; Xi, Y.; Liu, M.F.; Zhang, M.; Li, B. CircANKRD17 promotes glycolysis by inhibiting miR-143 in breast cancer cells. J. Cell Physiol. 2023, 238, 2765–2777. [Google Scholar] [CrossRef]
- Liao, M.; Yao, D.; Wu, L.; Luo, C.; Wang, Z.; Zhang, J.; Liu, B. Targeting the Warburg effect: A revisited perspective from molecular mechanisms to traditional and innovative therapeutic strategies in cancer. Acta Pharm. Sin. B 2024, 14, 953–1008. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Li, W.; Qiu, L.; Zhang, X.; Zhang, L.; Miyagishi, M.; Zhao, H.; Wu, S.; Kasim, V. The p52-ZER6/G6PD axis alters aerobic glycolysis and promotes tumor progression by activating the pentose phosphate pathway. Oncogenesis 2023, 12, 17. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Wei, M.; Li, W.; Zhao, H.; Kasim, V.; Wu, S. PBX3 promotes pentose phosphate pathway and colorectal cancer progression by enhancing G6PD expression. Int. J. Biol. Sci. 2023, 19, 4525–4538. [Google Scholar] [CrossRef] [PubMed]
- Pliszka, M.; Szablewski, L. Glucose Transporters as a Target for Anticancer Therapy. Cancers 2021, 13, 4184. [Google Scholar] [CrossRef]
- Fu, J.; Yu, S.; Zhao, X.; Zhang, C.; Shen, L.; Liu, Y.; Yu, H. Inhibition of TIGAR Increases Exogenous p53 and Cisplatin Combination Sensitivity in Lung Cancer Cells by Regulating Glycolytic Flux. Int. J. Mol. Sci. 2022, 23, 16034. [Google Scholar] [CrossRef]
- Wittke, C.I.; Cheung, E.C.; Athineos, D.; Clements, N.; Butler, L.; Hughes, M.; Morrison, V.; Watt, D.M.; Blyth, K.; Vousden, K.H.; et al. p53 and TIGAR promote redox control to protect against metabolic dysfunction-associated steatohepatitis. JHEP Rep. 2025, 7, 101397. [Google Scholar] [CrossRef]
- Abbaszadeh, Z.; Cesmeli, S.; Biray Avci, C. Crucial players in glycolysis: Cancer progress. Gene 2020, 726, 144158. [Google Scholar] [CrossRef]
- Abdel-Wahab, A.F.; Mahmoud, W.; Al-Harizy, R.M. Targeting glucose metabolism to suppress cancer progression: Prospective of anti-glycolytic cancer therapy. Pharmacol. Res. 2019, 150, 104511. [Google Scholar] [CrossRef]
- Su, Y.; Luo, Y.; Zhang, P.; Lin, H.; Pu, W.; Zhang, H.; Wang, H.; Hao, Y.; Xiao, Y.; Zhang, X.; et al. Glucose-induced CRL4(COP1)-p53 axis amplifies glycometabolism to drive tumorigenesis. Mol. Cell 2023, 83, 2316–2331.e7. [Google Scholar] [CrossRef] [PubMed]
- Chang, X.; Liu, X.; Wang, H.; Yang, X.; Gu, Y. Glycolysis in the progression of pancreatic cancer. Am. J. Cancer Res. 2022, 12, 861–872. [Google Scholar] [PubMed]
- Xu, Y.; Chen, W.; Liang, J.; Zeng, X.; Ji, K.; Zhou, J.; Liao, S.; Wu, J.; Xing, K.; He, Z.; et al. The miR-1185-2-3p-GOLPH3L pathway promotes glucose metabolism in breast cancer by stabilizing p53-induced SERPINE1. J. Exp. Clin. Cancer Res. 2021, 40, 47. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Xiong, C.; Wang, C.; Deng, J.; Zuo, Z.; Wu, H.; Xiong, J.; Wu, X.; Lu, H.; Hao, Q.; et al. p53-responsive CMBL reprograms glucose metabolism and suppresses cancer development by destabilizing phosphofructokinase PFKP. Cell Rep. 2023, 42, 113426. [Google Scholar] [CrossRef]
- Chao, C.H.; Wang, C.Y.; Wang, C.H.; Chen, T.W.; Hsu, H.Y.; Huang, H.W.; Li, C.W.; Mai, R.T. Mutant p53 Attenuates Oxidative Phosphorylation and Facilitates Cancer Stemness through Downregulating miR-200c-PCK2 Axis in Basal-Like Breast Cancer. Mol. Cancer Res. 2021, 19, 1900–1916. [Google Scholar] [CrossRef]
- Tighanimine, K.; Nabuco Leva Ferreira Freitas, J.A.; Nemazanyy, I.; Bankole, A.; Benarroch-Popivker, D.; Brodesser, S.; Dore, G.; Robinson, L.; Benit, P.; Ladraa, S.; et al. A homoeostatic switch causing glycerol-3-phosphate and phosphoethanolamine accumulation triggers senescence by rewiring lipid metabolism. Nat. Metab. 2024, 6, 323–342. [Google Scholar] [CrossRef]
- Zhao, J.; Zhou, X.; Chen, B.; Lu, M.; Wang, G.; Elumalai, N.; Tian, C.; Zhang, J.; Liu, Y.; Chen, Z.; et al. p53 promotes peroxisomal fatty acid beta-oxidation to repress purine biosynthesis and mediate tumor suppression. Cell Death Dis. 2023, 14, 87. [Google Scholar] [CrossRef]
- Wu, T.; Qu, Y.; Xu, S.; Wang, Y.; Liu, X.; Ma, D. SIRT6: A potential therapeutic target for diabetic cardiomyopathy. Faseb J. 2023, 37, e23099. [Google Scholar] [CrossRef]
- Jiang, Y.; Han, Q.; Zhao, H.; Zhang, J. The Mechanisms of HBV-Induced Hepatocellular Carcinoma. J. Hepatocell. Carcinoma 2021, 8, 435–450. [Google Scholar] [CrossRef]
- Abukwaik, R.; Vera-Siguenza, E.; Tennant, D.; Spill, F. p53 Orchestrates Cancer Metabolism: Unveiling Strategies to Reverse the Warburg Effect. Bull. Math. Biol. 2024, 86, 124. [Google Scholar] [CrossRef]
- Han, C.W.; Jeong, M.S.; Jang, S.B. Influence of the interaction between p53 and ZNF568 on mitochondrial oxidative phosphorylation. Int. J. Biol. Macromol. 2024, 275, 133314. [Google Scholar] [CrossRef]
- Nagano, H.; Hashimoto, N.; Nakayama, A.; Suzuki, S.; Miyabayashi, Y.; Yamato, A.; Higuchi, S.; Fujimoto, M.; Sakuma, I.; Beppu, M.; et al. p53-inducible DPYSL4 associates with mitochondrial supercomplexes and regulates energy metabolism in adipocytes and cancer cells. Proc. Natl. Acad. Sci. USA 2018, 115, 8370–8375. [Google Scholar] [CrossRef]
- Bergeaud, M.; Mathieu, L.; Guillaume, A.; Moll, U.M.; Mignotte, B.; Le Floch, N.; Vayssiere, J.L.; Rincheval, V. Mitochondrial p53 mediates a transcription-independent regulation of cell respiration and interacts with the mitochondrial F(1)F0-ATP synthase. Cell Cycle 2013, 12, 2781–2793. [Google Scholar] [CrossRef]
- Guo, M.; Zhuang, Y.; Wu, Y.; Zhang, C.; Cheng, X.; Xu, D.; Zhang, Z. The cell fate regulator DACH1 modulates ferroptosis through affecting P53/SLC25A37 signaling in fibrotic disease. Hepatol. Commun. 2024, 8, e0396. [Google Scholar] [CrossRef]
- Polonsky, K.S. The past 200 years in diabetes. N. Engl. J. Med. 2012, 367, 1332–1340. [Google Scholar] [CrossRef]
- Vigneri, R.; Sciacca, L.; Vigneri, P. Rethinking the Relationship between Insulin and Cancer. Trends Endocrinol. Metab. 2020, 31, 551–560. [Google Scholar] [CrossRef]
- Strycharz, J.; Drzewoski, J.; Szemraj, J.; Sliwinska, A. Is p53 Involved in Tissue-Specific Insulin Resistance Formation? Oxid. Med. Cell Longev. 2017, 2017, 9270549. [Google Scholar] [CrossRef] [PubMed]
- Yan, Z.; Miao, X.; Zhang, B.; Xie, J. p53 as a double-edged sword in the progression of non-alcoholic fatty liver disease. Life Sci. 2018, 215, 64–72. [Google Scholar] [CrossRef] [PubMed]
- Kung, C.P.; Murphy, M.E. The role of the p53 tumor suppressor in metabolism and diabetes. J. Endocrinol. 2016, 231, R61–R75. [Google Scholar] [CrossRef] [PubMed]
- Yahagi, N.; Shimano, H.; Matsuzaka, T.; Najima, Y.; Sekiya, M.; Nakagawa, Y.; Ide, T.; Tomita, S.; Okazaki, H.; Tamura, Y.; et al. p53 Activation in adipocytes of obese mice. J. Biol. Chem. 2003, 278, 25395–25400. [Google Scholar] [CrossRef]
- Feng, T.; Zhang, H.; Zhou, Y.; Zhu, Y.; Shi, S.; Li, K.; Lin, P.; Chen, J. Roles of posttranslational modifications in lipid metabolism and cancer progression. Biomark. Res. 2024, 12, 141. [Google Scholar] [CrossRef]
- Cruz-Gil, S.; Fernandez, L.P.; Sanchez-Martinez, R.; Gomez de Cedron, M.; Ramirez de Molina, A. Non-Coding and Regulatory RNAs as Epigenetic Remodelers of Fatty Acid Homeostasis in Cancer. Cancers 2020, 12, 2890. [Google Scholar] [CrossRef] [PubMed]
- Sanford, J.D.; Franklin, D.; Grois, G.A.; Jin, A.; Zhang, Y. Carnitine o-octanoyltransferase is a p53 target that promotes oxidative metabolism and cell survival following nutrient starvation. J. Biol. Chem. 2023, 299, 104908. [Google Scholar] [CrossRef] [PubMed]
- Koundouros, N.; Poulogiannis, G. Reprogramming of fatty acid metabolism in cancer. Br. J. Cancer 2020, 122, 4–22. [Google Scholar] [CrossRef] [PubMed]
- Nie, G.; Chen, S.; Song, Q.; Zou, D.; Li, M.; Tang, X.; Deng, Y.; Huang, B.; Yang, M.; Lv, G.; et al. DHX33 mediates p53 to regulate mevalonate pathway gene transcription in human cancers. Biochim. Biophys. Acta Gen. Subj. 2024, 1868, 130547. [Google Scholar] [CrossRef]
- Chu, P.Y.; Tzeng, Y.T.; Tsui, K.H.; Chu, C.Y.; Li, C.J. Downregulation of ATP binding cassette subfamily a member 10 acts as a prognostic factor associated with immune infiltration in breast cancer. Aging 2022, 14, 2252–2267. [Google Scholar] [CrossRef]
- Moon, S.H.; Huang, C.H.; Houlihan, S.L.; Regunath, K.; Freed-Pastor, W.A.; Morris, J.P.t.; Tschaharganeh, D.F.; Kastenhuber, E.R.; Barsotti, A.M.; Culp-Hill, R.; et al. p53 Represses the Mevalonate Pathway to Mediate Tumor Suppression. Cell 2019, 176, 564–580.e19. [Google Scholar] [CrossRef]
- Nakayama, A.; Yokoyama, M.; Nagano, H.; Hashimoto, N.; Yamagata, K.; Murata, K.; Tanaka, T. Mechanism of Mutant p53 Using Three-Dimensional Culture on Breast Cancer Malignant Phenotype via SREBP-Dependent Cholesterol Synthesis Pathway. J. Endocr. Soc. 2021, 5, A1026. [Google Scholar] [CrossRef]
- Yu, H.; Li, M.; He, R.; Fang, P.; Wang, Q.; Yi, Y.; Wang, F.; Zhou, L.; Zhang, Y.; Chen, A.; et al. Major Vault Protein Promotes Hepatocellular Carcinoma Through Targeting Interferon Regulatory Factor 2 and Decreasing p53 Activity. Hepatology 2020, 72, 518–534. [Google Scholar] [CrossRef]
- Azar, S.; Udi, S.; Drori, A.; Hadar, R.; Nemirovski, A.; Vemuri, K.V.; Miller, M.; Sherill-Rofe, D.; Arad, Y.; Gur-Wahnon, D.; et al. Reversal of diet-induced hepatic steatosis by peripheral CB1 receptor blockade in mice is p53/miRNA-22/SIRT1/PPARalpha dependent. Mol. Metab. 2020, 42, 101087. [Google Scholar] [CrossRef]
- Pawlicka, K.; Henek, T.; Uhrik, L.; Hernychova, L.; Padariya, M.; Faktor, J.; Makowiec, S.; Vojtesek, B.; Goodlett, D.; Hupp, T.; et al. Misincorporations of amino acids in p53 in human cells at artificially constructed termination codons in the presence of the aminoglycoside Gentamicin. Front. Genet. 2024, 15, 1407375. [Google Scholar] [CrossRef]
- Sniegowski, T.; Korac, K.; Bhutia, Y.D.; Ganapathy, V. SLC6A14 and SLC38A5 Drive the Glutaminolysis and Serine-Glycine-One-Carbon Pathways in Cancer. Pharmaceuticals 2021, 14, 216. [Google Scholar] [CrossRef]
- Tran, A.P.; Tralie, C.J.; Reyes, J.; Moosmuller, C.; Belkhatir, Z.; Kevrekidis, I.G.; Levine, A.J.; Deasy, J.O.; Tannenbaum, A.R. Long-term p21 and p53 dynamics regulate the frequency of mitosis events and cell cycle arrest following radiation damage. Cell Death Differ. 2023, 30, 660–672. [Google Scholar] [CrossRef]
- Humpton, T.J.; Hock, A.K.; Maddocks, O.D.K.; Vousden, K.H. p53-mediated adaptation to serine starvation is retained by a common tumour-derived mutant. Cancer Metab. 2018, 6, 18. [Google Scholar] [CrossRef]
- Li, Y.; Jiang, B.; Wang, R.; Wang, J.; Li, Y.; Bao, Y. Synergistic effects of tanshinone IIA and andrographolide on the apoptosis of cancer cells via crosstalk between p53 and reactive oxygen species pathways. Pharmacol. Rep. 2020, 72, 400–417. [Google Scholar] [CrossRef]
- Chang, H.W.; Lee, M.; Lee, Y.S.; Kim, S.H.; Lee, J.C.; Park, J.J.; Nam, H.Y.; Kim, M.R.; Han, M.W.; Kim, S.W.; et al. p53-dependent glutamine usage determines susceptibility to oxidative stress in radioresistant head and neck cancer cells. Cell Signal 2021, 77, 109820. [Google Scholar] [CrossRef] [PubMed]
- Montironi, C.; Chen, Z.; Derks, I.A.M.; Cretenet, G.; Krap, E.A.; Eldering, E.; Simon-Molas, H. Metabolic signature and response to glutamine deprivation are independent of p53 status in B cell malignancies. iScience 2024, 27, 109640. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhou, J.; Dong, L.; Zhu, L.; Ye, Y. Unveiling the impact of glutathione (GSH) and p53 gene deletion on tumor cell metabolism by amino acid and proteomics analysis. J. Gastrointest. Oncol. 2024, 15, 1002–1019. [Google Scholar] [CrossRef] [PubMed]
- Hosea, R.; Duan, W.; Meliala, I.T.S.; Li, W.; Wei, M.; Hillary, S.; Zhao, H.; Miyagishi, M.; Wu, S.; Kasim, V. YY2/BUB3 Axis promotes SAC Hyperactivation and Inhibits Colorectal Cancer Progression via Regulating Chromosomal Instability. Adv. Sci 2024, 11, e2308690. [Google Scholar] [CrossRef]
- Cao, W.; Tan, X.; Li, X.; Wang, Y.; Zhai, Y.; Zhang, Z.; Yuan, J.; Song, W. MIS18BP1 promotes bladder cancer cell proliferation and growth via inactivating P53 signaling pathway. Med. Oncol. 2025, 42, 156. [Google Scholar] [CrossRef]
- Kollareddy, M.; Martinez, L.A. Distinct Classes of Flavonoids and Epigallocatechin Gallate, Polyphenol Affects an Oncogenic Mutant p53 Protein, Cell Growth and Invasion in a TNBC Breast Cancer Cell Line. Cells 2021, 10, 797. [Google Scholar] [CrossRef]
- Li, W.J.; Wang, Y.; Liu, R.; Kasinski, A.L.; Shen, H.; Slack, F.J.; Tang, D.G. MicroRNA-34a: Potent Tumor Suppressor, Cancer Stem Cell Inhibitor, and Potential Anticancer Therapeutic. Front. Cell Dev. Biol. 2021, 9, 640587. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, W.; Demidova, E.V.; Serrao, S.; ValizadehAslani, T.; Rosen, G.; Arora, S. RRM2B Is Frequently Amplified Across Multiple Tumor Types: Implications for DNA Repair, Cellular Survival, and Cancer Therapy. Front. Genet. 2021, 12, 628758. [Google Scholar] [CrossRef] [PubMed]
- Chi, Y.; Remsik, J.; Kiseliovas, V.; Derderian, C.; Sener, U.; Alghader, M.; Saadeh, F.; Nikishina, K.; Bale, T.; Iacobuzio-Donahue, C.; et al. Cancer cells deploy lipocalin-2 to collect limiting iron in leptomeningeal metastasis. Science 2020, 369, 276–282. [Google Scholar] [CrossRef] [PubMed]
- Maio, N.; Zhang, D.L.; Ghosh, M.C.; Jain, A.; SantaMaria, A.M.; Rouault, T.A. Mechanisms of cellular iron sensing, regulation of erythropoiesis and mitochondrial iron utilization. Semin. Hematol. 2021, 58, 161–174. [Google Scholar] [CrossRef]
- Joachim, J.H.; Mehta, K.J. Hepcidin in hepatocellular carcinoma. Br. J. Cancer 2022, 127, 185–192. [Google Scholar] [CrossRef]
- Dong, Y.N.; Mercado-Ayon, E.; Coulman, J.; Flatley, L.; Ngaba, L.V.; Adeshina, M.W.; Lynch, D.R. The Regulation of the Disease-Causing Gene FXN. Cells 2024, 13, 1040. [Google Scholar] [CrossRef]
- Tang, L.J.; Zhou, Y.J.; Xiong, X.M.; Li, N.S.; Zhang, J.J.; Luo, X.J.; Peng, J. Ubiquitin-specific protease 7 promotes ferroptosis via activation of the p53/TfR1 pathway in the rat hearts after ischemia/reperfusion. Free Radic. Biol. Med. 2021, 162, 339–352. [Google Scholar] [CrossRef]
- Yilmaz, D.; Tharehalli, U.; Paganoni, R.; Knoop, P.; Gruber, A.; Chen, Y.; Dong, R.; Leithauser, F.; Seufferlein, T.; Leopold, K.; et al. Iron metabolism in a mouse model of hepatocellular carcinoma. Sci. Rep. 2025, 15, 2180. [Google Scholar] [CrossRef]
- Sung, Y.; Yu, Y.C.; Han, J.M. Nutrient sensors and their crosstalk. Exp. Mol. Med. 2023, 55, 1076–1089. [Google Scholar] [CrossRef] [PubMed]
- Higaki, K.; Aiba, S.; Shimoyama, T.; Omatsu, Y.; Nagasawa, T. Universal fibroblasts across tissues can differentiate into niche cells for hematopoietic stem cells. Cell Rep. 2025, 44, 115620. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Lee, H.Y. Ginseng-derived compounds as potential anticancer agents targeting cancer stem cells. J. Ginseng Res. 2024, 48, 266–275. [Google Scholar] [CrossRef] [PubMed]
- Zhong, H.; Zhou, S.; Yin, S.; Qiu, Y.; Liu, B.; Yu, H. Tumor microenvironment as niche constructed by cancer stem cells: Breaking the ecosystem to combat cancer. J. Adv. Res. 2025, 71, 279–296. [Google Scholar] [CrossRef]
- Marzagalli, M.; Fontana, F.; Raimondi, M.; Limonta, P. Cancer Stem Cells-Key Players in Tumor Relapse. Cancers 2021, 13, 376. [Google Scholar] [CrossRef]
- Li, W.; Huang, C.; Qiu, L.; Tang, Y.; Zhang, X.; Zhang, L.; Zhao, H.; Miyagishi, M.; Kasim, V.; Wu, S. p52-ZER6/IGF1R axis maintains cancer stem cell population to promote cancer progression by enhancing pro-survival mitophagy. Oncogene 2024, 43, 2115–2131. [Google Scholar] [CrossRef]
- Cao, L.; Fang, H.; Yan, D.; Wu, X.M.; Zhang, J.; Chang, M.X. CD44a functions as a regulator of p53 signaling, apoptosis and autophagy in the antibacterial immune response. Commun. Biol. 2022, 5, 889. [Google Scholar] [CrossRef]
- Guo, S.; Zheng, S.; Liu, M.; Wang, G. Novel Anti-Cancer Stem Cell Compounds: A Comprehensive Review. Pharmaceutics 2024, 16, 1024. [Google Scholar] [CrossRef]
- Lee, Y.K.; Heo, H.H.; Kim, N.; Park, U.H.; Youn, H.; Moon, E.Y.; Kim, E.J.; Um, S.J. Tumor antigen PRAME is a potential therapeutic target of p53 activation in melanoma cells. Bmb Rep. 2024, 57, 299–304. [Google Scholar] [CrossRef]
- Marrone, S.; Alomari, A.A.; Mastronardi, L. Current relation between Durante-Conheim theory and radiation resistance. Commentary on “Embryonic stem cell-like subpopulations are present within Schwannoma”. J. Clin. Neurosci. 2024, 124, 169–171. [Google Scholar] [CrossRef]
- Jo, H.; Shim, K.; Jeoung, D. Potential of the miR-200 Family as a Target for Developing Anti-Cancer Therapeutics. Int. J. Mol. Sci. 2022, 23, 5881. [Google Scholar] [CrossRef]
- Singh, S.K.; Chen, N.M.; Hessmann, E.; Siveke, J.; Lahmann, M.; Singh, G.; Voelker, N.; Vogt, S.; Esposito, I.; Schmidt, A.; et al. Antithetical NFATc1-Sox2 and p53-miR200 signaling networks govern pancreatic cancer cell plasticity. EMBO J. 2015, 34, 517–530. [Google Scholar] [CrossRef]
- Ghatak, D.; Das Ghosh, D.; Roychoudhury, S. Cancer Stemness: p53 at the Wheel. Front. Oncol. 2020, 10, 604124. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, N.; So, T.; Miyata, T.; Yoshimatsu, T.; Nakano, R.; Oyama, T.; Matsunaga, W.; Gotoh, A. Triple-negative expression (ALDH1A1-/CD133-/mutant p53-) cases in lung adenocarcinoma had a good prognosis. Sci. Rep. 2022, 12, 1473. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Chen, Q.; Tan, C.; Su, M.; Min, L.; Ling, L.; Zhou, J.; Zhu, T. ZEB family is a prognostic biomarker and correlates with anoikis and immune infiltration in kidney renal clear cell carcinoma. BMC Med. Genomics 2024, 17, 153. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.M.; Zhang, J.G.; Zhang, X.; Li, Q. Targeting cancer stem cells for reversing therapy resistance: Mechanism, signaling, and prospective agents. Signal Transduct. Target. Ther. 2021, 6, 62. [Google Scholar] [CrossRef]
- Cao, X.; Hou, J.; An, Q.; Assaraf, Y.G.; Wang, X. Towards the overcoming of anticancer drug resistance mediated by p53 mutations. Drug Resist. Updat. 2020, 49, 100671. [Google Scholar] [CrossRef]
- Neophytou, C.M.; Trougakos, I.P.; Erin, N.; Papageorgis, P. Apoptosis Deregulation and the Development of Cancer Multi-Drug Resistance. Cancers 2021, 13, 4363. [Google Scholar] [CrossRef]
- Zhang, K.X.; Ip, C.K.; Chung, S.K.; Lei, K.K.; Zhang, Y.Q.; Liu, L.; Wong, V.K.W. Drug-resistance in rheumatoid arthritis: The role of p53 gene mutations, ABC family transporters and personal factors. Curr. Opin. Pharmacol. 2020, 54, 59–71. [Google Scholar] [CrossRef]
- Wang, J.; Liu, D.; Sun, Z.; Ye, T.; Li, J.; Zeng, B.; Zhao, Q.; Rosie Xing, H. Autophagy augments the self-renewal of lung cancer stem cells by the degradation of ubiquitinated p53. Cell Death Dis. 2021, 12, 98. [Google Scholar] [CrossRef]
- Wei, H.; Wang, H.; Wang, G.; Qu, L.; Jiang, L.; Dai, S.; Chen, X.; Zhang, Y.; Chen, Z.; Li, Y.; et al. Structures of p53/BCL-2 complex suggest a mechanism for p53 to antagonize BCL-2 activity. Nat. Commun. 2023, 14, 4300. [Google Scholar] [CrossRef]
- Haghighi, Z.M.S.; Tabatabaei, T.; Rafigh, M.; Karampour, R.; Babaei, F.; Amjad, Z.S.; Payandeh, M.; Roozgari, M.; Bayat, M.; Doroudian, M.; et al. Human papillomavirus maybe is a critical player in the regulation of chemoresistance related factors (P53, Rb, TWIST, Bcl-2, Bcl-XL, c-IAP2, cytochrome C, and caspase 3) in breast cancer. Pathol. Res. Pract. 2023, 248, 154653. [Google Scholar] [CrossRef]
- Zhu, G.; Pan, C.; Bei, J.X.; Li, B.; Liang, C.; Xu, Y.; Fu, X. Mutant p53 in Cancer Progression and Targeted Therapies. Front. Oncol. 2020, 10, 595187. [Google Scholar] [CrossRef] [PubMed]
- Shu, S.; Li, Z.; Liu, L.; Ying, X.; Zhang, Y.; Wang, T.; Zhou, X.; Jiang, P.; Lv, W. HPV16 E6-Activated OCT4 Promotes Cervical Cancer Progression by Suppressing p53 Expression via Co-Repressor NCOR1. Front. Oncol. 2022, 12, 900856. [Google Scholar] [CrossRef] [PubMed]
- Olivares-Urbano, M.A.; Grinan-Lison, C.; Marchal, J.A.; Nunez, M.I. CSC Radioresistance: A Therapeutic Challenge to Improve Radiotherapy Effectiveness in Cancer. Cells 2020, 9, 1651. [Google Scholar] [CrossRef] [PubMed]
- Synoradzki, K.J.; Bartnik, E.; Czarnecka, A.M.; Fiedorowicz, M.; Firlej, W.; Brodziak, A.; Stasinska, A.; Rutkowski, P.; Grieb, P. TP53 in Biology and Treatment of Osteosarcoma. Cancers 2021, 13, 4284. [Google Scholar] [CrossRef]
- Song, B.; Wang, J.; Ren, Y.; Su, Y.; Geng, X.; Yang, F.; Wang, H.; Zhang, J. Butein inhibits cancer cell growth by rescuing the wild-type thermal stability of mutant p53. Biomed. Pharmacother. 2023, 163, 114773. [Google Scholar] [CrossRef]
- Phan, T.T.T.; Truong, N.V.; Wu, W.G.; Su, Y.C.; Hsu, T.S.; Lin, L.Y. Tumor suppressor p53 mediates interleukin-6 expression to enable cancer cell evasion of genotoxic stress. Cell Death Discov. 2023, 9, 340. [Google Scholar] [CrossRef]
- Chen, X.; Zhang, T.; Su, W.; Dou, Z.; Zhao, D.; Jin, X.; Lei, H.; Wang, J.; Xie, X.; Cheng, B.; et al. Mutant p53 in cancer: From molecular mechanism to therapeutic modulation. Cell Death Dis. 2022, 13, 974. [Google Scholar] [CrossRef]
- Blondy, S.; David, V.; Verdier, M.; Mathonnet, M.; Perraud, A.; Christou, N. 5-Fluorouracil resistance mechanisms in colorectal cancer: From classical pathways to promising processes. Cancer Sci. 2020, 111, 3142–3154. [Google Scholar] [CrossRef]
- Alalem, M.; Bhosale, M.; Ranjan, A.; Yamamoto, S.; Kaida, A.; Nishikawa, S.; Parrales, A.; Farooki, S.; Anant, S.; Padhye, S.; et al. Mutant p53 Depletion by Novel Inhibitors for HSP40/J-Domain Proteins Derived from the Natural Compound Plumbagin. Cancers 2022, 14, 4187. [Google Scholar] [CrossRef]
- Yang, Y.; Yuan, H.; Zhao, L.; Guo, S.; Hu, S.; Tian, M.; Nie, Y.; Yu, J.; Zhou, C.; Niu, J.; et al. Targeting the miR-34a/LRPPRC/MDR1 axis collapse the chemoresistance in P53 inactive colorectal cancer. Cell Death Differ. 2022, 29, 2177–2189. [Google Scholar] [CrossRef]
- do Patrocinio, A.B.; Rodrigues, V.; Guidi Magalhaes, L. P53: Stability from the Ubiquitin-Proteasome System and Specific 26S Proteasome Inhibitors. Acs Omega 2022, 7, 3836–3843. [Google Scholar] [CrossRef] [PubMed]
- Santarelli, R.; Curra, P.; Di Crosta, M.; Gonnella, R.; Gilardini Montani, M.S.; Cirone, M. Changes in Lysine Methylation Contribute to the Cytotoxicity of Curcumin in Colon Cancer Cells. Molecules 2025, 30, 335. [Google Scholar] [CrossRef] [PubMed]
- Yu, R.; Hang, Y.; Tsai, H.I.; Wang, D.; Zhu, H. Iron metabolism: Backfire of cancer cell stemness and therapeutic modalities. Cancer Cell Int. 2024, 24, 157. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.; Jeong, H.S.; Hwang, S.Y.; Lee, Y.G.; Kang, Y.J. ABCB1 confers resistance to carboplatin by accumulating stem-like cells in the G2/M phase of the cell cycle in p53(null) ovarian cancer. Cell Death Discov. 2025, 11, 132. [Google Scholar] [CrossRef]
- Mencattini, A.; Di Giuseppe, D.; Comes, M.C.; Casti, P.; Corsi, F.; Bertani, F.R.; Ghibelli, L.; Businaro, L.; Di Natale, C.; Parrini, M.C.; et al. Discovering the hidden messages within cell trajectories using a deep learning approach for in vitro evaluation of cancer drug treatments. Sci. Rep. 2020, 10, 7653. [Google Scholar] [CrossRef]
- Tu, S.M.; Guo, C.C.; Chow, D.S.; Zacharias, N.M. Stem Cell Theory of Cancer: Implications for Drug Resistance and Chemosensitivity in Cancer Care. Cancers 2022, 14, 1548. [Google Scholar] [CrossRef]
- Anish Ruban, S.; Raj, F.J.; Thangaraj, P. Phytochemical intervention in BCRP-driven cancer drug resistance: A comprehensive review. Biochim. Biophys. Acta Rev. Cancer 2025, 1880, 189349. [Google Scholar] [CrossRef]
- Al-Shamma, S.A.; Zaher, D.M.; Hersi, F.; Abu Jayab, N.N.; Omar, H.A. Targeting aldehyde dehydrogenase enzymes in combination with chemotherapy and immunotherapy: An approach to tackle resistance in cancer cells. Life Sci. 2023, 320, 121541. [Google Scholar] [CrossRef]
- Kim, H.S.; Grimes, S.M.; Chen, T.; Sathe, A.; Lau, B.T.; Hwang, G.H.; Bae, S.; Ji, H.P. Direct measurement of engineered cancer mutations and their transcriptional phenotypes in single cells. Nat. Biotechnol. 2024, 42, 1254–1262. [Google Scholar] [CrossRef]
- Hashim, H.O.; Al-Shuhaib, M.B. Exploring the Potential and Limitations of PCR-RFLP and PCR-SSCP for SNP Detection: A Review. J. Appl. Biotechnol. Rep. 2019, 6, 137–144. [Google Scholar] [CrossRef]
- Shen, C.C.; Cheng, W.Y.; Lee, C.H.; Dai, X.J.; Chiao, M.T.; Liang, Y.J.; Hsieh, W.Y.; Mao, T.F.; Lin, G.S.; Chen, S.R.; et al. Both p53 codon 72 Arg/Arg and pro/Arg genotypes in glioblastoma multiforme are associated with a better prognosis in bevacizumab treatment. BMC Cancer 2020, 20, 709. [Google Scholar] [CrossRef]
- Zhang, L.; Gao, W.; Keohavong, P. Analysis of Mutations in K-ras and p53 Genes in Sputum and Plasma Samples. Methods Mol. Biol. 2020, 2102, 373–394. [Google Scholar] [CrossRef]
- Timmaraju, V.A.; Finkelstein, S.D.; Levine, J.A. Analytical Validation of Loss of Heterozygosity and Mutation Detection in Pancreatic Fine-Needle Aspirates by Capillary Electrophoresis and Sanger Sequencing. Diagnostics 2024, 14, 514. [Google Scholar] [CrossRef]
- Chrystoja, C.C.; Diamandis, E.P. Whole genome sequencing as a diagnostic test: Challenges and opportunities. Clin. Chem. 2014, 60, 724–733. [Google Scholar] [CrossRef]
- Sikkema-Raddatz, B.; Johansson, L.F.; de Boer, E.N.; Almomani, R.; Boven, L.G.; van den Berg, M.P.; van Spaendonck-Zwarts, K.Y.; van Tintelen, J.P.; Sijmons, R.H.; Jongbloed, J.D.; et al. Targeted next-generation sequencing can replace Sanger sequencing in clinical diagnostics. Hum. Mutat. 2013, 34, 1035–1042. [Google Scholar] [CrossRef]
- Rossi, D.; Khiabanian, H.; Spina, V.; Ciardullo, C.; Bruscaggin, A.; Fama, R.; Rasi, S.; Monti, S.; Deambrogi, C.; De Paoli, L.; et al. Clinical impact of small TP53 mutated subclones in chronic lymphocytic leukemia. Blood 2014, 123, 2139–2147. [Google Scholar] [CrossRef] [PubMed]
- Campo, E.; Cymbalista, F.; Ghia, P.; Jager, U.; Pospisilova, S.; Rosenquist, R.; Schuh, A.; Stilgenbauer, S. TP53 aberrations in chronic lymphocytic leukemia: An overview of the clinical implications of improved diagnostics. Haematologica 2018, 103, 1956–1968. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Cabrera, J.M.; Del Valle, J.; Feliubadalo, L.; Pineda, M.; Gonzalez, S.; Campos, O.; Cuesta, R.; Brunet, J.; Serra, E.; Capella, G.; et al. Screening of CNVs using NGS data improves mutation detection yield and decreases costs in genetic testing for hereditary cancer. J. Med. Genet. 2022, 59, 75–78. [Google Scholar] [CrossRef] [PubMed]
- Hussen, B.M.; Abdullah, S.T.; Salihi, A.; Sabir, D.K.; Sidiq, K.R.; Rasul, M.F.; Hidayat, H.J.; Ghafouri-Fard, S.; Taheri, M.; Jamali, E. The emerging roles of NGS in clinical oncology and personalized medicine. Pathol. Res. Pract. 2022, 230, 153760. [Google Scholar] [CrossRef] [PubMed]
- Pecoraro, V.; Mandrioli, J.; Carone, C.; Chio, A.; Traynor, B.J.; Trenti, T. The NGS technology for the identification of genes associated with the ALS. A systematic review. Eur. J. Clin. Invest. 2020, 50, e13228. [Google Scholar] [CrossRef] [PubMed]
- Bartels, S.; Grote, I.; Wagner, M.; Boog, J.; Schipper, E.; Reineke-Plaass, T.; Kreipe, H.; Lehmann, U. Concordance in detection of microsatellite instability by PCR and NGS in routinely processed tumor specimens of several cancer types. Cancer Med. 2023, 12, 16707–16715. [Google Scholar] [CrossRef] [PubMed]
- Cui, C.; Shu, W.; Li, P. Fluorescence In situ Hybridization: Cell-Based Genetic Diagnostic and Research Applications. Front. Cell Dev. Biol. 2016, 4, 89. [Google Scholar] [CrossRef]
- Mincherton, T.I.; Lam, S.J.; Clarke, S.E.; Hui, H.Y.L.; Malherbe, J.A.J.; Chuah, H.S.; Sidiqi, M.H.; Fuller, K.A.; Erber, W.N. Imaging flow cytometric detection of del(17p) in bone marrow and circulating plasma cells in multiple myeloma. Int. J. Lab. Hematol. 2024, 46, 495–502. [Google Scholar] [CrossRef]
- Tang, Z.; Kanagal-Shamanna, R.; Tang, G.; Patel, K.; Medeiros, L.J.; Toruner, G.A. Analytical and clinical performance of chromosomal microarrays compared with FISH panel and conventional karyotyping in patients with chronic lymphocytic leukemia. Leuk. Res. 2021, 108, 106616. [Google Scholar] [CrossRef]
- Nathan, C.A.; Khandelwal, A.R.; Wolf, G.T.; Rodrigo, J.P.; Makitie, A.A.; Saba, N.F.; Forastiere, A.A.; Bradford, C.R.; Ferlito, A. TP53 mutations in head and neck cancer. Mol. Carcinog. 2022, 61, 385–391. [Google Scholar] [CrossRef]
- Sung, Y.N.; Kim, D.; Kim, J. p53 immunostaining pattern is a useful surrogate marker for TP53 gene mutations. Diagn. Pathol. 2022, 17, 92. [Google Scholar] [CrossRef]
- Robles, A.I.; Harris, C.C. Clinical outcomes and correlates of TP53 mutations and cancer. Cold Spring Harb. Perspect. Biol. 2010, 2, a001016. [Google Scholar] [CrossRef]
- Nenutil, R.; Smardova, J.; Pavlova, S.; Hanzelkova, Z.; Muller, P.; Fabian, P.; Hrstka, R.; Janotova, P.; Radina, M.; Lane, D.P.; et al. Discriminating functional and non-functional p53 in human tumours by p53 and MDM2 immunohistochemistry. J. Pathol. 2005, 207, 251–259. [Google Scholar] [CrossRef]
- Brar, N.; Lawrence, L.; Fung, E.; Zehnder, J.L.; Greenberg, P.L.; Mannis, G.N.; Zhang, T.Y.; Gratzinger, D.; Oak, J.; Silva, O.; et al. p53 immunohistochemistry as an ancillary tool for rapid assessment of residual disease in TP53-mutated acute myeloid leukemia and myelodysplastic syndromes. Am. J. Clin. Pathol. 2024, 162, 269–281. [Google Scholar] [CrossRef]
- Fitzpatrick, M.J.; Boiocchi, L.; Fathi, A.T.; Brunner, A.M.; Hasserjian, R.P.; Nardi, V. Correlation of p53 immunohistochemistry with TP53 mutational status and overall survival in newly diagnosed acute myeloid leukaemia. Histopathology 2022, 81, 496–510. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Pol, S.; Ma, L.; Ohgami, R.S.; Arber, D.A. Immunohistochemistry for p53 is a useful tool to identify cases of acute myeloid leukemia with myelodysplasia-related changes that are TP53 mutated, have complex karyotype, and have poor prognosis. Mod. Pathol. 2017, 30, 382–392. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.; Scalia, J.; Legare, R.; Quddus, M.R.; Sung, C.J. Immunohistochemical findings and clinicopathological features of breast cancers with pathogenic germline mutations in Non-BRCA genes. Hum. Pathol. 2024, 146, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Biatta, C.M.; Paudice, M.; Greppi, M.; Parrella, V.; Parodi, A.; De Luca, G.; Cerruti, G.M.; Mammoliti, S.; Caroti, C.; Menichini, P.; et al. The fading guardian: Clinical relevance of TP53 null mutation in high-grade serous ovarian cancers. Front. Immunol. 2023, 14, 1221605. [Google Scholar] [CrossRef]
- Vermij, L.; Leon-Castillo, A.; Singh, N.; Powell, M.E.; Edmondson, R.J.; Genestie, C.; Khaw, P.; Pyman, J.; McLachlin, C.M.; Ghatage, P.; et al. p53 immunohistochemistry in endometrial cancer: Clinical and molecular correlates in the PORTEC-3 trial. Mod. Pathol. 2022, 35, 1475–1483. [Google Scholar] [CrossRef]
- Shahbandi, A.; Nguyen, H.D.; Jackson, J.G. TP53 Mutations and Outcomes in Breast Cancer: Reading beyond the Headlines. Trends Cancer 2020, 6, 98–110. [Google Scholar] [CrossRef]
- Fan, Z.; Zhang, Q.; Feng, L.; Wang, L.; Zhou, X.; Han, J.; Li, D.; Liu, J.; Zhang, X.; Zuo, J.; et al. Genomic landscape and prognosis of patients with TP53-mutated non-small cell lung cancer. Ann. Transl. Med. 2022, 10, 188. [Google Scholar] [CrossRef]
- Andersson, J.; Larsson, L.; Klaar, S.; Holmberg, L.; Nilsson, J.; Inganas, M.; Carlsson, G.; Ohd, J.; Rudenstam, C.M.; Gustavsson, B.; et al. Worse survival for TP53 (p53)-mutated breast cancer patients receiving adjuvant CMF. Ann. Oncol. 2005, 16, 743–748. [Google Scholar] [CrossRef]
- Petitjean, A.; Achatz, M.I.; Borresen-Dale, A.L.; Hainaut, P.; Olivier, M. TP53 mutations in human cancers: Functional selection and impact on cancer prognosis and outcomes. Oncogene 2007, 26, 2157–2165. [Google Scholar] [CrossRef]
- Landsburg, D.J.; Morrissette, J.J.; Nasta, S.D.; Barta, S.K.; Schuster, S.J.; Svoboda, J.; Chong, E.A.; Bagg, A. TP53 mutations predict for poor outcomes in patients with newly diagnosed aggressive B-cell lymphomas in the current era. Blood Adv. 2023, 7, 7243–7253. [Google Scholar] [CrossRef]
- Canale, M.; Andrikou, K.; Priano, I.; Cravero, P.; Pasini, L.; Urbini, M.; Delmonte, A.; Crino, L.; Bronte, G.; Ulivi, P. The Role of TP53 Mutations in EGFR-Mutated Non-Small-Cell Lung Cancer: Clinical Significance and Implications for Therapy. Cancers 2022, 14, 1143. [Google Scholar] [CrossRef] [PubMed]
- Sinn, M.; Sinn, B.V.; Treue, D.; Keilholz, U.; Damm, F.; Schmuck, R.; Lohneis, P.; Klauschen, F.; Striefler, J.K.; Bahra, M.; et al. TP53 Mutations Predict Sensitivity to Adjuvant Gemcitabine in Patients with Pancreatic Ductal Adenocarcinoma: Next-Generation Sequencing Results from the CONKO-001 Trial. Clin. Cancer Res. 2020, 26, 3732–3739. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Abro, B.; Campbell, A.; Ding, Y. TP53 mutations in myeloid neoplasms: Implications for accurate laboratory detection, diagnosis, and treatment. Lab. Med. 2024, 55, 686–699. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Bai, J.; Li, W.; Su, Z.; Cheng, X. Advancements in p53-Based Anti-Tumor Gene Therapy Research. Molecules 2024, 29, 5315. [Google Scholar] [CrossRef]
- Zhang, W.W.; Li, L.; Li, D.; Liu, J.; Li, X.; Li, W.; Xu, X.; Zhang, M.J.; Chandler, L.A.; Lin, H.; et al. The First Approved Gene Therapy Product for Cancer Ad-p53 (Gendicine): 12 Years in the Clinic. Hum. Gene Ther. 2018, 29, 160–179. [Google Scholar] [CrossRef]
- Qi, L.; Li, G.; Li, P.; Wang, H.; Fang, X.; He, T.; Li, J. Twenty years of Gendicine(R) rAd-p53 cancer gene therapy: The first-in-class human cancer gene therapy in the era of personalized oncology. Genes. Dis. 2024, 11, 101155. [Google Scholar] [CrossRef]
- Batir, M.B.; Sahin, E.; Cam, F.S. Evaluation of the CRISPR/Cas9 directed mutant TP53 gene repairing effect in human prostate cancer cell line PC-3. Mol. Biol. Rep. 2019, 46, 6471–6484. [Google Scholar] [CrossRef]
- Zhan, H.; Xie, H.; Zhou, Q.; Liu, Y.; Huang, W. Synthesizing a Genetic Sensor Based on CRISPR-Cas9 for Specifically Killing p53-Deficient Cancer Cells. Acs Synth. Biol. 2018, 7, 1798–1807. [Google Scholar] [CrossRef]
- Mirgayazova, R.; Khadiullina, R.; Chasov, V.; Mingaleeva, R.; Miftakhova, R.; Rizvanov, A.; Bulatov, E. Therapeutic Editing of the TP53 Gene: Is CRISPR/Cas9 an Option? Genes. 2020, 11, 704. [Google Scholar] [CrossRef]
- Newman, A.; Starrs, L.; Burgio, G. Cas9 Cuts and Consequences; Detecting, Predicting, and Mitigating CRISPR/Cas9 On- and Off-Target Damage: Techniques for Detecting, Predicting, and Mitigating the On- and off-target Effects of Cas9 Editing. Bioessays 2020, 42, e2000047. [Google Scholar] [CrossRef]
- Alfason, L.; Li, W.; Altaf, F.; Wu, S.; Kasim, V. Resuscitating the Guardian: Current Progress in p53-Based Anti-Tumor Therapy. Onco Therapeutics 2021, 8, 51–92. [Google Scholar] [CrossRef]
- Silva, J.L.; Lima, C.G.S.; Rangel, L.P.; Ferretti, G.D.S.; Pauli, F.P.; Ribeiro, R.C.B.; da Silva, T.B.; da Silva, F.C.; Ferreira, V.F. Recent Synthetic Approaches towards Small Molecule Reactivators of p53. Biomolecules 2020, 10, 635. [Google Scholar] [CrossRef]
- Ceder, S.; Eriksson, S.E.; Liang, Y.Y.; Cheteh, E.H.; Zhang, S.M.; Fujihara, K.M.; Bianchi, J.; Bykov, V.J.N.; Abrahmsen, L.; Clemons, N.J.; et al. Correction: Mutant p53-reactivating compound APR-246 synergizes with asparaginase in inducing growth suppression in acute lymphoblastic leukemia cells. Cell Death Dis. 2022, 13, 672. [Google Scholar] [CrossRef]
- Synnott, N.C.; O’Connell, D.; Crown, J.; Duffy, M.J. COTI-2 reactivates mutant p53 and inhibits growth of triple-negative breast cancer cells. Breast Cancer Res. Treat. 2020, 179, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Rippin, T.M.; Bykov, V.J.; Freund, S.M.; Selivanova, G.; Wiman, K.G.; Fersht, A.R. Characterization of the p53-rescue drug CP-31398 in vitro and in living cells. Oncogene 2002, 21, 2119–2129. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Liu, W.; Zhang, L.; Zhang, J. Targeting mutant p53 stabilization for cancer therapy. Front. Pharmacol. 2023, 14, 1215995. [Google Scholar] [CrossRef] [PubMed]
- Tanveer, M.A.; Rashid, H.; Nazir, L.A.; Archoo, S.; Shahid, N.H.; Ragni, G.; Umar, S.A.; Tasduq, S.A. Trigonelline, a plant derived alkaloid prevents ultraviolet-B-induced oxidative DNA damage in primary human dermal fibroblasts and BALB/c mice via modulation of phosphoinositide 3-kinase-Akt-Nrf2 signalling axis. Exp. Gerontol. 2023, 171, 112028. [Google Scholar] [CrossRef]
- Li, D.; Marchenko, N.D.; Moll, U.M. SAHA shows preferential cytotoxicity in mutant p53 cancer cells by destabilizing mutant p53 through inhibition of the HDAC6-Hsp90 chaperone axis. Cell Death Differ. 2011, 18, 1904–1913. [Google Scholar] [CrossRef]
- Parrales, A.; Ranjan, A.; Iyer, S.V.; Padhye, S.; Weir, S.J.; Roy, A.; Iwakuma, T. DNAJA1 controls the fate of misfolded mutant p53 through the mevalonate pathway. Nature cell biology 2016, 18, 1233–1243. [Google Scholar] [CrossRef]
- Tong, X.; Xu, D.; Mishra, R.K.; Jones, R.D.; Sun, L.; Schiltz, G.E.; Liao, J.; Yang, G.Y. Identification of a druggable protein-protein interaction site between mutant p53 and its stabilizing chaperone DNAJA1. J. Biol. Chem. 2021, 296, 100098. [Google Scholar] [CrossRef] [PubMed]
- Ferretti, G.D.S.; Quarti, J.; Dos Santos, G.; Rangel, L.P.; Silva, J.L. Anticancer Therapeutic Strategies Targeting p53 Aggregation. Int. J. Mol. Sci. 2022, 23, 11023. [Google Scholar] [CrossRef] [PubMed]
- Kwan, K.; Castro-Sandoval, O.; Gaiddon, C.; Storr, T. Inhibition of p53 protein aggregation as a cancer treatment strategy. Curr. Opin. Chem. Biol. 2023, 72, 102230. [Google Scholar] [CrossRef] [PubMed]
- Garufi, A.; Pistritto, G.; Cirone, M.; D’Orazi, G. Reactivation of mutant p53 by capsaicin, the major constituent of peppers. J. Exp. Clin. Cancer Res. 2016, 35, 136. [Google Scholar] [CrossRef]
- Meng, X.; Gao, J.Z.; Gomendoza, S.M.T.; Li, J.W.; Yang, S. Recent Advances of WEE1 Inhibitors and Statins in Cancers With p53 Mutations. Front. Med. 2021, 8, 737951. [Google Scholar] [CrossRef]
- Gupta, A.; Shah, K.; Oza, M.J.; Behl, T. Reactivation of p53 gene by MDM2 inhibitors: A novel therapy for cancer treatment. Biomed. Pharmacother. 2019, 109, 484–492. [Google Scholar] [CrossRef]
- Zhao, S.; Liu, X.; Luo, R.; Jian, Z.; Xu, C.; Hou, Y.; Liu, X.; Zhang, P. USP38 functions as an oncoprotein by downregulating the p53 pathway through deubiquitination and stabilization of MDM2. Cell Death Differ. 2025, 32, 1128–1141. [Google Scholar] [CrossRef]
- Chessari, G.; Hardcastle, I.R.; Ahn, J.S.; Anil, B.; Anscombe, E.; Bawn, R.H.; Bevan, L.D.; Blackburn, T.J.; Buck, I.; Cano, C.; et al. Structure-Based Design of Potent and Orally Active Isoindolinone Inhibitors of MDM2-p53 Protein-Protein Interaction. J. Med. Chem. 2021, 64, 4071–4088. [Google Scholar] [CrossRef]
- Li, W.F.; Alfason, L.; Huang, C.; Tang, Y.; Qiu, L.; Miyagishi, M.; Wu, S.R.; Kasim, V. p52-ZER6: A determinant of tumor cell sensitivity to MDM2-p53 binding inhibitors. Acta Pharmacol. Sin. 2023, 44, 647–660. [Google Scholar] [CrossRef]
- Uddin, M.B.; Roy, K.R.; Hill, R.A.; Roy, S.C.; Gu, X.; Li, L.; Zhang, Q.J.; You, Z.; Liu, Y.Y. p53 missense mutant G242A subverts natural killer cells in sheltering mouse breast cancer cells against immune rejection. Exp. Cell Res. 2022, 417, 113210. [Google Scholar] [CrossRef]
- Peng, L.; Zhou, L.; Li, H.; Zhang, X.; Li, S.; Wang, K.; Yang, M.; Ma, X.; Zhang, D.; Xiang, S.; et al. Hippo-signaling-controlled MHC class I antigen processing and presentation pathway potentiates antitumor immunity. Cell Rep. 2024, 43, 114003. [Google Scholar] [CrossRef] [PubMed]
- Levine, A.J. Improving T cell killing and understanding senescence: Possible roles for TP53 in cancer immunotherapy. Proc. Natl. Acad. Sci. USA 2024, 121, e2402533121. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Cao, J.; Topatana, W.; Juengpanich, S.; Li, S.; Zhang, B.; Shen, J.; Cai, L.; Cai, X.; Chen, M. Targeting mutant p53 for cancer therapy: Direct and indirect strategies. J. Hematol. Oncol. 2021, 14, 157. [Google Scholar] [CrossRef] [PubMed]
- Manohar, S.M.; Joshi, K.S. Molecular pharmacology of multitarget cyclin-dependent kinase inhibitors in human colorectal carcinoma cells. Expert. Opin. Ther. Targets 2023, 27, 251–261. [Google Scholar] [CrossRef]
- Monks, A.; Harris, E.D.; Vaigro-Wolff, A.; Hose, C.D.; Connelly, J.W.; Sausville, E.A. UCN-01 enhances the in vitro toxicity of clinical agents in human tumor cell lines. Investig. New Drugs 2000, 18, 95–107. [Google Scholar] [CrossRef]
- Kravchenko, J.E.; Ilyinskaya, G.V.; Komarov, P.G.; Agapova, L.S.; Kochetkov, D.V.; Strom, E.; Frolova, E.I.; Kovriga, I.; Gudkov, A.V.; Feinstein, E.; et al. Small-molecule RETRA suppresses mutant p53-bearing cancer cells through a p73-dependent salvage pathway. Proc. Natl. Acad. Sci. USA 2008, 105, 6302–6307. [Google Scholar] [CrossRef]
- Zawacka-Pankau, J.E. The Undervalued Avenue to Reinstate Tumor Suppressor Functionality of the p53 Protein Family for Improved Cancer Therapy-Drug Repurposing. Cancers 2020, 12, 2717. [Google Scholar] [CrossRef]
- Di Agostino, S.; Cortese, G.; Monti, O.; Dell’Orso, S.; Sacchi, A.; Eisenstein, M.; Citro, G.; Strano, S.; Blandino, G. The disruption of the protein complex mutantp53/p73 increases selectively the response of tumor cells to anticancer drugs. Cell Cycle 2008, 7, 3440–3447. [Google Scholar] [CrossRef]
- Ling, X.; Cao, S.; Cheng, Q.; Keefe, J.T.; Rustum, Y.M.; Li, F. A novel small molecule FL118 that selectively inhibits survivin, Mcl-1, XIAP and cIAP2 in a p53-independent manner, shows superior antitumor activity. PLoS ONE 2012, 7, e45571. [Google Scholar] [CrossRef]
- Ling, X.; Xu, C.; Fan, C.; Zhong, K.; Li, F.; Wang, X. FL118 induces p53-dependent senescence in colorectal cancer cells by promoting degradation of MdmX. Cancer Res. 2014, 74, 7487–7497. [Google Scholar] [CrossRef]
- Yerlikaya, A.; Okur, E.; Ulukaya, E. The p53-independent induction of apoptosis in breast cancer cells in response to proteasome inhibitor bortezomib. Tumour Biol. 2012, 33, 1385–1392. [Google Scholar] [CrossRef] [PubMed]
- Czuczman, M.; Gruber, E.; Hosking, P.; Olejniczak, S.H.; Khubchandani, S.; Hernandez-Ilizaliturri, F.J. The BH3-Mimetic Obatoclax (GX15-070) Posses a Dual-Mechanism of Action and Induces Both Apoptosis and Autophagy-Dependent Cell Death of B Cell Non-Hodgkin’s Lymphoma (B-NHL) Cells. Blood 2008, 112, 605. [Google Scholar] [CrossRef]
- Akhoundova, D.; Rubin, M.A. Clinical application of advanced multi-omics tumor profiling: Shaping precision oncology of the future. Cancer Cell 2022, 40, 920–938. [Google Scholar] [CrossRef] [PubMed]
- Puzio-Kuter, A.M.; Xu, L.; McBrayer, M.K.; Dominique, R.; Li, H.H.; Fahr, B.J.; Brown, A.M.; Wiebesiek, A.E.; Russo, B.M.; Mulligan, C.L.; et al. Restoration of the Tumor Suppressor Function of Y220C-Mutant p53 by Rezatapopt, a Small-Molecule Reactivator. Cancer Discov. 2025, 15, 1159–1179. [Google Scholar] [CrossRef] [PubMed]
- Bykov, V.J.; Wiman, K.G. Mutant p53 reactivation by small molecules makes its way to the clinic. Febs Lett. 2014, 588, 2622–2627. [Google Scholar] [CrossRef]
- Roszkowska, K.A.; Piecuch, A.; Sady, M.; Gajewski, Z.; Flis, S. Gain of Function (GOF) Mutant p53 in Cancer-Current Therapeutic Approaches. Int. J. Mol. Sci. 2022, 23, 13287. [Google Scholar] [CrossRef]
- Boustani, J.; Lecoester, B.; Baude, J.; Latour, C.; Adotevi, O.; Mirjolet, C.; Truc, G. Anti-PD-1/Anti-PD-L1 Drugs and Radiation Therapy: Combinations and Optimization Strategies. Cancers 2021, 13, 4893. [Google Scholar] [CrossRef]
- Yap, T.A.; Parkes, E.E.; Peng, W.; Moyers, J.T.; Curran, M.A.; Tawbi, H.A. Development of Immunotherapy Combination Strategies in Cancer. Cancer Discov. 2021, 11, 1368–1397. [Google Scholar] [CrossRef]
- Loh, S.N. Follow the Mutations: Toward Class-Specific, Small-Molecule Reactivation of p53. Biomolecules 2020, 10, 303. [Google Scholar] [CrossRef]
- Zhang, L.; Zhao, F.; Li, W.; Song, G.; Kasim, V.; Wu, S. The Biological Roles and Molecular Mechanisms of Long Non-Coding RNA MEG3 in the Hallmarks of Cancer. Cancers 2022, 14, 6032. [Google Scholar] [CrossRef]
- Greshock, J.; Lewi, M.; Hartog, B.; Tendler, C. Harnessing Real-World Evidence for the Development of Novel Cancer Therapies. Trends Cancer 2020, 6, 907–909. [Google Scholar] [CrossRef]
- Schulz-Heddergott, R.; Stark, N.; Edmunds, S.J.; Li, J.; Conradi, L.C.; Bohnenberger, H.; Ceteci, F.; Greten, F.R.; Dobbelstein, M.; Moll, U.M. Therapeutic Ablation of Gain-of-Function Mutant p53 in Colorectal Cancer Inhibits Stat3-Mediated Tumor Growth and Invasion. Cancer Cell 2018, 34, 298–314.e7. [Google Scholar] [CrossRef]
- Cooks, T.; Pateras, I.S.; Tarcic, O.; Solomon, H.; Schetter, A.J.; Wilder, S.; Lozano, G.; Pikarsky, E.; Forshew, T.; Rosenfeld, N.; et al. Mutant p53 prolongs NF-kappaB activation and promotes chronic inflammation and inflammation-associated colorectal cancer. Cancer Cell 2013, 23, 634–646. [Google Scholar] [CrossRef]
- Di Minin, G.; Bellazzo, A.; Dal Ferro, M.; Chiaruttini, G.; Nuzzo, S.; Bicciato, S.; Piazza, S.; Rami, D.; Bulla, R.; Sommaggio, R.; et al. Mutant p53 reprograms TNF signaling in cancer cells through interaction with the tumor suppressor DAB2IP. Mol. Cell 2014, 56, 617–629. [Google Scholar] [CrossRef]
- Cooks, T.; Pateras, I.S.; Jenkins, L.M.; Patel, K.M.; Robles, A.I.; Morris, J.; Forshew, T.; Appella, E.; Gorgoulis, V.G.; Harris, C.C. Mutant p53 cancers reprogram macrophages to tumor supporting macrophages via exosomal miR-1246. Nat. Commun. 2018, 9, 771. [Google Scholar] [CrossRef]
- Solomon, H.; Dinowitz, N.; Pateras, I.S.; Cooks, T.; Shetzer, Y.; Molchadsky, A.; Charni, M.; Rabani, S.; Koifman, G.; Tarcic, O.; et al. Mutant p53 gain of function underlies high expression levels of colorectal cancer stem cells markers. Oncogene 2018, 37, 1669–1684. [Google Scholar] [CrossRef]
- Zhao, Y.; Li, Y.; Sheng, J.; Wu, F.; Li, K.; Huang, R.; Wang, X.; Jiao, T.; Guan, X.; Lu, Y.; et al. P53-R273H mutation enhances colorectal cancer stemness through regulating specific lncRNAs. J. Exp. Clin. Cancer Res. 2019, 38, 379. [Google Scholar] [CrossRef]
- Weissmueller, S.; Manchado, E.; Saborowski, M.; Morris, J.P.t.; Wagenblast, E.; Davis, C.A.; Moon, S.H.; Pfister, N.T.; Tschaharganeh, D.F.; Kitzing, T.; et al. Mutant p53 drives pancreatic cancer metastasis through cell-autonomous PDGF receptor beta signaling. Cell 2014, 157, 382–394. [Google Scholar] [CrossRef]
- Vaughan, C.A.; Singh, S.; Windle, B.; Yeudall, W.A.; Frum, R.; Grossman, S.R.; Deb, S.P.; Deb, S. Gain-of-Function Activity of Mutant p53 in Lung Cancer through Up-Regulation of Receptor Protein Tyrosine Kinase Axl. Genes Cancer 2012, 3, 491–502. [Google Scholar] [CrossRef]
- Ali, A.; Wang, Z.; Fu, J.; Ji, L.; Liu, J.; Li, L.; Wang, H.; Chen, J.; Caulin, C.; Myers, J.N.; et al. Differential regulation of the REGgamma-proteasome pathway by p53/TGF-beta signalling and mutant p53 in cancer cells. Nat. Commun. 2013, 4, 2667. [Google Scholar] [CrossRef]
- Singh, S.; Vaughan, C.A.; Frum, R.A.; Grossman, S.R.; Deb, S.; Palit Deb, S. Mutant p53 establishes targetable tumor dependency by promoting unscheduled replication. J. Clin. Investig. 2017, 127, 1839–1855. [Google Scholar] [CrossRef]
- Jiang, L.; Kon, N.; Li, T.; Wang, S.J.; Su, T.; Hibshoosh, H.; Baer, R.; Gu, W. Ferroptosis as a p53-mediated activity during tumour suppression. Nature 2015, 520, 57–62. [Google Scholar] [CrossRef]
- Polotskaia, A.; Xiao, G.; Reynoso, K.; Martin, C.; Qiu, W.G.; Hendrickson, R.C.; Bargonetti, J. Proteome-wide analysis of mutant p53 targets in breast cancer identifies new levels of gain-of-function that influence PARP, PCNA, and MCM4. Proc. Natl. Acad. Sci. USA 2015, 112, E1220–E1229. [Google Scholar] [CrossRef]
- Zhu, J.; Sammons, M.A.; Donahue, G.; Dou, Z.; Vedadi, M.; Getlik, M.; Barsyte-Lovejoy, D.; Al-awar, R.; Katona, B.W.; Shilatifard, A.; et al. Gain-of-function p53 mutants co-opt chromatin pathways to drive cancer growth. Nature 2015, 525, 206–211. [Google Scholar] [CrossRef]
- Lisek, K.; Campaner, E.; Ciani, Y.; Walerych, D.; Del Sal, G. Mutant p53 tunes the NRF2-dependent antioxidant response to support survival of cancer cells. Oncotarget 2018, 9, 20508–20523. [Google Scholar] [CrossRef]
- Ali, A.; Shah, A.S.; Ahmad, A. Gain-of-function of mutant p53: Mutant p53 enhances cancer progression by inhibiting KLF17 expression in invasive breast carcinoma cells. Cancer Lett. 2014, 354, 87–96. [Google Scholar] [CrossRef]
- Ghosh, M.; Saha, S.; Bettke, J.; Nagar, R.; Parrales, A.; Iwakuma, T.; van der Velden, A.W.M.; Martinez, L.A. Mutant p53 suppresses innate immune signaling to promote tumorigenesis. Cancer Cell 2021, 39, 494–508.e5. [Google Scholar] [CrossRef]
- Ubertini, V.; Norelli, G.; D’Arcangelo, D.; Gurtner, A.; Cesareo, E.; Baldari, S.; Gentileschi, M.P.; Piaggio, G.; Nistico, P.; Soddu, S.; et al. Mutant p53 gains new function in promoting inflammatory signals by repression of the secreted interleukin-1 receptor antagonist. Oncogene 2015, 34, 2493–2504. [Google Scholar] [CrossRef]
- Liu, D.S.; Duong, C.P.; Haupt, S.; Montgomery, K.G.; House, C.M.; Azar, W.J.; Pearson, H.B.; Fisher, O.M.; Read, M.; Guerra, G.R.; et al. Inhibiting the system x(C)(-)/glutathione axis selectively targets cancers with mutant-p53 accumulation. Nat. Commun. 2017, 8, 14844. [Google Scholar] [CrossRef]
- Tanaka, N.; Zhao, M.; Tang, L.; Patel, A.A.; Xi, Q.; Van, H.T.; Takahashi, H.; Osman, A.A.; Zhang, J.; Wang, J.; et al. Gain-of-function mutant p53 promotes the oncogenic potential of head and neck squamous cell carcinoma cells by targeting the transcription factors FOXO3a and FOXM1. Oncogene 2018, 37, 1279–1292. [Google Scholar] [CrossRef]
- Xu, Y.; Zhang, Q.; Miao, C.; Dongol, S.; Li, Y.; Jin, C.; Dong, R.; Li, Y.; Yang, X.; Kong, B. CCNG1 (Cyclin G1) regulation by mutant-P53 via induction of Notch3 expression promotes high-grade serous ovarian cancer (HGSOC) tumorigenesis and progression. Cancer Med. 2019, 8, 351–362. [Google Scholar] [CrossRef]

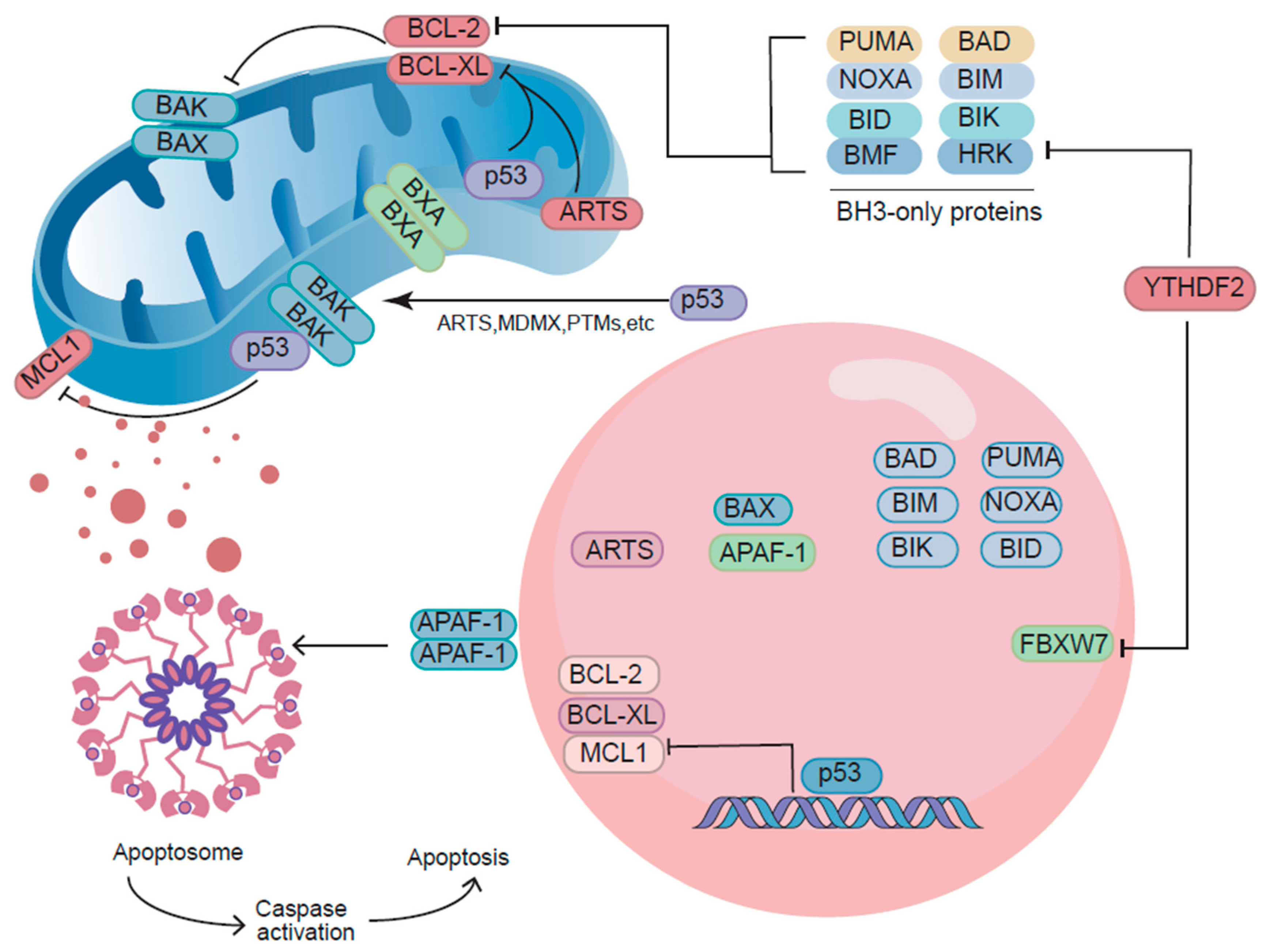

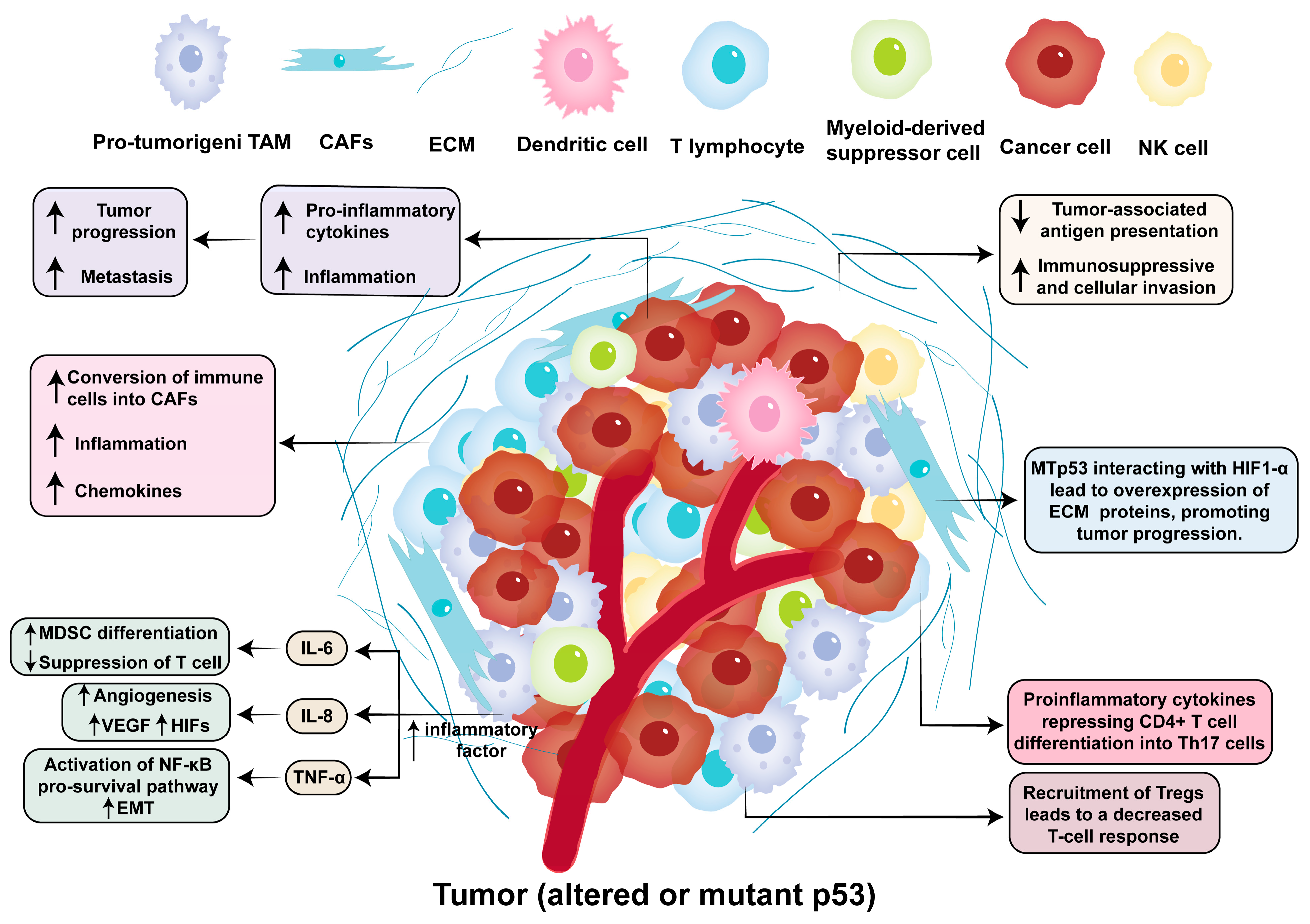
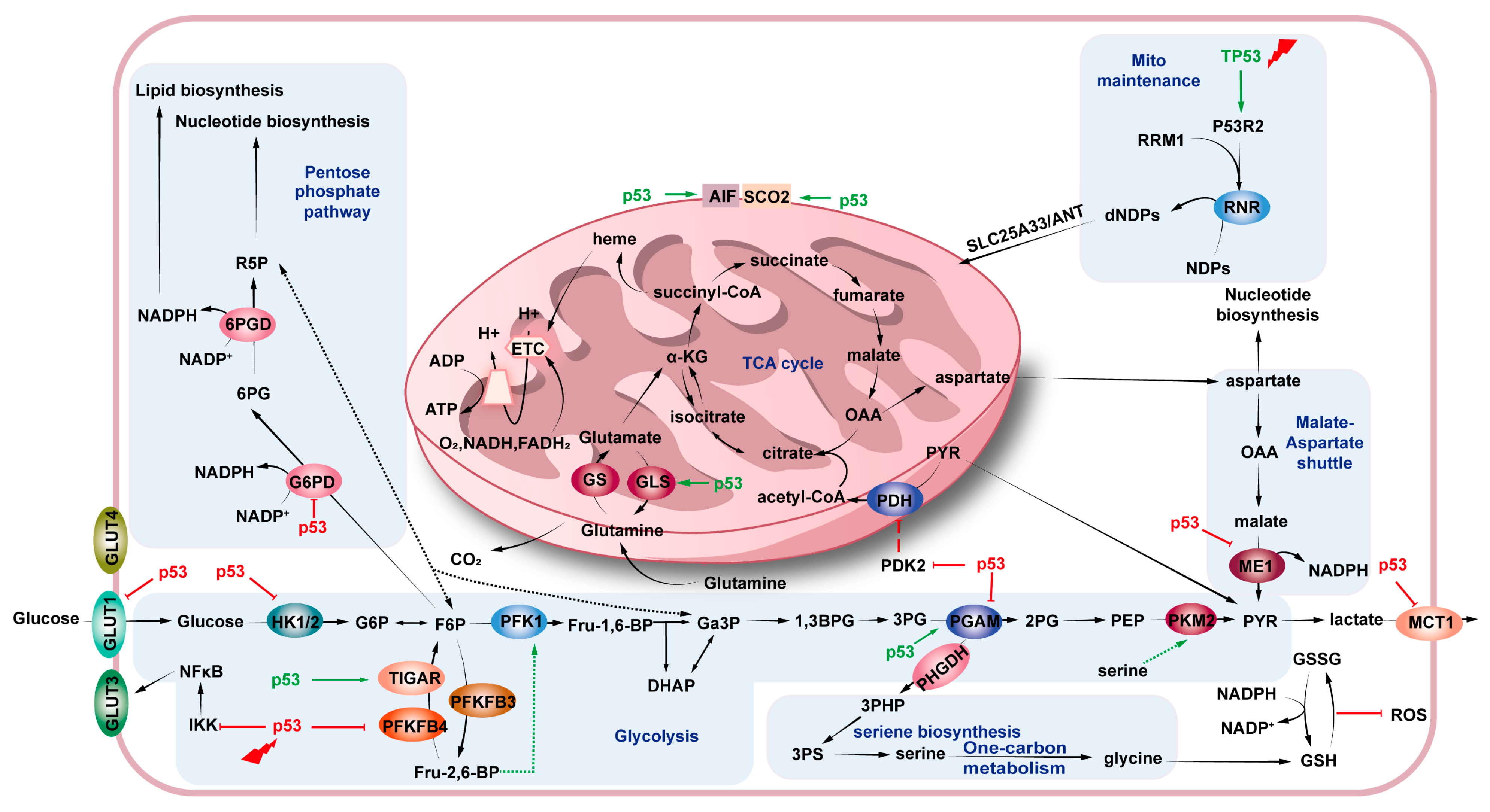
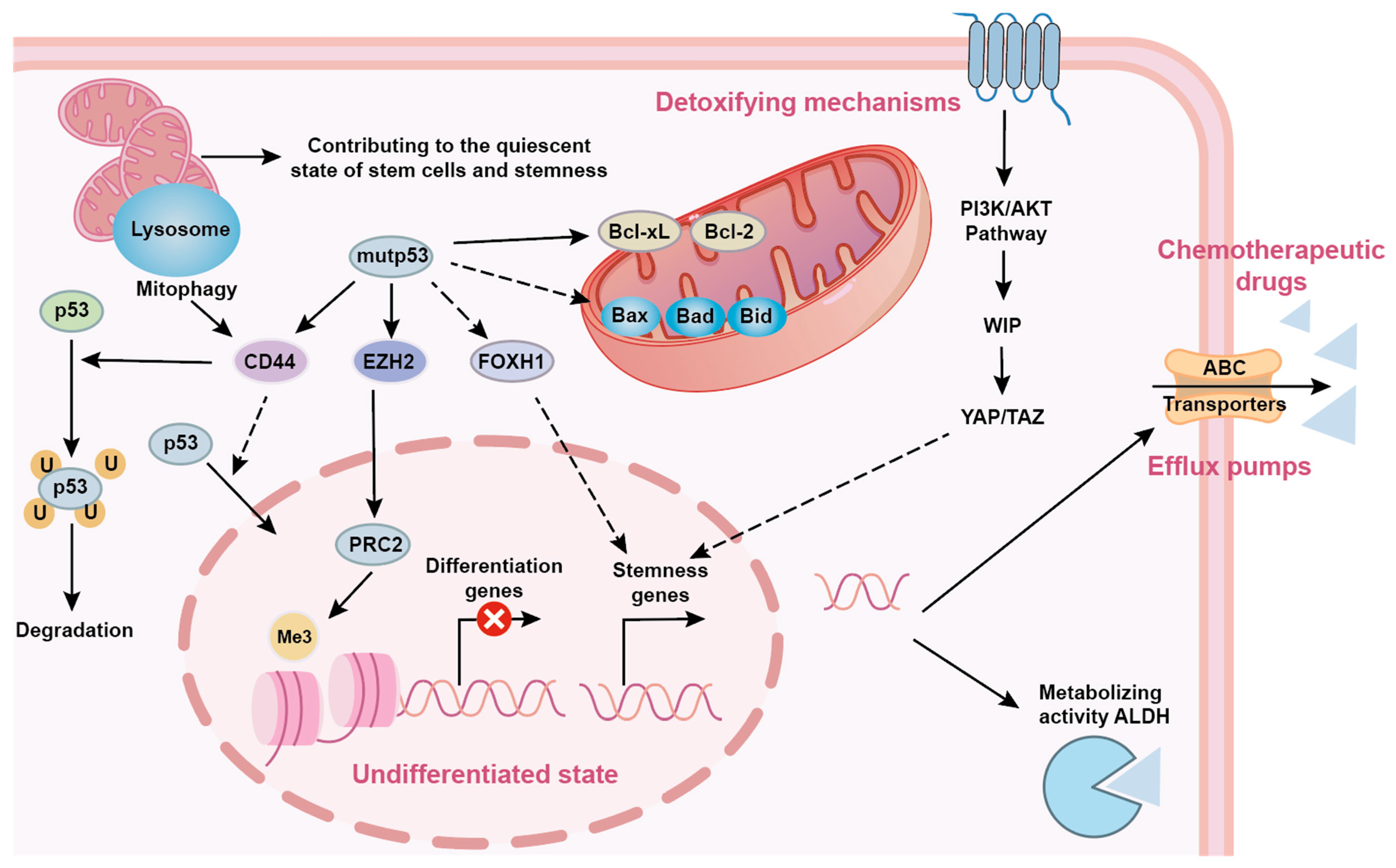
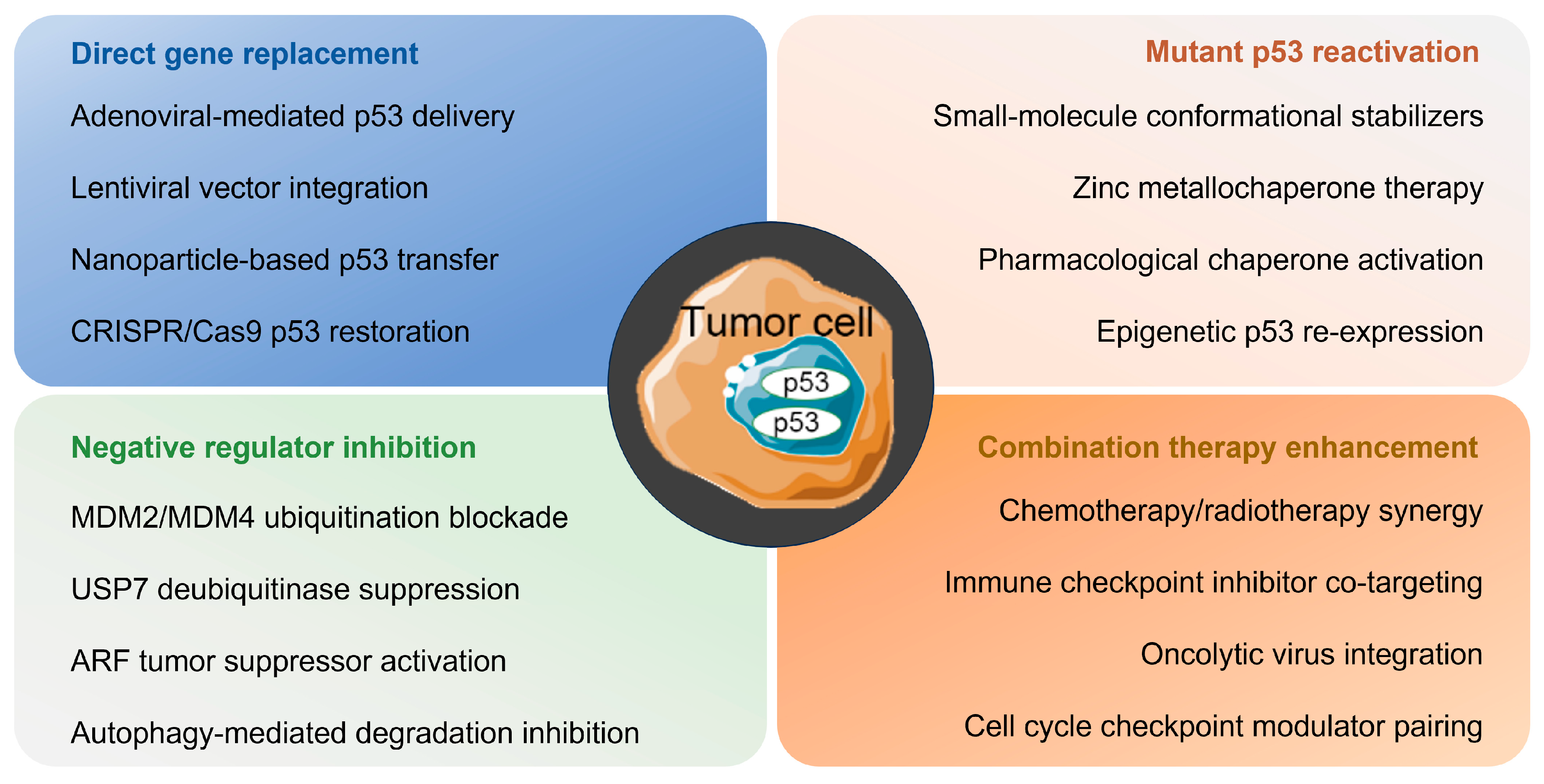

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cancer Type | p53 Mutation | Consequence | Reference |
|---|---|---|---|
| Colorectal cancer | R248Q | Hyperactivation of Jak2/Stat3 signaling to enhance tumor size, diversity, and aggressiveness. | [302] |
| R273H/R273H | Increased secretion of inflammatory cytokines through sustained activation of NF-κB exacerbates chronic inflammation and invasion. | [303] | |
| R273H | Enhancement of invasiveness by promoting NF-κB activation or inhibiting the activation of ASK1/JNK by TNFα. | [304] | |
| R248W | Release of miR-1246-containing exosomes to promote the formation of uniquely reprogrammed macrophage populations. | [305] | |
| R273H/P309S | Enhancing tumor CSC marker-dependent chemoresistance and tumorigenesis (specifically ALDH1A1) through transcriptional modulation. | [306] | |
| R273H | Promote tumor CSCs marker expression to enhance tumor migration, invasion and chemotherapy resistance. | [307] | |
| R172H/R273H | Deregulates transcriptional repression of PDGFRβ by disrupting the p73/NF-Y complex, thereby promoting autonomous tumor cell invasion and metastasis. | [308] | |
| Lung cancer | R175H/R175H/W | Indirect activation of the Axl promoter upregulates its expression to enhance tumor proliferation and migration. | [309] |
| R175H/R248W/R282W/R273H | Enhancement of the REGγ-20S proteasome pathway in cancer cells, thereby affecting drug resistance and cell proliferation. | [310] | |
| R172H | Stabilization of DNA replication forks by increasing excitation of DNA replication starting points promotes proliferation of genomically abnormal cells. | [311] | |
| 3KR | Regulation of cystine metabolism and iron-dead cells through the ability to inhibit SLC7A11 expression. | [312] | |
| Breast cancer | R273H/R280K/L194F | Non-sequence-specific DNA binding or recruitment via other DNA-binding proteins to participate in the regulation of chromatin structure. | [313] |
| R175H/R248Q/R248W/R249S/R273H | Promoting tumorigenesis and drug resistance by activating oncogenic transcription in an indirect manner. | [314] | |
| R175H/R280K/R273H | Enhanced survival and migration of cancer cells dependent on TXN upregulation and TXN/HMOX1 imbalance. | [315] | |
| R280K/R282W | Obstruction of KLF17 binding to target gene promoter repression leads to EMT repressor genes thereby enhancing invasion. | [316] | |
| R172H | Attenuates TBK1-dependent activation of IRF3, thereby promoting tolerance to cytoplasmic DNA and immune evasion. | [317] | |
| Colon adenocarcinoma | R273H | Inhibition of the expression of the naturally occurring anti-inflammatory cytokine sIL-1Ra to promote tumor malignancy. | [318] |
| Oesophageal cancer | R273H/R175H | Binding to NRF2 directly promotes the accumulation of ROS in cancer cells and triggers death. | [319] |
| Head and neck squamous cell carcinoma | G245D/C238F/R175H | Promotes tumor invasion by inhibiting AMPK phosphorylation and deregulating FOXM1 transcriptional repression by FOXO3a. | [320] |
| Ovarian cancer | R135H | Upregulation of CCNG1 by inducing Notch3 expression promotes EMT, tumor metastasis and cisplatin resistance. | [321] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, B.; Maimaitijiang, A.; Nuerbiyamu, D.; Su, Z.; Li, W. The Multifaceted Role of p53 in Cancer Molecular Biology: Insights for Precision Diagnosis and Therapeutic Breakthroughs. Biomolecules 2025, 15, 1088. https://doi.org/10.3390/biom15081088
Xu B, Maimaitijiang A, Nuerbiyamu D, Su Z, Li W. The Multifaceted Role of p53 in Cancer Molecular Biology: Insights for Precision Diagnosis and Therapeutic Breakthroughs. Biomolecules. 2025; 15(8):1088. https://doi.org/10.3390/biom15081088
Chicago/Turabian StyleXu, Bolong, Ayitila Maimaitijiang, Dawuti Nuerbiyamu, Zhengding Su, and Wenfang Li. 2025. "The Multifaceted Role of p53 in Cancer Molecular Biology: Insights for Precision Diagnosis and Therapeutic Breakthroughs" Biomolecules 15, no. 8: 1088. https://doi.org/10.3390/biom15081088
APA StyleXu, B., Maimaitijiang, A., Nuerbiyamu, D., Su, Z., & Li, W. (2025). The Multifaceted Role of p53 in Cancer Molecular Biology: Insights for Precision Diagnosis and Therapeutic Breakthroughs. Biomolecules, 15(8), 1088. https://doi.org/10.3390/biom15081088